
Cohesin Proteins Promote Ribosomal RNA Production and Protein Translation in Yeast and Human Cells
Cohesin is a protein complex known for its essential role in chromosome segregation. However, cohesin and associated factors have additional functions in transcription, DNA damage repair, and chromosome condensation. The human cohesinopathy diseases are thought to stem not from defects in chromosome segregation but from gene expression. The role of cohesin in gene expression is not well understood. We used budding yeast strains bearing mutations analogous to the human cohesinopathy disease alleles under control of their native promoter to study gene expression. These mutations do not significantly affect chromosome segregation. Transcriptional profiling reveals that many targets of the transcriptional activator Gcn4 are induced in the eco1-W216G mutant background. The upregulation of Gcn4 was observed in many cohesin mutants, and this observation suggested protein translation was reduced. We demonstrate that the cohesinopathy mutations eco1-W216G and smc1-Q843Δ are associated with defects in ribosome biogenesis and a reduction in the actively translating fraction of ribosomes, eiF2α-phosphorylation, and 35S-methionine incorporation, all of which indicate a deficit in protein translation. Metabolic labeling shows that the eco1-W216G and smc1-Q843Δ mutants produce less ribosomal RNA, which is expected to constrain ribosome biogenesis. Further analysis shows that the production of rRNA from an individual repeat is reduced while copy number remains unchanged. Similar defects in rRNA production and protein translation are observed in a human Roberts syndrome cell line. In addition, cohesion is defective specifically at the rDNA locus in the eco1-W216G mutant, as has been previously reported for Roberts syndrome. Collectively, our data suggest that cohesin proteins normally facilitate production of ribosomal RNA and protein translation, and this is one way they can influence gene expression. Reduced translational capacity could contribute to the human cohesinopathies.
Published in the journal:
. PLoS Genet 8(6): e32767. doi:10.1371/journal.pgen.1002749
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002749
Summary
Cohesin is a protein complex known for its essential role in chromosome segregation. However, cohesin and associated factors have additional functions in transcription, DNA damage repair, and chromosome condensation. The human cohesinopathy diseases are thought to stem not from defects in chromosome segregation but from gene expression. The role of cohesin in gene expression is not well understood. We used budding yeast strains bearing mutations analogous to the human cohesinopathy disease alleles under control of their native promoter to study gene expression. These mutations do not significantly affect chromosome segregation. Transcriptional profiling reveals that many targets of the transcriptional activator Gcn4 are induced in the eco1-W216G mutant background. The upregulation of Gcn4 was observed in many cohesin mutants, and this observation suggested protein translation was reduced. We demonstrate that the cohesinopathy mutations eco1-W216G and smc1-Q843Δ are associated with defects in ribosome biogenesis and a reduction in the actively translating fraction of ribosomes, eiF2α-phosphorylation, and 35S-methionine incorporation, all of which indicate a deficit in protein translation. Metabolic labeling shows that the eco1-W216G and smc1-Q843Δ mutants produce less ribosomal RNA, which is expected to constrain ribosome biogenesis. Further analysis shows that the production of rRNA from an individual repeat is reduced while copy number remains unchanged. Similar defects in rRNA production and protein translation are observed in a human Roberts syndrome cell line. In addition, cohesion is defective specifically at the rDNA locus in the eco1-W216G mutant, as has been previously reported for Roberts syndrome. Collectively, our data suggest that cohesin proteins normally facilitate production of ribosomal RNA and protein translation, and this is one way they can influence gene expression. Reduced translational capacity could contribute to the human cohesinopathies.
Introduction
Cohesin is a protein complex that binds to chromosomes from the time of their replication until their segregation. Cohesin creates cohesion between two sister chromatids in order to ensure their correct segregation upon division at the metaphase to anaphase transition. In addition to its essential role in chromosome segregation, the cohesin complex and its accessory factors have been shown to play roles in chromosome condensation, DNA damage repair and gene regulation. The cohesin complex is composed of four subunits: Smc1, Smc3, Mcd1/Scc1/Rad21, and Scc3/Irr1. The complex is loaded onto chromosomes by the Scc2-Scc4 complex [1], [2], [3]. In order to establish cohesion between sisters, Eco1 acetylates the Smc3 subunit of the complex [4], [5], [6]. Pds5 is required for maintenance of cohesion in G2/M [7], [8]. Cohesion is dissolved at the metaphase to anaphase transition when sisters are separated to opposite poles for inclusion in new daughter cells.
Heterozygous mutations in Smc1, Smc3 and Scc2/Nipped-B/NIPBL have been associated with the human disease Cornelia de Lange syndrome (CdLS) [9], [10], [11], [12]. Homozygous mutation of ESCO2 (yeast ECO1) is associated with the human disease Roberts syndrome [13]. The human diseases, referred to as the cohesinopathies, are perplexing since the developmental defects suggest that the primary dysfunction is transcription, rather than chromosome segregation [14]. Metaphase chromosomes from Roberts syndrome patients show “heterochromatic repulsion,” which refers to regions of “puffing” at heterochromatic regions around the centromeres and nucleolar organizers (rDNA) [15].
In order to better understand the molecular underpinning of the human diseases, and to further explore the cohesin network, we constructed yeast strains bearing mutations analogous to those associated with human disease [16]. Our yeast strains are haploid, so they do not genocopy the disease state. However, characterization of the cellular defects associated with the mutations may still be informative. Previous characterization of these strains revealed very few defects in chromosome segregation or the location of cohesin binding, but interestingly, two mutants (eco1-W216G and scc2-D730V) had defects in nucleolar morphology, induction of the GAL2 gene, and chromosome condensation. Three mutant strains exhibited cohesion defects at 37°C (eco1-W216G, smc1-Q843Δ, and scc2-D730V). The eco1-W216G mutation disrupts the acetyltransferase activity of the protein toward Smc3 and is lethal at 37°C [17], [18]. Scc2 has recently been shown to participate not only in cohesin loading, but also in condensin loading [19]. Despite cohesion defects at 37°C, the growth of the scc2-D730V and smc1-Q843Δ mutants appears nearly normal.
To further characterize the mutants, we carried out gene expression profiling in rich medium and at various times following amino acid starvation. The gene expression pattern of the eco1-W216G mutant showed changes in over 1600 genes while the scc2-D730V mutant had essentially a wild-type gene expression profile. Under rich medium conditions, the gene expression profile of the eco1-W216G mutant suggested that protein translation was inhibited. By directly testing protein synthesis and ribosome biogenesis, we confirmed that translation was reduced. Strikingly, ribosomal RNA (rRNA) transcripts were significantly reduced in eco1-W216G and smc1-Q843Δ mutants. Since ribosome assembly is regulated at the level of rRNA [20], this could affect ribosome biogenesis. Cohesion was specifically reduced at the rDNA in the eco1-W216G mutant, reminiscent of the heterochromatic repulsion observed in Roberts syndrome. Importantly, protein synthesis and ribosomal RNA production were reduced in a human Roberts syndrome cell line, very similar to our yeast mutants. Taken together, our results suggest that cohesin proteins may normally promote production of ribosomal RNAs.
Results
Hundreds of genes are differentially expressed in the eco1-W216G mutant
Given the hypothesis that mutations in cohesin can affect gene expression, we undertook gene expression profiling of three strains: 1) wild-type (WT), 2) scc2-D730V, and 3) eco1-W216G. We selected conditions under which we expected many transcriptional changes to maximize the likelihood of finding transcriptional differences in the mutants. Cultures growing in log phase in rich YPD medium (time 0) were transferred to medium lacking amino acids and samples were collected for analysis at 15, 35, and 55 minutes. Three independent cultures were analyzed for each strain background. mRNA was extracted, purified, labeled, and used for hybridization to Affymetrix microarrays (Yeast Genome 2.0) to examine gene expression.
To compare each mutant directly to WT, ratios were formed between each mutant and WT for each time point. Contrasts were created using limma to average replicates and determine p-values for each difference. After adjusting the p-values for multiple hypothesis testing, a set of genes was selected on the basis of adjusted p-values of less than 0.001 from any time point in either mutant/WT comparison. The result was that 1659 genes differed in expression, 1657 for eco1-W216G and 2 for scc2-D730V. Hierarchical clustering of the 1657 genes revealed the expression pattern in the eco1-W216G mutant was highly disrupted relative to the other two strains (Figure 1A). The number of genes up or down regulated in mutant/WT by at least 1.4 fold, with p-values of less than 0.05, for each timepoint is shown in Figure 1B. The lack of disruption in the scc2-D730V mutant background is notable since scc2-D730V and eco1-W216G mutant strains both have similar levels of chromosome decondensation and disrupted nucleolar morphology [16]. These results suggest that the scc2-D730V defects are not sufficient to cause major changes in gene expression.
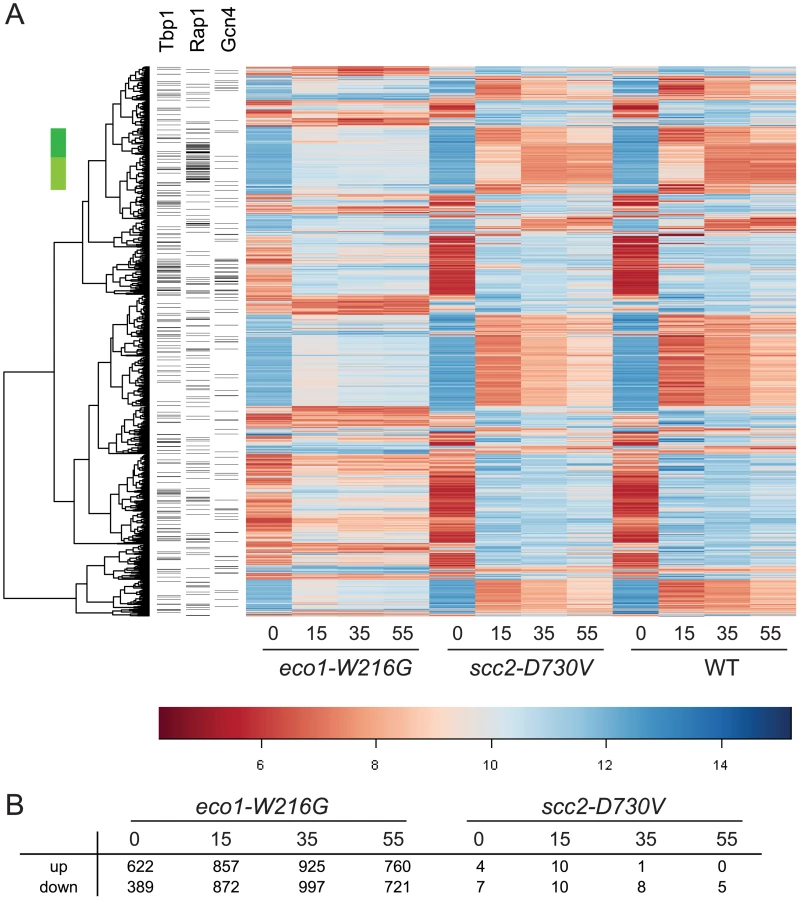
We have previously reported that the clustering of tDNA adjacent to the nucleolus is disrupted in both the scc2-D730V and eco1-W216G mutant strains [16]. This clustering has been associated with the silencing of genes adjacent to tDNAs, a phenomenon referred to as tDNA gene mediated silencing [21]. We analyzed whether expression of the genes adjacent to tDNAs were misregulated in the mutants relative to WT. We found no evidence that genes adjacent to tDNAs were differentially regulated in the mutants (Figure S1), suggesting that control of gene expression via tDNA clustering is not a wide-spread phenomenon, although there still may be individual cases of gene regulation via this mechanism. Our results are consistent with previous findings showing that mutations in RNA pol III, which disrupt tDNA clustering, do not disrupt the expression of neighboring genes [22].
We performed a GO analysis on the genes differentially expressed (both up and down) with an adjusted p value less than 0.005 at time 0 (639 genes) and 15 minutes (627 genes) in the eco1-W216G mutant as compared to WT [23]. At time 0 we found that many of the differentially expressed genes are involved in glutamate metabolic processes, TCA cycle, cell wall organization, and acetyl-CoA metabolism (Table S1). Glutamate and glutamine are donors of amino groups for the biosynthesis of nucleotides, amino acids, and other nitrogen containing compounds. When the gene expression profile in rich medium for the eco1-W216G strain was compared to a variety of stress response profiles [24], it most closely matched nitrogen starvation. At the 15 minute timepoint, the enriched GO terms are almost all related to ribosome biogenesis, including biogenesis of ribosomal proteins and processing of RNAs needed for ribosome assembly (Table S1).
The transcriptional activator Gcn4 is upregulated in cohesin mutants
The gene expression data was further analyzed to determine whether the genes that were misregulated in the eco1-W216G mutant had any enrichment for particular transcription factor binding sites in their promoter regions. In the promoters of genes that were upregulated at the time 0 timepoint, we found a significant enrichment for Gcn4 and Tbp1/Spt15 binding sites (Figure 2A). Gcn4 is a transcriptional activator that activates the expression of many classes of genes, including stress and amino acid biosynthesis genes. Tbp1/Spt15, or TATA binding protein, is an evolutionarily conserved general transcription factor that interacts with other factors to form transcription preinitiation complexes at promoters. SNO1 and SNZ1 have been reported to be upregulated in pol III mutants [22] in a Gcn4-dependent manner [25]. These genes were found to be upregulated in the microarray data. The misregulation of SNO1 and SNZ1 was confirmed by RT-qPCR (SNO1, 3-fold, SNZ1, 9-fold, eco1-W216G at time 0, Figure S2). In the promoters of genes that were downregulated at time 0 there were fewer than average Gcn4 and Tbp1/Spt15 binding sites.

In the promoters of genes that were differentially expressed at the 15 minute timepoint, there was a significant enrichment for Rap1 binding sites. One group of these genes spans two clusters (Figure 1A, green bar); most of these genes are involved in ribosome biogenesis (adj p<0.001). Rap1 (Repressor Activator Protein) regulates the transcription of many ribosomal protein genes [26]. When cells are starved for amino acids, they normally repress genes involved in ribosome biogenesis [24]. While these genes were repressed once amino acid starvation was initiated in all three strain backgrounds, the genes were more weakly repressed in the eco1-W216G background. The reason for this is currently unclear, but may be related to the baseline ribosome defect in this strain (see below).
Gcn4 is a transcriptional activator that is normally translated only when cells encounter stress or nutritional starvation [27]. Surprisingly, the enrichment for Gcn4 binding sites in the promoters of genes upregulated in the eco1-W216G mutant at time 0 suggested that Gcn4 was activating the transcription of its normal target genes in the eco1-W216G mutant when cultures were growing in rich medium, prior to amino acid starvation. Although many Gcn4 target genes were induced in the eco1-W216G mutant background under rich growth conditions, the mRNA corresponding to Gcn4 was unchanged in the mutants relative to WT (see microarray data GEO GSE27235). Gcn4 contains an unusual leader sequence with four short ORFs (uORFs). One level at which Gcn4 is regulated is translation; translation of the Gcn4 mRNA occurs when ribosomes become processive due to limiting pools of GTP. For this reason, Gcn4 has been used extensively as a reporter for ribosome function [27], [28].
We used a Gcn4-lacZ reporter (p180) to determine whether β-galactosidase levels were elevated in the cohesinopathy mutants in the W303a strain background. We found a 4-fold elevation in β-galactosidase activity in the eco1-W216G strain as compared to a WT strain (Figure 2B). The cohesinopathy mutant smc1-Q843Δ also showed elevated β-galactosidase activity in this assay, while the scc2-D730V showed a very mild elevation. We also analyzed the β-galactosidase levels in two additional eco1 alleles. We previously reported that eco1-H53Y, eco1-W216G, and eco1-ack represent an allelic series (strongest to weakest) with respect to both cohesion as measured by a 1 spot-2 spot assay, and DNA damage sensitivity [17]. Stronger cohesion defects and DNA damage sensitivity were correlated with higher levels of β-galactosidase activity. Defects in cohesion have been previously noted at 37°C for the eco1-W216G, smc1-Q843Δ, and scc2-D730V strains [16] and the degree of defect correlates with the β-galactosidase activity observed.
We previously showed that deletion of RAD61/WPL1 rescued the growth of the eco1-W216G mutant at 37°C but did not rescue the X-ray sensitivity [17]. While β-galactosidase levels in the eco1-W216G rad61 double mutant were lower than the eco1-W216G single mutant, they remained higher than WT, suggesting that some defect persists. Deletion of RAD61 has been shown to partially rescue the cohesion defect of an eco1-1 mutant [4].
We further tested whether the increase in β-galactosidase activity was dependent on the presence of uORF4 in the Gcn4 promoter using an additional reporter construct. p226 has only uORF4. The deletion of the first 3 uORFs results in very minimal translational control [29]. The elevation in β-galactosidase activity remained with uORF4 for eco1-W216G (Figure 2C), but the level was reduced compared to the p180 reporter, as expected if translational control is contributing to the elevation.
We also analyzed the β-galactosidase levels in the cohesinopathy mutants in the BY4742/S288C strain background, as well as scc2-4 and pds5-2 mutants (Figure 2D). All mutants except scc2-D730V showed elevated levels of β-galactosidase compared to a WT control. We conclude that mutations in many different cohesin associated genes and in two different strain backgrounds can give rise to elevated levels of β-galactosidase activity expressed from the Gcn4 promoter.
We measured Gcn4 protein levels directly by Western blotting. The eco1-W216G mutant strain has a higher level of Gcn4 than a wild-type strain when grown in rich medium (Figure 2E), consistent with the results from the reporter assay and the gene expression data.
Protein translation is impaired in the cohesinopathy strains
Given that high levels of Gcn4 can indicate a defect in protein translation, we tested whether protein translation was impaired in the cohesinopathy mutants. An evolutionarily conserved indicator of translational inhibition is the phosphorylation of elongation initiation factor 2α (eiF2α) [27], [30]. Phosphorylation of eiF2α inhibits the exchange of GDP for GTP in the ternary complex, blocking translation. We used Western blotting to measure the levels of total eif2α and the phosphorylated fraction. We found a 3-fold, 2.4-fold, and 1.9 fold increase in phosphorylated Eif2α in the eco1-W216G, smc1-Q843Δ, and scc2-D730V lysates, respectively (Figure 3A).

Since defects in translation could slow growth, we monitored growth in our cohesinopathy strains in rich medium (YPD+CSM) at 30°C. The eco1-W216G mutation confers a strong growth defect in the W303a background (p<0.0001). However, the growth of the scc2-D730V and smc1-Q843Δ mutant strains was not significantly different from WT (Figure 3B). Since mutations in cohesin or its regulators could cause chromosomal instability, we verified that our strains (Table S2) are not aneuploid (Figure S3).
Since growth can be affected by many different factors, we decided to analyze protein translation using more direct measures. To analyze ribosomes directly, we used sucrose gradients in combination with fractionation (Figure 3C). The ratio of polyribosomes to 80S indicates the active translating fraction. The 80S peak will consist of ribosomes without an associated mRNA or “vacant” ribosomes as well as some with an mRNA (monosomes). In theory, the polysome to 80S ratio becomes smaller with initiation defects, while it becomes larger with elongation defects [31]. The ratio of polysomes to 80S in WT, smc1-Q843Δ, and eco1-W216G, respectively, was 1.78, 1.23, and 0.89, consistent with a translation initiation defect in the mutants. The decrease in actively translating ribosomes could indicate a defect in protein synthesis.
In order to further measure protein translation, we used 35S-methionine incorporation to quantify protein synthesis. We found ∼50% reduction in incorporation in the eco1-W216G mutant and a ∼20% reduction in the smc1-Q843Δ mutant relative to the WT strain (Figure 3D). Collectively these results are consistent with the idea that the smc1-Q843Δ and eco1-W216G mutants support lower levels of protein synthesis.
The ribosome profiles suggested that initiation was limiting in the mutants. To test whether initiation was defective in the eco1-W216G strain, we transformed it with a plasmid carrying the ternary complex (eif2α, β, and γ, and tRNA-fMet) [32]. Overexpression of the ternary complex could reduce β-galactosidase levels expressed from the Gcn4 promoter if the high levels were due to poor translation initiation. We found that the plasmid reduced β-galactosidase levels in the eco1-W216G strain background (Figure 3E), consistent with a defect in the initiation of translation.
Ribosome biogenesis is impaired in the cohesinopathy strains
In order to further analyze the production of ribosomes in the scc2-D730V, smc1-Q843Δ and eco1-W216G mutants, we transformed them with plasmids that contain GFP reporters for the assembly of the 40S (Rps2-GFP) and 60S (Rpl25-GFP) components of the ribosome. In WT cells these proteins are mainly found evenly distributed in the cytoplasm. However, if there is an assembly and/or export defect, this is visualized as an accumulation of the GFP protein in the nucleus or nucleolus [33], [34]. We collected images of our mutants transformed with these reporters and we observed the accumulation of both reporter proteins in the smc1-Q843Δ and eco1-W216G mutants (Figure 4A and 4B). To further quantify this effect we developed a cytometric approach that allowed us to monitor at least 10,000 cells per sample. When the peak GFP fluorescence was measured, the smc1-Q843Δ and eco1-W216G mutants had higher mean fluorescence for both the 40S and 60S reporters, while the scc2-D730V mutant showed a mild phenotype for the 40S reporter but no increase in fluorescence for the 60S reporter (Figure 4C and 4D). To further analyze the data we generated the cumulative distribution function for each sample (not shown), and then we calculated the distance between biological replicates and between mutant and WT using a KS test (see Materials and methods). These distances are depicted as a box plot with an associated p value to indicate whether the distance from WT is statistically significant (Figure 4E and 4F). In summary, both the 40S and 60S subunits of the ribosome exhibit assembly/export defects in both the smc1-Q843Δ and eco1-W216G mutants, with a more severe defect observed in the eco1-W216G mutant.

Ribosomal RNA production is reduced in cohesin mutants
We noticed from the microarray data that RNA polymerase I dependent ribosomal RNA (35S transcript) was downregulated approximately 4-fold in the eco1-W216G mutant (median p value 0.01, median adjusted p = 0.07) and 2-fold in the scc2-D730V mutant (median p value 0.12, median adjusted p = 0.40) in rich medium (Figure 5A). We note that transcripts corresponding to RNA polymerase I subunits appear to be unaffected in the transcription profile of the eco1-W216G mutant, suggesting downregulation of RNA Polymerase I is not causing the reduction in the 35S transcript. Notably, ribosomal RNA has been shown to be a limiting factor for ribosome assembly [20]. Since ribosomal protein genes showed no significant differences in transcription between the eco1-W216G mutant and WT in rich medium (Figure 1), we speculated that the ribosome defect was not due to a lack of proteins needed to make ribosomes, but possibly due to the low levels of rRNA.

Because rRNA constitutes ∼60% of the RNA being made by actively growing cells, 3H-uridine incorporation is commonly used to measure total rRNA synthesis. To further test the new production of rRNA, we pulsed with 3H-uridine and measured incorporation into RNA. In the eco1-W216G and smc1-Q843Δ mutants, there is less incorporation in 5 minutes in an equal number of cells (Figure 5B), indicating that these mutants produce less rRNA in this time frame. These experiments were carried out in the BY4742 background and the growth in SD-ura at 30°C was measured (Figure 5C). In log phase, which is when the labeling is performed, only the eco1-W216G mutant showed slower growth. We carried out a similar labeling experiment with the eco1-W216G mutant in the W303a background and obtained similar levels of incorporation (Figure S4A). In this background, growth is much more severely affected (Figure S4B). Thus, while the eco1-W216G mutation confers different growth defects in different strain backgrounds, the effect on total rRNA production appears to be similar, suggesting growth may not perfectly correlate with rRNA production.
RNA polymerase I produces the 35S transcript that is then processed into the 25S, 18S, and 5.8S transcripts and further modified by methylation and pseudouridylation. To measure the production of methylated rRNA, we used incorporation of 3H-methyl-methionine. Total RNA was isolated from equal numbers of cells following a 5 minute pulse labeling and a chase with cold methionine. Equal amounts of RNA were electrophoresed on a formaldehyde agarose gel and visualized with ethidium bromide (Figure 5D). Following exposure to film, the bands were excised and radioactivity was measured. We found that the eco1-W216G mutant produced 8–10% of WT levels of the methylated 25S and 18S transcripts and the smc1-Q843Δ mutant produced 18–28% of WT levels (Figure 5D). The growth curves for the mutants in SD-met at 30°C are shown (Figure 5E). Thus, while new production of total rRNA appears to be reduced approximately 2-fold in both mutants, the methylated form of the 25S and 18S transcripts is produced at a 10-fold lower level in the eco1-W216G mutant as compared to a 4-fold lower level in the smc1-Q843Δ mutant. The difference in production of total rRNA versus the processed and modified forms suggests that both initial production and subsequent processing are defective in the mutants, with a more severe defect in the eco1-W216G mutant. The fact that both 25S (60S rRNA component) and 18S (40S rRNA component) transcripts are affected in both mutants is consistent with the result that both 40S and 60S biogenesis are affected in both mutants.
A W303a strain bearing the eco1-W216G mutation does not grow at 33°C and cohesion is compromised at 37°C. Cohesion defects have been correlated with growth defects, and so it might be assumed that errors in chromosome segregation cause the lethality associated with mutations in cohesin. However, the scc2-D730V and smc1-Q843Δ strains have cohesion defects at 37°C, but can grow [16] (Figure S4C), suggesting precocious sister separation does not necessarily cause lethality. We tested whether transcription by RNA polymerase II of the 35S transcript from a galactose-inducible promoter would rescue growth of the eco1-W216G strain at 33°C. This plasmid allowed a partial rescue of the growth defect, suggesting some portion of the defect may be due to limiting levels of rRNA (Figure 5F). We further tested how much the rRNA levels increase in galactose medium and we found that the increase was a modest 40–50% (Figure 5G). However, this increase is similar in degree to the decrease in labeling observed with 3H-uridine, suggesting this increase should be sufficient to make up the difference. To explain the partial rescue we point out that 1) the morphology of the nucleolus is disrupted in the mutant, so even with more rRNA ribosome biogenesis may still be impaired, 2) the endogenous rDNA locus may still have defects associated with it, for instance, if there is difficulty with its replication, this defect will not be corrected by providing more rRNA and 3) at the elevated temperature there may be so little Eco1 function that other chromosomal processes such as chromosome segregation have become severely affected. A high copy plasmid with the 35S transcript produced from the normal promoter provides no rescue (data not shown). Overall our results suggest that some mutations in cohesin are associated with defects in 25S and 18S production.
The eco1-W216G and smc1-Q843Δ mutations are associated with fewer transcripts from a single repeat
Many different cohesin mutations confer elevation in β-galactosidase levels from the Gcn4 leader sequence, suggesting the elevation is related to defects in chromosome cohesion. However, mutations in the cohesin network have been shown to affect both chromosome condensation [35] and DNA damage repair [36]. Both the eco1-W216G and scc2-D730V mutations confer defects in chromosome condensation and nucleolar morphology, but importantly, the smc1-Q843Δ strain does not share these defects [16]. This suggests that aberrant chromosome condensation and nucleolar morphology are not the primary cause of the reduction in rDNA transcription.
However, since condensation can affect segregation of the rDNA we decided to further examine whether the cohesinopathy mutations disrupted rDNA segregation. At the metaphase to anaphase transition, chromosomes segregate, followed by segregation of the rDNA. The segregation of the rDNA is dependent on condensin and decatenation [37]. Since the rDNA is silenced during anaphase [38], a longer anaphase could potentially account for a reduction in transcription. To measure rDNA segregation, we used yeast strains tagged with Net1-GFP (rDNA marker) and Spc42-mCherry (spindle pole body marker). The duration of rDNA separation can be calculated by the timing of the start of spindle elongation (sudden increase in the distance between the two SPBs) to fully separated Net1-GFP. In wild-type cells, rDNA separation takes an average of 6.5 minutes. We found no significant difference in the kinetics of rDNA segregation in any of the mutants (Figure 6A). Thus, delayed rDNA segregation during anaphase cannot account for the slow growth or the transcriptional defects at the rDNA.

The number of rDNA repeats can expand and contract, controlled by recombination. We considered the possibility that contraction of the rDNA was limiting transcription. We monitored the copy number of the rDNA using qPCR. To demonstrate that our assay can detect differences in copy number, we used strains containing 20, 40, 80, and 110 copies of rDNA, as estimated by pulsed field gel electrophoresis [39]. We found that the number of rDNA repeats was not significantly different from WT in the scc2-D730V and eco1-W216G mutants in either the BY4742 or W303 backgrounds. Copy number was also examined in a smc1-Q843Δ strain and found to be normal (data not shown). This result suggests reduced copy number cannot account for reduced transcription (Figure 6B).
The rDNA is especially susceptible to genotoxic stress. It is estimated that the rDNA incurs several DSBs per S phase which result in an average of 3.6 Holliday junctions [40]. Cohesin is known to bind to the rDNA [41], [42] and the eco1-W216G mutation decreases cohesin binding at the rDNA as measured by ChIP approximately 2-fold [16]. Since cohesion is important for the resolution of DNA damage, we hypothesized that the decrease in transcription at the rDNA in some cohesin mutants might be related to an inability to efficiently resolve recombination intermediates due to defective damage induced cohesion. We used Southern blot analysis to measure whether DSBs accumulate at the rDNA. The level of DSBs in cohesin mutants and a WT strain was similar, indicating unresolved DSBs do not accumulate at the rDNA in the cohesin mutant strains (Figure 6C). Thus, failure to repair the locus cannot account for the transcriptional defect.
A normal yeast cell contains 100–150 copies of the 9.1 kb rDNA repeat, about half of which are actively transcribed and half are inactive. The cell can regulate the number of active repeats and the rate of transcription since in a 20 or 40 copy strain, all the repeats are active and the rRNA is present at normal levels [39]. rDNA repeats can be differentiated by their different chromatin structures and accessibility to cross-linking by psoralen followed by Southern blot [43]. Inactive or closed gene copies contain nucleosomes and are therefore less accessible to psoralen, and migrate faster on a gel following crosslinking whereas active or open gene copies are devoid of nucleosomes and are more accessible to psoralen, and migrate slower following crosslinking [43]. To verify the method, we used a strain with 40 copies and found few closed repeats, as previously reported (data not shown) [39]. We examined whether the mutations in cohesin were affecting the fraction of open repeats. We found no reproducible change in open repeats in the cohesin mutants relative to a WT control strain, at least in asynchronous culture (Figure 6D). Thus, a steady state increase in closed repeats does not appear to account for the decrease in transcription.
Given that the copy number and fraction of open rDNA repeats do not seem to be affected in the cohesin mutants, we sought to further understand the reduction in rRNA we observed by microarray and metabolic labeling. We used a FISH assay in which a unique sequence is inserted into the 5′ end of one 35S gene (Figure 6E) [44]. The transcription of this sequence can be monitored with a fluorescent probe in individual cells to indicate the dynamics of transcription in the population. We integrated three different cohesinopathy mutations into this strain and monitored transcription. We found that transcripts made from this single repeat were present at significantly lower levels in the smc1-Q843Δ and eco1-W216G strains, but not in the scc2-D730V strain. Thus, when a single repeat is monitored, less rRNA is made from this repeat.
Lower production of rRNAs could potentially be explained by 1) reduced copy number, 2) fewer transcriptionally active repeats, or 3) reduced RNA production from active repeats. Collectively our data suggests that mutations in ECO1 and SMC1 can be associated with production of fewer transcripts from the open fraction of rDNA repeats. Interestingly, the smc1-Q843Δ and eco1-W216G mutations were associated with a ∼2-fold reduction in rRNA using either the 3H-uridine labeling method to detect total rRNA or FISH to detect a single repeat. However, the eco1-W216G mutant showed a 10-fold reduction in the production of the methylated rRNA while the smc1-Q843Δ mutant showed a 4-fold reduction. This difference correlates well with the degree of defect in protein synthesis and ribosome biogenesis. We speculate that due to the disruption in nucleolar morphology in the eco1-W216G mutant [16] that processing and modification of the 35S transcript as well as ribosome assembly and export might be more severely affected than in the smc1-Q843Δ mutant, with the outcome that translation and growth are more affected.
We have previously measured cohesion at three loci in the eco1-W216G mutant. We observed a 15% reduction at an arm locus, a 9% reduction at a telomere locus, and an 8% reduction at a pericentric locus relative to a WT strain, and no defect in chromosome transmission [16], [17]. However, when we measured cohesion using strains with lacO repeats integrated adjacent the rDNA [39], cohesion is reduced ∼25% in the eco1-W216G background in the 50 copy strain (Figure 6F). Thus, Eco1 acetyltransferase activity is differentially required for genomic and ribosomal DNA cohesion. The mechanism for this is currently unclear and will require more investigation. However, we speculate that the decrease in cohesion at the rDNA is related to the reduced transcription at this locus.
Furthermore, the specific defect in cohesion at a heterochromatic region is reminiscent of the heterochromatic repulsion observed in cells from Roberts syndrome patients [45].
Human Roberts syndrome fibroblasts display similar physiology to yeast
Given that the eco1-W216G mutation is associated with reduced protein translation and rRNA production in budding yeast, we decided to investigate whether a human cell line bearing the same mutation displays similar physiology. We used 35S-methionine labeling to measure protein synthesis in 1) a Roberts syndrome fibroblast cell line, 2) a version of the cell line that has been corrected with a wild-type copy of ESCO2 [45] and 3) a normal fibroblast line. We found that the Roberts syndrome cells incorporated methionine at about 50% the level as the corrected line or a normal fibroblast line (Figure 7A), very similar to the observations in yeast (Figure 3D). Furthermore, we measured the incorporation of 3H-uridine as an indicator of ribosomal RNA synthesis. We found that the rate of incorporation in the Roberts syndrome cells is about 50% the level as the corrected line or a normal fibroblast line (Figure 7B), very similar to the observation in yeast (Figure 5B). Finally, we examined the polysome profile in the Roberts cells. We find that the polysome to 80S ratio is lower in the Roberts cells relative to the corrected line (Figure 7C), similar to the observation in yeast (Figure 3C). Thus, it appears that protein synthesis and ribosomal RNA production are reduced in human Roberts syndrome fibroblasts, and suggests that the findings in yeast are relevant to the human disease.

Discussion
Several groups working in fish, flies, mouse, and humans have shown that cohesin associated mutations or reductions in cohesin associated genes result in hundreds of small alterations in gene expression, and a few cases of big changes in gene expression [46], [47], [48], [49]. These changes in gene expression are thought to cause the human cohesinopathies. However, the mechanism by which mutations in cohesin-associated genes alter gene expression has been elusive. Cohesin together with CTCF [50] or mediator [51] may facilitate gene looping and communication between promoters and enhancers [52] to influence transcription by RNA polymerase II. Cohesin may also act directly at certain loci in an activating manner [47], [49] or a repressive manner [53] to regulate transcription by RNA polymerase II . In this report, we suggest a key locus at which cohesin proteins may influence transcription is the ribosomal DNA. Misregulation at this locus can affect the transcription of hundreds of genes as translation is affected.
The elevation in Gcn4 targets suggested that this transcriptional activator was induced in the eco1-W216G strain, and further suggested that translation would be repressed. Analysis using a Gcn4-lacZ transgene revealed that mutations in Pds5, Scc2, Eco1, and Smc1 all showed an increase in expression, consistent with the idea that cohesion defects correlate with reduced protein translation. Also consistent with our findings, inactivation of Mcd1/Rad21 in budding yeast in G1 was shown to affect the expression of 29 genes, including genes involved in rRNA maturation and ribosome biogenesis [54]. The differential effect of the eco1-W216G mutation on cohesion at the rDNA is notable since the rDNA in budding yeast has many properties of heterochromatin and lack of cohesion specifically in heterochromatic regions, including the rDNA, is a hallmark of Roberts syndrome. Thus, a cohesion deficit at the rDNA is common to both our yeast model and Roberts syndrome cells. The local cohesion defect at the rDNA in the eco1-W216G mutant is associated with the production of fewer 35S RNA products and reduced translation. A human Roberts fibroblast line displays similar physiology to our yeast mutant in that both protein synthesis and ribosomal RNA production are impaired, suggesting yeast may provide a good model for these particular defects.
We have characterized three different cohesinopathy mutants in yeast which have overlapping sets of defects. The mutation with the strongest phenotype is eco1-W216G, which confers defects in nucleolar morphology, DNA damage response, growth, condensation, gene expression, ribosome biogenesis and rRNA production (this work, [16], [17]). The smc1-Q843Δ mutant shares the defects in ribosome biogenesis and rRNA production, albeit less severe, and without much effect on growth. If one extrapolates to multicellular organisms, one can imagine that the developmental outcomes for the RBS allele could be more severe compared to the SMC1 CdLS allele. This proposal is consistent with observations made in zebrafish in which ESCO2 and RAD21 depletion were compared and ESCO2 depletion was uniquely associated with poor cell proliferation and cell death [55]. The scc2-D730V allele does not have the same effect on protein synthesis as the SMC1 and ECO1 mutations, instead exhibiting defects in nucleolar morphology and chromosome condensation. These defects could potentially be explained by the requirement for Scc2 for condensin loading [19]. The scc2-D730V mutation in the W303a background does show weak elevation in β-galactosidase activity from the Gcn4 leader sequence and eif2α-phosphorylation, and a weak 40S biogenesis defect. The scc2-D730V mutation may affect some aspect of chromosome biology that we do not currently understand or cannot be fully evaluated in budding yeast. We note that the scc2-4 mutation causes more severe defects in yeast (Figure 2 and data not shown). A future challenge will be to achieve a molecular understanding of how different mutations in different proteins can lead to similar disease outcomes.
In RBS both copies of ESCO2 have lost function, but CdLS is most often caused by a single mutant copy of SCC2/NIPBL. However, SMC1 is on the X chromosome in humans and the cases of CdLS associated with the smc1-Q843Δ allele have been in males with a sole mutant copy [10]. Thus, both our haploid yeast and human patients express only mutant copies of ECO1/ESCO2 or SMC1. In contrast, the evaluation of the scc2-D730V allele in haploid yeast does not genocopy the human disease since there would be an additional WT copy of SCC2/NIPBL present. It may be important to model haploinsufficiency to understand how the SCC2/NIPBL mutations cause disease.
Since defects in protein translation affect cell growth and division, protein translation can affect size. The reports of small size in a mouse model [48] and human CdLS patients [14] are consistent with our hypothesis that mutations in cohesin can generate a deficit in ribosome function. In a report on gene expression in Drosophila cells depleted for Nipped-B or Rad21 (CdLS model), nearly all ribosomal protein and aminoacyl-tRNA synthetase transcripts are reduced [46]. In addition, expression of Myc, p53 and Mdm2 are altered by depletion of Rad21 and Nipped-B in humans [47], mouse [48], flies and zebrafish [49]. These targets are known to be regulated by ribosome biogenesis [56]. When these data are taken in context of our current report, they collectively suggest that reduced translational capacity may contribute to the developmental defects associated with the cohesinopathies.
How do cohesin proteins facilitate rRNA production? Our data suggests that transcription from a given repeat is reduced in the eco1-W216G strain, rather than there being fewer open repeats or reduced copy number. One mechanism by which we can imagine cohesin contributing to transcription is through gene looping, which might facilitate reloading of RNA Polymerase I from the 3′ to 5′ end of the 35S transcript. Loops at the rDNA have been reported [57] and cohesin binds flanking each repeat in a pattern that would enable looping [41]. Other possibilities include cohesin promoting some other aspect of rDNA metabolism such as replication fork speed [58] or nucleolar organization that in turn facilitates the production of the rRNA transcripts. In any case, the lower levels of ribosomal RNA as measured in both yeast and human are associated with decreased protein synthesis. In future work it will be important to further examine the mechanism by which cohesin proteins promote production of rRNA. Coupling protein synthesis capacity to chromosome metabolism might provide the cell with a useful feedback loop for regulating proliferation.
Materials and Methods
β-galactosidase assays
Wild type and cohesin mutant strains transformed with the plasmids p180 (pGCN4 URA3 lacZ CEN) having all four μORFs or p226 (with only the fourth μORF) [29] were grown at 30°C to an A600 of ∼0.8 under repressive conditions (overnight growth in SD-ura then shifted to YPD+CSM till desired absorbance is reached). The cells were pelleted and protein extracts were made. β-galactosidase activity was measured following standardized protocols using ONPG (o-nitrophenyl-β-D-galactopyranoside) as the substrate. We note that the level of β-galactosidase activity is very sensitive to the growth protocol used [27].
Microarray methods
Concentration and quality of RNA were determined by spectrophotometer and Agilent bioanalyzer analysis (Agilent Technologies, Inc., Palo Alto, CA). For array analysis, labeled mRNA was prepared from 300 ng of total RNA using the MessageAmp III RNA Amplification kit (Applied Biosystems/Ambion, Austin, TX) according to the manufacturer's specifications. Array analysis was performed using Affymetrix GeneChip Yeast Genome 2.0 Arrays processed with the GeneChip Fluidics Station 450 and scanned with a GeneChip Scanner 3000 7G using standard protocols. Resulting CEL files were analyzed using RMA [59] and limma [60] in the R statistical environment. Affymetrix GeneChip data are available at GEO under accession number GSE27235.
Motif identification and analysis for Gcn4, Tbp1, and Rap1 were based on presence or absence calls for each binding site within the region of the annotated gene start site and 400 bp upstream. Presence of the Gcn4 motif was determined by a match to the sequence TGA(C/G)TC(T/A). The Tbp1 and Rap1 matches were determined using the TRANSFAC [61] matrices F$TBP_Q6 and F$Rap1_C and the MATCH program [62]. The score cut-off profiles for Tbp1 and Rap1 were minFP and minFN, respectively, using TRANSFAC version 2009.3. Sequences, microarray probe mapping, and gene annotations were from Ensembl 56. P-values for the gene set indicated were determined using the hypergeometric test of all protein-coding genes.
Polysome analysis
100 ml of yeast culture was grown to an OD600 of 0.8 and treated with 100 µg/ml of cycloheximide for 10 mins on ice before centrifugation. After centrifugation the cell pellets were washed twice and resuspended in lysis buffer (10 mM Tris-HCl pH 7.5, 100 mM NaCl, 30 mM MgCl2, 100 ug/ml cycloheximide, 0.2 mg/ml heparin in DEPC). The cells were lysed in the cold by bead beating and the lysate (10 OD units) was loaded on top of an 11 ml 7–47% sucrose gradient in 15 mM Tris-Cl pH 7.4, 140 mM NH4Cl and 7.8 mM MgOAc-4H2O centrifuged at 36,000 rpm for 3 h. The gradients were fractionated and OD254 was monitored using an ISCO UV-6 monitor [63].
Polysome quantitation
Polysome quantitation was done by both Image J and Mathematica, version 7.0. A common baseline was chosen and the area under the peaks was calculated with the Image J software. For Mathematica, TIFF images from the instrument were read using the Import function. The signal intensity was isolated from the image using the ImageSubtract function of the non-signal colors. The signal plot was scaled and shifted along the y-axis to position the baseline (x = 0) at the lowest signal level of the plot. Boundary regions were selected manually by zooming onto the image and recording the x-axis coordinate of extremal point. The total area between the selected boundaries and above the baseline was calculated using the Take and Total functions.
Growth curves
Growth curves were collected in triplicate for each genotype at time intervals of 15 min. We used a Tecan Infinite M200 Pro machine. Due to the non-linearity between optical density (OD) and cell number at higher cell densities, the measured Tecan ODs were converted to ‘real’ ODs using the calibration function ‘real OD’ = −1.0543+12.2716×measured OD [64]. The maximum slope was determined for each curve from 12 consecutive points and the statistical significance between slopes was calculated using a t test.
GFP measurements using cytometry
We used flow cytometry to quantify the peak GFP fluorescence in WT and mutant cells. By measuring the digitized pulse height from the B1 detector (525/50 emission), the maximum GFP intensity of each cell could be ascertained. For each sample approximately 10,000 cells were measured. WT and mutant strains that did not bear the GFP-plasmid showed no significant difference in their maximum fluorescence intensities, indicating they have similar levels of intrinsic fluorescence (autofluorescence); however, some mutant strains expressing a GFP tagged ribosomal subunit had on average a higher maximum GFP intensity than WT cells expressing the same fluorescent tag. Since the distribution of fluorescence intensity among GFP positive cells was non-Gaussian, we used the Kolmogorov-Smirnov (KS) statistic to characterize the distribution differences, which quantifies the distance between empirical cumulative distribution functions of two samples. Using this statistic, we can calculate distances between biological replicates (same genotype) and distances between samples with different genotypes. In this way we can determine whether the average KS-distance between the WT and mutant samples is significantly greater than between replicates (same genotype) using a t test.
Western blotting
Overnight cultures of yeast cells were diluted in YPD+CSM to an OD of 0.1 and grown to an OD of approximately 0.8. Cells were then pelleted by centrifugation and washed in PBS. Cells were lysed with glass beads in buffer containing 10 mM Tris, pH 7.4, 100 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM NaF, 20 mM Na4P2O7, 2 mM Na3VO4, 0.1% SDS, 0.5% sodium deoxycholate, 1% Triton-X 100, 10% glycerol, and 1 mM PMSF protease inhibitor cocktail (Sigma). Equal amounts of protein were loaded for each sample after quantification by Bradford assay. Protein samples were electrophoresed on a 12% SDS-polyacrylamide and transferred to nitrocellulose filters. The immunoblots were probed for phospho-specific eIF2α (Ser-51) (Cell Signalling #9721). Total eIF2α was measured with rabbit polyclonal antibody (a gift from T. Dever). eIF2α was visualized by HRP-conjugated anti-rabbit IgG.
Metabolic labeling—RNA
Methods for RNA labeling were derived from a previous report [65]. For the experiment in Figure 5B, triplicate cultures of BY4742, eco1-W216G and smc1-Q843Δ carrying pRS316 were grown in SD-Ura medium to exponential phase (OD600∼0.3). 3H-uridine (5 µCi) was mixed with 500 µL of each culture and incubated at 30°C for 5 min with aeration. Then samples were treated with 2.5 mL of 10% trichloroacetic acid (TCA) with 2.5 mg/ml of uridine. After filtration through a 25 mm glass filter, each membrane was washed with 5% TCA, dried, and counted in a Beckman LS 6500 multipurpose scintillation counter. For labeling with 3H-methylmethionine (Figure 5D), we grew cells in SD-met medium and pulse-labeled for 5 min with 20 µCi/mL 3H-methylmethionine followed by a 5 min chase with cold methionine. RNA was prepared from 1×10∧7 cells. 8 µl of each sample was run on a 1.2% formaldehyde agarose gel, transferred to a Gene Screen membrane, and detected by autoradiography. Individual RNA species (25S and 18S) were excised from the blot (together with nearby regions of the blot for assessment of background) and quantified with a scintillation counter.
Metabolic labeling—protein
Yeast strains were grown to mid-log phase (OD600∼0.5) at 30°C in medium containing dextrose (YPD+CSM). Cells were harvested and washed in PBS and resuspended in a similar volume of prewarmed methionine-minus medium containing dextrose (SD-met). Aliquots were taken from this culture (0.75 ml) for the zero time point. The medium was supplemented with 27.5 µCi of 35S-methionine and unlabeled methionine 1 mg/ml. At 15–20 mins intervals (0.75 ml) samples were withdrawn from an actively growing culture. The amount of 35S-methionine incorporated into proteins was then measured by an adaptation of the method of Kang and Hershey [66]. The cells were lysed in 1.8 N NaOH containing 0.2 M β-mercaptoethanol. Proteins were precipitated by the addition of hot 10% trichloroacetic acid. After centrifugation, the precipitate was washed twice in acetone. The precipitate was dissolved in 100 µl of 1% sodium dodecyl sulfate and heated at 95°C for 10 min. An aliquot of the SDS extract was counted in Ecoscint for 35S radioactivity in a liquid scintillation spectrometer to determine the amount of 35S-methionine incorporated into proteins.
Psoralen cross-linking
Psoralen cross-linking experiments were carried out as previously described [67] with the following modifications: 1.3% Tris-Taurine-EDTA (TTE) gels were run at 80 volts for 20 hours in 0.5× TTE, processed and transferred to Gene Screen membrane in 6× SSC. Hybridization with a 35S specific probe was carried out at 60°C and the membrane was exposed for 2 hours to a phosphorimager screen (GE/Amersham).
qPCR for aneuploidy
Genomic DNA was isolated from strains and used as a template for qPCR. For each chromosome arm, one locus, usually near the centromere, was monitored according to the method previously described [68].
FISH
Yeast cells were grown in CSM-URA at 30°C to an OD600 of 0.4. The cells were then fixed by adding formaldehyde to a final concentration of 4% (v/v) for 45 min at room temperature with shaking. After three washes with wash buffer (1.2 M sorbitol, 0.1 M potassium phosphate, pH 7.5), the cell wall was digested with 0.3 mg/ml zymolase in spheroplast buffer (1.2 M sorbitol, 0.1 M potassium phosphate, 10 mM vanadyl ribonucleoside complex, 0.06 mg/ml PMSF, 28 mM β-mercaptoethanol) at 37°C for 45 min. After digestion, the cells were washed three times with FISH wash buffer (30% formamide, 2×SCC). The cells were then hybridized in 30 µl hybridization solution containing 5 ng/µl DNA probe in 25% (v/v) formamide, 2×SCC, 1 mg/ml BSA (nuclease free), 10 mM vanadyl ribonucleoside complex, 0.5 mg/ml salmon sperm DNA and 0.1 g/ml dextran sulfate overnight at room temperature. Before imaging, cells were washed twice with FISH wash buffer for 30 min and then added to slides pre-coated with poly-L lysine.
The probe used for the FISH experiment is a synthesized DNA oligonucleotide modified from the previous publication [44]. The sequence of the oligonucleotide is 5′-CGGCRGGTAAGGGRTTCCATARAAACTCCTRAGGCCACGA-3′; the ‘R’s indicates an amino-dT replacing a regular dT where a fluorescein molecule was coupled. The probe was further purified by polyacrylamide gel purification to ensure that each amino-dT was coupled with a fluorescein molecule.
For counting RNA levels, it was first necessary to derive a calibration plot that relates intensity observed to RNA levels. This is required due to a wide range of RNA levels observed between different cells. In cells with more than ∼20 RNA, RNA spots overlap, making it impossible to distinguish individual RNAs. Extreme examples of this occur when cells have undergone recent ‘bursts’ in transcription (see Figure 6).
The general method previously developed was followed [44]. To generate a calibration plot, we acquired long-exposure z-stacks of RNA using the widefield module of a Zeiss-200 m that was also equipped with a Yokagawa CSU-10 spinning disc. For cells with few (generally less than 15) RNAs, it was possible to use the long-exposure images to count single RNAs. After counting RNAs in these cells, we switched to the confocal set-up and acquired a confocal z-stack as described below. This iteration allowed for the generation of a calibration plot that related overall intensity of the sum projection of the confocal z-stack to the number of counted RNA per cell. We obtained a linear plot, with an intercept at ∼0, demonstrating that RNA per cell is linearly proportional to total RNA, and thus total intensity per cell from the confocal data can be used to measure total RNA per cell even in cells where density is too high to distinguish single RNAs.
To acquire RNA per cell for groups of where the range is between 0 and ∼300, it was necessary to develop a system where it was possible to obtain fluorescence from cells with few (1 to 10) RNA, but yet not saturate the camera with cells possessing up to hundreds of RNA. We acquired 30 z-slices with spacing 0.3 microns. A background was subtracted for each slice, and then a sum projection was applied. Total intensity per cell was compared to the linear, extrapolated calibration plot to generate RNA per cell. A sum-projection of a non-background subtracted z-series was able to detect the location of cells where RNA levels were very low, eliminating the risk of missing low RNA-possessing cells with the background-subtracted analysis. We note that this method generated a distribution of RNA per cell that matched very closely the published result for the same strain [44].
Emission from the confocal z-slices was collected through a 500–550 nm bp filter onto a Hamamatsu C9100-13 EMCCD. A 488 nm laser line was used to excite the flourescein tagged FISH probe.
Supporting Information
Zdroje
1. CioskRShirayamaMShevchenkoATanakaTTothA 2000 Cohesin's binding to chromosomes depends on a separate complex consisting of Scc2 and Scc4 proteins. Mol Cell 5 243 254
2. GillespiePJHiranoT 2004 Scc2 couples replication licensing to sister chromatid cohesion in Xenopus egg extracts. Curr Biol 14 1598 1603
3. TakahashiTSYiuPChouMFGygiSWalterJC 2004 Recruitment of Xenopus Scc2 and cohesin to chromatin requires the pre-replication complex. Nat Cell Biol 6 991 996
4. Ben-ShaharTRHeegerSLehaneCEastPFlynnH 2008 Eco1-dependent cohesin acetylation during establishment of sister chromatid cohesion. Science 321 563 566
5. ZhangJShiXLiYKimBJJiaJ 2008 Acetylation of Smc3 by Eco1 is required for S phase sister chromatid cohesion in both human and yeast. Mol Cell 31 143 151
6. RowlandBDRoigMBNishinoTKurzeAUluocakP 2009 Building sister chromatid cohesion: smc3 acetylation counteracts an antiestablishment activity. Mol Cell 33 763 774
7. HartmanTSteadKKoshlandDGuacciV 2000 Pds5p is an essential chromosomal protein required for both sister chromatid cohesion and condensation in Saccharomyces cerevisiae. J Cell Biol 151 613 626
8. PanizzaSTanakaTHochwagenAEisenhaberFNasmythK 2000 Pds5 cooperates with cohesin in maintaining sister chromatid cohesion. Curr Biol 10 1557 1564
9. DeardorffMAKaurMYaegerDRampuriaAKorolevS 2007 Mutations in cohesin complex members SMC3 and SMC1A cause a mild variant of cornelia de Lange syndrome with predominant mental retardation. Am J Hum Genet 80 485 494
10. MusioASelicorniAFocarelliMLGervasiniCMilaniD 2006 X-linked Cornelia de Lange syndrome owing to SMC1L1 mutations. Nat Genet 38 528 530
11. KrantzIDMcCallumJDeScipioCKaurMGillisLA 2004 Cornelia de Lange syndrome is caused by mutations in NIPBL, the human homolog of Drosophila melanogaster Nipped-B. Nat Genet 36 631 635
12. TonkinETWangTJLisgoSBamshadMJStrachanT 2004 NIPBL, encoding a homolog of fungal Scc2-type sister chromatid cohesion proteins and fly Nipped-B, is mutated in Cornelia de Lange syndrome. Nat Genet 36 636 641
13. VegaHWaisfiszQGordilloMSakaiNYanagiharaI 2005 Roberts syndrome is caused by mutations in ESCO2, a human homolog of yeast ECO1 that is essential for the establishment of sister chromatid cohesion. Nat Genet 37 468 470
14. LiuJKrantzID 2008 Cohesin and human disease. Annu Rev Genomics Hum Genet 9 303 320
15. SchuleBOviedoAJohnstonKPaiSFranckeU 2005 Inactivating mutations in ESCO2 cause SC phocomelia and Roberts syndrome: no phenotype-genotype correlation. Am J Hum Genet 77 1117 1128
16. GardSLightWXiongBBoseTMcNairnAJ 2009 Cohesinopathy mutations disrupt the subnuclear organization of chromatin. Journal of Cell Biology 187 455 462
17. LuSGoeringMGardSXiongBMcNairnAJ 2010 Eco1 is important for DNA damage repair in S. cerevisiae. Cell Cycle 9
18. XiongBLuSGertonJL 2010 Hos1 is a lysine deacetylase for the Smc3 subunit of cohesin. Curr Biol 20 1660 1665
19. D'AmbrosioCSchmidtCKKatouYKellyGItohT 2008 Identification of cis-acting sites for condensin loading onto budding yeast chromosomes. Genes Dev 22 2215 2227
20. LaferteAFavryESentenacARivaMCarlesC 2006 The transcriptional activity of RNA polymerase I is a key determinant for the level of all ribosome components. Genes Dev 20 2030 2040
21. WangLHaeuslerRAGoodPDThompsonMNagarS 2005 Silencing near tRNA genes requires nucleolar localization. J Biol Chem 280 8637 8639
22. ConesaCRuotoloRSoularuePSimmsTADonzeD 2005 Modulation of yeast genome expression in response to defective RNA polymerase III-dependent transcription. Mol Cell Biol 25 8631 8642
23. BerrizGFBeaverJECenikCTasanMRothFP 2009 Next generation software for functional trend analysis. Bioinformatics 25 3043 3044
24. GaschAPSpellmanPTKaoCMCarmel-HarelOEisenMB 2000 Genomic expression programs in the response of yeast cells to environmental changes. Mol Biol Cell 11 4241 4257
25. NatarajanKMeyerMRJacksonBMSladeDRobertsC 2001 Transcriptional profiling shows that Gcn4p is a master regulator of gene expression during amino acid starvation in yeast. Mol Cell Biol 21 4347 4368
26. LiebJDLiuXBotsteinDBrownPO 2001 Promoter-specific binding of Rap1 revealed by genome-wide maps of protein-DNA association. Nat Genet 28 327 334
27. DeverTE 1997 Using GCN4 as a reporter of eIF2 alpha phosphorylation and translational regulation in yeast. Methods 11 403 417
28. HinnebuschAG 2005 Translational regulation of GCN4 and the general amino acid control of yeast. Annu Rev Microbiol 59 407 450
29. MuellerPPHarashimaSHinnebuschAG 1987 A segment of GCN4 mRNA containing the upstream AUG codons confers translational control upon a heterologous yeast transcript. Proc Natl Acad Sci U S A 84 2863 2867
30. DeverTEFengLWekRCCiganAMDonahueTF 1992 Phosphorylation of initiation factor 2 alpha by protein kinase GCN2 mediates gene-specific translational control of GCN4 in yeast. Cell 68 585 596
31. LeeBUdagawaTSinghCRAsanoK 2007 Yeast phenotypic assays on translational control. Methods Enzymol 429 105 137
32. AsanoKKrishnamoorthyTPhanLPavittGDHinnebuschAG 1999 Conserved bipartite motifs in yeast eIF5 and eIF2Bepsilon, GTPase-activating and GDP-GTP exchange factors in translation initiation, mediate binding to their common substrate eIF2. EMBO J 18 1673 1688
33. HurtEHannusSSchmelzlBLauDTollerveyD 1999 A novel in vivo assay reveals inhibition of ribosomal nuclear export in ran-cycle and nucleoporin mutants. J Cell Biol 144 389 401
34. LiZLeeIMoradiEHungNJJohnsonAW 2009 Rational extension of the ribosome biogenesis pathway using network-guided genetics. PLoS Biol 7 e1000213 doi:10.1371/journal.pbio.1000213
35. GuacciVKoshlandDStrunnikovA 1997 A direct link between sister chromatid cohesion and chromosome condensation revealed through the analysis of MCD1 in S. cerevisiae. Cell 91 47 57
36. SjogrenCNasmythK 2001 Sister chromatid cohesion is required for postreplicative double-strand break repair in Saccharomyces cerevisiae. Curr Biol 11 991 995
37. D'AmbrosioCKellyGShirahigeKUhlmannF 2008 Condensin-dependent rDNA decatenation introduces a temporal pattern to chromosome segregation. Curr Biol 18 1084 1089
38. Clemente-BlancoAMayan-SantosMSchneiderDAMachinFJarmuzA 2009 Cdc14 inhibits transcription by RNA polymerase I during anaphase. Nature 458 219 222
39. IdeSMiyazakiTMakiHKobayashiT 2010 Abundance of ribosomal RNA gene copies maintains genome integrity. Science 327 693 696
40. ZouHRothsteinR 1997 Holliday junctions accumulate in replication mutants via a RecA homolog-independent mechanism. Cell 90 87 96
41. LalorayaSGuacciVKoshlandD 2000 Chromosomal addresses of the cohesin component Mcd1p. J Cell Biol 151 1047 1056
42. GlynnEFMegeePCYuHGMistrotCUnalE 2004 Genome-Wide Mapping of the Cohesin Complex in the Yeast Saccharomyces cerevisiae. PLoS Biol 2 e259 doi:10.1371/journal.pbio.0020259
43. DammannRLucchiniRKollerTSogoJM 1993 Chromatin structures and transcription of rDNA in yeast Saccharomyces cerevisiae. Nucleic Acids Res 21 2331 2338
44. TanRZvan OudenaardenA 2010 Transcript counting in single cells reveals dynamics of rDNA transcription. Mol Syst Biol 6 358
45. van der LelijPGodthelpBCvan ZonWvan GosligaDOostraAB 2009 The cellular phenotype of Roberts syndrome fibroblasts as revealed by ectopic expression of ESCO2. PLoS ONE 4 e6936 doi:10.1371/journal.pone.0006936
46. SchaafCAMisulovinZSahotaGSiddiquiAMSchwartzYB 2009 Regulation of the Drosophila Enhancer of split and invected-engrailed gene complexes by sister chromatid cohesion proteins. PLoS ONE 4 e6202 doi:10.1371/journal.pone.0006202
47. LiuJZhangZBandoMItohTDeardorffMA 2009 Transcriptional dysregulation in NIPBL and cohesin mutant human cells. PLoS Biol 7 e1000119 doi:10.1371/journal.pbio.1000119
48. KawauchiSCalofALSantosRLopez-BurksMEYoungCM 2009 Multiple organ system defects and transcriptional dysregulation in the Nipbl(+/−) mouse, a model of Cornelia de Lange Syndrome. PLoS Genet 5 e1000650 doi:10.1371/journal.pgen.1000650
49. RhodesJMBentleyFKPrintCGDorsettDMisulovinZ 2010 Positive regulation of c-Myc by cohesin is direct, and evolutionarily conserved. Dev Biol
50. WendtKSPetersJM 2009 How cohesin and CTCF cooperate in regulating gene expression. Chromosome Res 17 201 214
51. KageyMHNewmanJJBilodeauSZhanYOrlandoDA 2010 Mediator and cohesin connect gene expression and chromatin architecture. Nature
52. RollinsRAMorcilloPDorsettD 1999 Nipped-B, a Drosophila homologue of chromosomal adherins, participates in activation by remote enhancers in the cut and Ultrabithorax genes. Genetics 152 577 593
53. FayAMisulovinZLiJSchaafCAGauseM 2011 Cohesin selectively binds and regulates genes with paused RNA polymerase. Curr Biol 21 1624 1634
54. SkibbensRVMarzillierJEastmanL 2010 Cohesins coordinate gene transcriptions of related function within Saccharomyces cerevisiae. Cell Cycle 9
55. MonnichMKurigerZPrintCGHorsfieldJA 2011 A zebrafish model of Roberts syndrome reveals that Esco2 depletion interferes with development by disrupting the cell cycle. PLoS ONE 6 e20051 doi:10.1371/journal.pone.0020051
56. NarlaAEbertBL 2010 Ribosomopathies: human disorders of ribosome dysfunction. Blood 115 3196 3205
57. MayanMAragonL 2010 Cis-interactions between non-coding ribosomal spacers dependent on RNAP-II separate RNAP-I and RNAP-III transcription domains. Cell Cycle 9 4328 4337
58. TerretM-ESherwoodRRahmanSQinJJallepalliPV 2009 Cohesin acetylation speeds the replication fork. Nature 462 231 234
59. IrizarryRAHobbsBCollinFBeazer-BarclayYDAntonellisKJ 2003 Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 4 249 264
60. SmythGK 2004 Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol 3 Article3
61. MatysVKel-MargoulisOVFrickeELiebichILandS 2006 TRANSFAC and its module TRANSCompel: transcriptional gene regulation in eukaryotes. Nucleic Acids Res 34 D108 110
62. KelAEGosslingEReuterICheremushkinEKel-MargoulisOV 2003 MATCH: A tool for searching transcription factor binding sites in DNA sequences. Nucleic Acids Res 31 3576 3579
63. RamirezMWekRCHinnebuschAG 1991 Ribosome association of GCN2 protein kinase, a translational activator of the GCN4 gene of Saccharomyces cerevisiae. Mol Cell Biol 11 3027 3036
64. HillenmeyerMEEricsonEDavisRWNislowCKollerD 2010 Systematic analysis of genome-wide fitness data in yeast reveals novel gene function and drug action. Genome Biol 11 R30
65. ZhangYSikesMLBeyerALSchneiderDA 2009 The Paf1 complex is required for efficient transcription elongation by RNA polymerase I. Proc Natl Acad Sci U S A 106 2153 2158
66. KangHAHersheyJW 1994 Effect of initiation factor eIF-5A depletion on protein synthesis and proliferation of Saccharomyces cerevisiae. J Biol Chem 269 3934 3940
67. SmithJSBoekeJD 1997 An unusual form of transcriptional silencing in yeast ribosomal DNA. Genes Dev 11 241 254
68. PavelkaNRancatiGZhuJBradfordWDSarafA 2010 Aneuploidy confers quantitative proteome changes and phenotypic variation in budding yeast. Nature 468 321 325
69. WangBDEyreDBasraiMLichtenMStrunnikovA 2005 Condensin binding at distinct and specific chromosomal sites in the Saccharomyces cerevisiae genome. Mol Cell Biol 25 7216 7225
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2012 Číslo 6
Nejčtenější v tomto čísle
- Rumors of Its Disassembly Have Been Greatly Exaggerated: The Secret Life of the Synaptonemal Complex at the Centromeres
- The NSL Complex Regulates Housekeeping Genes in
- Tipping the Balance in the Powerhouse of the Cell to “Protect” Colorectal Cancer
- Interplay between Synaptonemal Complex, Homologous Recombination, and Centromeres during Mammalian Meiosis