
Eleven Candidate Susceptibility Genes for Common Familial Colorectal Cancer
Hereditary factors are presumed to play a role in one third of colorectal cancer (CRC) cases. However, in the majority of familial CRC cases the genetic basis of predisposition remains unexplained. This is particularly true for families with few affected individuals. To identify susceptibility genes for this common phenotype, we examined familial cases derived from a consecutive series of 1514 Finnish CRC patients. Ninety-six familial CRC patients with no previous diagnosis of a hereditary CRC syndrome were included in the analysis. Eighty-six patients had one affected first-degree relative, and ten patients had two or more. Exome sequencing was utilized to search for genes harboring putative loss-of-function variants, because such alterations are likely candidates for disease-causing mutations. Eleven genes with rare truncating variants in two or three familial CRC cases were identified: UACA, SFXN4, TWSG1, PSPH, NUDT7, ZNF490, PRSS37, CCDC18, PRADC1, MRPL3, and AKR1C4. Loss of heterozygosity was examined in all respective cancer samples, and was detected in seven occasions involving four of the candidate genes. In all seven occasions the wild-type allele was lost (P = 0.0078) providing additional evidence that these eleven genes are likely to include true culprits. The study provides a set of candidate predisposition genes which may explain a subset of common familial CRC. Additional genetic validation in other populations is required to provide firm evidence for causality, as well as to characterize the natural history of the respective phenotypes.
Published in the journal:
. PLoS Genet 9(10): e32767. doi:10.1371/journal.pgen.1003876
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1003876
Summary
Hereditary factors are presumed to play a role in one third of colorectal cancer (CRC) cases. However, in the majority of familial CRC cases the genetic basis of predisposition remains unexplained. This is particularly true for families with few affected individuals. To identify susceptibility genes for this common phenotype, we examined familial cases derived from a consecutive series of 1514 Finnish CRC patients. Ninety-six familial CRC patients with no previous diagnosis of a hereditary CRC syndrome were included in the analysis. Eighty-six patients had one affected first-degree relative, and ten patients had two or more. Exome sequencing was utilized to search for genes harboring putative loss-of-function variants, because such alterations are likely candidates for disease-causing mutations. Eleven genes with rare truncating variants in two or three familial CRC cases were identified: UACA, SFXN4, TWSG1, PSPH, NUDT7, ZNF490, PRSS37, CCDC18, PRADC1, MRPL3, and AKR1C4. Loss of heterozygosity was examined in all respective cancer samples, and was detected in seven occasions involving four of the candidate genes. In all seven occasions the wild-type allele was lost (P = 0.0078) providing additional evidence that these eleven genes are likely to include true culprits. The study provides a set of candidate predisposition genes which may explain a subset of common familial CRC. Additional genetic validation in other populations is required to provide firm evidence for causality, as well as to characterize the natural history of the respective phenotypes.
Introduction
Colorectal cancer (CRC) (MIM 114500) is a major cancer type, with over one million new cases diagnosed worldwide each year. It is the third most common malignancy [1], and the second most common cause of cancer mortality [2]. Inherited factors are estimated to play a crucial role in at least one third of all CRC cases [3]. However, high-penetrance mutations in known CRC predisposing genes, such as the mismatch repair (MMR) genes, APC, MUTYH (MYH), SMAD4, BMPR1A, STK11/LKB1, PTEN, AXIN2, POLE, and POLD1 explain only around 5% of these cases [4]–[6].
There are a few examples of rare variants in CRC predisposing genes conferring moderate or low carrier risk, such as APC (I1307K) [7], BLM [8] and GALNT12 [9]. Of these, the APC I1307K variant has been most extensively studied and occurs almost exclusively in the Ashkenazi Jewish population [7]. In addition to these, genome-wide association (GWA) studies have identified common low-penetrance variants at approximately 20 genomic loci associated with CRC susceptibility. However, the identified common variants at these loci exert only a modest effect on CRC risk [10]–[12].
Unknown variants of moderate or low penetrance are likely to explain at least part of the missing heritability in CRC. CRC families with few affected individuals are an attractive patient group to search for such genetic factors, but tools for such work have been poor. These families are relatively common but too small for linkage analyses, and the culprit variants are likely to be too diverse and rare to be detected in GWA studies. One approach has been to study the additive contribution of low-penetrance variants on familial risk. A previous study has estimated that ten known low-penetrance CRC variants collectively explain around 9% of the variance in familial risk [13]. Advances in sequencing technologies have made exome sequencing a feasible approach to search for rare coding variants of varying penetrance. In this study, we aimed at identifying variants predisposing to common familial CRC by performing exome sequencing on 96 independent familial CRC cases derived from a consecutive collection of unselected patients. Here, familial CRC is characterized as having at least one first-degree relative diagnosed with CRC; indeed the great majority of the 96 familial cases displayed only one first-degree relative with CRC. All patients were from Finland, known for its relatively homogenous population [14], [15]. This empowers the analysis since affected individuals are more likely to share ancestral predisposition mutations and haplotypes, stemming from a handful of founders. To our knowledge, this is the largest effort to date where exome sequencing has been applied to familial forms of cancer to identify novel predisposing genes.
Results
Exome sequencing
Exome sequencing analysis was performed on germline DNA from 96 independent familial CRC cases. The clinical and histopathological features of the cases are summarized in Table 1 and in more detail in Table S1. The average read depth attained for target regions was 43 and at least 86% of the captured target regions were covered by four or more sequence reads for all the samples. We identified a total of 76,487 nonsynonymous variants in the exome data (Figure 1). Sequence data were first evaluated for known predisposing genes (MLH1, MSH2, MSH6, PMS2, APC, MUTYH, SMAD4, BMPR1A, LKB1/STK11, PTEN, AXIN2, POLE, and POLD1). No clear pathogenic mutations were found in these genes. The following missense variants were identified (not confirmed by Sanger sequencing); MSH6 c.2800G>C p.D934H, and PTEN c.1016C>A P339Q. However, the patients did not present typical clinical phenotypes; in the case of the MSH6 variant the tumor did not display microsatellite instability and in the case of the PTEN variant patient records revealed no features suggestive of Cowden syndrome (MIM 158350). Thus, these variants remain of unknown clinical significance.


Identification of colorectal cancer predisposing variants
We hypothesized that predisposing germline variants would likely be rare in the general population, and predicted to truncate the protein product. We therefore filtered the data to prioritize such variants (Figure 1). First, variants had to be protein truncating with putative loss-of-function alteration; including nonsense, frameshift (insertion and deletion) or splice-site variants (IVS +1, +2, −1, and −2). A total number of 3,654 truncating variants were found in the exome data. Second, variants were excluded if present in the 1000 Genomes Project [16] or population matched exome control data (n = 212) at minor allele frequency (MAF)>0.001. After control filtering, 2,090 truncating variants remained. Third, genes with truncating variants in more than one familial CRC case were selected for further analysis. There were a total of 588 such variants of which 422 were frameshift, 115 nonsense, and 51 splice site variants (Figure 1). Frameshift variants were grossly overrepresented in the list of truncating variants due to sequencing artifacts. Finally, manual filtering was performed on all variants to further remove artifacts due to duplicated regions, mapping errors, and systematic sequence specific errors. The filtering procedure resulted in a shortlist of 29 genes with 46 truncating variants. These were subsequently validated by Sanger sequencing (Figure 1).
Sanger sequencing was successful for all amplicons, and 23 truncating variants in 18 genes were confirmed. Of these seven were frameshift, 12 nonsense, and four splice-site variants. To further exclude neutral polymorphisms, the confirmed variants were screened in 310 Finnish population matched controls, of which approximately two-thirds were also regionally matched. Variants with MAF>0.001 in the overall discovery phase control set (including Finnish control exome data and Sanger sequenced controls) were excluded (Figure 1).
In total, we identified 11 candidate predisposing genes with 14 truncating germline variants in at least two familial CRC cases (Table 2); UACA, SFXN4, TWSG1, PSPH, NUDT7, ZNF490, PRSS37, CCDC18, PRADC1, MRPL3, and AKR1C4. A summary of all these variants and respective frequencies are presented in Table 2. Gene descriptions and proposed functions of the identified genes are listed in Table S2. Typically, the same truncating variant was detected in several patients. However, three genes harbored two different types of truncating germline variants (Table 2). Nine genes showed truncating variants in 2/96 familial cases. Two genes had truncating variants in 3/96 cases; UACA (uveal autoantigen with coiled-coil domains and ankyrin repeats) (3/96, 3.1%) and SFXN4 (sideroflexin 4) (3/96, 3.1%). In UACA, p.Q1116X was identified in two out of 96 familial cases and present in 522 Finnish population matched controls with a MAF of 0.001. UACA p.R1292X was found in one out of 96 cases and the variant was not found in controls (Figure 2). In SFXN4, three out of 96 cases had c.32delC. This variant had a MAF of 0.001 in population matched controls. None of the other identified truncating variants were identified in population matched controls, except for c.389_390insA in PSPH which was found in 1/502 controls (MAF 0.001). To further explore the frequency of these variants in controls, we referred to the Exome Variant Server (NHLBI GO Exome Sequencing Project (ESP), Seattle, WA, http://evs.gs.washington.edu/EVS/ [July 2013]). Three of the identified germline variants, SFXN4 c.32delC, NUDT7 c.111T>A, and PRSS37 c.176+1G>A, were reported, however, at a MAF of less than 0.0003.


The exome data was also searched for missense variants in the 11 candidate predisposition genes; five missense variants were observed in five genes (Table S3). All of the missense variants were present in one case only, except for p.Q83H in PSPH which was identified in two out of the 96 familial cases. None of the missense variants were predicted to have a damaging effect on the protein by either of the prediction programs used (Table S3). The identified missense variants were very rare in population matched controls (MAF<0.001).
Loss of heterozygosity analysis
Loss of heterozygosity (LOH) was examined in cancers of CRC cases with candidate predisposing germline variants (Figure 1). The following genes displayed LOH in at least one cancer: UACA, TWSG1, PSPH, and ZNF490 (Table 2). Seven LOH events were observed and all targeted the wild-type allele (P = 0.0078). In UACA three out of six examined tumors showed loss of the wild-type allele and in TWSG1 (twisted gastrulation protein homolog 1) both of the tumors showed loss of the wild-type allele (Figure 2).
Genotyping population matched cases and controls
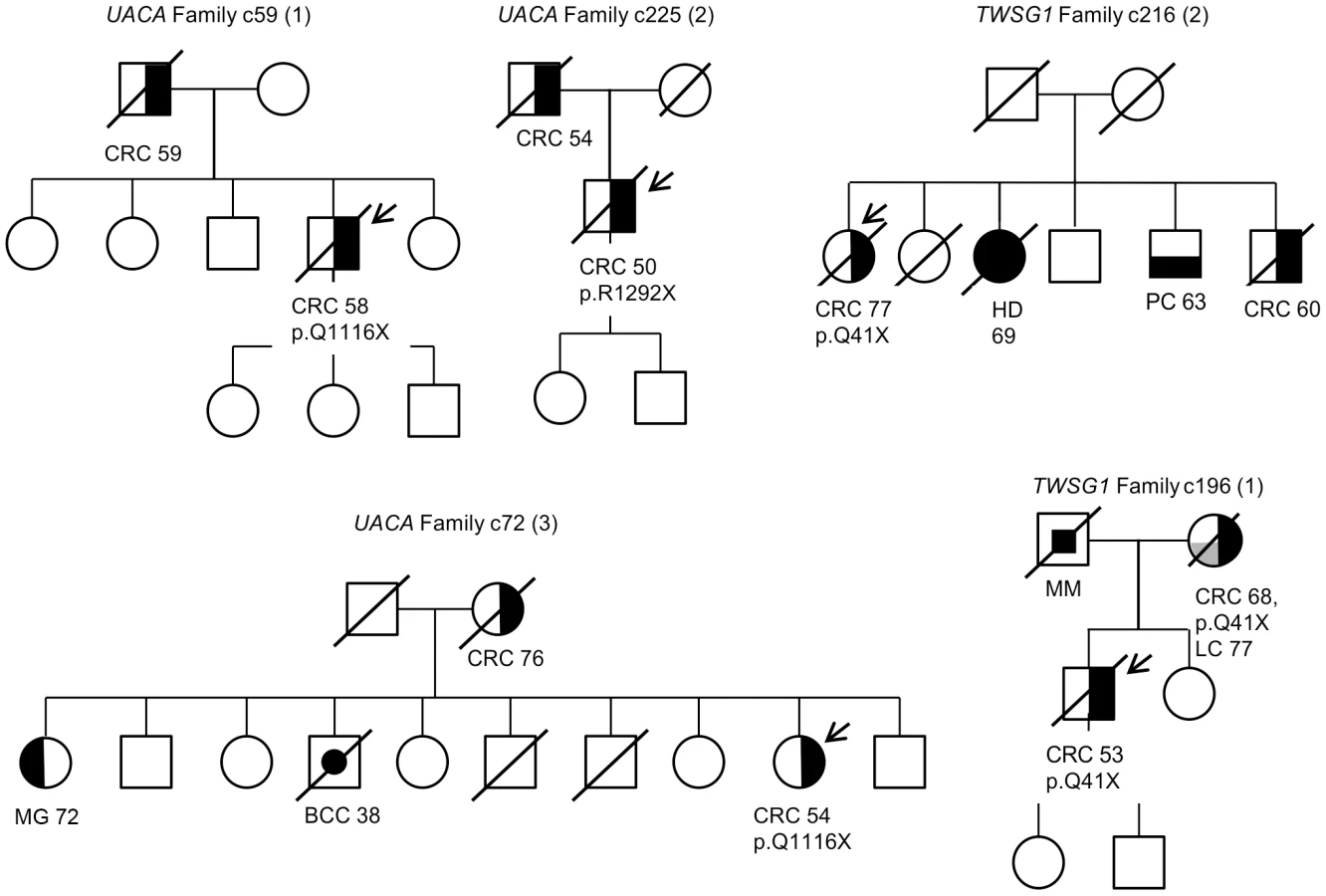
Variants in genes showing loss of the wild-type allele in tumor tissue were genotyped in an independent set of validation phase samples (Figure 1). This set included 954 Finnish population matched CRC cases and 586 Finnish population matched controls. UACA p.Q1116X was identified in two additional unrelated CRC cases and one control (Table 2). The ages at diagnosis were 67 and 58 years for the two cases. In the overall set of Finnish population matched controls used in this study, two out of 1,108 controls had UACA p.Q1116X (MAF = 0.0009). UACA p.R1292X was found in one additional case (diagnosis at the age of 61) and no controls were heterozygous for this variant. The variant p.R350X in ZNF490 was found in one additional case (diagnosed at the age of 58) and remained absent in controls (Table 2). TWSG1 p.Q41X was not present in any additional cases or controls. Genotyping was not successful for PSPH c.389_390insA. Next, LOH was analyzed in the tumors of the four additional cases with truncating variants (Table 2). One of the additional cases with UACA p.Q1116X showed LOH involving the wild-type allele (Figure 2).
Segregation
Segregation analysis of the identified truncating variants was performed for all the affected first degree relatives for whom samples were available. In total, segregation was analyzed in seven families for five of the identified truncating variants; c.32delC in SFXN4, p.Q41X in TWSG1, p.R350X in ZNF490, c.168+1G>A in PRADC1, and c.620delA in AKR1C4 (Figure 3 and Figure S1). The following variants showed segregation; c.32delC in SFXN4, c.168+1G>A in PRADC1, and c.620delA in AKR1C4. The variant p.Q41X in TWSG1 segregated in one family but not the other (Figure 3) and p.R350X in ZNF490 did not segregate (Figure S1).
Discussion
Exome sequencing is a powerful tool for discovering novel genetic variants that predispose to disease [17]. To examine the genetic basis of common familial CRC we exome sequenced 96 independent cases (Table 1) derived from a previously described population-based collection of patients [4], [18] and from an additional unselected collection (unpublished). To our knowledge, this is the largest effort to date where familial CRC has been studied by exome sequencing to identify novel CRC predisposing genes. Several strategies were applied to improve the power of gene discovery. First, a large set of familial CRC cases (at least one first-degree relative diagnosed with CRC) was utilized, negative for any known high penetrance CRC mutation. Second, the cases were from Finland, known for its isolated population with reduced genetic heterogeneity. Such isolated populations are enriched for rare founder variants, facilitating identification of disease genes [15]. Third, tumor tissue availability for all the CRC cases allowed for the assessment of somatic allelic imbalance, which gave important additional information related to pathogenicity of the variants. Fourth, genotyping of selected variants was performed in a set of validation phase population matched samples, consisting of 954 cases and 586 controls.
In total, we identified 11 novel candidate CRC susceptibility genes with rare truncating variants in two or three familial CRC cases; UACA, SFXN4, TWSG1, PSPH, NUDT7, ZNF490, PRSS37, CCDC18, PRADC1, MRPL3, and AKR1C4 (Table 2 and Table S2). They were absent or rare (MAF≤0.001) in the general population. The results fit with the “rare variant hypothesis” that proposes that a significant proportion of the missing heritability of complex diseases is due to a series of rare variants, each conferring a moderate increase in risk. Typically, such risk alleles function dominantly and independently [19], [20]. The “rare variant hypothesis” is strongly supported by evolutionary theory, which argues that variants that promote disease are selected against and are therefore rare. Another argument for the hypothesis comes from recent empirical population genetic data which shows that rare variants are enriched for deleterious mutations [21]. The question remains whether the identified candidate genes act as classical tumor suppressors with second hits or show alternative characteristics, such as haploinsufficiency or dominant-negative effects. Of the genes identified, four out of 11 showed loss of the wild-type allele in at least one tumor. In total, seven LOH events were observed and none showed loss of the mutant allele (P = 0.0078). This suggests that complete inactivation of these genes seems to be preferentially selected for in tumor evolution and that these germline variants are prime candidates for CRC susceptibility.
Perhaps the strongest candidate predisposition gene, in view of the LOH data and case frequency, was the apoptosis-associated gene UACA. Three of the 96 familial CRC cases were found to carry heterozygous truncating variants (p.Q1116X and p.R1292X) in UACA (Table 2). We performed genotyping to screen the variants in a set of validation phase samples. We identified three additional unrelated cases who were heterozygous for the variants encoding either p.Q1116X or p.R1292X. Second hits by LOH involving the germline wild-type allele were found in three of the six tumors (Figure 2). The average age of onset of CRC in the familial cases was 54 years (58, 54 and 50) (Figure 3), younger than the mean age of onset of 71 in familial cases without the UACA truncating variants (Table S1). UACA has recently been identified as a novel regulator of apoptosis. It is known to reside within the Apaf-1/procaspase-9 complex and regulate apoptosis activating factor (APAF-1). It also regulates the apoptotic pathway by controlling the activation of nuclear factor (NF)-κB [22]. In addition, UACA gene expression has been shown to be down-regulated in non-small cell lung carcinoma (MIM 211980) [23]. Taken together, the loss of UACA in cancer cells might result in altered activation of apoptotic pathways, ultimately promoting genesis of CRC.
Another gene of particular interest was TWSG1. The detected truncating germline variant (p.Q41X) was present in two familial CRC cases (2/96 cases) and completely absent in 1,039 Finnish population matched controls (Table 2). Loss of the remaining normal TWSG1 allele was observed in both tumors indicating that the gene might act as a classical tumor suppressor gene (Figure 2). The index case with Q41X in family 1 developed CRC at the age of 53 and segregation analysis showed that the variant was inherited from the affected mother (Figure 3). The mother had developed CRC at the age of 68 and lung cancer at the age of 77. The variant did not segregate with CRC in family 2. Rare risk alleles of moderate penetrance are usually over-represented in familial cases; however co-segregation of disease is not always observed [19]. Previous studies have shown TWSG1 to be a regulator of BMP-signaling [24]. It is known to act downstream of TGF-β, inducing SMAD2 phosphorylation and mediating DNA binding on Smad3/4 consensus sites [25]. TWSG1 functions in cellular pathways that are essential in genesis of CRC, however, its exact role in these pathways remains to be clarified.
In summary, exome sequencing is a well-justified strategy for discovering cancer predisposing variants. The identification of predisposing variants has substantial implications for disease risk assessment and surveillance in family members. Here, we identified eleven candidate predisposing genes with truncating variations in familial CRC. A key challenge is how to identify predisposing variants in the background of non-pathogenic polymorphisms. Screening the eleven genes in familial CRC cases representing different populations will be important to gain robust evidence for pathogenicity, as well as to characterize the natural history of the respective phenotypes. This information, then, can be translated into tools for cancer prevention and early diagnosis in individuals carrying true predisposition alleles.
Materials and Methods
Samples
This study was reviewed and approved by the Ethics Committee of the Hospital district of Helsinki and Uusimaa (HUS). Signed informed consent or authorization from the National Supervisory Authority for Welfare and Health was obtained for all the study participants.
Discovery phase samples
A population-based material of normal and tumor tissue from 1,042 CRC patients was collected between 1994 and 1998 from nine Finnish central hospitals [4], [18]. After 1998 sample collection continued from two of the nine hospitals (unpublished collection). From the additional series 472 CRCs and respective normal tissue samples were available. From these materials, familial CRC cases were selected according to the following criteria: (i) at least one CRC case in a first degree relative, (ii) negative for any known high penetrance CRC mutation, and (iii) availability of sufficient amount of DNA extracted from normal tissue (Figure 1). As part of previous efforts [4], [18], all cases had been tested for MSI, and in positive cases Sanger sequencing of MLH1 and MSH2 had been performed. Known polyposis syndromes could be excluded for all the familial cases based on medical and pathological reports. In total, 96 familial cases fulfilled the above mentioned criteria. Clinical characteristics are presented in Table 1 and in more detail in Table S1. Both germline DNA extracted from blood or normal colonic tissue and corresponding fresh-frozen tumour DNA were available. Information on histological tumor grade and Dukes stage was obtained from pathology reports. The discovery phase control set compromised the following non-overlapping collections; in-house Finnish population matched control exome data (n = 212) and DNA samples (n = 310) from population matched healthy individuals obtained from the Finnish Red Cross Blood Transfusion Service. Around two-thirds of the control DNA samples were regionally matched controls.
Validation phase samples
Finnish population matched cases (n = 954) were selected from a population-based material of 1,042 CRC patients [4], [18] and from an additional series of 472 Finnish CRC cases, based on DNA availability. The control DNA samples (n = 586) were from population matched healthy individuals and obtained from the Finnish Red Cross Blood Transfusion Service.
Exome sequencing
The Agilent SureSelect Human All Exon Kit v1 (Agilent, Santa Clara, CA, USA) was used to capture exomic regions. Paired end short reads were sequenced on either Illumina GAII or HiSeq platform (Illumina Inc., San Diego, CA, USA). Raw sequence data was received in FASTQ format and quality checked with FASTQC (http://www.bioinformatics.bbsrc.ac.uk/projects/fastqc). All exomes passed the quality control. 3′ ends with high adapter similarity were removed by an in-house script whereafter reads were mapped to the human reference genome GRCh37 by BWA (Burrow-Wheelers Aligner). Duplicates were removed with Picard Tools (http://picard.sourceforge.net) MarkDuplicates. Local realignment was done by Genome Analysis Toolkit (GATK) IndelRealigner to improve the detection of small insertions and deletions. The initial single nucleotide variant (SNV) and indel calls needed for creating the GATK realignment intervals were made using samtools mpileup and downloaded from the 1000 genomes project Phase I indel calls ([16]the August 2010 release). Final SNV and indel calls were made using the GATK UnifiedGenotyper with a low variant quality score threshold (1.0).
Exome data analysis
Exome data analysis was performed in “Rikurator”, an in-house visualization and comparative analysis tool (unpublished). The tool allowed for simultaneous analysis of all the 96 exomes and interactive quality/control filtering. The following quality filters were used: (i) variants had to have a quality score ≥50, (ii) coverage had to be ≥6, and (iii) the percentage of mutated reads had to be ≥30. Truncating variants, including nonsense, frameshifting insertion and deletion, or splice-site alteration IVS +1, +2, −1, and −2, were extracted. Data was control filtered against population matched exome control data (n = 212) and data from the 1000 Genomes Project (Phase 1 release) [16]. Variants were excluded if present in the 1000 Genomes Project or exome control data at MAF>0.001 (Figure 1). Genes with truncating variants present in at least 2/96 cases were studied further. Manual filtering was performed on all variants to further remove artifacts due to duplicated regions, mapping errors, and systematic errors. Systematic errors, both position specific and sequence specific, in high-throughput sequence data have been described previously by Meacham et al [26]. Finally, outputs were generated for Ensembl canonical transcripts (Ensembl build 37).
Sanger validation of the exome findings
Potential loss-of-function variants were verified by Sanger sequencing from DNA extracted from normal tissue samples. Sequencing primers were designed with the Primer3-program (http://frodo.wi.mit.edu/primer3/) using NCBI37/Hg19 as the reference sequence. The primer sequences can be found in Text S1. The fragments were amplified with the AmpliTaqGold enzyme (Applied Biosystems, Foster City, CA). The PCR products were purified using the ExoSAP-IT PCR purification kit (USB Corporation, Cleveland, OH, USA). Electrophoresis was run on a 3730xl DNA Analyzer (Applied Biosystems at Institute for Molecular Medicine Finland, FIMM). The sequencing reactions were performed utilizing the Big Dye Terminator v.3.1 kit (Applied Biosystems, Foster City, USA), Sanger sequencing was performed implementing the ABI3100×l technology (Applied Biosystems), and the sequence graphs were visualized with the Chromas – software (version 2.33, Technelysium Pty Ltd, Helensvale, Australia). The results were analyzed both manually and with the Mutation Surveyor –software (version v3,30, Softgenetics, State College, PA, USA). Confirmed truncating variants were Sanger sequenced in 310 Finnish population matched healthy controls, of whom about two-thirds were regionally matched. Sanger sequencing was performed as described above. All variants that had a MAF>0.001 in the discovery phase control set were excluded.
Loss of heterozygosity analysis
Sanger sequencing was also performed on DNA extracted from tumor tissue in cases carrying validated truncating variants. All tumors had been microscopically evaluated by a pathologist and all except one contained ≥50% of carcinoma tissue. Loss of heterozygosity was analyzed by comparing allelic ratios of tumor and respective normal tissue DNA, as previously described [27]. Peak heights were manually measured from sequence graphs based on which allelic ratios were calculated.
Genotyping of population matched cases and controls
Variants in genes showing loss of the wild-type allele in tumor tissue were genotyped in a set of validation phase samples, comprising 954 population matched CRC cases and 586 population matched controls. Genotyping was carried out by using the 7900HT Fast Real-Time PCR System (Applied Biosystems) and was performed at the Estonian Genome Center, University of Tartu. The variant p.Q41X in TWSG1 was genotyped using massARRAY iPLEX Gold (Sequenom, San Diego, CA) and performed at the Institute for Molecular Medicine Finland (FIMM), University of Helsinki. The genotyping conditions and primers utilized can be found in Text S1. Genotyping success rates were over 90% for all the variants, except for PSPH where the genotyping assay failed. All the variants identified by genotyping were further confirmed by Sanger sequencing.
Missense variants at candidate CRC loci
The exome data was searched for missense variants at the 11 candidate predisposition loci. The same filtering criteria were utilized as for truncating variants. The variants were excluded if present in Exome Variant Server (NHLBI GO Exome Sequencing Project (ESP), Seattle, WA, http://evs.gs.washington.edu/EVS/ [July 2013]) with MAF>0.001. The functional effects of the identified missense variants were predicted by SIFT (http://sift.jcvi.org/) and PolyPhen 2 (http://genetics.bwh.harvard.edu/pph2/).
Segregation of truncating variants
Archived Formalin-fixed, Paraffin-embedded (FFPE) tissue samples were ordered for first degree relatives with CRC whenever possible. Genomic DNA was extracted from all available FFPE samples. Sanger sequencing was performed on identified truncating variants to test for segregation. In total, segregation was analyzed in seven families for five of the identified truncating variants.
Statistics
One-tailed exact binominal test was used for P-value calculations.
Supporting Information
{kind=link}
Zdroje
1. ParkinDM, BrayF, FerlayJ, PisaniP (2005) Global cancer statistics, 2002. CA Cancer J Clin 55 : 74–108.
2. American Cancer Society (2013) Cancer Facts & Figures 2013. Available: http://www.cancer.org/acs/groups/content/epidemiologysurveilance/documents/document/acspc-036845.pdf. Accessed 26 July 2013.
3. LichtensteinP, HolmNV, VerkasaloPK, IliadouA, KaprioJ, et al. (2000) Environmental and heritable factors in the causation of cancer–analyses of cohorts of twins from sweden, denmark, and finland. N Engl J Med 343 : 78–85.
4. AaltonenLA, SalovaaraR, KristoP, CanzianF, HemminkiA, et al. (1998) Incidence of hereditary nonpolyposis colorectal cancer and the feasibility of molecular screening for the disease. N Engl J Med 338 : 1481–1487.
5. PallesC, CazierJB, HowarthKM, DomingoE, JonesAM, et al. (2012) Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat Genet 45 : 136–144.
6. PearsonP, FrancomanoC, FosterP, BocchiniC, LiP, et al. (1994) The status of online mendelian inheritance in man (OMIM) medio 1994. Nucleic Acids Res 22 : 3470–3473.
7. LakenSJ, PetersenGM, GruberSB, OddouxC, OstrerH, et al. (1997) Familial colorectal cancer in ashkenazim due to a hypermutable tract in APC. Nat Genet 17 : 79–83.
8. ClearySP, ZhangW, Di NicolaN, AronsonM, AubeJ, et al. (2003) Heterozygosity for the BLM(ash) mutation and cancer risk. Cancer Res 63 : 1769–1771.
9. GudaK, MoinovaH, HeJ, JamisonO, RaviL, et al. (2009) Inactivating germ-line and somatic mutations in polypeptide N-acetylgalactosaminyltransferase 12 in human colon cancers. Proc Natl Acad Sci U S A 106 : 12921–12925.
10. LubbeSJ, Di BernardoMC, BroderickP, ChandlerI, HoulstonRS (2012) Comprehensive evaluation of the impact of 14 genetic variants on colorectal cancer phenotype and risk. Am J Epidemiol 175 : 1–10.
11. DunlopMG, DobbinsSE, FarringtonSM, JonesAM, PallesC, et al. (2012) Common variation near CDKN1A, POLD3 and SHROOM2 influences colorectal cancer risk. Nat Genet 44 : 770–776.
12. DunlopMG, TenesaA, FarringtonSM, BallereauS, BrewsterDH, et al. (2013) Cumulative impact of common genetic variants and other risk factors on colorectal cancer risk in 42,103 individuals. Gut 62 : 871–881.
13. NiittymakiI, KaasinenE, TuupanenS, KarhuA, JarvinenH, et al. (2010) Low-penetrance susceptibility variants in familial colorectal cancer. Cancer Epidemiol Biomarkers Prev 19 : 1478–1483.
14. PeltonenL, PalotieA, LangeK (2000) Use of population isolates for mapping complex traits. Nat Rev Genet 1 : 182–190.
15. JakkulaE, RehnstromK, VariloT, PietilainenOP, PaunioT, et al. (2008) The genome-wide patterns of variation expose significant substructure in a founder population. Am J Hum Genet 83 : 787–794.
16. Genomes Project Consortium (2012) AbecasisGR, AutonA, BrooksLD, DePristoMA, et al. (2012) An integrated map of genetic variation from 1,092 human genomes. Nature 491 : 56–65.
17. KuCS, NaidooN, PawitanY (2011) Revisiting mendelian disorders through exome sequencing. Hum Genet 129 : 351–370.
18. SalovaaraR, LoukolaA, KristoP, KaariainenH, AhtolaH, et al. (2000) Population-based molecular detection of hereditary nonpolyposis colorectal cancer. J Clin Oncol 18 : 2193–2200.
19. FletcherO, HoulstonRS (2010) Architecture of inherited susceptibility to common cancer. Nat Rev Cancer 10 : 353–361.
20. BodmerW, BonillaC (2008) Common and rare variants in multifactorial susceptibility to common diseases. Nat Genet 40 : 695–701.
21. GibsonG (2012) Rare and common variants: twenty arguments. Nat Rev Genet 13 : 135–145.
22. LiuL, SakaiT, SanoN, FukuiK (2004) Nucling mediates apoptosis by inhibiting expression of galectin-3 through interference with nuclear factor kappaB signalling. Biochem J 380 : 31–41.
23. MoravcikovaE, KrepelaE, ProchazkaJ, RousalovaI, CermakJ, et al. (2012) Down-regulated expression of apoptosis-associated genes APIP and UACA in non-small cell lung carcinoma. Int J Oncol 40 : 2111–2121.
24. ChangC, HoltzmanDA, ChauS, ChickeringT, WoolfEA, et al. (2001) Twisted gastrulation can function as a BMP antagonist. Nature 410 : 483–487.
25. TzachanisD, LiL, LafuenteEM, BerezovskayaA, FreemanGJ, et al. (2007) Twisted gastrulation (tsg) is regulated by tob and enhances TGF-beta signaling in activated T lymphocytes. Blood 109 : 2944–2952.
26. MeachamF, BoffelliD, DhahbiJ, MartinDI, SingerM, et al. (2011) Identification and correction of systematic error in high-throughput sequence data. BMC Bioinformatics 12 : 451-2105-12-451.
27. TuupanenS, NiittymakiI, NousiainenK, VanharantaS, MecklinJP, et al. (2008) Allelic imbalance at rs6983267 suggests selection of the risk allele in somatic colorectal tumor evolution. Cancer Res 68 : 14–17.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2013 Číslo 10
Nejčtenější v tomto čísle
- A GDF5 Point Mutation Strikes Twice - Causing BDA1 and SYNS2
- Dominant Mutations in Identify the Mlh1-Pms1 Endonuclease Active Site and an Exonuclease 1-Independent Mismatch Repair Pathway
- Eleven Candidate Susceptibility Genes for Common Familial Colorectal Cancer
- The Histone H3 K27 Methyltransferase KMT6 Regulates Development and Expression of Secondary Metabolite Gene Clusters