
Limiting of the Innate Immune Response by SF3A-Dependent Control of MyD88 Alternative mRNA Splicing
Controlling infectious disease without inducing unwanted inflammatory disease requires proper regulation of the innate immune response. Thus, innate immunity needs to be activated when needed during an infection, but must be limited to prevent damage. To accomplish this, negative regulators of innate immunity limit the response. Here we investigate one such negative regulator encoded by an alternative splice form of MyD88. MyD88 mRNA exists in two alternative splice forms: MyD88L, a long form that encodes a protein that activates innate immunity by transducing Toll-like receptor (TLR) signals; and a short form that encodes a different protein, MyD88S, that inhibits the response. We find that MyD88S levels regulate the extent of inflammatory cytokine production in murine macrophages. MyD88S mRNA levels are regulated by the SF3A and SF3B mRNA splicing complexes, and these mRNA splicing complexes function with TLR signaling to regulate MyD88S production. Thus, the SF3A mRNA splicing complex controls production of a negative regulator of TLR signaling that limits the extent of innate immune activation.
Published in the journal:
. PLoS Genet 9(10): e32767. doi:10.1371/journal.pgen.1003855
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1003855
Summary
Controlling infectious disease without inducing unwanted inflammatory disease requires proper regulation of the innate immune response. Thus, innate immunity needs to be activated when needed during an infection, but must be limited to prevent damage. To accomplish this, negative regulators of innate immunity limit the response. Here we investigate one such negative regulator encoded by an alternative splice form of MyD88. MyD88 mRNA exists in two alternative splice forms: MyD88L, a long form that encodes a protein that activates innate immunity by transducing Toll-like receptor (TLR) signals; and a short form that encodes a different protein, MyD88S, that inhibits the response. We find that MyD88S levels regulate the extent of inflammatory cytokine production in murine macrophages. MyD88S mRNA levels are regulated by the SF3A and SF3B mRNA splicing complexes, and these mRNA splicing complexes function with TLR signaling to regulate MyD88S production. Thus, the SF3A mRNA splicing complex controls production of a negative regulator of TLR signaling that limits the extent of innate immune activation.
Introduction
The innate immune response plays a key role in fighting infection [1]. Innate immune signaling regulates the process of inflammation, which involves the secretion of cytokines and chemokines and the resulting recruitment and activation of innate and adaptive immune cells [1]. Thus, the activation of the innate immune response is an important positive step in the response to infection. However, an acutely overactive innate immune response or a chronically activated innate immune response can contribute to the pathogenesis of many diseases including sepsis, atherosclerosis, Crohn's disease, and cancer [2]–[5]. Thus it is critical that innate immunity be tightly controlled, activated when necessary and kept inactivate when not. A complex system of receptors and signal transduction molecules control the activation of the innate immune response induced by numerous stimuli. One key class of innate receptors is the Toll-like receptor (TLR) family; different TLRs recognize different pathogen-associated molecular patterns (PAMPs). For example, TLR4 is responsible for the response to lipopolysacharide (LPS) from Gram negative bacteria while TLR2 is responsible for the response to various lipopeptides present in Gram positive bacteria [6], [7].
In addition to the complex regulatory mechanism that controls innate immune activation, there are many negative regulators of innate immunity that limit the response, thereby limiting potential damage due to overactive or chronic inflammation [8]–[13]. Many such negative regulators of innate immunity have been described, including proteins that bind to and inactivate TLR signaling components [8]–[13]; deubiquitinases that inactivate ubiquitinated TLR signaling components [14]; microRNAs that regulate expression of TLR signaling genes [15]; and alternate mRNA splice forms of innate immunity genes, some of which are known to inhibit TLR signaling [16]–[22].
One such alternatively spliced TLR signaling gene is MyD88 [23]–[26]. MyD88 is a TLR signaling adaptor that acts downstream of most but not all TLRs, and that acts as a positive regulator of innate immunity [6], [7]. In response to stimulation with PAMPs, TLRs dimerize and recruit MyD88. This activates MyD88, which in turn recruits and activates several IL-1 receptor-associated kinases (IRAKs). This complex signal transduction cascade continues, ultimately leading to the production of inflammatory cytokines. MyD88 is encoded by an mRNA with five exons (long form or MyD88L). However, a shorter MyD88 mRNA (MyD88S) also has been described in both mice and humans [23]–[27]; this mRNA is missing the 135 base pair second exon. Overexpression studies have demonstrated that MyD88S is a negative regulator of TLR signaling that fails to recruit and phosphorylate the IRAKs [23], [24], [26]. MyD88S mRNA levels are drastically increased in monocytes from septic patients suggesting that alterations in MyD88 mRNA splicing could play an important role in human disease [27]. However, thus far, the MyD88S loss of function phenotype has not been reported, and the mechanisms controlling MyD88 alternative mRNA splicing have not been investigated.
Using RNAi screens in mouse macrophage cell lines, we previously identified two members of the SF3A mRNA splicing regulatory complex as regulators of the innate immune response to LPS [28]. The SF3A complex is composed of three proteins (SF3A1, SF3A2, and SF3A3) that interact with the U2 snRNP (small nuclear ribonucleoprotein) [29]. The U2 snRNP interacts with the pre-mRNA branch point near the 3′ splice site in pre-mRNA [30] and facilitates mRNA splicing in conjunction with the rest of the spliceosome [31]–[34]. All three SF3A subunits are required for mRNA splicing [29], [35]–[39].
Here we investigate the innate immune regulatory function of the SF3A mRNA splicing complex and the related splicing factor, SF3B1 which is part of the SF3B complex that functions at a similar step of mRNA splicing to SF3A [40]–[42]. We find that SF3A and SF3B are both required for a robust innate immune response to LPS and other TLR agonists. SF3A and SF3B do so in conjunction with TLR signaling, in part, by regulating the production of the alternate inhibitory splice form MyD88S. We also show that changing MyD88S mRNA levels can significantly impact the strength of the innate immune response.
Results
The SF3A mRNA splicing complex regulates the innate immune response
We previously found that inhibition of either Sf3a1 or Sf3a2 by RNAi strongly diminished LPS-induced inflammatory cytokine production in either of two mouse macrophage cell lines, RAW264.7 and J774A.1 [28]. To confirm that all three SF3A subunits (SF3A1, SF3A2, and SF3A3) regulated the LPS response, we used siRNAs to inhibit each Sf3a subunit in the RAW264.7 macrophage cell line and then monitored LPS-induced production of inflammatory cytokines. Inhibition of each Sf3a subunit led to a strong decrease in IL-6 protein production and a more moderate decrease in TNFα protein production (Figure 1A, B). The effect on IL-6 roughly correlated with the extent of knockdown of each subunit as measured by qPCR; siRNA directed against either Sf3a1 or Sf3a3 exhibited the strongest gene knockdown (Figure 1C) and the strongest IL-6 phenotypes (Figure 1A). The effect on IL-6 occurred at both the protein (Figure 1A) and mRNA level, as the amount of IL-6 mRNA also was strongly diminished by Sf3a1 or Sf3a3 inhibition (Figure 1D). In prior control experiments using RNAi in mouse macrophage cell lines, we found that inhibition of control genes such as TLR4 and MyD88 led to a stronger IL-6 than TNFα phenotype [43], suggesting that the differing extent of inhibition of IL-6 and TNFα production when SF3A is inhibited could be due to differences in the effect of SF3A or could be due to differing sensitivities of the two cytokine promoters to perturbation of signaling.

Because SF3A regulates mRNA splicing, an essential process, it was formally possible that the defects in LPS-induced cytokine production were caused by an overall non-specific decrease in mRNA splicing or fitness caused by Sf3a inhibition. However, as outlined below, several experiments argue against this possibility and argue for some specificity. First, other macrophage functions were not affected by Sf3a inhibition, including phagocytosis of FITC-labeled E. coli particles (Figure 1E). Second, cell viability was not altered by Sf3a inhibition (Figure 1F). Third, tests of several other mRNA splicing events indicated that many other mRNA splicing events were not affected when Sf3a1 was inhibited. For example, we monitored βactin mRNA levels when Sf3a1 was inhibited by RNAi, comparing expression by qPCR using primers that both annealed to exon 4 or primers that annealed to exons 3 and 4 (thus crossing intron 3); we observed no significant difference between the two primer sets (Figure S1). Similarly, using primers bracketing intron 6 of the Macf1 gene (another innate immune regulator [28]), we observed no change when Sf3a1 was inhibited by RNAi (101±13% of control, mean±SEM). Finally, we also monitored alternative mRNA splicing of a TLR regulatory gene known to be alternatively spliced, MD-2. MD-2 exists in two isoforms, MD-2, which along with TLR4 is involved in LPS recognition, and MD-2B, which lacks part of exon 3 and acts as a negative regulator of TLR4 signaling [21]. We observed no significant alteration in production of MD-2 or MD-2B mRNA when Sf3a1 was inhibited by RNAi (Figure S2).
The mRNA splicing factor SF3B1 also regulates innate immunity
The fact that inhibition of any of the three Sf3a subunits diminished LPS-induced cytokine production suggested that the effect of SF3A on innate immunity was due to its mRNA splicing function. To further test this hypothesis, we used RNAi to inhibit the SF3B1 mRNA splicing factor. RNAi-mediated inhibition of Sf3b1 also strongly reduced LPS-induced IL-6 production (Figure 1G) and, as expected, diminished Sf3b1 mRNA levels (Figure 1H). Thus, both the SF3A and SF3B complexes are required for a robust innate immune response to LPS, as inhibition of either diminished that response.
The effects of SF3A1 and SF3B1 were not unique to mouse macrophages; we also found that RNAi-mediated inhibition of Sf3a1 or Sf3b1 greatly diminished LPS-induced IL-6 production in human THP1 differentiated macrophages while having only a moderate (SF3B1) or no (SF3A1) effect on viability in these cells (Figure S3).
As a second method to confirm that SF3B1 was required for LPS-induced cytokine production, we used a known pharmacological inhibitor of SF3B1, spliceostatin A (SSA) [44]–[46], to inhibit SF3B1 and then monitored LPS-induced cytokine production. SSA at doses greater than 3 ng/ml diminished overall survival of RAW264.7 cells; these high doses of SSA also diminished the cells' ability to phagocytose FITC-labeled E. coli particles (Figure S4). However, at lower SSA doses that did not diminish survival or phagocytic ability, treatment of RAW264.7 macrophages with SSA led to a profound decrease in LPS-induced IL-6 production (Figure 1I, S4). Thus, inhibition of SF3B1 by RNAi or with a pharmacological agent strongly inhibited the innate immune response to LPS.
To determine if these mRNA splicing regulators affected other innate immune stimuli, we also monitored the effect of SF3A1 and SF3B1 when cells were stimulated with the TLR2/1 agonist PAM3CSK4 [47]. Inhibition of either Sf3a1 or Sf3b1 by RNAi led to a strong decrease in IL-6 protein, IL-6 mRNA, and TNFα levels (Figure 2). The effect of SF3A1 and SF3B1 did not extent to all stimuli, however, as inhibition of these genes by RNAi did not inhibit the response to the TLR3 agonists poly(I:C) (Figure S5) or poly(A:U) (data not shown) [we monitored production of TNFα rather than IL-6 in these assays because poly(I:C) induced little IL-6].

TLR4 uses two signaling adaptors, MyD88 and TRIF, to control the response to LPS. SF3A1 and SF3B1 likely regulate MyD88 signaling, because they affect LPS and PAM3CSK4-induced production of IL-6 and TNFα. To determine if SF3B1 affects the TRIF-dependent arm of the TLR4 pathway, we monitored production of IFNβ by qPCR when SF3B1 was inhibited by SSA; we found that SSA treatment diminished both IL-6 and IFNβ production (Figure S6), indicating that SF3B1 has both MyD88-dependent and MyD88-independent effects downstream of TLR4.
SF3A1 acts upstream in the TLR4 signaling pathway
To investigate the relationship between SF3A1 and TLR signaling further, we used RNAi to inhibit Sf3a1 in cells that expressed activated TLR signaling components (Figure 3A). These activated constructs included a constitutively active IKK construct containing two mutations in the active site [IKK-2S177ES181E [48]] and an inducibly active MyD88 construct [49]. MyD88 was rendered inducibly active in a construct in which full length MyD88 is fused to the effector domain of subunit B of E. coli DNA gyrase [49]; treatment of cells with the antibiotic coumermycin [49] leads to dimerization of DNA gyrase B and thus dimerization and activation of MyD88. As a negative control, we also inhibited Sf3a1 by RNAi in macrophages expressing chloramphenicol acetyltransferase (CAT), which should not alter innate immunity.
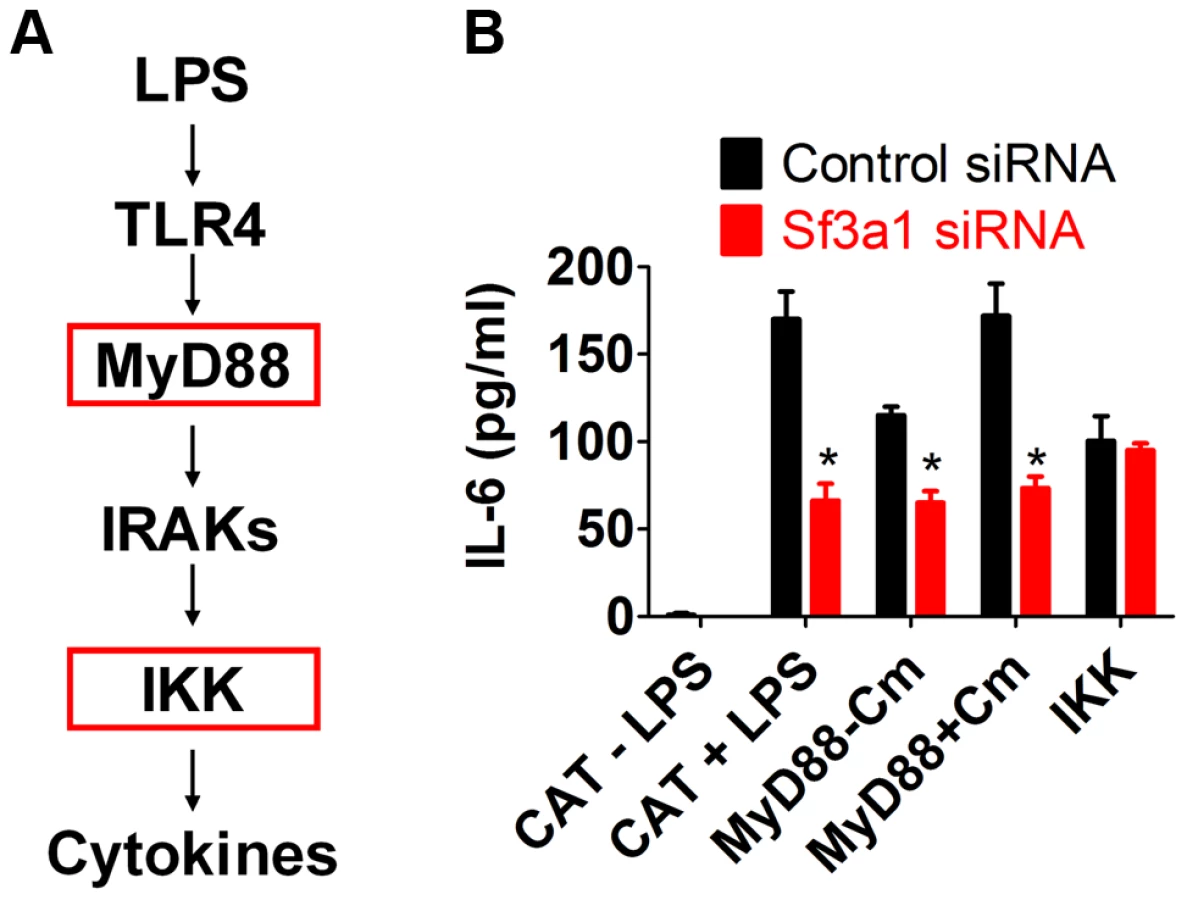
As expected, cells expressing the negative control protein CAT produced IL-6 in the presence but not the absence of LPS, and further inhibition of Sf3a1 in these cells led to a strong decrease in IL-6 production (Figure 3B). Cells overexpressing the MyD88-gyrB fusion produced IL-6 in the absence of LPS, and this was enhanced by the addition of coumermycin, as expected (Figure 3B). The MyD88-gyrB fusion expresses full length MyD88 protein. MyD88 mRNA levels were 162±24 (mean±SEM)-fold higher than normal (determined by qPCR) in cells overexpressing MyD88-gyrB, and this high level of full length MyD88 may be able to activate signaling even in the absence of the dimerizing agent. Nevertheless, Sf3a1 siRNA diminished IL-6 production when MyD88-gyrB was overexpressed (Figure 3B). However, Sf3a1 inhibition did not prevent IL-6 production induced by overexpression of the constitutively activated IKK construct (Figure 3B). These data are consistent with SF3A1 exerting its effect on TLR signaling pathways, acting downstream of MyD88 and upstream of IKK.
Inhibition of SF3A1 or SF3B1 enhances production of MyD88S
Because multiple SF3A subunits and SF3B1 all regulate LPS-induced cytokine production, we hypothesized that these mRNA splicing factors were regulating the splicing of a TLR-regulatory gene(s). Moreover, this gene likely acts downstream of MyD88 and upstream of IKK. One candidate that fits these data is MyD88 itself, as the alternate splice form MyD88S inhibits IRAK activation downstream of MyD88 and upstream of IKK [23], [24], [26]. We therefore chose to monitor MyD88 mRNA splicing by qPCR using primers that can distinguish between the five exon MyD88L splice form and the four exon MyD88S splice form (Figure 4A). For the qPCR studies, we detected MyD88L using a primer that spanned exons 2 and 3 and a second primer in exon 3 (Figure 4A). We detected MyD88S using a primer that spanned exons 1 and 3 and a second primer in exon 3 (Figure 4A). Inhibition of Sf3a1 in either the presence or absence of LPS did not alter MyD88L mRNA levels substantially but did increase the MyD88S shorter splice form (Figure 4B). Both LPS and Sf3a1 inhibition contributed to this increase in MyD88s mRNA (Figure 4B).

To test if SF3B1, like SF3A1, regulated MyD88S mRNA levels, we inhibited SF3B1 using either RNAi or SSA treatment and monitored MyD88L and MyD88S mRNA by qPCR. Inhibition of Sf3b1 by RNAi, like inhibition of Sf3a1, did not significantly alter MyD88L mRNA levels in the presence of LPS but did significantly increase MyD88S mRNA levels in the presence of LPS (Figure 4C). Similarly, inhibition of SF3B1 by SSA had at most a moderate effect on MyD88L mRNA levels while leading to a substantial increase in MyD88S mRNA levels (Figure 4D). As a control to confirm that not all siRNA treatments altered MyD88 mRNA splicing, we found that RNAi-mediated inhibition of the 26S proteasome subunit Psmd3 greatly diminished LPS-induced IL-6 production [43] (Figure S7A) without significantly altering production of MyD88L or MyD88S (Figure S7B,C).
The qPCR data indicated that one of the wild type functions of SF3A1 and SF3B1 is to inhibit production of MyD88S, as MyD88S levels are increased when either mRNA splicing factor is inhibited. To confirm these MyD88 mRNA qPCR data, we monitored MyD88L and MyD88S mRNA levels using semi-quantitative reverse transcription-PCR followed by agarose gel elecrophoresis. For these experiments, we used two sets of primers, one set designed to specifically monitor MyD88S levels and one set designed to monitor both MyD88L and MyD88S simultaneously (Figure 4A). For the MyD88S-specific primers, we used a reverse primer that spanned exons 3 to 1 and a forward primer that annealed to exon 1 (Figure 4A); these primers generate a 124 bp product when MyD88S is present. For primers that amplify both MyD88L and MyD88S, we used primers that annealed to exons 1 and 3 (Figure 4A); these generate a 370 bp band corresponding to MyD88L and a 235 bp band corresponding to MyD88S.
Using the MyD88S-specific RT-PCR primers (Figure 4A), we found that treatment of cells with either LPS or Sf3a1 siRNA alone had a fairly moderate effect on MyD88S mRNA levels (agarose gel in Figure 5A, quantitation from three independent experiments in Figure 5B). However, cells treated with both LPS and Sf3a1 siRNA exhibited a substantial increase in MyD88S mRNA (Figure 5A, B). As a control, we found that these treatments did not alter production of βactin (Figure 5A, C).

Using the reverse transcription primers that amplified both MyD88L and MyD88S simultaneously (Figure 4A), we were able to visualize MyD88L and MyD88S (Figure 5D). Consistent with the qPCR data, treatment of cells with LPS and Sf3a1 siRNA either alone or together did not significantly alter MyD88L mRNA levels (agarose gel in Figure 5D, quantitation from three independent experiments in Figure 5E). However, treatment of cells with LPS and Sf3a1 siRNA did significantly enhance MyD88S mRNA levels (Figure 5D, F and S8).
Changes in MyD88S levels can have a significant impact on LPS-induced cytokine production
The RT-PCR experiments using primer sets that amplified both MyD88L and MyD88S allowed us to estimate the ratio of MyD88L to MyD88S. In the absence of treatment, this ratio was roughly 20∶1; in the presence of LPS and Sf3a1 siRNA, the MyD88L to MyD88S ratio increased significantly to approximately 5∶1. This raised the question of whether this change in MyD88S was sufficient to overcome the greater MyD88L level and alter innate immunity significantly. To test this directly, we engineered a siRNA that spanned the exon 1–3 boundary in MyD88S (and would thus not be present in MyD88L) with the goal of designing an siRNA that specifically inhibits MyD88S but not MyD88L. We tested two siRNAs in this fashion that differed in their start location by only one base; one did not affect MyD88S levels significantly (not shown) but the other did, as described below.
Treatment of macrophages with the MyD88S-specific siRNA led to a roughly 75% decrease in MyD88S mRNA levels while having a more moderate effect on MyD88L mRNA (Figure 6A), indicating some degree of efficacy and specificity. As a control, treatment of cells with a pool of four siRNAs targeting total MyD88 led to an 80% decrease in both MyD88L and MyD88S levels, as expected (Figure 6A). We therefore used these siRNAs to monitor the effect of changes in total MyD88 or MyD88S levels on LPS-induced cytokine production. Inhibition of total MyD88 (MyD88L+MyD88S) decreased LPS-induced IL-6 production (Figure 6B), as expected. In contrast, inhibition of MyD88S using the MyD88S-specific siRNA led to significantly increased LPS-induced IL-6 production (Figure 6B). This near doubling in cytokine production was impressive as the knockdown of MyD88S using this siRNA was incomplete and there was also some effect on MyD88L, both of which could allow us to underestimate the true scope of the effect of MyD88S.

MyD88S mediates some of the effect of Sf3a1 inhibition on innate immunity
It was possible that all of the effects of SF3A1 on innate immunity could be mediated by changes in MyD88S levels; alternatively, it was possible that SF3A1 affects production of several regulators of TLR signaling including MyD88S. To distinguish between these possibilities, we treated cells with two siRNAs simultaneously: siRNA targeting Sf3a1 and siRNA targeting MyD88S. Inhibition of Sf3a1, as described above, strongly diminished LPS-induced IL-6 production (Figure 6C). Inhibition of Sf3a1 and MyD88S simultaneously partially restored IL-6 production (Figure 6C). Thus, at least some of the effect of Sf3a1 inhibition is mediated by increases in MyD88S levels. As a control, qPCR used to monitor gene knockdown demonstrated that the various siRNA treatments were acting as expected (Figure S9). However, the small effect of the MyD88S-specific siRNA on MyD88L makes it difficult to determine with certainty if this incomplete rescue is due to limitations of the RNAi experiment or because other TLR regulators are also affected by SF3A1. Regardless, it is clear that changes in MyD88S levels account for at least part of the effect of SF3A1 on innate immunity.
Discussion
MyD88S modulates the extent of the innate immune response
MyD88S has been observed in multiple mouse and human cells, cell lines, and tissues [23]–[25], [27], [50], [51]. Prior studies investigating MyD88S used overexpression or reintroduction strategies to demonstrate that MyD88S was a TLR signaling inhibitor that functioned by inhibiting IRAK phosphorylation [23], [24], [26]. By developing a siRNA that targets the unique splice junction in MyD88S, we have now been able to demonstrate in a MyD88S loss-of-function experiment that MyD88S is, as demonstrated in the published overexpression studies, an inhibitor of the innate immune response. Moreover, we show that while there is far more MyD88L than MyD88S in macrophages, changes in the amount of the minor MyD88S splice form are sufficient to overcome the much larger pool of MyD88L and affect inflammatory cytokine production. This is consistent with published overexpression data that indicate that MyD88S is able to inhibit a large pool of MyD88L [24].
The SF3A/SF3B mRNA splicing complexes regulate the innate immune response in part by regulating MyD88S levels
We have found that inhibition of the SF3A or SF3B complexes by RNAi or with a pharmacological agent leads to a strong decrease in production of the pro-inflammatory cytokine IL-6 and a more moderate decrease in production of the pro-inflammatory cytokine TNFα without affecting macrophage viability or phagocytosis. Other reports indicate that RNAi-mediated knockdown of Sf3a subunits can affect cell survival in HeLa cells [38]; moreover, a knockout in Sf3b1 in mice is lethal, although heterozygous Sf3b1 mice develop largely normally [52]. The difference in our data may be due to incomplete but still very strong knockdown in macrophages or other cell-type-specific differences.
One of the innate immune targets of SF3A and SF3B is production of the alternate splice form of MyD88, MyD88S (Figure 7). The spliceosome is a large multi-subunit protein and RNA complex that facilitates intron removal in two catalytic steps. First, the 5′ splice site is cleaved resulting in the formation of a lariat structure. Second, the 3′ splice site is cleaved and the exons are ligated together [29], [32], [33]. The SF3A and SF3B complexes in conjunction with the U2 snRNP bind to the branch site near the 3′ splice site to facilitate mRNA splicing [30], [35]–[42], . These mRNA splicing complexes play an important role in proper 3′ splice site recognition; this is evidenced by the frequent observation of altered mRNA splicing observed when SF3B1 is inhibited or mutated [44], [54]–[56]; in many cases, inappropriate exon skipping is observed. This phenomenon of altered splicing and exon skipping is also consistent with the effect of SF3A1 or SF3B1 inhibition on MyD88S production; MyD88S is produced if the 3′ splice site at the end of intron 1 is skipped and the 3′ splice site at the end of intron 2 is used instead (Figure 7). It is intriguing to speculate that the MyD88 splice site choice evolved to be exquisitely sensitive to cellular conditions because of its functional significance, and may be a key point of regulation to limit inflammation.

The SF3A/SF3B mRNA splicing complexes likely also affect other genes to modulate innate immunity
While our data indicate the importance of MyD88S in mediating the innate immune effects of SF3A/SF3B, our data also suggest that other regulator(s) of TLR signaling also are affected by these mRNA splicing regulators. Inhibition of MyD88S using our MyD88S-specific siRNA is only able to partially rescue the innate immune defect caused by inhibition of Sf3a1. It is unclear if this incomplete rescue is due to limitations of the RNAi experiment or because another gene(s) are also regulated by SF3A1, although we favor the latter possibility. Several other pieces of data argue that SF3A/SF3B are affecting other genes besides MyD88 to regulate innate immunity, including the observation that SF3B1 inhibition can affect LPS-induced IFNβ production. Moreover, we did not observe significant inhibition of IRAK1 activation when SF3a1 is inhibited (data not shown); while the regulation of IRAK1 by MyD88S has not been studied in a loss-of-function context, these data also are consistent with the possibility that SF3a1 regulates innate immunity using both MyD88S-dependent and independent means.
mRNA splicing factors, MyD88S, and human disease
By inhibiting MyD88S using a MyD88S-specific siRNA, we demonstrated a significant increase in inflammatory cytokine production. Even a moderate increase in inflammation could have a significant impact on disease. Adib-Conquy et al. [27] observed that there was a roughly 10-fold increase in MyD88S levels in monocytes from patients with sepsis, which could explain the immunosuppressed phenotype of these cells. The SF3B1 and SF3A1 mRNA splicing factors have also been implicated in the pathogenesis of numerous hematologic malignancies. In particular, Sf3b1 mutations are prevalent in a wide range of hematologic malignancies, associated with 5% of acute myeloid leukemia cases, 10–15% of chronic lymphocytic leukemia cases, and 60–80% of the myelodysplastic syndrome subtype Refractory Anemia with Ring Sideroblasts (MDS-RARS) [57]–[68]. Chronic inflammation has been implicated in the pathogenesis of many solid and hematologic malignancies [4], [69]–[73]; conceivably, altered innate immunity signaling could be one of the factors involved in the pathogenesis of these malignancies.
The relationship between TLR signaling, MyD88, and SF3A1 function
MyD88S inhibits TLR signaling at the level of the IRAK kinases [23], [24], [26] (Figure 7). We have now demonstrated that the SF3A and SF3B complexes inhibit production of this alternative MyD88S splice form (Figure 7). Prolonged LPS exposure is reported to enhance MyD88S production [24], and our data also indicates that LPS can enhance MyD88S levels (Figure 7). It is possible that SF3A1 could act either in parallel to or downstream of TLR signaling to control MyD88S production.
How might the TLR pathway interact with the SF3A mRNA splicing complex? Some intriguing published protein interaction studies raised the possibility that MyD88 itself could directly regulate the SF3A complex to modulate its own mRNA splicing. Human SF3A3 has been reported to bind to MyD88 in liver cells using an immunoprecipitation-mass spectrometry assay [74]. The C. elegans SF3A1 ortholog has been reported to bind to the sole C. elegans MyD88 family member TIR-1 in a yeast 2 hybrid assay [75]. TIR-1 is most homologous to mammalian SARM, a negative regulator of mammalian innate immunity [76], [77], but functionally behaves most like MyD88, in that it is required for host defense to many classes of pathogens and acts upstream of the p38 MAPK pathway [78]–[80]. Finally, Drosophila SF1 (another mRNA splicing factor that interacts with the U2 snRNP) has been reported to interact with Drosophila MyD88 in a yeast two hybrid assay [81]. While the SF3A complex functions in the nucleus to control mRNA splicing, it is assembled in the cytoplasm [82], [83] and could be available for modification by MyD88, at least transiently. Future studies will be needed to determine the biological significance of this interaction. However, these data raise the possibility of a MyD88-mediated negative feedback loop that ensures that innate immunity is self limiting (Figure 7); this could be relevant to sepsis, cancer, and the myriad of other diseases with an inflammatory component [84].
Materials and Methods
RNAi experiments
RNAi using RAW264.7 cells was performed largely as described [43]. In brief, pools of siRNA duplexes (Dharmacon) targeting the indicated genes were transfected into the mouse macrophage cell line RAW264.7 using the Amaxa 96-well nucleofector shuttle. Cells were then either plated in 100,000 cells per well in 96-well format for subsequent ELISA assays or 200,000 cells/well in 24-well format for subsequent qPCR analysis. The MyD88S-specific siRNA sequence was 5′-GAAGTCGCGCATCGGACAA-3′. We also tested a second siRNA that was shifted one base pair 3′; this siRNA did not significantly inhibit MyD88S mRNA levels. Negative control siRNAs were Dharmacon's non-targeting siRNA pool #1 and in some cases non-targetting siRNA #1. Twenty-four hours after siRNA transfection, cells were stimulated with the indicated PAMPs for six hours. Doses used were 20 ng/ml LPS (List Biological Labs), 1.5 µg/ml PAM3CSK4 (Invivogen), or 6 µg/ml poly(I:C) (Invivogen). Several different assays were then performed. In some cases, cell supernatants were collected and cytokine production was monitored by ELISA (R&D Biosystems). Viability of the remaining adherent cells was monitored using fluorescein diacetate, which is cleaved into a fluorescent form in intact live cells [85]. In other experiments, RNAi-treated cells were analyzed for their ability to phagocytose FITC-labeled E. coli particles using the Vybrant Phagocytosis assay kit (Molecular Probes). In other experiments, RNAi-treated cells were lysed in RLT buffer (Qiagen) and used to prepare RNA for qPCR or RT-PCR.
In the experiments using two different siRNAs at the same time in RAW264.7 cells, the siRNAs were transfected simultaneously with an equal volume of each. In these dual transfection experiments, when only a single siRNA was used, the volume of siRNA was made up with an equal volume of Dharmacon non-targetting siRNA pool #1.
RNAi in the human THP1 cell line was performed by first transfecting pools of siRNAs targeting either SF3A1 or SF3B1 (and control non-targeting siRNA pool) using the Amaxa 96-well nucleofector shuttle. Cells were then plated at 200,000 cells per well in 96-well format. 8 hours later, 50 ng/ml phorbol 12-myristate 13-acetate (PMA) was added. 24 hours later, the cells were exposed to 50 ng/ml LPS for six hours, and then IL-6 production and viability were monitored.
Treatment of cells with both siRNA and overexpression plasmids
In the experiments in which both plasmids and siRNAs were delivered to RAW264.7 macrophages, cells were first transfected in 12 well-format with the indicated plasmids (1.6 µg each) using 0.7% (v/v) Fugene HD (Roche) for transfection. 24 hours later, cells were transfected with siRNA in 96 well-format using the Amaxa system. 24 hours later, cells were exposed to either of 0.1 µM Coumermycin A (Sigma) or LPS for six hours as indicated, and then cytokine production was monitored by ELISA. Transfection efficiency using Fugene HD was roughly 40% (determined using a plasmid expressing GFP and fluorescence microscopy) and was not affected by the different siRNA treatments.
qPCR and RT-PCR to monitor mRNA levels
RNA for analysis was prepared by lysing cells in RLT buffer and using the Qiagen RNAeasy kit for RNA purification. qPCR was performed on an ABI 7900 using the Qiagen Quantitect SYBR-green assay kit. Data was normalized using βactin as a control. Because we were monitoring the effect of known mRNA splicing regulators, we tested primers that were entirely internal to exon 4 and primers that crossed intron 3; both gave similar results in all experiments.
mRNA levels were also analyzed in semi-quantitative fashion using RT-PCR followed by agarose gel electrophoresis. 500 ng total RNA prepared as described above was first subjected to reverse transcription using Superscript III reverse transcriptase (Invitrogen). The reverse transcribed cDNA was then subjected to PCR using Taq DNA polymerase (Invitrogen), PCR products were subjected to electrophoresis in 2% agarose gels, and images were captured using a UV box and a CCD camera.
The sequences of all primers are listed in Table S1.
We were unable to visualize MyD88S protein by western blot using three different commercial sources of MyD88 antisera; presumably, this is reflective of the large ratio of MyD88L∶MyD88S that we observed using RT-PCR (Figure 5D). This also is consistent with prior published studies in which MyD88S protein could be monitored by western blot in RAW264.7 cells in which the entire pool of MyD88L is artificially converted to MyD88S using antisense oligonucleotides [86], but not in otherwise wild-type cells [24].
Use of Spliceostatin A (SSA) to inhibit SF3B1
Cells were treated with the indicated doses of SSA (diluted in methanol from a 100 µg/ml stock in methanol) for six hours. Methanol was kept at less than <0.5% total volume, was matched in control cells, and didn't alter cytokine production or viability. After the six hour SSA exposure, LPS was added to the media containing SSA for an additional six hours. Then viability, phagocytosis, cytokine production, and various mRNA levels, were analyzed as outlined above.
Statistical analyses
All data are from a minimum of three biological replicates. Data were graphed and analyzed using Graphpad Prism 5. Statistically significant differences in all experiments were considered p<0.05 and were determined using t-tests. Bands on agarose gels were quantified using Image J [87] and subsequently analyzed for significance in Graphpad Prism 5. While we only display representative images of agarose gels from one experiment, the analyzed data and statistics are from three independent experiments.
Supporting Information
Zdroje
1. Kaufmann SHE, Medzhitov, Ruslan, Gordon, Siamon (2004) The innate immune response to infection. Washington D.C.: ASM Press.
2. ChaudhuriN, DowerSK, WhyteMK, SabroeI (2005) Toll-like receptors and chronic lung disease. Clin Sci (Lond) 109 : 125–133.
3. CookDN, PisetskyDS, SchwartzDA (2004) Toll-like receptors in the pathogenesis of human disease. Nat Immunol 5 : 975–979.
4. GrivennikovSI, GretenFR, KarinM (2010) Immunity, inflammation, and cancer. Cell 140 : 883–899.
5. TakedaK, AkiraS (2005) Toll-like receptors in innate immunity. Int Immunol 17 : 1–14.
6. KawaiT, AkiraS (2010) The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol 11 : 373–384.
7. TakeuchiO, AkiraS (2010) Pattern recognition receptors and inflammation. Cell 140 : 805–820.
8. KondoT, KawaiT, AkiraS (2012) Dissecting negative regulation of Toll-like receptor signaling. Trends Immunol 33 : 449–458.
9. LangT, MansellA (2007) The negative regulation of Toll-like receptor and associated pathways. Immunol Cell Biol 85 : 425–434.
10. LiewFY, XuD, BrintEK, O'NeillLA (2005) Negative regulation of toll-like receptor-mediated immune responses. Nature Reviews Immunology 5 : 446–458.
11. LuYC, YehWC, OhashiPS (2008) LPS/TLR4 signal transduction pathway. Cytokine 42 : 145–151.
12. MurrayPJ, SmaleST (2012) Restraint of inflammatory signaling by interdependent strata of negative regulatory pathways. Nat Immunol 13 : 916–924.
13. WangJ, HuY, DengWW, SunB (2009) Negative regulation of Toll-like receptor signaling pathway. Microbes Infect 11 : 321–327.
14. SunSC (2008) Deubiquitylation and regulation of the immune response. Nat Rev Immunol 8 : 501–511.
15. AlamMM, O'NeillLA (2011) MicroRNAs and the resolution phase of inflammation in macrophages. Eur J Immunol 41 : 2482–2485.
16. GrayP, MichelsenKS, SiroisCM, LoweE, ShimadaK, et al. (2010) Identification of a novel human MD-2 splice variant that negatively regulates Lipopolysaccharide-induced TLR4 signaling. J Immunol 184 : 6359–6366.
17. HardyMP, O'NeillLA (2004) The murine IRAK2 gene encodes four alternatively spliced isoforms, two of which are inhibitory. J Biol Chem 279 : 27699–27708.
18. IwamiKI, MatsuguchiT, MasudaA, KikuchiT, MusikacharoenT, et al. (2000) Cutting edge: naturally occurring soluble form of mouse Toll-like receptor 4 inhibits lipopolysaccharide signaling. J Immunol 165 : 6682–6686.
19. LeemanJR, GilmoreTD (2008) Alternative splicing in the NF-kappaB signaling pathway. Gene 423 : 97–107.
20. LynchKW (2004) Consequences of regulated pre-mRNA splicing in the immune system. Nat Rev Immunol 4 : 931–940.
21. OhtaS, BahrunU, TanakaM, KimotoM (2004) Identification of a novel isoform of MD-2 that downregulates lipopolysaccharide signaling. Biochem Biophys Res Commun 323 : 1103–1108.
22. WellsCA, ChalkAM, ForrestA, TaylorD, WaddellN, et al. (2006) Alternate transcription of the Toll-like receptor signaling cascade. Genome Biol 7: R10.
23. BurnsK, JanssensS, BrissoniB, OlivosN, BeyaertR, et al. (2003) Inhibition of interleukin 1 receptor/Toll-like receptor signaling through the alternatively spliced, short form of MyD88 is due to its failure to recruit IRAK-4. Journal of Experimental Medicine 197 : 263–268.
24. JanssensS, BurnsK, TschoppJ, BeyaertR (2002) Regulation of interleukin-1 - and lipopolysaccharide-induced NF-kappaB activation by alternative splicing of MyD88. Curr Biol 12 : 467–471.
25. JanssensS, BurnsK, VercammenE, TschoppJ, BeyaertR (2003) MyD88S, a splice variant of MyD88, differentially modulates NF-kappaB - and AP-1-dependent gene expression. FEBS Lett 548 : 103–107.
26. Mendoza-BarberaE, Corral-RodriguezMA, Soares-SchanoskiA, VelardeM, MacieiraS, et al. (2009) Contribution of globular death domains and unstructured linkers to MyD88.IRAK-4 heterodimer formation: an explanation for the antagonistic activity of MyD88s. Biochem Biophys Res Commun 380 : 183–187.
27. Adib-ConquyM, AdrieC, FittingC, GattolliatO, BeyaertR, et al. (2006) Up-regulation of MyD88s and SIGIRR, molecules inhibiting Toll-like receptor signaling, in monocytes from septic patients. Crit Care Med 34 : 2377–2385.
28. De ArrasL, SengA, LackfordB, KeikhaeeM, BowermanB, et al. (2013) An evolutionarily conserved innate immunity protein interaction network. J Biol Chem 288 : 1967–1978.
29. KramerA, FerfogliaF, HuangCJ, MulhauptF, NesicD, et al. (2005) Structure-function analysis of the U2 snRNP-associated splicing factor SF3a. Biochem Soc Trans 33 : 439–442.
30. HodgesPE, BeggsJD (1994) RNA splicing. U2 fulfils a commitment. Curr Biol 4 : 264–267.
31. SperlingJ, AzubelM, SperlingR (2008) Structure and function of the Pre-mRNA splicing machine. Structure 16 : 1605–1615.
32. WahlMC, WillCL, LuhrmannR (2009) The spliceosome: design principles of a dynamic RNP machine. Cell 136 : 701–718.
33. RinoJ, Carmo-FonsecaM (2009) The spliceosome: a self-organized macromolecular machine in the nucleus? Trends Cell Biol 19 : 375–384.
34. CollinsLJ, KurlandCG, BiggsP, PennyD (2009) The modern RNP world of eukaryotes. J Hered 100 : 597–604.
35. BrosiR, GroningK, BehrensSE, LuhrmannR, KramerA (1993) Interaction of mammalian splicing factor SF3a with U2 snRNP and relation of its 60-kD subunit to yeast PRP9. Science 262 : 102–105.
36. NesicD, KramerA (2001) Domains in human splicing factors SF3a60 and SF3a66 required for binding to SF3a120, assembly of the 17S U2 snRNP, and prespliceosome formation. Mol Cell Biol 21 : 6406–6417.
37. RubySW, ChangTH, AbelsonJ (1993) Four yeast spliceosomal proteins (PRP5, PRP9, PRP11, and PRP21) interact to promote U2 snRNP binding to pre-mRNA. Genes Dev 7 : 1909–1925.
38. TanackovicG, KramerA (2005) Human splicing factor SF3a, but not SF1, is essential for pre-mRNA splicing in vivo. Mol Biol Cell 16 : 1366–1377.
39. WiestDK, O'DayCL, AbelsonJ (1996) In vitro studies of the Prp9.Prp11.Prp21 complex indicate a pathway for U2 small nuclear ribonucleoprotein activation. J Biol Chem 271 : 33268–33276.
40. DasBK, XiaL, PalandjianL, GozaniO, ChyungY, et al. (1999) Characterization of a protein complex containing spliceosomal proteins SAPs 49, 130, 145, and 155. Mol Cell Biol 19 : 6796–6802.
41. KramerA, GruterP, GroningK, KastnerB (1999) Combined biochemical and electron microscopic analyses reveal the architecture of the mammalian U2 snRNP. J Cell Biol 145 : 1355–1368.
42. WillCL, SchneiderC, ReedR, LuhrmannR (1999) Identification of both shared and distinct proteins in the major and minor spliceosomes. Science 284 : 2003–2005.
43. AlperS, LawsR, LackfordB, BoydWA, DunlapP, et al. (2008) Identification of innate immunity genes and pathways using a comparative genomics approach. ProcNatl Acad Sci USA 105 : 7016–7021.
44. CorrioneroA, MinanaB, ValcarcelJ (2011) Reduced fidelity of branch point recognition and alternative splicing induced by the anti-tumor drug spliceostatin A. Genes Dev 25 : 445–459.
45. KaidaD, MotoyoshiH, TashiroE, NojimaT, HagiwaraM, et al. (2007) Spliceostatin A targets SF3b and inhibits both splicing and nuclear retention of pre-mRNA. Nat Chem Biol 3 : 576–583.
46. RoybalGA, JuricaMS (2010) Spliceostatin A inhibits spliceosome assembly subsequent to prespliceosome formation. Nucleic Acids Res 38 : 6664–6672.
47. AliprantisAO, YangRB, MarkMR, SuggettS, DevauxB, et al. (1999) Cell activation and apoptosis by bacterial lipoproteins through toll-like receptor-2. Science 285 : 736–739.
48. MercurioF, ZhuH, MurrayBW, ShevchenkoA, BennettBL, et al. (1997) IKK-1 and IKK-2: cytokine-activated IkappaB kinases essential for NF-kappaB activation. Science 278 : 860–866.
49. HäckerH, RedeckeV, BlagoevB, KratchmarovaI, HsuLC, et al. (2006) Specificity in Toll-like receptor signalling through distinct effector functions of TRAF3 and TRAF6. Nature 439 : 204–207.
50. MaruyamaK, SuganoS (1994) Oligo-capping: a simple method to replace the cap structure of eukaryotic mRNAs with oligoribonucleotides. Gene 138 : 171–174.
51. SuzukiY, Yoshitomo-NakagawaK, MaruyamaK, SuyamaA, SuganoS (1997) Construction and characterization of a full length-enriched and a 5′-end-enriched cDNA library. Gene 200 : 149–156.
52. IsonoK, Mizutani-KosekiY, KomoriT, Schmidt-ZachmannMS, KosekiH (2005) Mammalian polycomb-mediated repression of Hox genes requires the essential spliceosomal protein Sf3b1. Genes Dev 19 : 536–541.
53. KramerA, MulhauserF, WersigC, GroningK, BilbeG (1995) Mammalian splicing factor SF3a120 represents a new member of the SURP family of proteins and is homologous to the essential splicing factor PRP21p of Saccharomyces cerevisiae. RNA 1 : 260–272.
54. AnM, HenionPD (2012) The zebrafish sf3b1b460 mutant reveals differential requirements for the sf3b1 pre-mRNA processing gene during neural crest development. Int J Dev Biol 56 : 223–237.
55. FanL, LagisettiC, EdwardsCC, WebbTR, PotterPM (2011) Sudemycins, novel small molecule analogues of FR901464, induce alternative gene splicing. ACS Chem Biol 6 : 582–589.
56. VisconteV, RogersHJ, SinghJ, BarnardJ, BupathiM, et al. (2012) SF3B1 haploinsufficiency leads to formation of ring sideroblasts in myelodysplastic syndromes. Blood 120 : 3173–3186.
57. DammF, KosmiderO, Gelsi-BoyerV, RennevilleA, CarbucciaN, et al. (2012) Mutations affecting mRNA splicing define distinct clinical phenotypes and correlate with patient outcome in myelodysplastic syndromes. Blood 119 : 3211–3218.
58. DammF, TholF, KosmiderO, KadeS, LoffeldP, et al. (2012) SF3B1 mutations in myelodysplastic syndromes: clinical associations and prognostic implications. Leukemia 26 : 1137–1140.
59. EllisMJ, DingL, ShenD, LuoJ, SumanVJ, et al. (2012) Whole-genome analysis informs breast cancer response to aromatase inhibition. Nature 486 : 353–360.
60. MakishimaH, VisconteV, SakaguchiH, JankowskaAM, Abu KarS, et al. (2012) Mutations in the spliceosome machinery, a novel and ubiquitous pathway in leukemogenesis. Blood 119 : 3203–3210.
61. MalcovatiL, PapaemmanuilE, BowenDT, BoultwoodJ, Della PortaMG, et al. (2011) Clinical significance of SF3B1 mutations in myelodysplastic syndromes and myelodysplastic/myeloproliferative neoplasms. Blood 118 : 6239–6246.
62. PapaemmanuilE, CazzolaM, BoultwoodJ, MalcovatiL, VyasP, et al. (2011) Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N Engl J Med 365 : 1384–1395.
63. PatnaikMM, LashoTL, HodnefieldJM, KnudsonRA, KetterlingRP, et al. (2012) SF3B1 mutations are prevalent in myelodysplastic syndromes with ring sideroblasts but do not hold independent prognostic value. Blood 119 : 569–572.
64. QuesadaV, CondeL, VillamorN, OrdonezGR, JaresP, et al. (2012) Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nat Genet 44 : 47–52.
65. RossiD, BruscagginA, SpinaV, RasiS, KhiabanianH, et al. (2011) Mutations of the SF3B1 splicing factor in chronic lymphocytic leukemia: association with progression and fludarabine-refractoriness. Blood 118 : 6904–6908.
66. VisconteV, MakishimaH, JankowskaA, SzpurkaH, TrainaF, et al. (2012) SF3B1, a splicing factor is frequently mutated in refractory anemia with ring sideroblasts. Leukemia 26 : 542–545.
67. WangL, LawrenceMS, WanY, StojanovP, SougnezC, et al. (2011) SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. N Engl J Med 365 : 2497–2506.
68. YoshidaK, SanadaM, ShiraishiY, NowakD, NagataY, et al. (2011) Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 478 : 64–69.
69. KarinM, GretenFR (2005) NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol 5 : 749–759.
70. LiQ, WithoffS, VermaIM (2005) Inflammation-associated cancer: NF-kappaB is the lynchpin. Trends Immunol 26 : 318–325.
71. OhshimaH, TatemichiM, SawaT (2003) Chemical basis of inflammation-induced carcinogenesis. Arch Biochem Biophys 417 : 3–11.
72. StarczynowskiDT, KarsanA (2010) Innate immune signaling in the myelodysplastic syndromes. Hematol Oncol Clin North Am 24 : 343–359.
73. TsanMF (2006) Toll-like receptors, inflammation and cancer. Semin Cancer Biol 16 : 32–37.
74. SchusterTB, CostinaV, FindeisenP, NeumaierM, Ahmad-NejadP (2011) Identification and functional characterization of 14-3-3 in TLR2 signaling. J Proteome Res 10 : 4661–4670.
75. LiS, ArmstrongCM, BertinN, GeH, MilsteinS, et al. (2004) A map of the interactome network of the metazoan C. elegans. Science 303 : 540–543.
76. CartyM, GoodbodyR, SchroderM, StackJ, MoynaghPN, et al. (2006) The human adaptor SARM negatively regulates adaptor protein TRIF-dependent Toll-like receptor signaling. Nat Immunol 7 : 1074–1081.
77. PengJ, YuanQ, LinB, PanneerselvamP, WangX, et al. (2010) SARM inhibits both TRIF - and MyD88-mediated AP-1 activation. Eur J Immunol 40 : 1738–1747.
78. CouillaultC, PujolN, ReboulJ, SabatierL, GuichouJF, et al. (2004) TLR-independent control of innate immunity in Caenorhabditis elegans by the TIR domain adaptor protein TIR-1, an ortholog of human SARM. Nat Immunol 5 : 488–494.
79. LiberatiNT, FitzgeraldKA, KimDH, FeinbaumR, GolenbockDT, et al. (2004) Requirement for a conserved Toll/interleukin-1 resistance domain protein in the Caenorhabditis elegans immune response. Proc Natl Acad Sci U S A 101 : 6593–6598.
80. MuhammedM, FuchsBB, WuMP, BregerJ, ColemanJJ, et al. (2012) The role of mycelium production and a MAPK-mediated immune response in the C. elegans-Fusarium model system. Med Mycol 50 : 488–496.
81. GuruharshaKG, RualJF, ZhaiB, MintserisJ, VaidyaP, et al. (2011) A protein complex network of Drosophila melanogaster. Cell 147 : 690–703.
82. HuangCJ, FerfogliaF, RaleffF, KramerA (2011) Interaction domains and nuclear targeting signals in subunits of the U2 small nuclear ribonucleoprotein particle-associated splicing factor SF3a. J Biol Chem 286 : 13106–13114.
83. NesicD, TanackovicG, KramerA (2004) A role for Cajal bodies in the final steps of U2 snRNP biogenesis. J Cell Sci 117 : 4423–4433.
84. HunterP (2012) The inflammation theory of disease. The growing realization that chronic inflammation is crucial in many diseases opens new avenues for treatment. EMBO Rep 13 : 968–970.
85. Fernandez-Botran R, Větvička V (2001) Methods in Cellular Immunology. Boca Raton: CRC Press. p. 8.
86. VickersTA, ZhangH, GrahamMJ, LemonidisKM, ZhaoC, et al. (2006) Modification of MyD88 mRNA splicing and inhibition of IL-1beta signaling in cell culture and in mice with a 2′-O-methoxyethyl-modified oligonucleotide. J Immunol 176 : 3652–3661.
87. SchneiderCA, RasbandWS, EliceiriKW (2012) NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9 : 671–675.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2013 Číslo 10
Nejčtenější v tomto čísle
- A GDF5 Point Mutation Strikes Twice - Causing BDA1 and SYNS2
- Dominant Mutations in Identify the Mlh1-Pms1 Endonuclease Active Site and an Exonuclease 1-Independent Mismatch Repair Pathway
- Eleven Candidate Susceptibility Genes for Common Familial Colorectal Cancer
- The Histone H3 K27 Methyltransferase KMT6 Regulates Development and Expression of Secondary Metabolite Gene Clusters