
Incompatibility and Competitive Exclusion of Genomic Segments between Sibling Species
The extent and nature of genetic incompatibilities between incipient races and sibling species is of fundamental importance to our view of speciation. However, with the exception of hybrid inviability and sterility factors, little is known about the extent of other, more subtle genetic incompatibilities between incipient species. Here we experimentally demonstrate the prevalence of such genetic incompatibilities between two young allopatric sibling species, Drosophila simulans and D. sechellia. Our experiments took advantage of 12 introgression lines that carried random introgressed D. sechellia segments in different parts of the D. simulans genome. First, we found that these introgression lines did not show any measurable sterility or inviability effects. To study if these sechellia introgressions in a simulans background contained other fitness consequences, we competed and genetically tracked the marked alleles within each introgression against the wild-type alleles for 20 generations. Strikingly, all marked D. sechellia introgression alleles rapidly decreased in frequency in only 6 to 7 generations. We then developed computer simulations to model our competition results. These simulations indicated that selection against D. sechellia introgression alleles was high (average s = 0.43) and that the marker alleles and the incompatible alleles did not separate in 78% of the introgressions. The latter result likely implies that most introgressions contain multiple genetic incompatibilities. Thus, this study reveals that, even at early stages of speciation, many parts of the genome diverge to a point where introducing foreign elements has detrimental fitness consequences, but which cannot be seen using standard sterility and inviability assays.
Published in the journal:
. PLoS Genet 8(6): e32767. doi:10.1371/journal.pgen.1002795
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002795
Summary
The extent and nature of genetic incompatibilities between incipient races and sibling species is of fundamental importance to our view of speciation. However, with the exception of hybrid inviability and sterility factors, little is known about the extent of other, more subtle genetic incompatibilities between incipient species. Here we experimentally demonstrate the prevalence of such genetic incompatibilities between two young allopatric sibling species, Drosophila simulans and D. sechellia. Our experiments took advantage of 12 introgression lines that carried random introgressed D. sechellia segments in different parts of the D. simulans genome. First, we found that these introgression lines did not show any measurable sterility or inviability effects. To study if these sechellia introgressions in a simulans background contained other fitness consequences, we competed and genetically tracked the marked alleles within each introgression against the wild-type alleles for 20 generations. Strikingly, all marked D. sechellia introgression alleles rapidly decreased in frequency in only 6 to 7 generations. We then developed computer simulations to model our competition results. These simulations indicated that selection against D. sechellia introgression alleles was high (average s = 0.43) and that the marker alleles and the incompatible alleles did not separate in 78% of the introgressions. The latter result likely implies that most introgressions contain multiple genetic incompatibilities. Thus, this study reveals that, even at early stages of speciation, many parts of the genome diverge to a point where introducing foreign elements has detrimental fitness consequences, but which cannot be seen using standard sterility and inviability assays.
Introduction
Explaining the present-day biological diversity requires an understanding of the speciation process. While we typically cannot observe speciation, we can ask to what extent are species incompatible if brought together to form hybrids. The founders of the Modern Synthesis typically argued that even the most recently diverged species accumulate enough fitness differences such that no large part of the genome can be shared between them (e.g. [1]–[3]). This view of speciation argues that lots of loci with a wide range of effects on fitness should characterize the speciation process. E. Mayr championed a “genetic revolutions” version of this view, arguing that once separated from gene flow, most of the genome will undergo rapid coadaptive change, resulting in widespread fitness differences during speciation [3], [4]. As a result, the Biological Species Concept (BSC) has historically emphasized the cohesiveness of the species, where most of the genome diverges as a single biological unit and the evolution of isolating barriers play a central role in protecting its “integrity” [2], [3], [5]. On the other hand, if adaptive functional divergence involves a limited number of loci, much of the genome could still penetrate across the species boundary during incipient stages of speciation. This is often described as the “genic view” of speciation and is argued to be especially applicable when speciation occurs with gene flow (i.e. parapatric and sympatric modes of speciation, [6]–[8]; see Figure 1 in [6]).

Recently, several studies have attempted to look for the so-called “genomic islands of speciation” (e.g. [9]–[12], see review in [13]). These assume that speciation with gene flow has occurred and that it will homogenize the genome except for a few genes involved in reproductive isolation and differential adaptation [6], [8], [14]. While earlier studies found support for the “islands” of speciation (e.g. [9]–[11]), more recent comprehensive genome-wide screens are revealing a different picture [15]–[17]. Rather than having small genomic islands surrounded by mostly undifferentiated genomes, these incipient and sympatric races show widespread genomic differentiation, either being randomly distributed across the genome or clustered in the so-called “genomic continents” such as inversions or particular chromosomes (see [15] for discussion).
Other studies focus on identifying “speciation genes” that underlie reproductive isolation between closely related species (see [5] for review). Historically, these studies have been interested in determining how many loci are involved in reproductive isolation [18]–[23], and elucidating their identity and their evolution [24]–[31]. The great majority of these studies focus on the more easily measurable effects of sterility and inviability of hybrids. Many such sterility and inviability factors differentiating closely related species have been identified (see reviews in [5], pg. 302; [32]).
While both approaches have made important contributions to understanding the genetics of speciation in nature, neither addresses the degree to which two genomes are genetically incompatible. Genome-wide scans show us the extent of sequence divergence across whole genomes, but they say nothing about whether these divergent sites carry fitness or functional consequences. Studies that search for speciation genes concentrate a priori on such effects as hybrid sterility and inviability, but ignore the rest of the genome for other fitness and functional differences between species. Perhaps genetic studies of natural hybrid zones and hybrid fitness come closest to estimating the true extent of genetic incompatibilities between incipient species (e.g. [33], [34]). Results from hybrid zones suggest that many fitness-related genes may differentiate genomes of even incipient races or recently diverged sibling species [33], [35], [36]. However, little has been done to determine whether these incompatibilities are associated with sterility or inviability effects or contain other fitness detriments. Further, the hybrid studies cannot identify specific genomic regions responsible for incompatibilities or determine the strength of selection associated with each of these genetic incompatibilities. Exploring these questions in a laboratory setting using genetic introgressions provides the best means to estimate the basic parameters of genetic incompatibilities on a genome-wide level.
To approach this general question, the present paper focuses on recently diverged sibling species Drosophila simulans and D. sechellia. Molecular evidence indicates that they have diverged only about 250,000 years ago and thus represent fairly early stages of speciation [37]. For instance, these species have accumulated partial, but incomplete premating isolation and still produce fertile hybrid females in F1 and subsequent generations [18], [38]. These sibling species have most likely speciated allopatrically; D. simulans likely evolving on the African continent, while D. sechellia has remained an island endemic to the Seychelles archipelago in the Indian Ocean [39], [40]. Today, both species can be found in the Seychelles archipelago, but seem to occupy different islands [39]. Thus, we address our main question about genome-wide incompatibilities in a relatively young pair of taxa where whole-genomes were likely able to diverge without being impeded by substantial gene flow.
To determine the extent and nature of genetic incompatibilities between D. simulans and D. sechellia, we have introgressed random genetic segments from D. sechellia into a D. simulans genome. We first ask if these random introgressions contain measurable sterility and/or inviability effects. If some of these introgressions do not show sterility or inviability, we can then ask whether these regions are selectively neutral upon introgression or whether they carry other deleterious fitness effects after long-term genetic competition experiments. If these random genomic introgressions turn out to be selectively neutral, this would indicate that genomic incompatibilities are typically restricted to previously described genes associated with such effects as sterility and inviability (e.g. see [32]). However, if we find that most introgressions placed into a foreign genetic background experience strong fitness reduction and are selected out of the host population, it would imply that we are fundamentally underestimating the extent and possibly the type of fitness differences that accumulate between species. Thus our paper highlights the need to incorporate competition and other selection experiments to accurately test theories related to “genomic islands of speciation”.
Results
No detectable fertility differences between introgression lines and the wild-type D. simulans line
The present study utilized 12 recombinant introgression lines (RILs; henceforth referred to as “introgression lines” for short) from Stuart J. Macdonald, Isabel Colson and David B. Goldstein (Oxford University). Briefly, each line was made by genetically introgressing D. sechellia chromosomal fragments into a D. simulans genetic background (for a detailed description of the construction of these lines see Materials and Methods). The introgressions were made homozygous by single-pair sib-mating for 18 generations, and 41 microsatellite genetic markers across X, 2nd, and 3rd chromosomes were used to map the regions of the sechellia introgressions. As those introgression lines have been maintained in Goldstein's laboratory for several years, we therefore tested whether each line was homozygous for the expected sechellia introgression (henceforth referred to as “confirmed lines”) or whether it did not contain the sechellia marker allele (henceforth referred to as “unconfirmed lines”; see Figure S1). The latter lines may have lost the introgression by stochastic or other processes during their years of maintenance. In total, 9 lines were confirmed to carry sechellia introgressions and 3 lines failed to show introgressions.
To test whether the created introgression lines had any obvious inviability and/or sterility factors, we assayed overall fertility of each introgression line and compared it to the fertility of the experimental simulans strain that was used as the genetic background of introgressions (Table 1). Our results showed that while the introgression experiment clearly increased the variance in fertility among introgression lines (one-way ANOVA: F = 9.05, d.f. = 11, p<0.0001), the average fertility among lines was nearly identical to that of the experimental simulans strain (Table 1). Further, there was no evidence of significant fertility reduction in any of the lines studied using the posthoc Tukey-Kramer HSD test (Table 1). There was also no trend in fertility reduction among our introgression lines, with five out of the eight tested introgression lines actually having higher fertility than the experimental simulans strain (Table 1). Similarly, crosses between different introgression lines and between their F1 progeny either resulted in non-significant differences in fertility from the simulans strain or higher fertility relative to the simulans strain (see Tables S2 and S3). Therefore, we conclude that the present sechellia alleles placed in a simulans genetic background did not generate any detectable inviability and/or sterility effects. We can begin to address our main question as to whether these sechellia introgressions are equally fit to wild-type simulans alleles in a simulans genetic background.

Competition tests between introgressions and wild-type segments reveal repeated declines of D. sechellia alleles
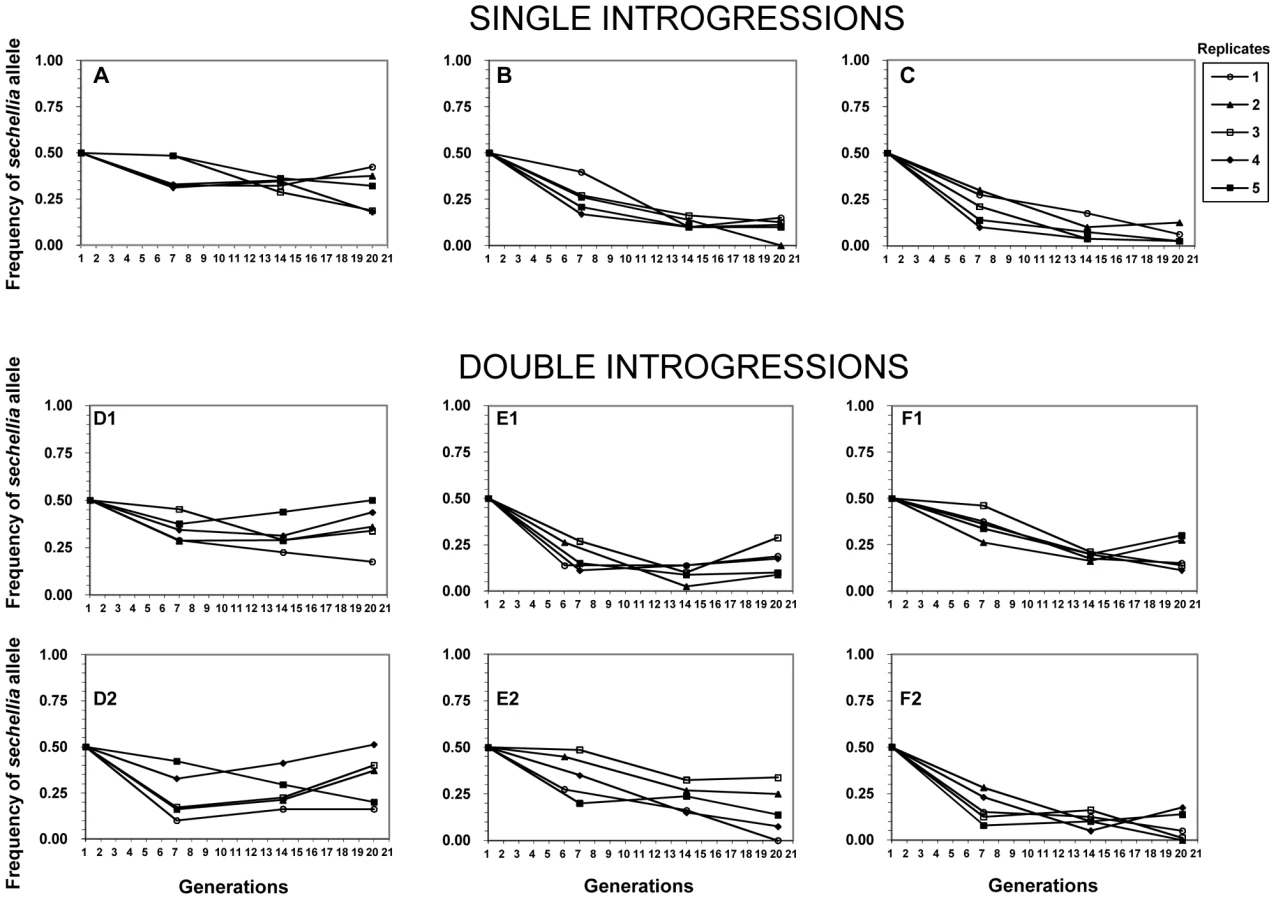
To test whether the introgressions had any other deleterious fitness affects, we set up 6 independent competition experiments to determine the evolutionary fate of sechellia alleles in a simulans genetic background. For each competition experiment, we crossed two different introgression lines (see Methods for details). Combining the introgression lines together allowed us to control for any non-intentional effects of the introgression procedure (e.g. to control for different levels of inbreeding). Competition crosses were of two types: The first set of experiments crossed a line containing a single confirmed introgression with another line that did not show evidence of the introgression. Thus in this experiment, sechellia alleles were competing with the wild-type simulans alleles at only a single genomic region (henceforth referred to as “single-introgression experiment”; shown in Figure 1A–1C as black blocks). The second type of experiment crossed two lines, each containing a unique confirmed introgressed region on either the 2nd or 3rd chromosome (henceforth referred to as “double-introgression experiment”; Figure 1D–1F). This allowed us to see if the introgressions on the 2nd and 3rd chromosomes interact when each competed against the wild-type simulans alleles (see below).
Our competition experiments revealed highly unexpected results based on the above lack of difference in fertility between introgression lines and the wild-type simulans line. We found that all of the sechellia marker alleles sharply decreased in their frequencies relative to the wild-type simulans alleles by generations six and seven (Figure 2). We found that from the starting 50% frequency, the sechellia marker alleles dropped to a range of 38% to 17% among different experiments. After this initial drop, the frequencies of sechellia alleles either: 1) kept further declining, 2) remained relatively unchanged or 3) actually increased over time in subsequent generations. This striking observation resulted in several conclusions. First, it showed that introgressed segments of sechellia into a simulans background does indeed carry strong deleterious fitness consequences. Second, it showed that these fitness effects cannot be detected by standard fertility measures above and were only revealed through long-term competition experiments. Third, it indicated that the marker alleles we were tracking either remained genetically linked to the deleterious alleles at surrounding fitness loci or became independent over time from these deleterious alleles due to recombination.
No detectable fitness epistasis between introgressed D. sechellia segments in a D. simulans background
We then tested whether the frequency declines of sechellia alleles are affected by having one or two confirmed introgressions during the competition experiment (i.e. single introgressions versus double introgressions). Because the two sechellia introgressed segments are on different chromosomes, these are expected to assort independently during competition. We found that particular sechellia marker alleles that were either in the presence of a single or a double introgression had nearly identical frequency declines after six or seven generations (t test: single avg. freq.gen.6/7 = 0.284, double avg. freq. gen.6/7 = 0.276; F = 0.048, p = 0.866; see also Figure 2). Similarly, after 20 generations of competition, both types of introgression designs showed very similar sechellia marker frequencies (t test: single avg. freq.gen.20 = 0.158, double avg. freq. gen.20 = 0.214; F = 1.58, p = 0.216; Figure 2). Thus we did not detect any significant differences between single and double introgressions on fitness.
Finally, we tested whether there is linkage disequilibrium between 2nd and 3rd chromosome marker alleles in double introgression experiments (experiments D, E, and F in Figure 1). Except for few cases in experiment D, experimental populations did not deviate significantly from linkage equilibrium (p>0.05; Table S1). Thus using this approach we failed to detect evidence for epistasis between 2nd and 3rd chromosome sechellia introgressions. In total, these results suggest that the observed fitness reduction during competition is not a consequence of combining two introgressions on 2nd and 3rd chromosome together. This implies that each sechellia segment is negatively epistatically interacting with the simulans genetic background on its own.
Computer simulations suggest that multiple incompatibilities exist within each introgressed segment
To estimate the intensity of selection against sechellia alleles and the recombination rate between the marker and the surrounding fitness loci, we performed multiple-generation, computer simulations using maximum likelihood approaches. We assumed that each microsatellite marker is neutral and is linked at a recombination distance of c to a single deleterious allele with selection coefficient s and dominance h. All other aspects of the competition experiment, such as experimental population sizes, recombination only in females, etc., were simulated accordingly (see Materials and Methods for details). Because our main interest is to estimate c (recombination rate; ranging from 0 to 0.5) and s (selection against sechellia allele; ranging from 0 to 1), we manipulated the dominance parameter, h. These estimates were not meant to precisely estimate s and c, but to give an idea of the scale of these values necessary to produce the observed marker allele frequency declines. The h parameter was assumed to equal either: 0, 0.5, 0.9 or 1. Thus, we allowed sechellia allele to become increasingly dominant over the simulans allele from complete recessivity (h = 0) to complete dominance (h = 1). In reality, it is not known which dominance best characterizes the sechellia-simulans allelic relationship, but as we will see below, our results are robust to changes in the dominance parameter.
Maximum likelihood estimates of s and c were obtained by comparing the observed D. sechellia marker frequencies to computer-generated distributions based on simulations of introgression lines. Table 2 summarizes the simulation results based on contour plots in Figure S2. Interestingly, we found that 7 of the 9 (78%) maximum likelihood estimates of c have values that are very close to 0, corresponding to very small physical distances (Table 2). This result has two possible interpretations: First, it may imply that the marker and fitness locus happen to be very close to each other in 78% of the experiments. Second, rather than a single fitness locus per segment (as our simulation model assumed), the introgressed segments may be carrying multiple fitness loci with deleterious effects, thus preventing the single marker from recombining away from multiple deleterious interactions. Given that our markers were chosen randomly and that the sechellia segments are fairly large (Figure 1, Figure S1), the chance that each randomly chosen marker locus happened to be so close to a single fitness locus with a deleterious effect seems very low. Instead these results most likely suggest that the introgressed sechellia segments probably carry multiple deleterious fitness alleles in a simulans background. Table 2 also shows that varying the dominance parameter, h, does not change the major results of the simulations, with essentially presence or absence of positive recombination across different lines.

Finally, our computer simulations revealed that selection coefficients against sechellia alleles must be strong in order to explain the observed evolutionary changes (Table 2). On average, the selection intensity against sechellia alleles was s = 0.43 with a range of 0.28 to 0.65. It can also be seen that the estimated selection coefficients were negatively correlated with the dominance of sechellia alleles (R2 = 0.89, F = 27.4, p = 0.034). This result is in general agreement with expectation of Haldane' sieve [41], since if alleles are more recessive, in order to explain the observed frequency declines, they must have stronger selection coefficients (note however that we are dealing with negative selection rather than positive as in [41]). However, even under completely dominant assumption, the selection strength against sechellia alleles is on average still high (s = 0.37; Table 2). In total, our simulations indicated that multiple incompatibilities likely exist within the great majority of our introgressed segments and that these factors have substantial negative fitness consequences that cannot be detected by standard fertility tests above.
Weak evidence for reduced mating success is responsible for declines of D. sechellia alleles from D. simulans genetic background
Determining exactly why sechellia alleles declined in frequency in our competition experiments is beyond the scope of this paper. However, we did perform one additional experiment focusing on whether introgression lines have reduced mating success relative to the original simulans strain (see Materials and Methods for details). These results showed that individuals (combined males and females) from 6 out of 8 (75%) introgression lines did indeed have lower relative mating success compared to individuals from the simulans strain (Table 3). While suggestive, this result is not statistically significant (sign test: one-tailed p = 0.14). On average, simulans individuals comprised 53% of the total matings relative to 47% of the introgression individuals, which did turn out to be slightly significant (Wilcoxon test: χ2 = 6.4, p<0.011). It is particularly the introgression males that are strongly outcompeted by simulans males (a 12% differential in fitness; Wilcoxon test: χ2 = 10.6, p<0.0011). Introgression females have the same mating success as simulans females (Wilcoxon test: χ2 = 0.03, p = 0.87; Table 3). Unfortunately, performing such an experiment does not allow us to adequately control for different overall levels of inbreeding between our introgression lines and our simulans line, a factor known to influence mating behavior in Drosophila (e.g. [42]). Thus, presently, we cannot conclude that mating behavior differences were responsible for the observed inferiority of sechellia alleles in a simulans background (see Discussion for additional possibilities).

Discussion
The “genic view” of speciation typically states that genomic introgression may readily occur except for rare reproductive isolation genes (see Figure 1 in [6]). However, E. Mayr and other founders of the Modern Synthesis typically viewed genomes of different species as tightly cohesive units that become largely impenetrable to gene flow during and after speciation events (see [2], [3], [5]). Recent array and whole-genome sequencing technologies are revealing that even between incipient races, nucleotide sequence divergence is often extensive across genomes [15], [16], [17]. However, to unambiguously determine which view of speciation is closer to reality, one needs to study genome-wide genetic incompatibilities between different races and species. To approach this seminal question, we performed an introgression study in order to assess genomic fitness divergence between relatively young and most likely allopatrically diverged sibling species Drosophila sechellia and D. simulans. Our paper for the first time demonstrates that genome-wide genetic incompatibilities between young sibling species are already fairly extensive. We found that all of our 9 random introgressed genetic segments from sechellia into a simulans genome carried negative fitness consequences when competed for multiple generations against wild-type simulans alleles. While some of the marker alleles were able to partially recombine away from the unidentified genetic incompatibilities, our simulations showed that most markers were likely surrounded by multiple such incompatibility factors within each introgressed segment.
Our results come closest to hybrid zone studies that estimate the number of genes involved in hybrid fitness problems by using spatial clinal information in the zone of contact (see [33]). Results from hybrids zones agree with our experimental observations that least several hundred fitness-related genes may differentiate genomes of even incipient races or recently diverged sibling species [33],[35],[36]. Similarly, a recent study of Rhagoletis host races [15] suggests that a significant amount of the genome is experiencing divergent selection under natural field conditions, consistent with our experimental results. Finally, this work appears to be also largely consistent with studies that measure various aspects of hybrid fitness under natural conditions [34], [43], [44], [45]. These studies have recently documented that hybrid fitness compared to parental individuals is particularly affected by competitive conditions [34]. Both our experimental work and these studies are suggesting that we may be fundamentally underestimating the extent of fitness divergence that lead to incompatibilities between incipient and sibling species.
Why are genetic incompatibilities extensive at this early stage of speciation?
Adaptive evolution within species largely rests on the basic parameters of genetic architecture of fitness-related traits [3], [46], [47], [48], [49], [50]. Such parameters as the level of genetic interactions (epistasis), the number of genes and their effects and the pleiotropic byproduct of genes will determine how much fitness and functional divergence is expected between species. If most phenotypes and developmental systems are governed by complex genetic architectures, whose genes are organized into epistatic networks that also have pleiotropic effects, we would expect that even incipient species would exhibit a multitude of fitness and functional differences between their genomes that cannot be easily broken down by subsequent gene flow [3], [6], [18], [51], [52]. This highly co-adaptive view of speciation was strongly favored by E. Mayr who even suggested that speciation will sometimes lead to veritable “genetic revolutions” due to the large-scale reorganization of allelic selective pressures as a result of new independent mutations and a change in epistatic interactions between new and existing alleles in each isolated population [3]. However, if most fitness-related traits and developmental systems are governed by few loci of additive and non-pleiotropic major effect, then it is conceivable that incipient speciation would only involve a handful of divergent loci with the rest of the genome being highly penetrable to gene flow [6]. The fact that we observed genetic incompatibilities with every random genetic introgression from D. sechellia into D. simulans suggests that the genetic basis of speciation is likely to be highly polygenic and epistatic between these young species.
What explains the observed fitness inferiority of introgressed regions?
Our competition results are particularly striking because we showed that while these introgressions are viable and fertile on their own, they nevertheless rapidly decline in frequency when they compete against wild-type alleles for multiple generations. We studied two obvious components of fitness that could have been potentially involved in the inferiority of D. sechellia introgressions. These included both premating (mating success) and postmating (fertility) assays in our introgression lines (D. simulans background+D. sechellia introgressed segment) relative to the experimental D. simulans strain. Our results did not detect significant fertility effects of introgression since we initially showed that fertility is not lower in the introgression lines compared to the D. simulans strain. This finding indicates that the observed competitive exclusion of D. sechellia introgressions is unlikely a result of “weak” sterility and/or inviability factors since these would have generated lower fertility in introgression lines. Therefore, the cause of D. sechellia introgression inferiority is likely to be in other components of fitness.
We also used multiple-choice mating trials to assess relative mating success of introgression lines against D. simulans strain. While individuals from introgression lines had a tendency to have lower mating success compared to the D. simulans line, this trend was not significant. Moreover, we could not control for inbreeding effects on mating success with this approach. Taken together, these assays could not identify a clear mechanism by which D. sechellia alleles were outcompeted from the D. simulans genetic background in our experiments. At this point we can only speculate that other as of yet unknown aspects of fitness particularly involved in soft-selection or competitive ability must be responsible for these fitness incompatibilities between these genomes.
Will more incipient and sympatric cases support the “genic view” of speciation?
What is presently unclear is which biogeographical conditions of speciation will facilitate the rapid accumulation of genetic incompatibilities. In our work we have shown that fitness incompatibilities are fairly extensive between 250,000 year old allopatric sibling species. Because these species most likely diverged in allopatry, their genomes are expected to have accumulated incompatibilities at more or less homogeneous rates over time without much gene flow [6]. Will younger sibling species also show similar patterns? Will parapatric or sympatric modes of speciation favor a more limited accumulation of genetic incompatibilities than what we have observed? While earlier studies of sequence divergence using small number of markers generally found “genomic islands of speciation” (e.g. [9], [11]), more recent analyses of incipient parapatric and sympatric forms show more extensive sequence differentiation [15]–[17]. However, it is still largely unknown whether any of these sequence differences will translate to fitness divergence and genetic incompatibilities (but see [15]).
Future work will gain further insights into the evolution of genetic incompatibilities by extending our genetic competition experiments to even more incipient cases of speciation and those that have likely speciated with gene flow. This appears to be a more accurate way to assess which view of speciation is likely to be correct. It will also determine under which circumstances extensive genetic incompatibilities accumulate between two genomes. Follow-up studies may also reveal the causes of non-sterility and non-inviability genetic incompatibilities that are likely to be observed in such long-term competition experiments.
Materials and Methods
Introgression lines
The recombinant introgression lines (RILs) were kindly provided by Stuart J. Macdonald, Isabel Colson and David B. Goldstein (Oxford University). The construction and genotype checking of these introgression lines are briefly described here. D. simulans females from the “sim132” (European Drosophila Stock Centre, Umeå) line were crossed to D. sechellia males from the “sec S9” (Mid-America Drosophila Stock Center), and the resultant F1 females were backcrossed to D. simulans males. The subsequent F2 males were individually crossed to either three simulans females (P cross, P) or three F1 females (H cross, H) and further made homozygous by single-pair sib-mating for 18 generations (SJ Macdonald, pers. comm.). Figure S1 illustrates the genotype for each introgression lines based on the information provided by SJ Macdonald.
In total, 41 microsatellite markers, i.e., 8, 16, and 17 markers on the X, 2nd and 3rd chromosomes respectively, with an average interval of about 8 cM [53] are used in the initial genotyping. There are much fewer introgression fragments with smaller sizes on the X chromosome compared to the two autosomes (SJ Macdonald, pers. Comm.). Only 3 of the 12 lines (6H, 16H, and 94P) carry a small X chromosomal introgression (Figure S1). We therefore focused on the two autosomes for the competition experiments.
Before all experiments, we genotyped these 12 introgression lines by using one microsatellite marker per introgressed segment and found 9 lines (6H, 12H, 25H, 28H, 60H, 29P, 62P, 78P, and 94P) showed the expected sechellia alleles (these lines are referred to as “confirmed lines”; see Figure S1 for specific location of each marker in each confirmed line). The other three lines (16H, 37P, and 129P) showed no evidence of sechellia alleles at the genotyped locus (Figure S1). Nevertheless, these lines may still carry some parts of the sechellia introgression that could not be assessed by our genotyping. Therefore we will refer to the latter three lines as “unconfirmed lines”.
Fertility assay of introgression lines
To see if these introgressions had any obvious viability and/or sterility effects, we assayed the overall fertility of each introgression line relative to the original wild-type D. simulans line without introgressions. This was done by measuring the number of offspring produced by each introgression line and comparing it to the fertility of the D. simulans strain. All fertility assays were performed by setting up 10 replicates of three pairs of males and females in small vials for each tested line. We allowed the mating pairs to lay eggs for 15 days, at which point all adults were cleared. We then counted the number of F1 progeny to determine fertility. To test for significance, we first confirmed that the fertility data did not significantly deviate from normal distribution using a Goodness of Fit test (Shapiro-Wilk test: W = 0.987, p = 0.8666). We then analyzed the whole dataset using a one-way ANOVA. To determine which specific introgression lines were significantly different from each other and from the wild-type simulans strain, we used a Tukey-Kramer HSD test that takes into account multiple testing. All tests were performed in JMP software (SAS).
Establishing six populations for fitness competition experiments
To determine if there were any other fitness effects of sechellia alleles in a simulans genetic background, we performed a multi-generational competition experiment lasting twenty generations. We set up six independent competition crosses between different introgression lines: (A) 37P×25H, (B) 78P×16H, (C) 6H×129P, (D) 62P×29P, (E) 94P×28H and (F) 12H×60H. Combining the introgression lines together allowed us to control for any non-intentional effects of the introgression procedure (i.e. all lines entering the competition experiment went through the same introgression procedure). The detailed procedures of the cross are as follows: (62P×29P as an example): 50 virgin females of 62P were crossed to 50 males of 29P and 50 virgin females of 29P were crossed to 50 males of 62P. The resultant F1 progeny of the two bottles were mixed and allowed to lay eggs to produce a large number of F2 progeny, which were transferred to five bottle replicates. There were approximately 300–500 flies in each bottle. The flies were allowed to lay eggs for 4 days and then collected in 100% ethanol. For the next generation, when enough flies (300–500) emerged, they were transferred to a new bottle. The same procedure applied to other crosses. In total, we set up five replicates for each one of the six distinct populations. The population sizes were kept at 300–500 for each replicate. All experiments were done at 22±1°C with a 12 hr–12 hr light–dark cycle.
Measuring allele frequency of the six populations
Samples of around 40 flies were taken from each bottle at generations 6 or 7, 14, and 20. For each introgression segment, we examined one microsatellite marker in that region. The microsatellite markers are: A: DMU25686 (cytological position: 93F14); B: DRODORSAL (36C8); C: DROGPAD (47A9); D1: AC005732 (cytological position 24C9); D2: DMRHO (62A2); E1: DMMP20 (49F13); E2: DMCATHPO (75E1); F1: DS00361 (54B5); F2: DMU43090 (99D5) [53], [54]. From the genotyping results, allele frequencies were calculated for each bottle replicate and for whole experiment sets.
Maximum likelihood estimates of the selection coefficients
We developed an individual-based computer simulation model of our competition experiments performed above. The purpose of the simulation was to manipulate the presumed selection pressures against sechellia alleles relative to simulans alleles, the dominance of sechellia alleles' fitness effects and the recombination rate between selected loci and marker loci. The goal was to determine which combination of selection pressures and recombination rates was best at explaining our observed results.
The simulations tracked either D. simulans or D. sechellia alleles at both the marker loci and the selected loci for each individual in the population. Each simulation involved 10,000 iterations with fixed values of selection coefficient (s), recombination distance (c), and dominance (h). Each iteration ran for 20 generations with population sizes fixed at 150 males and 150 females each generation. Each generation was divided into the following stages: selection, recombination (females only), and reproduction. In the selection stage: individuals homozygous for the D. simulans allele all survived, heterozygous individuals survived with probability = hs, and individuals homozygous for the D. sechellia alleles survived with probability = s. Reproduction began with recombination in females which occurred with probability = c. During random mating, male and female haplotypes were randomly selected from the population to make 150 males and 150 females for the next generation.
Sampling took place in generations 7, 14, and 20 where 20 males and females were removed from the population after selection and before random mating. The allele frequencies of the D. simulans alleles at the marker loci were recorded from these sampled flies. The simulation ran for 10,000 iterations. Distributions were created for each of the three sampled generations (7, 14, and 20). The allele frequencies were assigned to one of 40 bins (0–.025, .025–.05, …, .975–1), and bin counts were incremented for each iteration. Observed allele frequencies for each marker were then compared to the three distributions. For a given generation, all of the bins containing observed frequencies were added together and divided by the total number of iterations. The log of this ratio was treated as a likelihood estimate for the parameters s, c, and h for that marker. Simulations were run for all combinations of c = 0.0001, 0.001, 0.01, 0.1, 0.2, 0.3, 0.4, and 0.5; s = 0.1, 0.2, 0.3, 0.4, 0.5, 0.6, 0.7, 0.8, and 0.9; and h = 0, 0.5, 0.9, and 1. The Maximum likelihood estimate for each marker was the set of s, c, and h that yielded the highest likelihood value.
Linkage disequilibrium analyses
Linkage disequilibrium (LD) analyses were carried out for double introgression experiments (experiments D, E, and F in Figure 1). The null hypothesis is that genotypes at one locus assort independently from genotypes at the other locus. The exact test for the LD was performed by using M. Raymond and F. Rousset's GENEPOP software package (http://genepop.curtin.edu.au/genepop_op2.html). We performed this probability test using a Markov chain with parameters of dememorization number = 1000, number of batches = 100 and number of iterations per batch = 1000.
Mating behavior assays of introgression lines in competition with the wild-type strain
To assay whether introgressed segments caused a reduction in the mating success of their individuals relative to wild-type D. simulans genotype, we applied a multiple-choice mating experiment design similar to [55]. All flies were fed red or green colored food 14–18 hours prior to the experiment. The food coloration alternated between replications and had no effect on D. simulans mating choice (data not shown). Experiments were started within one hour after the beginning of the light cycle and conducted at 22±1°C. Sixty 4-day-old virgin flies from each sex of D. simulans S132 line and the introgression line were simultaneously released into a Plexiglas cage (14.5″ L×10″ H×9.5″ W) with fly food in a 14-cm diameter Petri dish. The copulating pairs were aspirated out of the cage for identification by the food coloring in the guts. We let the mating trials run for 1 hour or until 60 matings (50% of possible copulations) had occurred, whichever came first (as recommended by [56] to avoid bias). Several comparisons were replicated at least three times to determine overall reliability in the mating behavior. We then calculated the percentage of matings by D. simulans individuals relative to introgression line individuals and also the relative percentage of matings by each sex of each line.
Supporting Information
Zdroje
1. DobzhanskyT 1937 Genetics and the origin of species New York Columbia University Press 364
2. MayrE 1959 Where are we? Cold Spring Harbor Symp Quant Biol 24 1 14
3. MayrE 1963 Animal species and evolution Cambridge Belknap Press 811
4. MayrE 1954 Change of genetic environment and evolution. HuxleyJHardyACFordEB Evolution as a process London Allen & Unwin 157 180
5. CoyneJOrrA 2004 Speciation Sunderland Sinauer Associates, Inc 545
6. WuCI 2001 The genic view of the process of speciation. J Evol Biol 14 851 865
7. BartonNH 2008 The effect of a barrier to gene flow on patterns of geographic variation. Genet Res 90 139 149
8. ViaS 2009 Natural selection in action during speciation. Proc Natl Acad Sci USA 106 9939 9946
9. TurnerTLHahnMWNuzhdinSV 2005 Genomic islands of speciation in Anopheles gambiae. PLoS Biol 3 e285 doi:10.1371/journal.pbio.0030285
10. HarrB 2006 Genomic islands of differentiation between house mouse subspecies. Genome Res 16 730 737
11. YatabeYKaneNCScotti-SaintagneCRiesebergLH 2007 Rampant gene exchange across a strong reproductive barrier between the annual sunflowers, Helianthus annuus and H. petiolaris. Genetics 175 1883 1893
12. NosilPFunkDJOrtiz-BarrientosD 2009 Divergent selection and heterogeneous genomic divergence. Mol Ecol 18 375 402
13. ButlinRK 2010 Population genomics and speciation. Genetica 138 409 418
14. FederJLNosilP 2010 The efficacy of divergence hitchhiking in generating genomic islands during ecological speciation. Evolution 64 1729 1747
15. MichelAPSimSPowellTHTaylorMSNosilP 2010 Widespread genomic divergence during sympatric speciation. Proc Natl Acad Sci USA 107 9724 9729
16. LawniczakMKNEmrichSJHollowayAKRegierAPOlsonM 2010 Widespread divergence between incipient Anopheles gambiae species revealed by whole genome sequences. Science 330 512 514
17. YukilevichRTurnerTLAokiFNuzhdinSVTrueJR 2010 Patterns and processes of genome-wide divergence between North American and African Drosophila melanogaster. Genetics 186 219 239
18. WuCIPalopoliMF 1994 Genetics of postmating reproductive isolation in animals. Annu Rev Genet 28 283 308
19. TrueJRWeirBSLaurieCC 1996 A genome-wide survey of hybrid incompatibility factors by the introgression of marked segments of Drosophila mauritiana chromosomes into Drosophila simulans. Genetics 142 819 837
20. NaveiraHMasideX 1998 The genetics of hybrid male sterility in Drosophila. HowardDJBerlocherSH Endless forms: species and speciation Oxford Oxford University Press 330 338
21. TaoYChenSHartlDLLaurieCC 2003 Genetic dissection of hybrid incompatibilities between Drosophila simulans and D. mauritiana. I. Differential accumulation of hybrid male sterility effects on the X and autosomes. Genetics 164 1383 1397
22. PresgravesDC 2003 A fine-scale genetic analysis of hybrid incompatibilities in Drosophila. Genetics 163 955 972
23. MaslyJPPresgravesDC 2007 High-resolution genome-wide dissection of the two rules of speciation in Drosophila. PLoS Biol 5 e243 doi:10.1371/journal.pbio.0050243
24. TingCTTsaurSCWuMLWuCI 1998 A rapidly evolving homeobox at the site of a hybrid sterility gene. Science 282 1501 1504
25. WittbrodtJAdamDMalitschekBMauelerWRaulfF 1989 Novel putative receptor tyrosine kinase encoded by the melanoma-inducing Tu locus in Xiphophorus. Nature 341 415 421
26. BarbashDASiinoDFTaroneAMRooteJ 2003 A rapidly evolving MYB-related protein causes species isolation in Drosophila. Proc Natl Acad Sci USA 100 5302 5307
27. BarbashDAAwadallaPTaroneAM 2004 Functional divergence caused by ancient positive selection of a Drosophila hybrid incompatibility locus. PLoS Biol 2 e142 doi:10.1371/journal.pbio.0020142
28. PresgravesDCBalagopalanLAbmayrSMOrrHA 2003 Adaptive evolution drives divergence of a hybrid inviability gene between two species of Drosophila. Nature 423 715 719
29. BrideauNJFloresHAWangJMaheshwariSWangX 2006 Two Dobzhansky-Muller genes interact to cause hybrid lethality in Drosophila. Science 314 1292 1295
30. MaslyJPJonesCDNoorMALockeJOrrHA 2006 Gene transposition as a cause of hybrid sterility in Drosophila. Science 313 1448 1450
31. PhadnisNOrrHA 2009 A single gene causes both male sterility and segregation distortion in Drosophila hybrids. Science 323 376 379
32. JohnsonNA 2010 Hybrid incompatibility genes: remnants of a genomic battlefield? Trends Genet 26 317 325
33. BartonNHGaleKS 1993 Genetic analysis of hybrid zones. HarrisonRG Hybrid zones and the evolutionary process Oxford Oxford University Press 13 45
34. MercerKLWyseDLShawRG 2006 Effects of competition on fitness of wild and crop-wild hybrid sunflower from a diversity of wild populations and crop lines. Evolution 60 2044 2055
35. BartonNHHewittGM 1981 Hybrid zones and speciation. AtchleyWRWoodruffD Evolution and Speciation Cambridge Cambridge University Press 109 145
36. RiesebergLHWhittonJGardnerK 1999 Hybrid zones and the genetic architecture of a barrier to gene flow between two sunflower species. Genetics 152 713 727
37. McDermottSRKlimanRM 2008 Estimation of isolation times of the island species in the Drosophila simulans complex from multilocus DNA sequence data. PLoS ONE 3 e2442 doi:10.1371/journal.pone.0002442
38. LachaiseDDavidJRLemeunierFTsacasLAshburnerM 1986 The reproductive relationships of Drosophila sechellia with D. mauritiana, D. simulans, and D. melanogaster from the Afrotropical region. Evolution 40 262 271
39. LachaiseDCariouMLDavidJRLemeunierFTsacasL 1988 Historical biogeography of the Drosophila melanogaster species subgroup. Evol Biol 22 159 225
40. LachaiseDSilvainJF 2004 How two Afrotropical endemics made two cosmopolitan human commensals: the Drosophila melanogaster-D. simulans palaeogeographic riddle. Genetica 120 17 39
41. HaldaneJBS 1924 A mathematical theory of natural and artificial selection, Part I. Trans Camb Philos Soc 23 19 41
42. SharpPM 1984 The effect of Inbreeding on competitive male-mating ability in Drosophila melanogaster. Genetics 106 601 612
43. SpencerLJSnowAA 2001 Fecundity of transgenic wild-crop hybrids of Cucurbita pepo (Cucurbitaceae): implications for crop-to-wild gene flow. Heredity 86 694 702
44. StewartCNJrHalfhillMDWarwickSI 2003 Transgene introgression from genetically modified crops to their wild relatives. Nat Rev Gen 4 806 817
45. SongZPLuBRWangBChenJK 2004 Fitness estimation through performance comparison of F1 hybrids with their parental species Oryza rufipogon and O. sativa. Ann Bot 93 311 316
46. WrightS 1932 The roles of mutation, inbreeding, crossbreeding and selection in evolution. Proc Sixth Inter Congr Genet 1 356 366
47. GavriletsS 2004 Fitness landscapes and the origin of species Princeton Princeton University Press 432
48. CarterAJHermissonJHansenTF 2005 The role of epistatic gene interactions in the response to selection and the evolution of evolvability. Theor Popul Biol 68 179 196
49. OrrHA 2006 The population genetics of adaptation on correlated fitness landscapes: the block model. Evolution 60 1113 1124
50. YukilevichRLachanceJAokiFTrueJR 2008 Long-term adaptation of epistatic genetic networks. Evolution 62 2215 2235
51. GavriletsS 2000 Waiting time to parapatric speciation. Proc Biol Sci 267 2483 2492
52. WadeMJ 2002 A gene's view of epistasis, selection and speciation. J Evol Biol 15 337 346
53. MacdonaldSJGoldsteinDB 1999 A quantitative genetic analysis of male sexual traits distinguishing the sibling species Drosophila simulans and D. sechellia. Genetics 153 1683 1699
54. ColsonIMacDonaldSJGoldsteinDB 1999 Microsatellite markers for interspecific mapping of Drosophila simulans and D. sechellia. Mol Ecol 8 1951 1955
55. WuCIHollocherHBegunDJAquadroCFXuY 1995 Sexual isolation in Drosophila melanogaster: a possible case of incipient speciation. Proc Natl Acad Sci USA 92 2519 2523
56. CasaresPCarracedoMCRioBDPiñeiroRGarcía-FlórezL 1998 Disentangling the effects of mating propensity and mating choice in Drosophila. Evolution 52 126 133
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2012 Číslo 6
Nejčtenější v tomto čísle
- Rumors of Its Disassembly Have Been Greatly Exaggerated: The Secret Life of the Synaptonemal Complex at the Centromeres
- The NSL Complex Regulates Housekeeping Genes in
- Tipping the Balance in the Powerhouse of the Cell to “Protect” Colorectal Cancer
- Interplay between Synaptonemal Complex, Homologous Recombination, and Centromeres during Mammalian Meiosis