
Reduced Lentivirus Susceptibility in Sheep with Mutations
Visna/Maedi, or ovine progressive pneumonia (OPP) as it is known in the United States, is an incurable slow-acting disease of sheep caused by persistent lentivirus infection. This disease affects multiple tissues, including those of the respiratory and central nervous systems. Our aim was to identify ovine genetic risk factors for lentivirus infection. Sixty-nine matched pairs of infected cases and uninfected controls were identified among 736 naturally exposed sheep older than five years of age. These pairs were used in a genome-wide association study with 50,614 markers. A single SNP was identified in the ovine transmembrane protein (TMEM154) that exceeded genome-wide significance (unadjusted p-value 3×10−9). Sanger sequencing of the ovine TMEM154 coding region identified six missense and two frameshift deletion mutations in the predicted signal peptide and extracellular domain. Two TMEM154 haplotypes encoding glutamate (E) at position 35 were associated with infection while a third haplotype with lysine (K) at position 35 was not. Haplotypes encoding full-length E35 isoforms were analyzed together as genetic risk factors in a multi-breed, matched case-control design, with 61 pairs of 4-year-old ewes. The odds of infection for ewes with one copy of a full-length TMEM154 E35 allele were 28 times greater than the odds for those without (p-value<0.0001, 95% CI 5–1,100). In a combined analysis of nine cohorts with 2,705 sheep from Nebraska, Idaho, and Iowa, the relative risk of infection was 2.85 times greater for sheep with a full-length TMEM154 E35 allele (p-value<0.0001, 95% CI 2.36–3.43). Although rare, some sheep were homozygous for TMEM154 deletion mutations and remained uninfected despite a lifetime of significant exposure. Together, these findings indicate that TMEM154 may play a central role in ovine lentivirus infection and removing sheep with the most susceptible genotypes may help eradicate OPP and protect flocks from reinfection.
Published in the journal:
. PLoS Genet 8(1): e32767. doi:10.1371/journal.pgen.1002467
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002467
Summary
Visna/Maedi, or ovine progressive pneumonia (OPP) as it is known in the United States, is an incurable slow-acting disease of sheep caused by persistent lentivirus infection. This disease affects multiple tissues, including those of the respiratory and central nervous systems. Our aim was to identify ovine genetic risk factors for lentivirus infection. Sixty-nine matched pairs of infected cases and uninfected controls were identified among 736 naturally exposed sheep older than five years of age. These pairs were used in a genome-wide association study with 50,614 markers. A single SNP was identified in the ovine transmembrane protein (TMEM154) that exceeded genome-wide significance (unadjusted p-value 3×10−9). Sanger sequencing of the ovine TMEM154 coding region identified six missense and two frameshift deletion mutations in the predicted signal peptide and extracellular domain. Two TMEM154 haplotypes encoding glutamate (E) at position 35 were associated with infection while a third haplotype with lysine (K) at position 35 was not. Haplotypes encoding full-length E35 isoforms were analyzed together as genetic risk factors in a multi-breed, matched case-control design, with 61 pairs of 4-year-old ewes. The odds of infection for ewes with one copy of a full-length TMEM154 E35 allele were 28 times greater than the odds for those without (p-value<0.0001, 95% CI 5–1,100). In a combined analysis of nine cohorts with 2,705 sheep from Nebraska, Idaho, and Iowa, the relative risk of infection was 2.85 times greater for sheep with a full-length TMEM154 E35 allele (p-value<0.0001, 95% CI 2.36–3.43). Although rare, some sheep were homozygous for TMEM154 deletion mutations and remained uninfected despite a lifetime of significant exposure. Together, these findings indicate that TMEM154 may play a central role in ovine lentivirus infection and removing sheep with the most susceptible genotypes may help eradicate OPP and protect flocks from reinfection.
Introduction
Visna/Maedi virus (VMV) and caprine arthritis encephalitis virus (CAEV) are small ruminant lentiviruses (SRLV) of the retroviridae family [1] that infect sheep and goats in major sheep producing countries worldwide. The exceptions are Iceland where VMV was eradicated after a 30-year effort [2], and Australia and New Zealand where VMV has not been reported in sheep but CAEV has been reported in goats [3], [4]. Once infected, seroconversion typically occurs within weeks to months and the infection is incurable. Sheep do not usually display signs of clinical disease in the first two years of infection. The first signs of disease are often loss of body condition and indurative mastitis (i.e., thin ewe syndrome and hard udder). When disease develops, severe clinical signs may include difficulty breathing, chronic wasting, loss of motor control, and arthritis. Ovine progressive pneumonia virus (OPPV) is a closely related North American counterpart to VMV and typically produces an interstitial pneumonia. Seroprevalence studies of U.S. sheep have shown that 36% of sheep operations have infected animals and 24% of all animals tested were seropositive [5]. The impact of subclinical OPPV infection is significant and includes detrimental effects on sheep production from breeding through weaning [6], [7], [8]. Considering that losses are cumulative during an animal's lifetime, the negative effects on ewe production and the sheep industry are substantial.
Natural transmission of ovine lentiviruses is primarily among adults, occurs most frequently after their first year [9], [10], [11], [12], [13], and is by the respiratory route [14], [15], [16]. In addition, some infections occur in lambs by ingestion of infected colostrum and milk [8], [17], [18], [19], [20], [21]. Ovine lentiviruses are macrophage-tropic but not T-lymphocyte-tropic and thus do not cause an immunodeficiency in sheep [22], [23], [24], [25], [26]. Persistence of ovine lentivirus in infected sheep is attributed to latent proviral DNA sequences integrated into the genome of a small fraction of monocytes circulating in the blood. Proviral DNA transcription and gene expression is suppressed until infected monocytes mature into macrophages as they migrate into the interstitial spaces of affected organs [27], [28]. Once in the target organs, infected macrophages initiate viral replication, which induces an inflammatory cascade that ultimately attracts more infected monocytes and other leukocytes. These lesions increase progressively, terminating in disease and eventual death.
Although there is no cure, the impact of disease can be reduced by lowering the prevalence. Voluntary SRLV control programs have been established in several European countries [29], [30], [31], [32], [33], [34]. OPP can be eradicated by testing and removing infected animals or by isolating lambs from seropositive dams at birth. The lambs are then raised on uninfected colostrum and milk, and maintained separately from seropositive sheep thereafter. Either of these methods may be used alone, or in combination, to break the cycle of transmission. However, an OPP-free flock is still susceptible to infection if exposed to other infected sheep or goats [35]. Thus, efforts to eradicate OPP and maintain infection-free status would be facilitated if replacement breeding stock were genetically resistant to lentivirus infections.
Examples of genetic resistance to lentivirus infection have been documented in human populations. Nearly all individuals who lack the lentivirus co-receptor CCR5 do not acquire human immunodeficiency virus (HIV) infection after significant exposure [36], [37], [38]. Moreover, an infected person receiving transplanted stem cells lacking CCR5 may be cured of HIV [39]. In the cases of VMV and OPPV, reports have suggested that host resistance to lentiviral infection may also occur in sheep [40], [41], [42], [43]. Significant breed effects on seroprevalence have also been observed in comingled flocks of purebred sheep, further indicating possible host genetic restriction [10], [12]. For example, in U.S. sheep the OPPV seroprevalence in purebred Finnsheep, Texel, and Suffolk was 77, 65, and 15%, respectively [12]. In Basque dairy-sheep, seroconversion was strongly associated with lifetime maternal VMV-serological status and was interpreted as evidence of genetic susceptibility [44].
The present article reports findings from a genome-wide association study (GWAS) that used naturally-exposed ewes, together with the International Sheep Genome Consortium SNP50 marker set, to test for genetic association with lentivirus infection. Ovine DNA sequence variation in a transmembrane protein gene (TMEM154) was associated with lentivirus infection. The ancestral TMEM154 allele encodes a 191 amino acid polypeptide with glutamate (E) at position 35 and is associated with infection susceptibility. A mutant TMEM154 allele encodes lysine (K) at position 35 allele and is associated with reduced susceptibility. Two deletion mutations were also observed in TMEM154, however there were not enough individuals with these deletions to test their effect. Together, these results suggest that TMEM154 may play a central role in ovine lentivirus biology.
Results
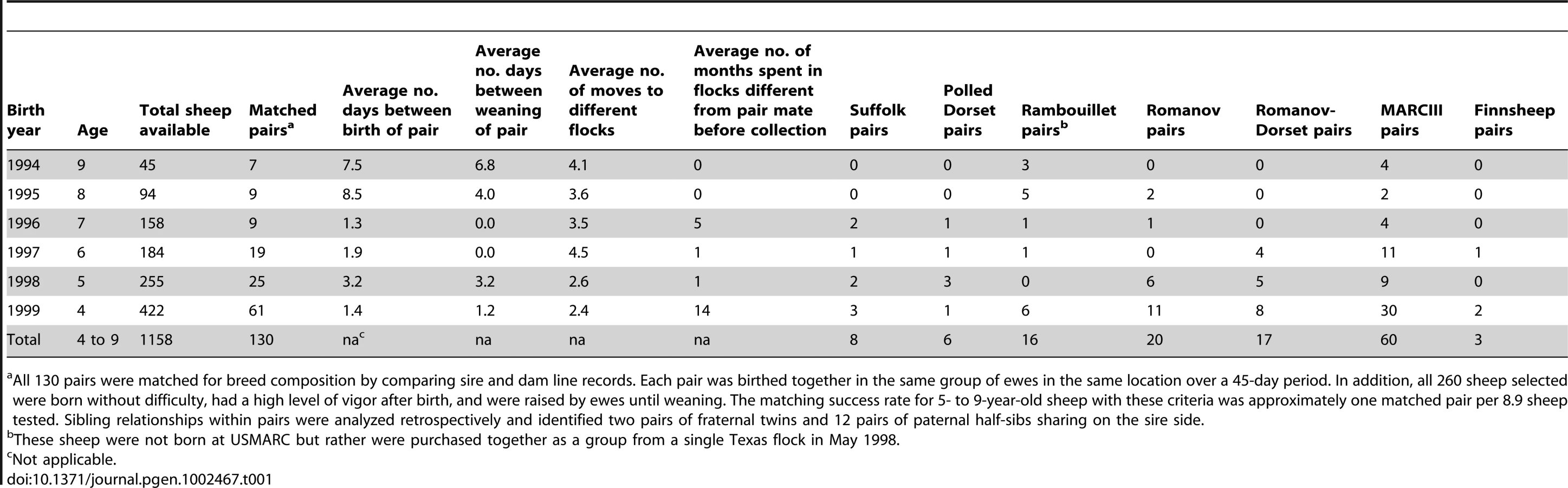
Identifying matched pairs of OPPV infected cases and uninfected controls
The presence of OPPV infection was tested with a competitive enzyme-linked immunosorbent assay (cELISA) in 3,545 breeding-age sheep from purebred and crossbred research flocks in South Central Nebraska, USA. This cELISA has high sensitivity (98.6%) and specificity (96.9%) in sheep naturally infected with OPPV [45]. Analysis by age class showed OPPV infection was lowest in 1-year-olds (8%), increased with age, and peaked at age 5 (43%, Figure 1A). From age 5 to 8 years, the number and proportion of OPPV-infected sheep declined in each year, indicating that the older infected sheep were leaving the flock at a faster rate than their uninfected flock mates. These results indicated that, by age 4, most sheep received sufficient OPPV exposure for infection to occur and that uninfected ewes appeared to have greater longevity in these flocks.

Although age is a risk factor for infection, seroprevalence varied widely within age class, depending on breed composition (Figure 1B). To examine the possibility that genetic risk factors may influence susceptibility to OPPV, matched case-control pairs consisting of infected and uninfected ewes were selected (Table 1). The strict matching criteria were intended primarily to reduce the variation in breed composition and OPPV exposure within each pair. The matching procedure identified 130 case-control pairs of 4 - to 9-year-old ewes (Table 1). These pairs were used in a two-stage design with the goal of reducing falsely positive marker associations and minimizing the number of costly genome-wide scans. For the genome-wide association phase of the study, 69 pairs of 5 - to 9-year-old ewes (white bars in Figure 1C) were evaluated first, while 61 matched pairs of 4-year-old ewes were held in reserve for verification of GWAS results.
GWAS for OPP risk factors
Single nucleotide polymorphisms (SNPs) in the Ovine SNP50 BeadChip array (n = 54,241) were scored in 69 matched case-control pairs and tested for association with OPPV. The experimental design was estimated to have a detectable relative risk of genetic association that ranged from two to six in dominant and co-dominant models of inheritance, depending on marker allele frequency, and the extent of linkage disequilibrium (LD) between a marker and a disease allele (Materials and Methods). Of the 54,241 SNPs tested, 50,614 had quality scores in the acceptable range as determined by clustering and genotype calling algorithms. A single SNP on chromosome 17 had an unadjusted p-value of 3.19×10−9 (OAR17_5388531; Figure 2A). This was highly significant compared to the significance threshold of 1×10−6 (i.e., a significance level of 0.05 divided by 50,614). Moreover, the Quantile-Quantile (Q-Q) plot showed no evidence of an inflated test statistic caused by population structure. The c/t SNP OAR17_5388531 was in intron 5 of an ovine gene homologous to the human TMEM154 gene on chromosome 4. The “c” allele of SNP OAR17_5388531 was on the sense strand of TMEM154, had a frequency of 0.257, and was associated with infected sheep. Another SNP (s46403) had the second lowest unadjusted p-value (2.22×10−6) and was on chromosome 13 in a gene similar to human angiopoietin 4 (ANGPT4). A third SNP (OAR17_5405721) had the third lowest unadjusted p-value (6.71×10−6) and was located in the 3′UTR of ovine TMEM154. The highly significant association of one SNP in ovine TMEM154, together with the third best SNP association being located in the same gene, suggested that a genetic risk factor associated with OPPV infection existed in this genomic interval. Subsequent efforts were directed towards characterizing the genomic region of ovine TMEM154, discovering additional polymorphisms, and testing them for association with infection.

Ovine TMEM154 DNA sequence assembly and SNP discovery
The complete sequence of the TMEM154 region was not available for sheep and thus was determined by identifying and sequencing four overlapping bacterial artificial chromosomes (BACs) spanning approximately 400 kb. A contiguous 78 kb region was assembled de novo and appeared to contain the complete TMEM154 gene region (Figure 2B, GenBank Accession HM355886). Other contigs from these BACs contained exons similar to human ARFIP1 and FBXW7. The ovine genes appeared to be in the same orientation and approximate positions as those in reported for ARFIP1, TMEM154, and FBXW7 on human chromosome 4 and cattle chromosome 17. Sanger sequencing of targeted genomic DNA fragments amplified by polymerase chain reaction (PCR) in the ARFIP/TMEM154/FBXW7 region revealed 128 additional SNPs in the 69 pairs of matched 5 - to 9-year-olds. However, SNPs associated with OPPV infection were observed only within the TMEM154 gene (Figure 2B). The results indicated that TMEM154, and not flanking genes, was the likely source of the association.
Analysis of ovine TMEM154 haplotypes encoding polypeptide isoforms
Although sequence variation in any number of gene elements can alter biological function, those that may directly affect the polypeptide sequence were evaluated first. The ovine TMEM154 genomic assembly contained seven TMEM154 exons encoding a 191 amino acid precursor protein (Figure 3A). The precursor protein contained a putative signal peptide at the N-terminus with a cleavage site predicted between positions 30 and 31 and resulted in a mature protein of 161 amino acids. The predicted mature ovine TMEM154 protein was 92.5, 67.3, and 53.8% identical with those of cattle, humans, and mice, respectively. The extracellular domain and signal peptide accounted for most of the amino acid sequence differences in these comparisons (83, 31, and 25% identity, respectively). The ovine intron/exon junctions were established by comparing the genomic sequences with those from a 1,012 bp reverse transcription (RT)-PCR fragment amplified from cDNA of contemporary animals. The ovine TMEM154 mRNA and exon structure was similar to those reported for cattle, human, and mice (data not shown). Although RNA samples were not available for the 69 pairs of case-control sheep, the transcript sequence was determined for 11 case-control pairs of contemporary sheep and seven other available sheep. In all 29 sheep tested, the expected full-length transcripts were observed and their sequences corresponded to those from genomic DNA. Thus, alternatively spliced TMEM154 transcripts did not explain the association observed with the SNP OAR17_5388531.

To evaluate whether amino acid sequence variation was encoded by ovine TMEM154, the exons were amplified from genomic DNA and sequenced for a panel of 234 animals that included all 69 matched case-control pairs and 96 rams representing common U.S. sheep breeds. In these 234 animals, five missense SNPs (T25I, D33N, E35K, T44M, N70I) and two frameshift deletion polymorphisms (R4AΔ, E82YΔ) were observed in the predicted signal peptide and the extracellular domain (exons 1 and 2, Figure 3A). Conversely, nonsynonymous SNPs and frameshift polymorphisms were not observed in exons 3 through 7 in any of these sheep. TMEM154 exons 1 and 2 were then considered as potential “hotspots” for coding polymorphisms, and these exons were sequenced for more than 5000 sheep from research populations, revealing one additional missense SNP (L14H). Combinations of the eight “coding” polymorphisms were observed on haplotypes encoding eight distinct precursor protein isoforms. Four haplotypes were predicted to encode full-length polypeptides with glutamate (E) at position 35 (Figure 3B, designated 2, 3, 9, and 11). The E35 allele was in strong LD with the “c” allele of OAR17_5388531 associated with infected cases (r2 = 0.98). Two haplotypes (designated 1 and 10) encoded full-length polypeptides with lysine (K) at position 35. The remaining haplotypes (4 and 6) had frameshift deletions predicted to cause premature termination of translation and loss of the putative membrane spanning and cytoplasmic domains of TMEM154.
Comparing polymorphic ovine TMEM154 amino acid residues with those in related mammalian species indicated that haplotype 3 was the most likely ancestral isoform in sheep. Thus, the ancestral ovine precursor protein isoform is inferred to be a 191 amino acid polypeptide with a negatively charged E35 residue. Haplotype comparisons between mammalian species also showed the E35 residue is highly conserved in mammals and the positively charged K35 residue of TMEM154 was not observed in other species analyzed (Figure 3B). A median-joining network of haplotypes encoding polypeptide isoforms of ovine TMEM154 showed that the two truncated isoforms were located on the distal branches of the tree (Figure 3C, haplotypes 4 and 6). Because the more recent haplotypes appeared to have evolved towards dysfunction and OPPV resistance, relationships presented in Figure 3C provide a framework for evaluating the potential role of TMEM154-encoded polypeptide isoforms in ovine lentivirus infection.
Analysis of TMEM154 haplotypes as risk factors for OPPV infection in matched cases and controls
We hypothesized that the more ancient full-length TMEM154 haplotypes encoding E35 were genetic risk factors for OPPV infection because these alleles were in strong LD with the “c” allele of OAR17_5388531. Thus, sheep with one copy of haplotype 2 or 3 were compared to those without. Because the experimental design included paired samples, the McNemar's test for two correlated proportions was used for the analysis [46]. Also, the 61 matched pairs of 4-year-old ewes were deployed at this stage for comparison with results obtained from the 69 matched pairs of 5 - to 9-year-old ewes. The dichotomous variable for this test was defined as having zero or one copy of the TMEM154 genetic risk factor (i.e., haplotype 2 or 3). In one discordant type, the infected case had one copy of a TMEM154 risk factor, but the uninfected control did not (Table 2 and Table S1). This type of discordant pair (n = 28) was consistent with the hypothesis. In the other discordant type, the infected case had zero copies of a TMEM154 risk factor, and the uninfected control had one copy. This type of discordant pair (n = 1) was inconsistent with the hypothesis. In the 61 matched pairs of 4-year-old ewes, the odds ratio of these discordant pairs indicated that ewes with one copy of a full-length TMEM154 E35 allele were 28 times more likely to be infected than those without (p-value<0.0001, 95% CI 5–1100). Compared to either SNP for OAR17_5388531 or E35K, the haplotype model for TMEM154 yielded more significant results (Table 2). In this study, the predictive value of having one versus two copies of TMEM154 risk factor alleles could not be determined because there were too few pairs of this type. Although an additive model was not excluded, one copy of the risk factor was significantly correlated with OPPV infection. If this were a diagnostic test for the 260 sheep in these 130 pairs, the predictive value (PV) for a positive test would be 85% with a sensitivity and specificity of 69% and 88%, respectively. These results were consistent with those of the GWAS and confirmed that TMEM154 haplotypes encoding polypeptide variants were associated with OPPV infection.

TMEM154 haplotypes 2 and 3 as risk factors for OPPV infection in multi-breed cohorts
Estimating the relative risk (RR) and PV of TMEM154 haplotype alleles 2 and 3 provides an indication of the potential impact of full-length E35 alleles in other affected flocks. A total of 2705 sheep, 3-years and older, were evaluated in nine cohort studies to test TMEM154 haplotype alleles 2 and 3 as risk factors for OPPV infection. These cohorts consisted of sheep from various breeds, ages, and production environments and were sampled over a span of seven years from research flocks in Nebraska and Idaho, and a private flock in Iowa. Animals were matched for gender and production environment in all cohorts; for three cohorts, animals were also matched for age at sampling. For each cohort, two-way contingency tables were used to analyze the relationship between the presence of TMEM154 haplotypes 2 or 3 and being infected with OPPV (Table S2 and Table S3). For all cohorts, the combined RR of OPPV infection for animals with TMEM154 haplotypes 2 or 3 was 2.85-times greater than those without (95% CI 2.36–3.43, p-value<0.0001). These cohort studies also confirmed that TMEM154 haplotype alleles 2 and 3 are significant risk factors for OPPV infection in various breeds, and may be associated with lentivirus infection in multiple geographic locations and environments.
Distribution of TMEM154 haplotype risk factors in sheep populations
The frequency of TMEM154 haplotype risk factors within breeds provides an indication of their potential susceptibility of OPPV in production environments similar to those described here. The combined frequencies of risk factor alleles 2 and 3 was highest in Texel (0.74) and lowest in Rambouillet (0.035, Table 3) and generally consistent with seroprevalence trends in the research flocks (Figure 1B). The most common truncated isoform of TMEM154 was encoded on haplotype 4, which was detected in Katahdin (0.15), Suffolk (0.13), Composites (0.033), Rambouillet (0.005), and Polypay (0.003). Overall, ovine TMEM154 haplotypes encoding polypeptide isoforms 1, 2, 3, and 4 accounted for more than 99% of the haplotypes observed.

Discussion
This report describes the discovery of an ovine gene that is associated with lentivirus infection in naturally-exposed U.S. sheep. In a GWAS with 50 k SNPs, one marker exceeded genome-wide significance and led to the identification of TMEM154 haplotypes predicted to encode altered peptide sequences. TMEM154 haplotypes 2 and 3 encode full-length polypeptides with E35 and appeared to be significant genetic risk factors for OPPV infection. Whether in matched pairs or cohorts, the presence of a TMEM154 haplotype encoding a full-length E35 polypeptide was predictive of OPPV infection. The ovine TMEM154 gene appears to be an OPPV susceptibility locus because the ancestral haplotype 3 was associated with infection. Thus, haplotype 1 (encoding a full-length K35 isoform) appears to be more recent and is associated with reduced susceptibility to OPPV infection. The two deletion mutations encoded on haplotypes 4 and 6 are also predicted to be more recent than haplotype 3 and indicate that TMEM154 may be under selection for reduced function.
The function of the TMEM154 protein has not yet been reported for any species and remains unknown. In humans, the most abundant TMEM154 mRNA was reported in CD19+ B cells and CD14+ monocytes with levels 15.8 - and 7.6-fold above the TMEM154 median, respectively (http://biogps.gnf.org). Expression of TMEM154 in cells of monocyte lineage is of interest because they are the target cells for OPPV infection in sheep. It is plausible that mutant ovine TMEM154 polypeptide isoforms have altered function and decrease OPPV susceptibility. For example, the non-conservative substitution of K35 for E35 was associated with a decrease in OPPV susceptibility in homozygous individuals. The E35 residue in TMEM154 was highly conserved among the 32 Mammalian species tested; the only other substitution for E35 was the negatively charged aspartate (D) residue in hedgehog and hyrax (Figure 3B). Additional evidence that loss of TMEM154 function may reduce OPPV susceptibility is derived from the existence of two severely truncated polypeptides encoded by TMEM154 haplotypes 4 and 6. Although there were not enough sheep with these haplotypes to test for association, the existence of two deletion mutations suggests that sheep without TMEM154 function may have a selective advantage when exposed to OPPV. The ovine TMEM154 protein does not appear to be essential for survival or reproduction because an 11-year-old purebred Suffolk ewe was identified in the Nebraska research flock with a homozygous genotype for TMEM154 haplotype 4. Both the genomic and cDNA sequences of this ewe were consistent with her being a TMEM154 “4,4 knockout” (data not shown). This ewe was kept year-round with OPP-infected ewes and lambed indoors in close quarters for 11 years, yet remained seronegative and in good condition prior to her death from an acute non-infectious illness. Multiple healthy TMEM154 “4,4 knockout” rams have also been identified in the Nebraska flock. The identification of multiple animals with the TMEM154 “4,4 knockout” genotype indicates that breeding natural TMEM154 knockouts is possible and a functional TMEM154 gene is apparently not required for sheep to thrive and reproduce.
Limitations and strengths of the study
Ovine gene regions including CCR5 and DRB1 were previously associated with OPP provirus levels in candidate gene studies [41], [42], [43]. However, the OvineSNP50 BeadChip marker density and distribution were not sufficient for evaluating whether CCR5 and DRB1 gene regions were associated with OPPV infection in the present study. The nearest SNPs flanking CCR5 (OAR19_55954161 and s65253) were approximately 20 kb from the coding region and had unadjusted p-values of 0.62 and 0.75, respectively. The degree of LD between the BeadChip SNP markers and those previously identified for CCR5 are unknown. The nearest SNPs flanking DRB1 (OAR20_ 26932949 and OAR20_27259292) were greater than 100 kb from the coding region and had unadjusted p-values of 0.77 and 0.21, respectively. For either gene, there were no markers within 1 Mb that had unadjusted p-values less than 0.01. We acknowledge that a GWAS with 50 k SNPs and 69 case-control pairs limits the detectable genetic risk factors to those with large effects. Thus, other genetic risk factors for OPP may exist and were missed for lack of power in our study. In addition, it is not known whether TMEM154 genetic risk factors are associated with specific OPPV strains such as those found in Nebraska, Idaho, and Iowa, USA. Strain differences, together with adverse production conditions like high animal density, indoor housing with poor ventilation, and moist climates, may enhance transmission and overcome any or all host genetic resistance. Thus, it is not known whether genetic variants of TMEM154 will be useful predictors of OPPV infection under other management and/or environmental conditions.
The success of the present GWAS study was dependent on seven key features of the research design, without which the allelic association may have escaped detection. The first important feature was the availability of a serological test with good sensitivity and specificity for correctly classifying infection status. Serological results from duplicate blinded samples tested in two laboratories indicated that less than 2% of the animals were inconsistently classified in the initial round of testing. Second, the matched case-control design with older sheep was a key feature for reducing variation in the management conditions, environment, breed composition, and pathogen exposure. The use of older sheep increased the chances that sufficient natural exposure had occurred so that a high proportion of susceptible individuals could become infected. Third, it was important to have sufficient numbers of older sheep to assemble enough matched pairs to detect an association. Of the older sheep tested, only 22% met the matching criteria. Fourth, the relatively diverse breed composition of the research flocks increased the likelihood that an association observed in the 69 matched pairs was not limited to one breed. Fifth, public availability of the OvineSNP50 BeadChip was essential to progress beyond a functional candidate gene approach for discovering gene-phenotype associations. TMEM154 had not previously been identified as a candidate gene in lentivirus biology for any species and thus would not have been considered. Sixth, the SNP marker spacing on the 50 k chip in the region of TMEM154 was fortuitous because a GWAS may have missed the TMEM154 association if the SNP density was lower or the distribution of SNPs happened to be less serendipitous. A higher density SNP chip would increase the chances of a marker SNP being in LD with a polymorphism that influences the trait of interest. A higher density SNP chip may also rule out the association of neighboring genes, and thereby narrow the region of focus. Seventh, TMEM154 in the study populations had three common haplotypes encoding polypeptide isoforms, two of which formed a risk factor group with a large effect. This was previously unknown and was determined by the evolutionary history of TMEM154 in these sheep. Nevertheless, this report demonstrates that a GWAS approach with 50 k SNPs and 69 matched case-control pairs was successful in sheep.
OPP infection in sheep without TMEM154 risk factor haplotypes
Although TMEM154 haplotype risk factors 2 and 3 were strongly associated with OPPV infection, some animals without these haplotypes were also infected. For example, 36 of 139 sheep with a 1,1 diplotype were seropositive in the pairs of matched case-control sheep. This is consistent with the concept that host genetic resistance is conditional. Many factors may contribute to a virus overcoming host genetic resistance including: a high viral dose during an exposure event, a long duration of repeated viral exposures, viral genetic adaptation to host defenses, and multiple routes by which infection may occur. In the latter case, other host-encoded genes may play significant roles. Thus, comparing the relative level of resistance conferred by various TMEM154 haplotypes, together with the identification of additional host genetic risk factors, will be important for developing flocks that are genetically resistant to lentivirus infections.
Materials and Methods
Ethics statement
Prior to their implementation, all animal procedures were reviewed and approved by the care and use committees at the United States Department of Agriculture (USDA), Agricultural Research Service (ARS) Meat Animal Research Center (USMARC) in Nebraska, the USDA, ARS, Sheep Experiment Station (USSES) in Idaho, and Washington State University in cooperation with the USDA, ARS, Animal Disease Research Unit (ADRU).
Animal sample collection and serologic testing
The USMARC (Nebraska) sheep population was sampled in 2003 (n = 3545) and used to select 69 matched case-control pairs of 5 - to 9-year-old ewes for the GWAS. The same population was also used to select 61 matched case-control pairs of 4-year-old ewes for analyzing TMEM154 haplotypes as risk factors for OPP infection. Animals not used in matched case-controls were used in unmatched cohort studies for validation as shown in Table S2. Animals were not members of more than one group. The USMARC sheep population is a relatively diverse flock with more than ten breeds representing genetic diversity for traits such as fertility, prolificacy, maternal ability, growth rate, carcass leanness, wool quality, mature weight and longevity [47].
The USMARC population was sampled again in 2010 and used to select a cohort of 280 ewes, 4 - to 5-year-old, and raised in similar conditions as those sampled in 2003. The purpose was to determine if the association of TMEM154 haplotypes with OPP infection was reproducible in animals sampled seven years later.
The USSES (Idaho) sheep population was sampled in 2004 and 2008 and used to select cohorts of 309 and 365 mature ewes, respectively. The purpose was to determine if an association of TMEM154 haplotypes with OPP infection was evident in another research flock that was geographically and historically distinct from the Nebraska flock. The USSES sheep population contains Columbia, Rambouillet, and Polypay breeds.
The private Polypay sheep flock (Iowa) was sampled in 2009 and used to select a cohort of 218 mature ewes. The purpose was to determine if an association of TMEM154 haplotypes with OPP infection was evident in a commercial flock distinct from those in Nebraska and Idaho. This commercial flock was chosen based on its availability.
Whole blood samples for serum fractionation and DNA extraction were drawn from the jugular vein into S-Monovette serum Z and EDTA KE 9 ml syringes, respectively (Sarstedt, Newton, NC, USA). Laboratory diagnosis for OPP was performed at the Washington Animal Disease Diagnostic Laboratory (Pullman, WA, USA) with a Caprine Arthritis Encephaltitis Virus (CAEV) competitive-inhibition ELISA (cELISA). This CAEV cELISA is applicable for the detection of OPPV antibodies in sheep [45], [48]. Briefly, this assay uses a proprietary monoclonal antibody derived from the fusion of goat splenocytes and mouse myeloma cells (VMRD, Inc., Pullman, WA, USA). This antibody is conjugated to horeseradish peroxide and is used to compete with serum antibodies for the CAEV antigen bound to the microtiter plate. Additional testing for OPP was performed at USMARC and ADRU with CAEV cELISA kits, according to manufacturer's instruction (VMRD, Inc., Pullman, WA, USA).
Statistical analysis
GWAS analyses
Sixty-nine pairs of ewes were selected from a total of 736 in the 5 - to 9-year-old age class. The OPPV seroprevalence of the 736 ewes was 43%. In dominant and co-dominant models, our GWAS design had a detectable RR of genetic association that ranged from two to six with 69 paired case-controls, 50,000 SNPs, a false-positive rate (alpha) of 0.05, and a false-negative rate (beta) of 0.1 (simulation data not shown [49]). In a co-dominant model of inheritance with a disease prevalence of 0.43, the minimum detectable RR was less than 2 for marker allele frequencies between 0.15 and 0.50, and LD values between 0.7 to 1.0. In a dominant model of inheritance, the minimum detectable RR ranged from 2 to 6 for conditions similar to those above. There were not enough matched pairs in this design to detect GWAS of recessively inherited disease risk alleles. SNP genotypes for the OvineSNP50 BeadChip DNA samples were measured and scored at GeneSeek Inc. (Lincoln, NE, USA), according to manufacturer's instructions (Illumina, Inc., San Diego, CA, USA). For determining the number of SNPs that performed reliably with the set of 138 ovine samples, a GenCall score greater than 0.7 was used as a cutoff and was determined by clustering and genotype calling algorithms provided by the manufacturer (Illumina, Inc., San Diego, CA USA). All single SNPs were analyzed for association with infection using PLINK v1.07 software [50], as described: http://pngu.mgh.harvard.edu/purcell/plink/.
Cohort analysis
The combined relative risk was assessed in sheep cohorts using the glimmix procedure of SAS 9.2 (SAS Institute, Cary, NC). The serological status of OPP was fit as the dependent variable in a Poisson model with a log link to give unbiased estimates of relative risk and slightly conservative (broad) confidence intervals [51]. Breed and risk/nonrisk diplotype status were treated as independent fixed variables, where the risk was defined as the presence of at least one TMEM154 haplotype allele 2 or 3. Age was included as a covariate and the cohort was treated as a random variable.
Genomic DNA sequencing of ovine TMEM154
Ovine BACs predicted to contain TMEM154 were identified from those mapped to the ovine draft genome sequence http://www.livestockgenomics.csiro.au/sheep/oar2.0.php). BACs were isolated from an arrayed 10–12× sheep BAC library (CHORI-243, [52]), cultured, and the BAC DNA was purified. The BACs were derived from the Texel ram used for the ovine genome sequencing project (USMARC animal no. 200118011). Pooled samples of the four BACs (CH243-492L14, 270 kb; CH243-229A18, 147 kb; CH243-363J1, 329 kb; and CH243-426G18, 279 kb) were sequenced by synthesis with conditions optimized for 600 bp read lengths, according to manufacturer's instructions (Roche Applied Sciences, Branford, CT, USA). DNA sequences were assembled de novo with Newbler software provided by the manufacturer and the contigs were evaluated and viewed with Consed [53]. Contig assembly made use of information and sequence available for cattle and sheep at the National Center for Biotechnology Information (NCBI) and International Sheep Genomics Consortium (ISGC), respectively. A 78 kb region of genomic DNA sequence containing the complete predicted TMEM154 gene was assembled with 70 k reads and 25 Mb of sequence. Four large contigs were manually joined with information derived from ovine mRNA sequences, and the annotated 78 kb sequence was deposited in GenBank (accession number HM355886).
Genotyping TMEM154 by sequencing genomic DNA and cDNA
Ovine TMEM154 exons were genotyped by Sanger sequencing of PCR fragments amplified from genomic DNA (Table S4). DNA extraction and genetic analyses were performed in a manner similarly to that previously described [47]. Briefly, a 1,000 bp PCR product containing each exon was sequenced in the 138 matched case-control sheep and 96 rams from a diverse panel of common U.S. sheep breeds (MARC Sheep Diversity Panel version 2.4) [47]. After scoring polymorphisms from these 234 sheep in all exons, a second round of nested PCR fragments were designed so that: 1) a 700 bp amplicon was fully nested within each previous 1,000 bp amplicon, and 2) the amplification primers for the 700 bp products did not bind to polymorphic sites discovered from sequencing the 1,000 bp on the genome (Table S4). The combined Sanger sequences from each animal were scored and recorded manually. More than 60 thousand tracefiles and 6.9 million genotypes from the present report are publicly available via the internet (http://cgemm.louisville.edu/USDA/index.html).
For mRNA transcript analysis, ovine blood (3 mL) was collected (Tempus Blood RNA tubes, Life Technologies Corporation, Carlsbad, CA, USA) and stored at −20°C prior to RNA extraction. Whole blood RNA was purified by centrifugation and filtration according to the manufacture's protocol (Tempus Spin RNA isolation kits, Life Technologies Corporation). RNA quantity and quality were determined spectophotometrically (ND-1000, NanoDrop Technologies, Inc., Wilmington, DE, USA; and Agilent 2100 Bioanalyzer (Agilent Technologies, Inc., Santa Clara, CA, USA). The complete TMEM154 mRNA coding region was amplified by PCR from cDNA (SuperScript III One-Step RT-PCR System, Platinum Taq High Fidelity, Invitrogen Corporation, Carlsbad, CA, USA). The 25 µL reactions contained 1× of the manufacturer's reagent cocktail, 0.2 µM each of the sense and antisense primers (Table S4), 0.5 µL SuperScript III RT/Platinum Taq High Fidelity Enzyme Mix, and 30–50 ng of total RNA. Reaction conditions were the following: 1 cycle of cDNA synthesis at 55 °C for 30 minutes followed by pre-denaturation at 94 °C for 2 minutes; 40 cycles of PCR amplification at 94 °C for 15 seconds, 58 °C for 30 seconds, 68 °C for 1 minute; and 1 cycle of final extension at 68 °C for 5 minutes. As a control for DNA contamination and any putative TMEM154 pseudogenes, duplicate sample reactions to those described above were subjected to PCR without preceding cDNA synthesis. Successful amplification of 1,012 bp fragments was monitored by gel electrophoresis. Amplicons were not observed in RT-PCR reactions lacking cDNA synthesis. Following an Exonuclease I digestion [54], TMEM154 RT-PCR amplicons were sequenced with dye-terminator chemistry and separated by capillary electrophoresis (ABI 3730, PE Applied Biosystems, Foster City, CA, USA). The oligonucleotide primers for PCR and sequencing are listed in Table S4. Sequences were analyzed for polymorphisms and scored manually with Phred and Phrap [55], [56], Polyphred (version 6.10) [57] and Consed software [53].
An artiodactyl species panel of DNAs similar to that described previously [58] was sequenced to provide an estimate of the likely ancestral state of the polymorphic ovine TMEM154 codons. This panel is composed primarily of species from the Pecoran clade, whose common ancestor dates to about 30 million years ago [59]. Oligonucleotide primers derived from ovine TMEM154 genomic sequences were used in PCR assays to amplify exons 1 and 2 and PCR products for both exons were produced for the following species: Wyoming bighorn sheep (Ovis canadensis, n = 7), American plains bison (Bison bison, n = 7), Alaskan caribou (Rangifer tarandus, n = 7) Wyoming elk (Cervus canadensis nelsoni, n = 7), Texas exotic red deer (Cervus elaphus, n = 2), Texas exotic fallow deer (Cervus dama, n = 1), gaur (Bos gaurus, n = 2), domestic goat (Capra hircus, n = 4), Arkansas exotic water buffalo (Bubalus bubalis, n = l), Wyoming mule deer (Odocoileus hemionus, n = 7), Wyoming white-tailed deer (Odocoileus virginianus, n = 5), Wyoming mountain goat (Oreamnos americanus n = 8), and Alaskan and Wyoming moose (Alces alces, n = 8), for a total of 66 non-ovine artiodactyl individuals. To ensure that amplified DNA sequences were not derived from spurious ovine DNA, only those sequences with distinctive species-associated nucleotide differences were included in the analysis. Proteins encoded by Pecoran species were more than 95% identical to that encoded by ovine TMEM154 haplotype 3.
Supporting Information
Zdroje
1. Büchen-OsmondC 2006 ICTVdB Management 00.061.1.06.008. Visna/maedi virus New York Columbia University
2. PeturssonG 1994 Experience with visna virus in Iceland. Ann N Y Acad Sci 724 43 49
3. GreenwoodPLNorthRNKirklandPD 1995 Prevalence, spread and control of caprine arthritis-encephalitis virus in dairy goat herds in New South Wales. Aust Vet J 72 341 345
4. MothaMXRalstonJC 1994 Evaluation of ELISA for detection of antibodies to CAEV in milk. Vet Microbiol 38 359 367
5. USDAAnimal and Plant Health Inspection Service VS, Centers for Epidemiology and Animal Health, editor 2003 Ovine Progressive Pneumonia: Awareness, Management, and Seroprevalence. Fort Collins
6. DohooIRHeaneyDPStevensonRGSamaghBSRhodesCS 1987 The effects of Maedi-Visna Virus infection on productivity in ewes. Preventive Veterinary Medicine 4 471 484
7. KeenJEHungerfordLLLittledikeETWittumTEKwangJ 1997 Effect of ewe ovine lentivirus infection on ewe and lamb productivity. Prev Vet Med 30 155 169
8. PeterhansEGreenlandTBadiolaJHarkissGBertoniG 2004 Routes of transmission and consequences of small ruminant lentiviruses (SRLVs) infection and eradication schemes. Vet Res 35 257 274
9. CutlipRCLehmkuhlHDBrogdenKASacksJM 1986 Breed susceptibility to ovine progressive pneumonia (maedi/visna) virus. Vet Microbiol 12 283 288
10. GatesNLWinwardLDGorhamJRShenDT 1978 Serologic survey of prevalence of ovine progressive pneumonia in Idaho range sheep. J Am Vet Med Assoc 173 1575 1577
11. HouwersDJVisscherAHDefizePR 1989 Importance of ewe/lamb relationship and breed in the epidemiology of maedi-visna virus infections. Res Vet Sci 46 5 8
12. KeenJEHungerfordLLWittumTEKwangJLittledikeET 1997 Risk factors for seroprevalence of ovine lentivirus in breeding ewe flocks in Nebraska, USA. Prev Vet Med 30 81 94
13. LeginagoikoaIDaltabuit-TestMAlvarezVArranzJJusteRA 2006 Horizontal Maedi-Visna virus (MVV) infection in adult dairy-sheep raised under varying MVV-infection pressures investigated by ELISA and PCR. Res Vet Sci 80 235 241
14. BlacklawsBABerriatuaETorsteinsdottirSWattNJde AndresD 2004 Transmission of small ruminant lentiviruses. Vet Microbiol 101 199 208
15. Broughton-NeiswangerLEWhiteSNKnowlesDPMouselMRLewisGS 2010 Non-maternal transmission is the major mode of ovine lentivirus transmission in a ewe flock: a molecular epidemiology study. Infect Genet Evol 10 998 1007
16. ReinaRBerriatuaELujanLJusteRSanchezA 2009 Prevention strategies against small ruminant lentiviruses: an update. Vet J 182 31 37
17. AlvarezVArranzJDaltabuit-TestMLeginagoikoaIJusteRA 2005 Relative contribution of colostrum from Maedi-Visna virus (MVV) infected ewes to MVV-seroprevalence in lambs. Res Vet Sci 78 237 243
18. Herrmann-HoesingLMPalmerGHKnowlesDP 2007 Evidence of proviral clearance following postpartum transmission of an ovine lentivirus. Virology 362 226 234
19. LerondelleCOuzroutR 1990 Expression of maedi-visna virus in mammary secretions of a seropositive ewe. Dev Biol Stand 72 223 227
20. PepinMVituCRussoPMornexJFPeterhansE 1998 Maedi-visna virus infection in sheep: a review. Vet Res 29 341 367
21. PreziusoSRenzoniGAllenTETacciniERossiG 2004 Colostral transmission of maedi visna virus: sites of viral entry in lambs born from experimentally infected ewes. Vet Microbiol 104 157 164
22. GendelmanHENarayanOKennedy-StoskopfSKennedyPGGhotbiZ 1986 Tropism of sheep lentiviruses for monocytes: susceptibility to infection and virus gene expression increase during maturation of monocytes to macrophages. J Virol 58 67 74
23. GendelmanHENarayanOMolineauxSClementsJEGhotbiZ 1985 Slow, persistent replication of lentiviruses: role of tissue macrophages and macrophage precursors in bone marrow. Proc Natl Acad Sci U S A 82 7086 7090
24. GorrellMDBrandonMRShefferDAdamsRJNarayanO 1992 Ovine lentivirus is macrophagetropic and does not replicate productively in T lymphocytes. J Virol 66 2679 2688
25. NarayanOWolinskyJSClementsJEStrandbergJDGriffinDE 1982 Slow virus replication: the role of macrophages in the persistence and expression of visna viruses of sheep and goats. J Gen Virol 59 345 356
26. ThormarH 2005 Maedi-visna virus and its relationship to human immunodeficiency virus. AIDS Rev 7 233 245
27. HaaseATStowringLNarayanPGriffinDPriceD 1977 Slow persistent infection caused by visna virus: role of host restriction. Science 195 175 177
28. PelusoRHaaseAStowringLEdwardsMVenturaP 1985 A Trojan Horse mechanism for the spread of visna virus in monocytes. Virology 147 231 236
29. BirontPDeluykerH 1985 Control programme for maedi/visna in Belgium. 123 126 Brussels, Belgium. 8076 EN. ECSC-EEC-EAEC 8076 EN. ECSC-EEC-EAEC
30. Hoff-JörgensenR 1985 Control programme for lenti-virus infections in Danish sheep and goats. 133 137 Brussels, Belgium. 8076 EN. ECSC-EEC-EAEC 8076 EN. ECSC-EEC-EAEC
31. HowersDJ 1985 Experimental maedi–visna control in the Netherlands. 291 295 Brussels, Belgium. 8076 EN. ECSC-EEC-EAEC 8076 EN. ECSC-EEC-EAEC
32. KrogsrudJ 1985 Control of maedi in Norway. 139 147 Brussels, Belgium. 8076 EN. ECSC-EEC-EAEC 8076 EN. ECSC-EEC-EAEC
33. SihvonenLNuotioLRikulaUHirvela-KoskiVKokkonenU 2000 Preventing the spread of maedi-visna in sheep through a voluntary control programme in Finland. Prev Vet Med 47 213 220
34. SyngeBARitchieCM 2010 Elimination of small ruminant lentivirus infection from sheep flocks and goat herds aided by health schemes in Great Britain. Vet Rec 167 739 743
35. GjersetBRimstadETeigeJSoetaertKJonassenCM 2009 Impact of natural sheep-goat transmission on detection and control of small ruminant lentivirus group C infections. Vet Microbiol 135 231 238
36. DeanMCarringtonMWinklerCHuttleyGASmithMW 1996 Genetic restriction of HIV-1 infection and progression to AIDS by a deletion allele of the CKR5 structural gene. Science 273 1856 1862
37. LiuRPaxtonWAChoeSCeradiniDMartinSR 1996 Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell 86 367 377
38. SamsonMLibertFDoranzBJRuckerJLiesnardC 1996 Resistance to HIV-1 infection in caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature 382 722 725
39. AllersKHutterGHofmannJLoddenkemperCRiegerK 2011 Evidence for the cure of HIV infection by CCR5{Delta}32/{Delta}32 stem cell transplantation. Blood 117 2791 2799
40. de la Concha-BermejilloABrodieSJMagnus-CorralSBowenRADeMartiniJC 1995 Pathologic and serologic responses of isogeneic twin lambs to phenotypically distinct lentiviruses. J Acquir Immune Defic Syndr Hum Retrovirol 8 116 123
41. Herrmann-HoesingLMWhiteSNMouselMRLewisGSKnowlesDP 2008 Ovine progressive pneumonia provirus levels associate with breed and Ovar-DRB1. Immunogenetics 60 749 758
42. LarruskainAMinguijonEGarcia-EtxebarriaKMorenoBArosteguiI 2010 MHC class II DRB1 gene polymorphism in the pathogenesis of Maedi-Visna and pulmonary adenocarcinoma viral diseases in sheep. Immunogenetics 62 75 83
43. WhiteSNMouselMRReynoldsJOLewisGSHerrmann-HoesingLM 2009 Common promoter deletion is associated with 3.9-fold differential transcription of ovine CCR5 and reduced proviral level of ovine progressive pneumonia virus. Anim Genet 40 583 589
44. BerriatuaEAlvarezVExtramianaBGonzalezLDaltabuitM 2003 Transmission and control implications of seroconversion to Maedi-Visna virus in Basque dairy-sheep flocks. Prev Vet Med 60 265 279
45. HerrmannLMCheeversWPMarshallKLMcGuireTCHuttonMM 2003 Detection of serum antibodies to ovine progressive pneumonia virus in sheep by using a caprine arthritis-encephalitis virus competitive-inhibition enzyme-linked immunosorbent assay. Clin Diagn Lab Immunol 10 862 865
46. McNemarQ 1947 Note on the sampling error of the difference between correlated proportions or percentages. Psychometrika 12 153 157
47. HeatonMPLeymasterKAKalbfleischTSFrekingBASmithTP 2010 Ovine reference materials and assays for prion genetic testing. BMC Vet Res 6 23
48. HerrmannLMCheeversWPMcGuireTCAdamsDSHuttonMM 2003 Competitive-inhibition enzyme-linked immunosorbent assay for detection of serum antibodies to caprine arthritis-encephalitis virus: diagnostic tool for successful eradication. Clin Diagn Lab Immunol 10 267 271
49. MenasheIRosenbergPSChenBE 2008 PGA: power calculator for case-control genetic association analyses. BMC Genet 9 36
50. PurcellSNealeBTodd-BrownKThomasLFerreiraMA 2007 PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81 559 575
51. McNuttLAWuCXueXHafnerJP 2003 Estimating the relative risk in cohort studies and clinical trials of common outcomes. Am J Epidemiol 157 940 943
52. DalrympleBPKirknessEFNefedovMMcWilliamSRatnakumarA 2007 Using comparative genomics to reorder the human genome sequence into a virtual sheep genome. Genome Biol 8 R152
53. GordonDAbajianCGreenP 1998 Consed: a graphical tool for sequence finishing. Genome Res 8 195 202
54. SmithTPGodtelRALeeRT M2000 PCR-based setup for high-throughput cDNA library sequencing on the ABI 3700™ automated DNA sequencer. Bio Techniques 29 698 700
55. EwingBGreenP 1998 Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res 8 186 194
56. EwingBHillierLWendlMCGreenP 1998 Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res 8 175 185
57. NickersonDATobeVOTaylorSL 1997 PolyPhred: automating the detection and genotyping of single nucleotide substitutions using fluorescence-based resequencing. Nucleic Acids Res 25 2745 2751
58. LaegreidWWHeatonMPKeenJEGrosseWMChitko-McKownCG 2002 Association of bovine neonatal Fc receptor alpha-chain gene (FCGRT) haplotypes with serum IgG concentration in newborn calves. Mamm Genome 13 704 710
59. MontgelardCCatzeflisFMDouzeryE 1997 Phylogenetic relationships of Artiodactyls and Cetaceans as deduced from the comparison of cytochrome b and 12S rRNA mitochondrial sequences. Mol Biol Evol 14 550 559
60. BendtsenJDNielsenHvon HeijneGBrunakS 2004 Improved prediction of signal peptides: SignalP 3.0. J Mol Biol 340 783 795
61. FlicekPAmodeMRBarrellDBealKBrentS 2011 Ensembl 2011. Nucleic Acids Res 39 D800 806
62. BandeltHJForsterPRohlA 1999 Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol 16 37 48
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2012 Číslo 1
Nejčtenější v tomto čísle
- Poly(ADP-Ribose) Polymerase 1 (PARP-1) Regulates Ribosomal Biogenesis in Nucleoli
- Microenvironmental Regulation by Fibrillin-1
- Parallel Mapping and Simultaneous Sequencing Reveals Deletions in and Associated with Discrete Inherited Disorders in a Domestic Dog Breed
- Two-Component Elements Mediate Interactions between Cytokinin and Salicylic Acid in Plant Immunity