
RIC-7 Promotes Neuropeptide Secretion
Secretion of neurotransmitters and neuropeptides is mediated by exocytosis of distinct secretory organelles, synaptic vesicles (SVs) and dense core vesicles (DCVs) respectively. Relatively little is known about factors that differentially regulate SV and DCV secretion. Here we identify a novel protein RIC-7 that is required for neuropeptide secretion in Caenorhabditis elegans. The RIC-7 protein is expressed in all neurons and is localized to presynaptic terminals. Imaging, electrophysiology, and behavioral analysis of ric-7 mutants indicates that acetylcholine release occurs normally, while neuropeptide release is significantly decreased. These results suggest that RIC-7 promotes DCV–mediated secretion.
Published in the journal:
. PLoS Genet 8(1): e32767. doi:10.1371/journal.pgen.1002464
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002464
Summary
Secretion of neurotransmitters and neuropeptides is mediated by exocytosis of distinct secretory organelles, synaptic vesicles (SVs) and dense core vesicles (DCVs) respectively. Relatively little is known about factors that differentially regulate SV and DCV secretion. Here we identify a novel protein RIC-7 that is required for neuropeptide secretion in Caenorhabditis elegans. The RIC-7 protein is expressed in all neurons and is localized to presynaptic terminals. Imaging, electrophysiology, and behavioral analysis of ric-7 mutants indicates that acetylcholine release occurs normally, while neuropeptide release is significantly decreased. These results suggest that RIC-7 promotes DCV–mediated secretion.
Introduction
Neurons secrete both neuropeptides and neurotransmitters. Neurotransmitters, such as acetylcholine (ACh), are secreted by exocytosis of small clear synaptic vesicles (SVs) whereas neuropeptide secretion is mediated by exocytosis of dense core vesicles (DCVs) [1], [2]. The mechanisms leading to DCV and SV exocytosis are similar in many respects. SVs and DCVs both undergo physical docking to the plasma membrane, requiring Munc18 and syntaxin for docking in both cases [3]–[7]. To become fusion competent, SVs and DCVs must both undergo a priming reaction, which is mediated by priming factors (e.g. Munc13 and CAPS) [8], [9]. Exocytosis of SVs and DCVs are both mediated by assembling complexes between vesicular and plasma membrane SNARE proteins [10], [11]. Finally, calcium-evoked fusion of SVs and DCVs are mediated by distinct calcium sensors, which are thought to be different synaptotagmin isoforms [12].
Beyond these similarities, DCVs and SVs exhibit many important differences. DCVs can be found all along the cell body, dendrites and axons of neurons whereas SVs cluster specifically at active zones of synapses [13]. SVs undergo repeated cycles of exo - and endocytosis at synapses, whereas neuropeptides are only packaged into nascent DCVs in the Golgi [14]. Consequently, DCVs cannot undergo local recycling in axons or dendrites. DCVs release their contents over long timescales (>50 ms) while SV exocytosis occurs more rapidly (<20 ms) [13], [15]. Exocytosis of SVs can be evoked by single action potentials while DCV release typically occurs after more prolonged or repeated depolarizations. These differences imply that different molecules are involved in SV and DCV secretion.
To date, very few proteins have been found that are specifically involved in the secretion of one or the other class of vesicles. UNC-31/CAPS (Calcium-dependent Activator Protein for Secretion) is proposed to promote priming of DCVs but not SVs [16]–[18]. However, a subsequent study showed compelling evidence for SV priming defects in CAPS1 and CAPS2 double knockout mice [19], implying that CAPS is also required for SV priming. Similarly, some studies propose that Munc13 primes SVs but not DCVs [18], while others find Munc13 mutants have exocytosis defects for both SVs and DCVs [20], [21]. C. elegans mutants lacking PKC-1, a PKCε ortholog, had significant defects in DCV release but little effect on SV release [20]. Identifying new genes that differentially regulate SV or DCV release will provide new insights into the mechanisms underlying these two forms of secretion.
In C. elegans, the acetylcholinesterase inhibitor aldicarb has been widely used to study neuromuscular signaling in live animals. Aldicarb treatment causes acetylcholine (ACh) to accumulate in the synaptic cleft at NMJs, resulting in an acute paralysis of treated animals. Mutations or RNAi treatments that reduce ACh release confer resistance to aldicarb-induced paralysis, whereas those that stimulate ACh secretion enhance aldicarb sensitivity [22], . We previously showed that neuropeptides also regulate aldicarb responsiveness [24], [25]. Inactivation of genes encoding proneuropeptide processing enzymes [for example, egl-3 prohormone convertase (PC2), egl-21 carboxypeptidase E (CPE), sbt-1 7B2, and nep-1 neprilysin], proneuropeptides (ins-22, ins-31, flp-1, nlp-12), neuropeptide receptors (fshr-1) all cause aldicarb resistance [24], [25]. These results suggested that new genes that are required for DCV secretion could be identified through screens for aldicarb resistant mutants.
In a screen for mutations that suppress the aldicarb hypersensitivity of dgk-1 diacylglycerol kinase (DAGK) mutants, we isolated a new allele of the ric-7 gene. Here we show that ric-7 encodes a novel nematode specific protein that is required for neuropeptide secretion.
Results
RIC-7 functions in cholinergic neurons for aldicarb responsiveness
To identify new genes required for neuromuscular function, we screened for mutations that suppress the aldicarb hypersensitivity defect of dgk-1 DAGK mutants. One suppressor (nu447) significantly decreased the aldicarb hypersensitivity of dgk-1 mutants. The nu447 mutation mapped close to the ric-7 gene, which was identified in prior screens for aldicarb resistant mutants [22]. We found that nu447 and ric-7(n2657) both correspond to mutations in F58E10.1 gene (Figure 1A), hereafter referred to as the ric-7 gene. The ric-7 locus encodes two isoforms (A and B) that differ only in their first exon. Orthologs of ric-7 are observed in several other nematodes, but homologous genes are not detected in other metazoans. The predicted RIC-7 protein does not contain any previously described structural domains.

Animals homozygous for nu447 or n2657 were resistant to the paralytic effects of aldicarb (Figure 1B). To determine whether RIC-7 functions in motor neurons for aldicarb responsiveness, we constructed a ric-7 transcriptional reporter. The resulting construct expressed GFP in many neurons, including both cholinergic and GABAergic motor neurons (Figure 2A–2B). Transgenes expressing either RIC-7A or B isoforms in all neurons (with the snb-1 promoter), and those expressing RIC-7B in cholinergic neurons (with the unc-17 promoter) rescued the aldicarb sensitivity defect of ric-7 mutants (Figure 1B). By contrast, expressing RIC-7B in GABAergic neurons (unc-25 promoter) or in muscles (myo-3 promoter) did not rescue the aldicarb phenotype of ric-7 mutants (Figure 1B). These results suggest that RIC-7 activity is required in cholinergic neurons for aldicarb responsiveness.

Animals lacking ric-7 also had decreased locomotion rates (Figure 1C). This locomotion defect was fully rescued by ric-7 transgenes expressed in all neurons (using the snb-1 promoter) whereas partial rescue was observed with transgenes expressed in cholinergic or GABAergic neurons (Figure 1C). The morphology of motor neuron axons and NMJs appeared superficially normal in ric-7 mutants (Figure 4C and data not shown), suggesting that these motor defects were unlikely to be caused by changes in neural development.
RIC-7 is targeted to presynaptic terminals
We expressed GFP-tagged RIC-7 constructs in the cholinergic DA neurons (using the unc-129 promoter). The RIC-7::GFP protein was localized in a punctate distribution in both cell bodies and dorsal cord axons. The majority of RIC-7 puncta co-localized with an SV marker (mCherry-tagged Endophilin) [26], suggesting that RIC-7 is targeted to synapses (Figure 2C). We also compared the distribution of GFP-tagged RIC-7 with a mCherry-tagged neuropeptide, NLP-21. Whereas RIC-7 showed partial overlap with endophilin, nearly complete co-localization was observed with NLP-21 (Figure 2C). Several results suggest that the co-localization of RIC-7 with DCVs was not mediated by physical association of RIC-7 with nascent DCVs. First, RIC-7 synaptic localization was not disrupted in unc-104 mutants (Figure 2D), which lack the KIF1A motor responsible for anterograde transport of SVs and DCVs. Thus, RIC-7 is not co-transported to synapses with immature DCVs or SVs. Second, RIC-7 lacks a predicted signal peptide sequence and GFP-tagged RIC-7 did not produce fluorescence in coelomocytes, both of which indicate that RIC-7 is not translocated into SVs or DCVs (data not shown). These results support the idea that RIC-7 functions in the cytoplasm at presynapses and could play a relatively direct role in regulating SV or DCV secretion.
Muscle responsiveness to ACh and GABA is unaltered in ric-7 mutants
We did several experiments to test the effects of RIC-7 on the responsiveness of body muscles to neuromuscular agonists (Figure 3). First, the sensitivity of ric-7 mutants to the paralytic effects of the nicotinic agonist levamisole was similar to that of wild type controls (Figure 3A). Second, we recorded body wall muscle currents evoked by application of ACh or the GABA agonist muscimol. In both cases, the amplitude of agonist-evoked current in ric-7 mutant body muscles was not significantly different from that observed in wild type controls (Figure 3B). Third, the fluorescent intensities of GFP-tagged ACR-16 ACh receptor (Figure 3C) and UNC-49 GABAA receptor (Figure 3D) puncta in the nerve cord were unaltered in ric-7 mutants, indicating that the abundance of post-synaptic receptors at NMJs was normal. Thus, the effect of RIC-7 on aldicarb responsiveness is unlikely to be caused by altered agonist responsiveness of body muscles.

ACh release occurs normally in ric-7 mutants
The aldicarb resistance phenotype observed in ric-7 mutants could be caused by decreased ACh secretion, increased GABA secretion, or decreased neuropeptide secretion, as aldicarb resistance would be expected in all three scenarios [25], [27], [28]. To assay ACh secretion more directly, we recorded excitatory post-synaptic currents (EPSCs) from body muscles (Figure 4). We found that the rate and amplitude of endogenous EPSCs, i.e. SV fusions evoked by the endogenous activity of motor neurons, in ric-7 mutants were similar to those found in wild type animals (Figure 4A). In addition, the amplitude of stimulus-evoked EPSCs in ric-7 mutants was not significantly different from wild type (Figure 4B). Therefore, baseline ACh secretion occurs normally at cholinergic NMJs in ric-7 mutants.

To further analyze the cholinergic NMJs, we examined the distribution of GFP-tagged Synaptobrevin (GFP::SNB-1) in motor neurons. Changes in the distribution of GFP::SNB-1 are correlated with changes in SV exo - and endocytosis. SNB-1 puncta intensity is correlated with the number of SVs at presynaptic elements [24], [29] whereas puncta density is a measure of synapse density. Neither the fluorescent intensity nor the density of SNB-1 puncta in the cholinergic axons of ric-7 mutants were significantly different from that observed in wild type controls (Figure 4C). Taken together, these results suggest that defects in baseline ACh secretion are unlikely to account for the aldicarb resistance of ric-7 mutants.
RIC-7 promotes neuropeptide secretion
Aldicarb resistance could also be caused by defects in neuropeptide signaling [25]; therefore, we next examined ric-7 mutants for changes in neuropeptide signaling. First, we analyzed the aldicarb responsiveness of ric-7 double mutants containing mutations in neuropeptide signaling components. If changes in neuropeptide action contribute to RIC-7's effects on aldicarb responsiveness, then we would expect that ric-7 mutations would occlude the effect of neuropeptide signaling mutations on aldicarb resistance. Consistent with this idea, ric-7 double mutants carrying mutations in either of two proneuropeptide processing enzymes (egl-21 CPE and egl-3 PC2) had aldicarb resistance that was similar to that observed in ric-7 single mutants (Figure 5). These results support the idea that RIC-7 regulation of neuropeptide secretion contributes to the effects of ric-7 mutations on aldicarb responsiveness.

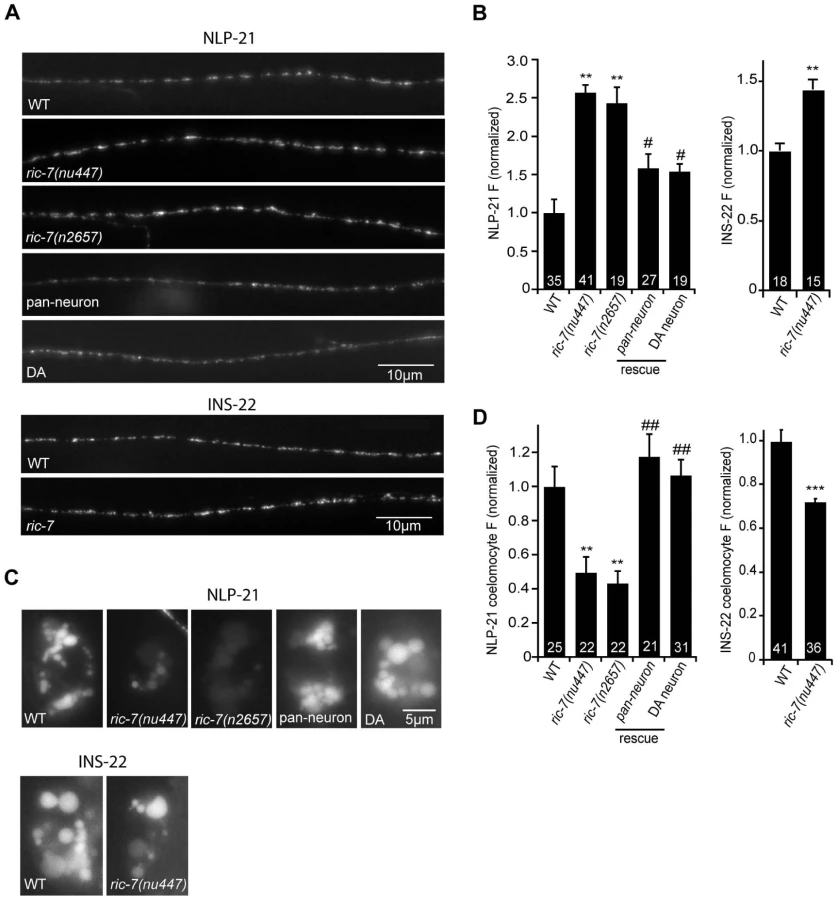
To further address the role of RIC-7 in neuropeptide secretion, we analyzed the secretion of YFP-tagged neuropeptides (Figure 6). For this analysis, we selected two proneuropeptides, NLP-21 and INS-22, which encode FMRFamide related peptides (FaRPs) and an insulin-like growth factor, respectively. When NLP-21::YFP or INS-22::YFP are expressed in the cholinergic DA motor neurons (using the unc-129 promoter), puntate fluorescence is detected in dorsal cord axons and in coelomocytes (Figure 6A and 6C). We previously showed that the axonal puncta fluorescence corresponds to secretory granules containing these proneuropeptides while the coelomocyte fluorescence corresponds to secreted neuropeptides that have been endocytosed [20], [30]. In ric-7 mutants, NLP-21 coelomocyte fluorescence was significantly decreased (∼50%, p<0.001) (Figure 6C–6D) whereas the NLP-21 puncta fluorescence intensity in dorsal cord axons was significantly increased (>2-fold, p<0.001) (Figure 6A–6B). The coelomocyte and axonal NLP-21 fluorescence defects were both rescued by ric-7 transgenes expressed in the DA neurons. Similar changes in axonal puncta fluorescence and coelomocyte fluorescence was observed for a second proneuropeptide (INS-22) in ric-7 mutants (Figure 6). These results suggest that ric-7 mutants have decreased neuropeptide secretion, which results in an accumulation of DCVs in motor axons.
RIC-7 is required for NLP-12–mediated enhancement of ACh release
Inactivation of NLP-21 and INS-22 by RNAi does not produce strong locomotion or aldicarb resistance defects [24]; consequently, NLP-21 and INS-22 are unlikely to be the only neuropeptides involved in the Ric-7 locomotion and aldicarb-resistance phenotypes. We recently identified NLP-12 as a neuropeptide that plays a critical role in regulating both locomotion rate and aldicarb-induced paralysis [31]. NLP-12 is expressed in a proprioceptive neuron (DVA) that is activated by body muscle contractions [32], [33]. If Ric-7 aldicarb resistance and locomotion defects are caused by decreased neuropeptide release, we would expect that NLP-12 secretion from DVA would be diminished in ric-7 mutants. We did several experiments to test this idea. First, we analyzed the effect of ric-7 mutations on secretion of YFP-tagged NLP-12 from DVA neurons (Figure 7A–7B). In wild type animals, aldicarb treatment significantly decreased NLP-12 puncta fluorescence in DVA axons (indicating increased NLP-12 secretion), which is most likely caused by activation of DVA stretch receptors by muscle contraction [31]. By contrast, in ric-7 mutants, aldicarb had no effect on NLP-12 puncta fluorescence, indicating that RIC-7 is required for aldicarb-evoked NLP-12 secretion.

Second, if the NLP-12 secretion defect contributes to the Ric-7 aldicarb resistance phenotype, we would expect that RIC-7 expression in DVA neurons would be sufficient to alter aldicarb responsiveness. Consistent with this idea, the aldicarb responsiveness of ric-7 mutants was significantly improved by transgenes expressing RIC-7 in DVA neurons (Figure 7C). This result is consistent with the preceding rescue data (Figure 1B) because the unc-17 promoter (which also rescued the Ric-7 aldicarb defect) is expressed in DVA neurons (data not shown). Collectively, these data suggest that proper aldicarb responsiveness requires RIC-7 function in multiple neuron classes because RIC-7 expression in a single cholinergic neuron (DVA) produced partial rescue of the aldicarb defect whereas complete rescue was obtained following expression in all cholinergic neurons (with the unc-17 promoter) (Figure 1B).
Third, we analyzed evoked ACh release following aldicarb treatment. In wild type animals, a 60 minute pre-treatment with aldicarb significantly increases the total synaptic charge occurring during an evoked response (Figure 7D–7E) [31]. This effect is eliminated in egl-3 PC2 mutants and in nlp-12 mutants [31]. Thus, aldicarb enhancement of evoked ACh release can be utilized to assess changes in endogenous NLP-12 secretion. Aldicarb's effect on evoked ACh release was also eliminated in ric-7 mutants (Figure 7D–7E). Collectively, these results strongly support the idea that RIC-7 acts in DVA neurons to promote secretion of endogenous NLP-12, and that this contributes to the Ric-7 aldicarb-resistance defect.
GABA transmission is altered in ric-7 mutants
Changes in GABA secretion could also contribute to the aldicarb resistance observed in ric-7 mutants [22], [27], [28]. Consistent with this idea, ric-7 mutants had defects in defecation behavior that are similar to those observed in mutants with decreased GABA transmission (Figure 8A). Contraction of the intestinal muscles during defecation (i.e. the expulsion step of the defecation motor program) is mediated by excitatory GABAergic input from defecation motor neurons [34]. We found that ric-7 mutants had a significant expulsion defect, which was rescued by ric-7 transgenes expressed in GABA neurons (Figure 8A). These results suggest that ric-7 mutants have a presynaptic defect at GABAergic NMJs involved in defecation.

To assay GABA release at ventral cord NMJs (which are involved in locomotion), we recorded inhibitory post-synaptic currents (IPSCs) from body muscles (Figure 8B). We found that ric-7 mutants had a wild type IPSC rate, while IPSC amplitudes were significantly increased (∼30%, p<0.001). These results suggest that GABA secretion still occurs in ric-7 mutants, albeit in a subtly altered form. Changes in IPSC amplitudes are often caused by changes in post-synaptic sensitivity, e.g. by changing GABA receptor abundance. However, neither the current evoked by applying an exogenous GABA agonist nor the abundance of GFP-tagged UNC-49 GABA-A receptors were altered in ric-7 mutants, suggesting that a post-synaptic defect was unlikely to account for the altered IPSC amplitude (Figure 3B, 3D). Consistent with a pre-synaptic defect, the distribution of GFP-tagged SNB-1 was altered in GABAergic neurons of ric-7 mutants. Although SNB-1 puncta intensity was unaltered, puncta were significantly wider (23% wider, p<0.001) and diffuse SNB-1 axon fluorescence was significantly increased in ric-7 mutants (Figure 8C). Finally, the IPSC and SNB-1 defects were both rescued by transgenes expressing RIC-7 in GABAergic motor neurons (Figure 8B–8C). Collectively, these results support the idea that ric-7 mutants have a pre-synaptic defect that increases GABA secretion, perhaps by increasing the amount of GABA packaged into each SV. Rescue of the IPSC defect (with ric-7 transgenes expressed in GABA neurons) failed to rescue the ric-7 aldicarb defect and weakly rescued the locomotion defect implying that the latter were not caused by altered GABA secretion (Figure 1C).
Mutations disrupting neuropeptide signaling exhibit defecation defects similar to ric-7 mutants [25], [35]. Prompted by these results, we wondered if the Ric-7 defecation and IPSC defects are caused by the neuropeptide secretion defect. In this scenario, we would expect that ric-7 mutations and mutations that impair proneuropeptide processing would not have additive effects on defecation behavior in double mutants. Contrary to this idea, the defecation defects observed in ric-7; egl-21 double mutants were significantly worse than those observed in either single mutant (Figure 8A). In addition, IPSC amplitudes were unaltered in egl-3; egl-21 doubles mutants (Figure S1), suggesting that decreased neuropeptide secretion is also unlikely to explain the Ric-7 IPSC defect. These results indicate that RIC-7 cell-autonomously regulates two forms of secretion, promoting neuropeptide secretion and inhibiting GABA secretion.
Discussion
Communication between neurons and their targets relies on two classes of neurosecretory vesicles, synaptic vesicles (SVs) and dense-core vesicles (DCVs). The mechanisms governing SV and DCV secretion share many properties (including related SNARE proteins, SNARE binding proteins, and calcium sensors). Despite these similarities, SV and DCV mediated secretion also have significant differences [1], which imply that some proteins will be selectively utilized in one or the other process. Because neuropeptides have dramatic effects on personality, behavior, and metabolism, there is significant interest in identifying molecules that selectively promote DCV secretion. Here we identify RIC-7 as a novel protein that is required for DCV secretion but has relatively subtle effects on SV secretion. Below we discuss the implications of these findings.
The ric-7 gene was identified in a screen for mutations conferring resistance to aldicarb induced paralysis. As RIC-7 lacks identified structural or functional motifs, the mechanisms underlying this behavioral defect were unclear. In principle, aldicarb resistance could arise from several alternative mechanisms, including: altered muscle responsiveness to ACh or GABA, decreased excitatory input from cholinergic motor neurons, increased hyperpolarizing input from GABA motor neurons, or decreased neuropeptide signaling. Our results strongly support the idea that Ric-7 aldicarb and locomotion defects arise from disruption of neuropeptide secretion.
Several results suggest that RIC-7 does not regulate muscle sensitivity to neuromuscular agonists. The currents evoked by applying exogenous ACh and GABA to body muscles, and the expression of nicotinic and GABA receptors in body muscles were both unaffected in ric-7 mutants. Furthermore, ric-7 aldicarb and locomotion defects were not corrected by transgenes expressing RIC-7 in body muscles, whereas rescue was observed for transgenes expressed in cholinergic neurons. Thus, RIC-7 neither acts in body muscles, nor regulates muscle sensitivity to agonists.
Other results indicate that RIC-7 is not required for baseline ACh secretion. In cholinergic motor neurons, the SV protein SNB-1 neither accumulated in synaptic puncta nor in the axonal membrane (as would occur in exocytosis and endocytosis mutants, respectively). The rate, amplitude, and kinetics of endogenous and evoked EPSCs were unaltered in ric-7 mutants. Thus, although RIC-7 expression in cholinergic neurons rescued the aldicarb and locomotion defects of ric-7 mutants, these defects are unlikely to arise from decreased baseline ACh secretion.
In GABA neurons, ric-7 mutants had a presynaptic defect that led to an apparent increase in GABA secretion. This presynaptic defect was manifest by an increase in amplitude of endogenous IPSCs in ric-7 mutants, while the IPSC rate was unaltered. Changes in IPSC amplitudes are often caused by changes in post-synaptic sensitivity, e.g. by changing GABA receptor abundance. However, neither the current evoked by applying an exogenous GABA agonist nor the abundance of GFP-tagged UNC-49 GABA-A receptors were altered in ric-7 mutants, suggesting that a post-synaptic defect was unlikely to account for the altered IPSC amplitude. Consistent with a pre-synaptic defect, SNB-1 axonal fluorescence was significantly increased in ric-7 mutant GABA motor neurons. The defecation, IPSC, and SNB-1 defects were all rescued by transgenes expressing RIC-7 in GABAergic motor neurons. Collectively, these results support the idea that ric-7 mutants have a pre-synaptic defect that increases GABA secretion. Rescue of the IPSC defect did not correlate with rescue of ric-7 aldicarb and locomotion defects, implying that the latter were not caused by altered GABA secretion. In principle, the IPSC defect could cause the Ric-7 defecation defect, for example if increased GABA secretion causes constitutive excitation of the intestinal muscles. Double mutant analysis suggests that decreased neuropeptide secretion is unlikely to account for the Ric-7 IPSC and defecation defects. Consequently, our results are most consistent with the idea that RIC-7 has direct cell autonomous effects on two forms of secretion, promoting neuropeptide secretion and inhibiting GABA secretion. Several other proteins are known to have distinct effects on different neurotransmitter systems. Mouse Munc13-1 knockouts drastically reduce glutamatergic transmission yet have little effect on GABA release [36]. Mouse ELKS2 knockouts have increased GABA release but unaltered glutamate release [37]. Finally, complexin promotes evoked release but inhibits tonic/spontaneous release [38], [39].
Several results suggest that the effects of RIC-7 on aldicarb sensitivity were caused by changes in neuropeptide secretion. First, mutations preventing neuropeptide processing (egl-21 CPE, egl-3 PC2) and ric-7 mutations did not have additive effects on aldicarb responsiveness in double mutants. Second, ric-7 mutants had greatly decreased secretion of YFP-tagged neuropeptides (NLP-21 and INS-22) from cholinergic motor neurons. Third, mCherry-tagged RIC-7 co-localized extensively with a DCV marker (NLP-21), implying that RIC-7 could play a relatively direct role in regulating neuropeptide release. Fourth, ric-7 mutants have decreased aldicarb-evoked secretion of NLP-12 from DVA neurons and lack aldicarb-induced potentiation of evoked ACh release (which is mediated by endogenous NLP-12). And fifth, restoring RIC-7 expression in DVA neurons was sufficient to partially rescue the Ric-7 aldicarb-resistance defect. Taken together, these results strongly support the idea that RIC-7 promotes secretion of endogenous neuropeptides, and that this function plays an important role in the Ric-7 aldicarb resistance and locomotion defects. The Ric-7 aldicarb resistance and locomotion defects are more severe than those observed in mutants lacking NLP-12 or those lacking EGL-3 PC2, indicating that additional RIC-7 functions also contribute to these phenotypes. These additional functions could include promoting secretion of other neuropeptides or novel RIC-7 functions that are not yet defined.
What aspect of DCV secretion is regulated by RIC-7? Decreased neuropeptide secretion in ric-7 mutants could reflect changes in any aspect of DCV biogenesis, transport, docking, priming, calcium-triggering, or fusion. Several results suggest that RIC-7 is not packaged into DCVs, and consequently cannot play a role in pro-neuropeptide processing. RIC-7 lacks a predicted signal peptide sequence, GFP-tagged RIC-7 is not secreted, and RIC-7 delivery to axons is not prevented in unc-104 KIF1A mutants. Collectively, these results suggest RIC-7 is cytoplasmic, and thus cannot play a direct role in neuropeptide processing. DCV biogenesis and transport also occur normally in ric-7 mutants, as neuropeptide fluorescence in motor axons was increased rather than decreased. These results suggest that RIC-7 regulates a step that occurs after DCV transport. Given the prominent colocalization of RIC-7 and DCV markers in axons, we speculate the RIC-7 could identify DCV release sites. Because the sequence of RIC-7 does not provide any clues as to its biochemical function, further experiments will be required to determine a more precise function for RIC-7. Understanding the mechanisms underlying RIC-7's functions will undoubtedly shed light on how DCVs assume their unique properties.
The mechanisms governing SV and DCV secretion share many properties, including: SNARE proteins, SNARE binding proteins (e.g. Munc13, Munc18, and CAPS), and calcium sensors (e.g. Synaptotagmin) [40]. These shared mechanisms are highly conserved across eukaryotic phylogeny, suggesting that these core exocytosis components comprise an ancient process. By contrast, RIC-7 orthologs are observed in other nematodes but homologous genes are not detected in other sequenced genomes. Interestingly, other putative DCV secretion factors have similar patterns of conservation. For example, the Rab27 effector granuphilin is conserved in mammals and flies but not in C. elegans, while a second Rab27 effector melanophilin is present in mammals but absent in flies and worms. These results suggest that the mechanisms distinguishing SV and DCV secretion evolved more recently than the more ancient shared secretion factors.
Materials and Methods
C. elegans strains and drug assays
Strains were maintained at 20°C as described [41]. The wild-type reference strain was N2 Bristol. Descriptions of allele lesions can be found at http://www.wormbase.org. The mutant strains used were: LGIV, egl-21(n476); LGV, egl-3(nr2090), ric-7 (nu447), ric-7(n2657). Acute aldicarb and levamisole assays were performed blind in triplicate on young adult worms as described [42]. The aldicarb (Chem Services) concentrations used ranged from 1 to 2 mM, the levamisole (Sigma) concentration was 200 µM. Locomotion was measured by transferring adults to plates containing fresh (16 h) lawns of HB101 bacteria, letting the worms recover for 30 min and counting the number of body bends of active worms in a 3 min interval.
ric-7 mapping and cloning
ric-7(nu447) was isolated in an EMS screen for suppressors of the aldicarb hypersensitive phenotype of dgk-1(nu62) mutants (D.S. and J.K., unpublished data). nu447 was mapped by small nucleotide polymorphism (SNP) mapping, based on its aldicarb resistance phenotype. Analysis of 16 nu447/CB4856 recombinants mapped nu447 to LGV, map unit 5∼6; Another 62 clones further positioned nu447 to 5.86∼6.00 m.u. Seven predicted genes were found in this interval with three of them previously characterized. Sequencing revealed that ric-7(nu447) contained a 830C/T (A277V) point mutation plus a 32 bp deletion (nucleotides 910–941) in F58E10.1b cDNA that shifts the reading frame, leading to a truncated RIC-7 protein that contains 305 amino acids. ric-7(n2657) contained a 621G/A (W207Stop) nonsense mutation in F58E10.1b cDNA. nu447 was backcrossed six times and used for most phenotypic analysis in this study.
Fluorescence microscopy and quantitative analysis
All quantitative imaging was done using a Olympus PlanAPO 100×1.4 NA objective and an ORCA100 CCD camera (Hamamatsu). Worms were immobilized with 30 mg/ml BDM (Sigma). Image stacks were captured and maximum intensity projections were obtained using Metamorph 7.1 software (Molecular Devices). GFP fluorescence was normalized to the absolute mean fluorescence of 0.5 mm FluoSphere beads (Molecular Probes). For dorsal cord imaging, young adult worms, in which the dorsal cords were oriented toward the objective, were imaged in the region midway between the posterior gonad bend at the tail. Line scans of dorsal cord fluorescence were analyzed in Igor Pro (WaveMetrics) using custom-written software [43], [44]. For coelomocyte imaging, the posterior coelomocyte was imaged in young adults [20]. Image stacks were captured and maximum intensity projections were obtained using Metamorph 7.1 software (Molecular Devices). For quantitation, the five brightest vesicles were analyzed for each coelomocyte and the mean fluorescence for each vesicle was logged. For each worm, coelomocyte fluorescence was calculated as the mean of the vesicle values in that animal. All p-values indicated were based on student t-tests.
Confocal images were taken using the Olympus FV1000 confocal microscope. Image stacks were captured, and maximum intensity projections were obtained using Metamorph 7.1 software (Universal Imaging).
Electrophysiology
Electrophysiology was done on dissected adults as previously described [45]. All recording conditions were as described previously [20]. For comparing average electrophysiological values, statistical significance was determined using the Mann-Whitney test or student's t test.
RIC-7 constructs and transgenes
Transgenic strains were generated by injecting either wild type or ric-7(nu447) mutants with the expression construct (10–25 ng/µl) mixed with the co-injection markers, KP#1338 (pttx-3::GFP), KP#1480 (pmyo-2::NLS-mCherry) or KP#1106 (pmyo-2::NLS-GFP), each at 10 ng/µl, using standard methods [46]. The co-localization experiments were done by co-injecting KP#1684 (pric-7::his-24 cDNA::wcherry) with KP#1685 (punc-30::NLS-GFP) or with KP#1686 (punc-17::NLS-GFP), respectively; or by injecting KP#1687 (punc-129::RIC-7 cDNA::GFP) into nuIs252 (Punc-129::mchry::PaGFP::unc-57), or into nuIs470 (punc-129::NLP-21::mCherry), respectively, all at a concentration of 25 ng/µl each. All constructs except KP#1684 were derivatives of pPD49.26 [47].
Two RIC-7 constructs rescued the ric-7(nu447) mutant aldicarb defect: KP#1680 (psnb-1::RIC-7), and KP#1681 (punc-17::RIC-7). Two RIC-7 constructs did not rescue the ric-7(nu447) mutant aldicarb defect: KP #1682(punc-25::RIC-7), and KP#1683(pmyo-3::RIC-7). All four constructs used F58E10.1b cDNA cloned with NheI and KpnI sites.
The previously reported lines used for imaging experiments described are nuIs152 (punc-129::GFP::SNB-1), nuIs183 (punc-129::NLP-21::VENUS), nuIs195 (punc-129::INS-22::VENUS) [20], [24], nuIs283 (pmyo-3::UNC-49::GFP) (J. Bai and J.K., unpublished), nuIs299 (pmyo-3::ACR-16::GFP) [48], nuIs444 (pnlp-12::NLP-12::VENUS) [31] and nuIs376 (punc-25::SNB-1::GFP).
Supporting Information
{kind=link}
Zdroje
1. BurgoyneRDMorganA 2003 Secretory granule exocytosis. Physiol Rev 83 581 632
2. SudhofTC 2004 The synaptic vesicle cycle. Annu Rev Neurosci 27 509 547
3. WeimerRMRichmondJEDavisWSHadwigerGNonetML 2003 Defects in synaptic vesicle docking in unc-18 mutants. Nat Neurosci 6 1023 1030
4. VoetsTToonenRFBrianECde WitHMoserT 2001 Munc18-1 promotes large dense-core vesicle docking. Neuron 31 581 591
5. HammarlundMPalfreymanMTWatanabeSOlsenSJorgensenEM 2007 Open syntaxin docks synaptic vesicles. PLoS Biol 5 e198
6. HammarlundMWatanabeSSchuskeKJorgensenEM 2008 CAPS and syntaxin dock dense core vesicles to the plasma membrane in neurons. J Cell Biol 180 483 491
7. de WitHCornelisseLNToonenRFVerhageM 2006 Docking of secretory vesicles is syntaxin dependent. PLoS One 1 e126
8. KlenchinVAMartinTF 2000 Priming in exocytosis: attaining fusion-competence after vesicle docking. Biochimie 82 399 407
9. MartinTF 2002 Prime movers of synaptic vesicle exocytosis. Neuron 34 9 12
10. JenaBP 2005 Cell secretion and membrane fusion. Domest Anim Endocrinol 29 145 165
11. SorensenJB 2005 SNARE complexes prepare for membrane fusion. Trends Neurosci 28 453 455
12. VoetsTMoserTLundPEChowRHGeppertM 2001 Intracellular calcium dependence of large dense-core vesicle exocytosis in the absence of synaptotagmin I. Proc Natl Acad Sci U S A 98 11680 11685
13. EdwardsRH 1998 Neurotransmitter release: variations on a theme. Curr Biol 8 R883 885
14. KimTGondre-LewisMCArnaoutovaILohYP 2006 Dense-core secretory granule biogenesis. Physiology (Bethesda) 21 124 133
15. BrunsDJahnR 1995 Real-time measurement of transmitter release from single synaptic vesicles. Nature 377 62 65
16. GrishaninRNKowalchykJAKlenchinVAAnnKEarlesCA 2004 CAPS acts at a prefusion step in dense-core vesicle exocytosis as a PIP2 binding protein. Neuron 43 551 562
17. BerwinBFloorEMartinTF 1998 CAPS (mammalian UNC-31) protein localizes to membranes involved in dense-core vesicle exocytosis. Neuron 21 137 145
18. SpeeseSPetrieMSchuskeKAilionMAnnK 2007 UNC-31 (CAPS) is required for dense-core vesicle but not synaptic vesicle exocytosis in Caenorhabditis elegans. J Neurosci 27 6150 6162
19. JockuschWJSpeidelDSiglerASorensenJBVaroqueauxF 2007 CAPS-1 and CAPS-2 are essential synaptic vesicle priming proteins. Cell 131 796 808
20. SieburthDMadisonJMKaplanJM 2007 PKC-1 regulates secretion of neuropeptides. Nat Neurosci 10 49 57
21. KangLHeZXuPFanJBetzA 2006 Munc13-1 is required for the sustained release of insulin from pancreatic beta cells. Cell Metab 3 463 468
22. MillerKGAlfonsoANguyenMCrowellJAJohnsonCD 1996 A genetic selection for Caenorhabditis elegans synaptic transmission mutants. Proc Natl Acad Sci U S A 93 12593 12598
23. NguyenMAlfonsoAJohnsonCDRandJB 1995 Caenorhabditis elegans mutants resistant to inhibitors of acetylcholinesterase. Genetics 140 527 535
24. SieburthDCh'ngQDybbsMTavazoieMKennedyS 2005 Systematic analysis of genes required for synapse structure and function. Nature 436 510 517
25. JacobTCKaplanJM 2003 The EGL-21 carboxypeptidase E facilitates acetylcholine release at Caenorhabditis elegans neuromuscular junctions. J Neurosci 23 2122 2130
26. BaiJHuZDittmanJSPymECKaplanJM 2010 Endophilin functions as a membrane-bending molecule and is delivered to endocytic zones by exocytosis. Cell 143 430 441
27. VashlishanABMadisonJMDybbsMBaiJSieburthD 2008 An RNAi screen identifies genes that regulate GABA synapses. Neuron 58 346 361
28. MullenGPMathewsEASaxenaPFieldsSDMcManusJR 2006 The Caenorhabditis elegans snf-11 gene encodes a sodium-dependent GABA transporter required for clearance of synaptic GABA. Mol Biol Cell 17 3021 3030
29. DittmanJKaplanJ 2006 Factors regulating the abundance and localization of Synaptobrevin in the plasma membrane. PNAS 103 11399 404
30. Ch'ngQSieburthDKaplanJM 2008 Profiling synaptic proteins identifies regulators of insulin secretion and lifespan. PLoS Genet 4 e1000283
31. HuZPymECBabuKVashlishan MurrayABKaplanJM 2011 A neuropeptide-mediated stretch response links muscle contraction to changes in neurotransmitter release. Neuron 71 92 102
32. JanssenTMeelkopELindemansMVerstraelenKHussonSJ 2008 Discovery of a cholecystokinin-gastrin-like signaling system in nematodes. Endocrinology 149 2826 2839
33. LiWFengZSternbergPWXuXZ 2006 A C. elegans stretch receptor neuron revealed by a mechanosensitive TRP channel homologue. Nature 440 684 687
34. JorgensenEM 2005 Gaba. WormBook 1 13
35. MahoneyTRLuoSRoundEKBraunerMGottschalkA 2008 Intestinal signaling to GABAergic neurons regulates a rhythmic behavior in Caenorhabditis elegans. Proc Natl Acad Sci U S A 105 16350 16355
36. AugustinIRosenmundCSudhofTCBrose 1999 Munc13-1 is essential for fusion competence of glutamatergic synaptic vesicles [In Process Citation]. Nature 400 457 461
37. KaeserPSDengLChavezAELiuXCastilloPE 2009 ELKS2alpha/CAST deletion selectively increases neurotransmitter release at inhibitory synapses. Neuron 64 227 239
38. MartinJAHuZFenzKMFernandezJDittmanJS 2011 Complexin has opposite effects on two modes of synaptic vesicle fusion. Curr Biol 21 97 105
39. HobsonRJLiuQWatanabeSJorgensenEM 2011 Complexin maintains vesicles in the primed state in C. elegans. Curr Biol 21 106 113
40. XuTXuP 2008 Searching for molecular players differentially involved in neurotransmitter and neuropeptide release. Neurochem Res 33 1915 1919
41. BrennerS 1974 The genetics of Caenorhabditis elegans. Genetics 77 71 94
42. LacknerMRNurrishSJKaplanJM 1999 Facilitation of synaptic transmission by EGL-30 Gqalpha and EGL-8 PLCbeta: DAG binding to UNC-13 is required to stimulate acetylcholine release. Neuron 24 335 346
43. BurbeaMDreierLDittmanJSGrunwaldMEKaplanJM 2002 Ubiquitin and AP180 regulate the abundance of GLR-1 glutamate receptors at postsynaptic elements in C. elegans. Neuron 35 107 120
44. DittmanJSKaplanJM 2006 Factors regulating the abundance and localization of synaptobrevin in the plasma membrane. Proc Natl Acad Sci U S A 103 11399 11404
45. RichmondJEJorgensenEM 1999 One GABA and two acetylcholine receptors function at the C. elegans neuromuscular junction. Nat Neurosci 2 791 797
46. MelloCCKramerJMStinchcombDAmbrosV 1991 Efficient gene transfer in C.elegans: extrachromosomal maintenance and integration of transforming sequences. Embo J 10 3959 3970
47. FireA 1997 Fire Vector Kit. In Addgene Inc Cambridge: USA
48. SimonDJMadisonJMConeryALThompson-PeerKLSoskisM 2008 The microRNA miR-1 regulates a MEF-2-dependent retrograde signal at neuromuscular junctions. Cell 133 903 915
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2012 Číslo 1
Nejčtenější v tomto čísle
- Poly(ADP-Ribose) Polymerase 1 (PARP-1) Regulates Ribosomal Biogenesis in Nucleoli
- Microenvironmental Regulation by Fibrillin-1
- Parallel Mapping and Simultaneous Sequencing Reveals Deletions in and Associated with Discrete Inherited Disorders in a Domestic Dog Breed
- Two-Component Elements Mediate Interactions between Cytokinin and Salicylic Acid in Plant Immunity