
The Bacterial Response Regulator ArcA Uses a Diverse Binding Site Architecture to Regulate Carbon Oxidation Globally
Despite the importance of maintaining redox homeostasis for cellular viability, how cells control redox balance globally is poorly understood. Here we provide new mechanistic insight into how the balance between reduced and oxidized electron carriers is regulated at the level of gene expression by mapping the regulon of the response regulator ArcA from Escherichia coli, which responds to the quinone/quinol redox couple via its membrane-bound sensor kinase, ArcB. Our genome-wide analysis reveals that ArcA reprograms metabolism under anaerobic conditions such that carbon oxidation pathways that recycle redox carriers via respiration are transcriptionally repressed by ArcA. We propose that this strategy favors use of catabolic pathways that recycle redox carriers via fermentation akin to lactate production in mammalian cells. Unexpectedly, bioinformatic analysis of the sequences bound by ArcA in ChIP-seq revealed that most ArcA binding sites contain additional direct repeat elements beyond the two required for binding an ArcA dimer. DNase I footprinting assays suggest that non-canonical arrangements of cis-regulatory modules dictate both the length and concentration-sensitive occupancy of DNA sites. We propose that this plasticity in ArcA binding site architecture provides both an efficient means of encoding binding sites for ArcA, σ70-RNAP and perhaps other transcription factors within the same narrow sequence space and an effective mechanism for global control of carbon metabolism to maintain redox homeostasis.
Published in the journal:
. PLoS Genet 9(10): e32767. doi:10.1371/journal.pgen.1003839
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1003839
Summary
Despite the importance of maintaining redox homeostasis for cellular viability, how cells control redox balance globally is poorly understood. Here we provide new mechanistic insight into how the balance between reduced and oxidized electron carriers is regulated at the level of gene expression by mapping the regulon of the response regulator ArcA from Escherichia coli, which responds to the quinone/quinol redox couple via its membrane-bound sensor kinase, ArcB. Our genome-wide analysis reveals that ArcA reprograms metabolism under anaerobic conditions such that carbon oxidation pathways that recycle redox carriers via respiration are transcriptionally repressed by ArcA. We propose that this strategy favors use of catabolic pathways that recycle redox carriers via fermentation akin to lactate production in mammalian cells. Unexpectedly, bioinformatic analysis of the sequences bound by ArcA in ChIP-seq revealed that most ArcA binding sites contain additional direct repeat elements beyond the two required for binding an ArcA dimer. DNase I footprinting assays suggest that non-canonical arrangements of cis-regulatory modules dictate both the length and concentration-sensitive occupancy of DNA sites. We propose that this plasticity in ArcA binding site architecture provides both an efficient means of encoding binding sites for ArcA, σ70-RNAP and perhaps other transcription factors within the same narrow sequence space and an effective mechanism for global control of carbon metabolism to maintain redox homeostasis.
Introduction
Maintaining redox balance is a crucial function for cell survival. Alteration of the cellular redox environment has been shown to affect a broad range of biological processes including energy metabolism [1]–[3], protein folding [4], signaling and stress responses [5]–[9]. Despite this, we have only a superficial understanding of how cells control redox homeostasis at a global level. Since the cellular redox environment is a reflection of many different redox couples [10], some of which are linked together through enzymatic reactions, an improved understanding of this process requires knowledge of how the redox state of each couple is controlled. One such important redox couple is NADH/NAD+, which plays a central role in catabolic pathways, shuttling electrons between donor and acceptor molecules and allowing cells to convert energy from various reduced substrates into cellular ATP. To ensure that catabolism proceeds, a balance between the rates of oxidation and reduction of NAD+ must be maintained. Many diverse regulatory mechanisms have evolved amongst different organisms to control the redox state of the NADH/NAD+ couple [6], [11]–[14]. In this study we investigated transcriptional inputs into this process by mapping the regulon of the transcription factor ArcA in Escherichia coli.
The ArcAB two component system, comprised of the membrane bound sensor kinase, ArcB, and the response regulator, ArcA, coordinates changes in gene expression in response to changes in the respiratory and fermentative state of the cell [15], [16]. This system is maximally activated in E. coli under anaerobic fermentative conditions when NADH from central metabolism is recycled to NAD+ by formation of the end products succinate, ethanol and lactate. The DNA binding activity of ArcA is regulated through reversible phosphorylation by ArcB [17], whose kinase activity is governed by the redox states of the ubiquinone and menaquinone pools [18]–[20] that are linked to the NADH/NAD+ redox couple through respiration. In the absence of O2, decreased flux through the aerobic respiratory chain lowers the ratio of oxidized to reduced quinones, stimulating ArcB kinase activity and transphosphorylation of ArcA [19]. Additionally, fermentation products have been shown to enhance the rate of ArcB autophosphorylation [21] and there is a positive correlation between the rate of fermentation and the levels of phosphorylated ArcA (ArcA-P) [16]. Thus, enzymatic linkage of the NADH/NAD+ couple to the oxidation state of the quinone pool and the production of fermentation products provides a link between the redox state of the NADH/NAD+ couple and the activity of the ArcAB system. Indeed, artificial perturbation of the NADH/NAD+ ratio has been shown to alter ArcA activity [22].
Consistent with the role of the ArcAB system in redox regulation, the majority of known ArcA targets in E. coli are associated with aerobic respiratory metabolism. Under anaerobic conditions, ArcA-P directly represses the operons encoding enzymes of the TCA cycle (gltA, icdA, sdhCDAB-sucABCD, mdh, lpdA) [23]–[27], and for the β-oxidation of fatty acids (fadH, fadBA, fadL, fadE, fadD, fadIJ) [25], lactaldehyde (aldA)/lactate oxidation (lldPRD) [24], [28], and glycolate/glyoxylate oxidation (glcC, glcDEFGBA) [29]. In contrast, ArcA-P activates the expression of operons encoding three enzymes that are important for adapting to microaerobic or anaerobic environments [cytochrome bd oxidase (cydAB) [24], pyruvate formate lyase (focA-pflB) [30] and hydrogenase 1 (hya) [31]]. However, gene expression profiling analyses indicate that the ArcA regulon is more complex than originally expected, including genes encoding a wide variety of functions outside of redox metabolism [32], [33]. Salmon et al. [33] and Liu et al. [32] each identified >350 genes that were differentially expressed when arcA was deleted. However, there was only a minimal overlap between these datasets and it is unclear how many of these genes are direct vs. indirect targets of ArcA. Thus, although ArcA plays a prominent role in the anaerobic repression of genes that encode enzymes for aerobic respiratory metabolism, the full extent of the ArcA regulon remains unclear, preventing a comprehensive understanding of its physiological role.
Despite the identification of several ArcA binding regions by footprinting, the sequence determinants for ArcA DNA binding are also not well understood. This is in large part due to the unusually long length (30–60 plus bp) [23], [24], [26], [28]–[30] and degenerate nature of these sequences, which makes bioinformatic searches challenging. Nevertheless, a 15-bp site consisting of two tandem direct repeats has been proposed as the ArcA recognition site [34]. A similar motif has been derived for Shewanella oneidensis ArcA based on binding energy measurements for every possible permutation of a 15-bp site [35]. However, a 15-bp site is insufficient to explain the extended footprints, raising the question of whether additional sequence conservation beyond 15 bp is important for ArcA DNA binding and transcriptional regulation.
To determine the in vivo binding locations of ArcA in E. coli under anaerobic fermentative growth conditions, we utilized chromatin immunoprecipitation followed by sequencing (ChIP-seq) or hybridization to a microarray (ChIP-chip). Bioinformatic analyses of sequences corresponding to ArcA-enriched regions were used to predict individual ArcA binding sites and to search for a binding motif that could explain the large ArcA footprints. Novel ArcA binding site architectures were then validated by DNase I footprinting. Additionally, gene expression profiling was performed in arcA+ and ΔarcA backgrounds to determine the effect of ArcA DNA binding on gene expression. This combination of genome-wide approaches provided insight into the mechanism of ArcA DNA binding and transcriptional regulation. These results also allowed us to identify additional operons under direct ArcA control, thereby providing a more complete understanding of the physiological role of ArcA in E. coli.
Results
Identification of the chromosomal binding locations of ArcA
We mapped 176 chromosomal ArcA binding regions (Table S1) across the genome of E. coli K-12 MG1655 during anaerobic fermentation of glucose using ChIP-chip and ChIP-seq (Figure 1). These sites include all but five of the 22 previously identified ArcA binding regions (uvrA/ssb [36], oriC [37], ptsG [38], rpoS [39] and sodA [40]; Figure 1); the absence of a binding region upstream of sodA is likely the result of Fur outcompeting ArcA from binding [40]. ArcA binding was also examined during aerobic respiration using ChIP-chip and as expected, revealed a pronounced decrease in site occupancy (Figure 1) except for a handful of peaks (e.g., ygjG and uxaB), which were not investigated further. As ArcA protein levels remained relatively constant between aerobic and anaerobic conditions (data not shown and [16]), the decrease in occupancy under aerobic conditions can be explained by decreased ArcA-P levels, resulting from the increase in the ratio of oxidized to reduced quinones [20].

ChIP-seq analysis provides improved resolution compared to ChIP-chip
Overall, there was good agreement between the ChIP-chip and ChIP-seq datasets (109 peaks in common). However, 15 regions identified by ChIP-chip were resolved into 32 binding regions (Table S2) using ChIP-seq and the CSDeconv peak deconvolution algorithm [41]. For example, compared to only one binding region resolved with ChIP-chip, three binding regions were identified upstream of cydA (Figure 2A) and two were identified within the divergent sdhC/gltA (Figure 2B) promoter region using ChIP-seq. Furthermore, the position of the peak calls with CSDeconv is consistent with the position of known ArcA binding sites mapped by DNase I footprinting within these promoters [24], [27] and 29 of these 32 regions contain a predicted ArcA binding site (Table S2). The correlation of footprinted sites and predicted sites with CSDeconv peak calls allowed us to establish that binding sites separated by as little as 76 bp (based on the CSDeconv-defined coordinate for each binding region) could be resolved. From this analysis, several novel closely spaced ArcA binding sites, e.g. three binding regions upstream of cyo and two binding regions upstream of nuo and pdhR-aceEF-lpdA, were identified. Thus, since ChIP-seq provided higher resolution identification of ArcA binding sites, this dataset was used for all other analyses.
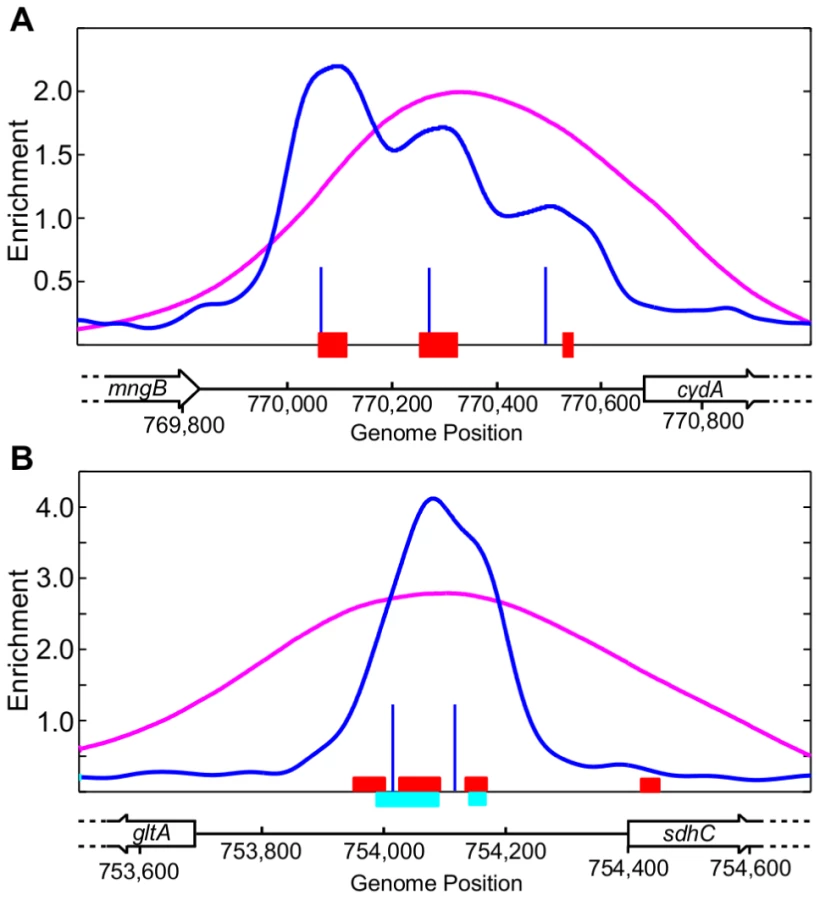
More than 50% of ArcA binding sites have additional DR elements beyond the ArcA box
DNase I footprinting experiments indicate that ArcA-P typically binds to long stretches of DNA (30–60+ bp) [23], [24], [26], [28]–[30]. However, the sequence determinants beyond a 15 bp direct repeat within these long stretches are not well understood. Using our high resolution binding regions, we searched for a common sequence recognition element [42], which identified a 18-bp sequence motif consisting of two direct repeat (DR) elements with a center to center (ctc) distance of 11 bp, close to the 10.5 bp per helical turn of B-form DNA, in nearly every (158 of 176) ArcA binding region (Figure 3A; Table S3). While this result extended the previously described ArcA box from 15 to 18 bp [34], we also found that many sites contained additional DR elements beyond the two DRs of the ArcA box. We then systematically searched the sequences surrounding each ArcA box with a 10-bp pair weight matrix (PWM), corresponding to a single DR element (Figure 3B), which revealed a diversity in the number and spacing of DR elements within ArcA binding sites. Although the largest class of binding sites contained just two DR elements at a ctc spacing of 11 bp (66), the majority of ArcA-binding sites (92) contain three to five DR elements predominantly at a ctc spacing of 11 bp (Figure 3C–D, Table S4).
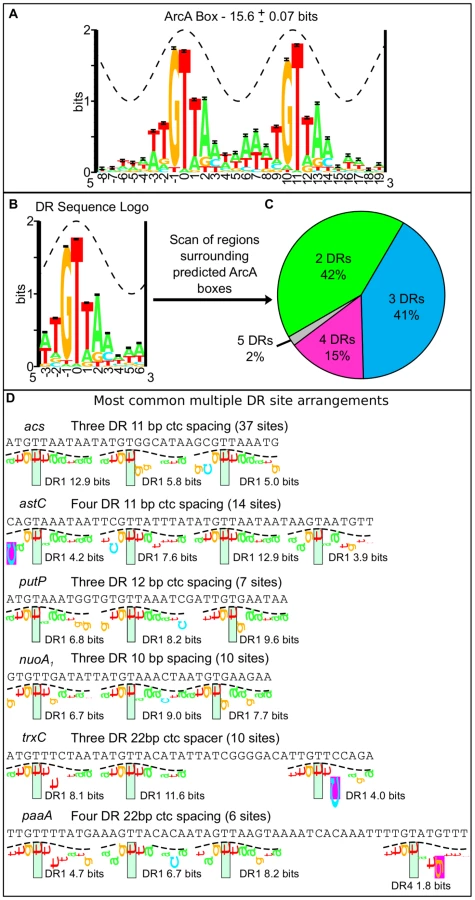
To validate the bioinformatic predictions, DNase I footprinting was performed for a representative set of promoters. Since the OmpR/PhoB family of response regulators is expected to dimerize upon phosphorylation [43], we hypothesized that ArcA would bind as two adjacent dimers to sites with three consecutive DR elements (e.g., icdA and acs), three DR elements at which the distal DR is separated from DR2 by approximately two helical turns (22 bp; e.g., trxC), or four consecutive DR elements (e.g., astC) and in each case, protect a region the size of four DRs (∼44 bp). As anticipated, ArcA-P protected a 44 bp region at the astC promoter (Figure 4A) and a 48 bp region at the trxC promoter (Figure 4B). In contrast, ArcA-P only protected 33 bp and 37 bp regions, respectively, at the icdA and acs promoters, which encompassed the three consecutive DR elements (Figure 4C–D). The result for icdA is in agreement with previous footprinting data [23].

Our footprinting data also suggested that the spacing between DR2 and DR3 is likely important for ArcA-P binding, because ArcA-P did not protect a predicted DR3 element in which the ctc distance between DR2 and DR3 contained an extra bp (12-bp spacing; putP); protection corresponded to only DR1 and DR2 (Figure 4E). A potential explanation of this result is that the increased spacer distance disrupted potential protein-protein contacts between ArcA dimers. Additionally, our footprinting data identified 57 bp and 60 bp ArcA-P-binding regions, respectively, at the paaA and phoH promoters, which spanned from three consecutive predicted DRs to a distal DR element spaced nearly two full helical turns away (22 bp) (Figure 4F–G). As expected, no footprints were detected with unphosphorylated ArcA (data not shown).
Unexpectedly, the ArcA-P footprint at the dctA promoter extended 50 bp downstream of the predicted two DR site (Figure 4H), although this extended region was less well protected. A bioinformatic search revealed a second, but weaker two DR site at the downstream end of this protected region on the opposite DNA strand but no DR elements in the intervening 24 bp region, suggesting that protein-protein contacts may compensate for the absence of identifiable sequence elements at this site. Altogether, these results suggest that the length of the ArcA-P footprint reflects the location of the outermost DR elements within the binding site. In addition, these data reveal plasticity in the architecture among ArcA binding sites with anywhere from two to five DR elements of differing predicted strength present at any given site.
The footprinting results also revealed interesting features about ArcA-P DNA binding. At acs and astC, all DR elements were occupied at the same ArcA-P concentration, whereas at icdA, paaA, phoH, and trxC, occupation of DR3 or DR4 required a higher concentration of ArcA-P. The difference in concentration dependent occupancy of the DR elements at the icdA and acs promoters likely reflects the fact that DR3 of acs is a better match to the ArcA DR element PWM than DR3 of icdA (5 bits versus 3 bits). Furthermore, the transition from an unbound to bound state occurred over a narrow range in ArcA-P concentration, suggesting that ArcA-P binding to DR sites is cooperative, although the apparent degree of cooperativity also varied from site to site. Cooperative binding was particularly striking at the acs and astC promoters and for the three DR region at the phoH promoter, for which saturation occurred with less than a four-fold increase in ArcA-P levels. Finally, we also found that the average sequence conservation of DR elements in predicted binding sites with two, three and four equally spaced DR elements decreases with an increasing number of repeats (Figure S1).
DNase I hypersensitive sites were observed at six of the tested promoters, suggesting that ArcA-P binding to multiple DR sites also results in a bend or kink in the DNA. However, the locations of these hypersensitive sites differed from site to site. For example, a hypersensitive site was observed within the spacer region between the 22-bp spaced DR element and the other DR elements at the trxC, paaA and phoH promoters, whereas hypersensitive sites were observed within DR1 and DR2 at the icdA promoter (+8 and +19). In contrast, hypersensitive sites were located upstream and downstream of the footprinted regions at the acs and astC promoters, respectively. Thus, the binding site architecture appears not only to dictate the length of ArcA-P binding sites, but also to affect the concentration dependence of site occupancy and the DNA structure at target operons. These variations in ArcA-P binding likely have important implications for global transcriptional regulation.
ArcA-P directly regulates the expression of 85 operons under anaerobic fermentative growth conditions
To determine which ArcA binding regions exert an effect on transcription, genome-wide mRNA expression profiles for wild type (WT) and ΔarcA strains were examined. In total, 229 differentially expressed operons (Table S5) were identified, 85 of which were associated with one or more of 88 ArcA binding regions (Text S1) and, thus, are directly regulated by ArcA (Figure 5, Table S6). More than half of the operons that we found to be regulated directly by ArcA have not been previously reported (Table S6) but consistent with previous studies, ArcA acted predominantly as a transcriptional repressor (Figure 1).

ArcA directly represses 74 operons
ArcA functions predominantly as a global repressor of pathways associated with the oxidation of non-glycolytic carbon sources. This includes all previously identified ArcA targets associated with central metabolism (e.g., the genes encoding pyruvate dehydrogenase, cytochrome o ubiquinol oxidase, NADH-quinone oxidoreductase I, and the enzymes of the TCA cycle) (Figure 6). In addition, ArcA repressed the genes encoding enzymes, transcriptional regulators, or transporters associated with short chain acid/aldehyde oxidation (aldA, lldPRD, acs-yjcH-actP, glcC, glcDEFGBA and fdoGHI; bolded operons have not been previously reported), amino acid and polyamine oxidation (puuA, puuDR, ygjG, potFGHI, astABCDE, argT-hisQMP, putA, putP), β-oxidation of fatty acids (fadH, fadBA, fadL, fadE, fadD, fadIJ, tesB), aromatic compound oxidation (hcaR, mhpR, feaR), other carbon oxidation pathways (betIBA, betT, ugpBAED, gcd, maeB) and peptide utilization (cstA). Other ArcA repressed targets include methionine sulfoxide reductase (msrB), thioredoxin 2 (trxC) [44], [45], a soluble pyridine nucleotide transhydrogenase (sthA) that reduces NAD+ with NADPH, and an ADP-sugar pyrophosphorylase (nudE) that could play a role in maintaining an optimal NADH/NAD+ ratio based on its ability to use NADH as a substrate [46]. Finally, an ArcA-regulated ribonucleoside transporter (nepI) and a nucleoside diphosphate kinase (ndk) could also function in NAD+ homeostasis via their functions in nucleotide metabolism [47].

A few repressed operons (9) encode proteins with functions not known to be associated with redox metabolism. This includes bssR and csgD, which encode transcription factors involved in biofilm formation and curli biosynthesis, respectively, and rsd, encoding a stationary phase induced anti-σ factor. Additionally, ArcA repressed outer membrane proteins (cirA, ompW), a potassium efflux system (kefGB-yheV), the ATPase component of the ClpAP protease (clpA), an ATP binding protein (phoH) and a methyl-galactoside ABC transporter (mgl). Although a rationalization for ArcA repression of each of these operons is not yet known, the control of mgl may be related to the report that E. coli K-12 is unable to grow fermentatively on galactose [48]. Finally, 13 repressed operons have only predicted or unknown function [47]; four are predicted membrane proteins, two are predicted transcriptional regulators (ydcI, yjiR), and two others are predicted to encode a dehydrogenase (yeiQ) and a fimbrial-like adhesin protein (yehD), respectively.
To gain insight into the mechanism of ArcA repression, we examined σ70 ChIP-seq data collected from growth conditions identical to those used with ArcA [49]. The vast majority (56/65) of ArcA-repressed promoters, exhibited a statistically significant reduction in σ70 peak height under anaerobic conditions compared to aerobic conditions, consistent with ArcA preventing RNA polymerase binding (Table S6). In agreement with this observation, correlation of the position of predicted ArcA binding sites with known σ70-dependent transcription start site (TSSs from EcoCyc [47] or [50]) indicated that the majority of repressed targets (52/66) with a confirmed TSS have an ArcA binding site that overlaps the region bound by σ70-RNAP (the TSS, the −35 element or the −10 elements; Figure 7A, Table S6). Eight promoter regions for ArcA-repressed operons did not exhibit a decrease in σ70 occupancy. Because these sites are located within divergently transcribed regions where the other operon is not affected by ArcA, σ70 occupancy may reflect only the adjacent non-ArcA-regulated promoter. In summary, the positioning of ArcA binding sites is consistent with the O2-dependent decrease in σ70 occupancy that is observed at nearly all ArcA-repressed operons, suggesting that ArcA likely represses transcription through promoter occlusion.

ArcA directly activates 11 operons
Analysis of the function of directly activated genes indicated a diversity of functions. This includes hydrogenase 1 (hyaABCDEF) [51], the ferrous iron transporter (feoABC), an oligopeptide ABC transporter (oppA), and the acid phosphatase transcriptional regulator (appY) that is involved in anaerobic gene regulation [52]. Our data also suggest a role for ArcA in the acid resistance response by activating operons encoding regulators of the glutamate dependent acid resistance system (gadE-mdtEF and gadXW) [53], [54], the arginine dependent acid resistance (adiC) system [55] and the resistance to organic acid stress (slp-dctR) [56]. The remaining ArcA-activated targets encode genes of unknown function (ybcW and ybfA) and a small regulatory RNA, fnrS [57], [58]. Although fnrS was not present on our microarrays, a previous study showed that ArcA is a coactivator of this sRNA [57].
Examination of the σ70 occupancy data indicated that there is a statistically significant change in σ70-RNAP occupancy under anaerobic conditions for nine of the 10 directly activated operons, consistent with ArcA functioning in activation of these operons (Table S6). However, both the position and orientation of the predicted ArcA binding site relative to the TSS for each operon is variable among activated targets (Figure 7B). Some binding sites are located downstream of the nearest mapped TSS, whereas others overlap the promoter elements or are located as far as 200–400 bp upstream of the TSS. Given this variable positioning and orientation of ArcA binding sites, it remains unclear whether ArcA can activate transcription by directly contacting σ70-RNAP as found with some OmpR/PhoB family members [59]–[61].
The direct regulon of ArcA extends beyond the 85 operons identified under our growth conditions
Many intergenic ArcA binding regions (76) were associated with operons that did not show an ArcA dependent change in gene expression in our studies. However, previous studies indicated that 13 operons are regulated by ArcA but under different growth conditions (Table S7). For example, cydAB expression is activated by ArcA under microaerobic growth conditions, when FNR repression is relieved [62]. Furthermore, many binding regions (31) are associated with operons that are poorly expressed under our growth conditions in both the arcA+ and ΔarcA strains (e.g., paa operon; Table S8). Since ArcA is predominantly a repressor of transcription, we hypothesized that these promoters were repressed by a second transcription factor or require a transcriptional activator and, therefore, growth under inducing conditions would be required to see an effect of ArcA binding on the transcription of these operons.
To test this idea, we constructed a paaA promoter-lacZ fusion and measured β-galactosidase activity in WT and ΔarcA strains supplemented with phenylacetate (PA) because the paaABCDEFGHIJK operon is known to be repressed by PaaX in the absence of PA [63]. In the presence of PA, ArcA strongly repressed paaA-lacZ expression under anaerobic conditions (23 Miller units for WT), whereas repression was relieved in a strain lacking ArcA (404 Miller units) or under aerobic conditions (294 and 372 Miller units for WT and ΔarcA, respectively), indicating that ArcA prevents induction of the paa operon under anaerobic conditions even when PA is present. Examination of regulatory data in EcoCyc [47] indicated that 11 other poorly expressed operons also are associated with other annotated activators or repressors (Table S8) that may contribute to synergistic regulation with ArcA. Furthermore, ChIP-chip experiments for other transcriptional repressors indicated that under our growth conditions, 15 targets are also bound by Fur, H-NS, or both [Beauchene and Kiley, personal communication; [49]] (Table S8). Thus, repression by Fur and H-NS may mask effects of ArcA. Altogether, these results indicate that ArcA repression likely serves as a secondary layer of control at many of these operons, ensuring that induction does not occur under anaerobic conditions even when the specific inducer is encountered. Thus, the 85 operons that show a change in expression under fermentative growth with glucose represent just a subset of the complete ArcA direct regulon.
The indirect regulon of ArcA may reflect a hierarchical mode of transcriptional regulation
Of the 229 operons regulated by ArcA, 145 lacked ArcA binding in vivo and have not been shown previously to be directly regulated by ArcA. To assess whether an ArcA binding site was missed by our ChIP analyses at any of these operons, we searched the intergenic region upstream of each operon using a cutoff of 15 bits (representing the average sequence conservation of the ArcA sequence logo). An ArcA binding site was identified upstream of only seven operons (acnA, prpR, folE, yibF, yigI, dcuC/crcA), indicating that the remaining 135 operons are likely regulated through an indirect mechanism. Since ArcA directly regulates the expression of 17 transcription factors, a hierarchical mode of regulation could, in part, explain the differential expression of some of these operons. Although not all of these transcription factors are expected or known to be active under our growth conditions, differential expression of nine operons can likely be traced to one of these transcription factors (Figure 8). For example, the expression of the AppY dependent appCBA-yccB operon [52] is decreased when arcA is deleted, presumably because of the decrease in appY activation by ArcA. In addition, four target operons (folE, gpmA, dld and eco) of the ArcA-activated sRNA, FnrS were upregulated in the arcA mutant [57], [58]. Finally, although we did not identify an ArcA binding site upstream of arcZ, the downregulation of sdaC (the most strongly repressed target of the ArcZ sRNA in S. enterica [64]) in the absence of arcA is consistent with ArcA-dependent activation of arcZ [65].

ArcA prevents the oxidation of non-fermentable carbon sources during fermentation
Examination of EcoCyc (v15.5) [47] for annotated dehydrogenase enzymes (MultiFun term BC-1), indicated that ArcA either directly or indirectly regulates 37 out of 40 non-glycolytic dehydrogenase enzymes that are favored in the direction of reducing equivalent formation and are not involved in biosynthetic or detoxification functions (Table S9). The carbon oxidation pathways and transporters associated with the substrates of each repressed dehydrogenase are displayed in Figure 6 and the majority of these pathways feed into the TCA cycle for further carbon oxidation. The scope of this repression strongly suggests that a major function of ArcA is to repress all genes encoding enzymes that oxidize non-fermentable carbon compounds, thus preventing the formation of excess reducing equivalents (e.g., NADH, FADH2 and quinols) that cannot be readily re-oxidized in the absence of respiration. Nevertheless, despite the extensive upregulation of dehydrogenase enzymes, ArcA mutants have only a small increase in doubling time from 90 to 105 min (Figure S2A) and only a minor alteration in the distribution of fermentation end products (Figure S2B–C). Succinate and ethanol production were marginally increased and decreased by equivalent amounts in a ΔarcA strain, respectively, and lactate was not a major fermentation product (Figure S2C). This suggests that the NADH/NAD+ ratio was not likely perturbed in our ΔarcA strain in agreement with previous results [66], [67].
Distinct functional roles for ArcA and FNR
Although ArcA and FNR are known to mediate widespread changes in gene expression during the transition from aerobic to anaerobic conditions, the extent of the regulatory overlap between these factors has not been established. Previous gene expression studies have suggested that there may be a large overlap between the genes regulated by ArcA and FNR in both E. coli [33] and S. enterica [68]. However, comparison of our dataset with that determined recently for FNR using identical growth conditions, suggests that there is little direct coregulation (Figure S3). Of the 37 operons that showed both FNR and ArcA dependent changes in expression, only seven are directly regulated by both ArcA and FNR. Rather, differential expression may result from an indirect effect of a fnr deletion on ArcA-P levels, which has been previously suggested to explain the FNR-dependent effect on sdhC and lldP expression [69]. An additional 12 operons show both ArcA and FNR binding in vivo but are differentially expressed in only one dataset (e.g., focA-pflB, cydAB). This minimal overlap in the direct regulons of ArcA-P and FNR suggests that these regulators occupy distinct functional roles in anaerobic gene regulation; the ArcA regulon is largely centered around the repression of aerobic carbon oxidation pathways while FNR appears to function as a more general activator of anaerobic gene expression [49]. Some coregulated operons encode enzymes that direct carbon flow towards either oxidative or fermentative metabolism (e.g., pdhR-aceEF-lpdA, focA-pflB, yfiD) while others encode principal components of the respiratory chain (e.g., nou, ndh, cydA). However, coregulation of other operons (e.g. bssR, ompW, ompC, oppA, ygjG, msrB) by ArcA and FNR is surprising and the physiological implications of this coregulation are unknown.
Discussion
By comparing ArcA binding in vivo with gene expression profiling data, we have greatly expanded the number of operons regulated by ArcA, leading to important insights into the physiological role, mechanism and sequence requirements for ArcA transcriptional regulation. Our analysis indicates that ArcA directly regulates the expression of nearly 100 operons and is predominantly a repressor of genes encoding proteins associated with carbon oxidation pathways. Furthermore, identification of binding sites upstream of many poorly expressed operons (e.g., paa) suggests that the direct regulon of ArcA could actually encompass as many as 150 operons. Additionally, our bioinformatic and DNase I footprinting analyses reveal a plasticity in the ArcA binding site architecture that likely has important implications for global regulation of carbon oxidation in E. coli.
ArcA is a global repressor of carbon oxidation pathways
Our finding that under anaerobic conditions, ArcA reprograms metabolism by either directly or indirectly repressing expression of nearly all pathways for carbon sources whose oxidation is coupled to aerobic respiration suggests a global mechanism for NAD+ sparing. This strategy would facilitate the preferential oxidation of the fermentable carbon source glucose and the sparing of NAD+ for glycolysis by recycling NADH to NAD+ via reductive formation of lactic acid, succinate and ethanol. Thus, ATP synthesis via substrate level phosphorylation is ensured and redox balance of NADH/NAD+ is maintained during anaerobic glucose fermentation. This function of ArcA exhibits parallels to carbon catabolite repression in that it is another mechanism for selective carbon source utilization in cells. Although carbon catabolite repression preferentially selects for glucose utilization over other sugars, ArcA reinforces glucose catabolism through the repression of non-glycolytic carbon oxidation pathways. By integrating signals from both respiratory and fermentative metabolism, which are both enzymatically linked to the NADH/NAD+ redox couple, the ArcAB two component system provides a means for E. coli to maintain the NADH/NAD+ ratio.
Despite the extensive upregulation of dehydrogenase enzymes in an arcA mutant, there was only a minor alteration in fermentation products. This result is in agreement with previous data, which also showed that the NADH/NAD+ ratio is not perturbed in strains lacking ArcA during fermentation [66], [67]. The ability of glucose fermenting cells to maintain redox balance in the absence of ArcA likely reflects thermodynamic and kinetic parameters that favor flux via glucose fermentation and the fact that although many dehydrogenases are upregulated, their substrates are not present preventing competition with glycolysis. Indeed, the activity of several dehydrogenases in cellular extracts was previously shown to be increased in an arcA mutant. However, the fact that the NADH/NAD+ ratio is altered in an arcB strain [70] may be explained by the additional roles of ArcB beyond regulating ArcA [39], [71].
Nevertheless, previous studies suggest that ArcA deficiencies may compromise growth more significantly under conditions that more closely parallel the natural habitats of E. coli. For example, an arcA mutant is defective in both survival during aerobic carbon starvation [72] and in colonization of the mouse intestine [73]. Increased NADH/NAD+ ratios have been observed in an arcA mutant during microaerobiosis [66], [67], which may contribute to the poor fitness of arcA mutants in the gut. Accordingly, it seems reasonable to conclude that this extensive repression of dehydrogenase enzymes by ArcA provides an evolutionary advantage for E. coli in its natural habitats where nutrient conditions are in flux and where many more growth substrates (i.e., both carbon sources and electron acceptors) could be encountered.
ArcA may repress and activate transcription solely by binding DNA
Surprisingly, very little in vitro data are available describing mechanisms of ArcA transcription regulation. Nevertheless, the location of the ArcA binding sites and the decrease in σ70 occupancy indicate that ArcA represses by occluding RNA polymerase binding like many repressors. However, the mechanism of activation is unlikely to occur through the direct recruitment of RNA Polymerase as observed with ArcA homologs OmpR [60], [61] and PhoB [59] since no conserved location or orientation of ArcA binding sites was evident. Rather, ArcA may increase transcription through an antirepression mechanism. In support of this notion, in vivo studies of hyaA [31], cydAB [74], appY [75] and yfiD [76] transcription suggest that ArcA activation occurs primarily through disruption of HNS (cydAB and appY), FNR (yfiD) or IscR (hyaA) binding. Furthermore, although the mechanism of ArcA activation of focA-pflB [77] and the PY promoter (from the conjugative resistance plasmid R1) [78] is unknown, DNA binding by ArcA alone appears insufficient for its transcriptional activation. In addition, binding of ArcA alone actually repressed transcription of ndh [79], despite the observation that ndh expression increased when arcA was deleted [32]. Although further in vitro experiments are necessary to investigate the activation mechanism, it seems plausible that ArcA functions solely by binding DNA and activates only indirectly when its binding interferes with the binding and repression by another transcriptional repressor.
Plasticity within the architecture of ArcA binding sites
The variation in the number, spacing, location and predicted strength of DR elements within the chromosomal ArcA binding regions suggests plasticity in the architecture of ArcA binding sites for either repressed or activated operons. Although the core of each site is an ArcA box containing two, 11-bp ctc spaced DR elements, the majority of binding sites contain an additional one to three DRs predominantly-spaced by approximately one or two turns of the helix of B-form DNA (11 bp or 22 bp ctc spacing). Multiple DR elements have also been observed for some promoters regulated by OmpR [80] and PhoB [59], [81], [82]. However, it is unclear how pervasive multiple repeat elements are for these regulators because the 41 genomic PhoB binding locations recently mapped by ChIP-chip were not searched for sequence elements beyond a single PhoB Box [83] and a conserved sequence motif was not identified within the majority of the 43 OmpR binding sites identified with ChIP-seq [84]. Although the three direct repeat binding site architecture represents a particularly novel finding for the OmpR/PhoB family of response regulators, at least one other example of a response regulator, ComA in B. subtilis, which binds three recognition elements (i.e., an inverted repeat and an additional half site) has been reported and all three elements were shown to be important for both DNA binding and transcriptional activation [85]. Whether the protection of only three DR elements by ArcA reflects binding by a dimer and monomer or two dimers, where the distal subunit is not bound sufficiently to protect sequences from DNase I cleavage, is not yet known.
Implications of binding site plasticity for global ArcA transcriptional regulation
Since the majority of ArcA binding sites overlap the σ70 promoter recognition elements, the plasticity of these cis-regulatory modules may provide an efficient means of encoding binding sites for ArcA, σ70-RNAP and perhaps other transcription factors within the same narrow sequence space. We propose that having binding sites with different architectures is also an effective mechanism for producing diverse transcriptional regulatory outputs. First, varying the number, strength or location of DR elements should modulate the extent of anaerobic repression. Second, embedding transcription factor binding sites within an ArcA binding site could either enhance or antagonize ArcA function. For example, the DR elements at the trxC, paaA and phoH promoters also overlap a binding site for a transcriptional activator (CRP for paaA [63], OxyR for trxC [45]) or a second promoter (P2 at phoH [86]), allowing additional regulatory control. Third, sites of varying affinities may also impact the sensitivity of promoters to the phosphorylation state of ArcA. For example, the different binding affinities of DR elements at the trxC, icdA, paaA and phoH promoters may allow the fine-tuning of expression in response to changing ArcA-P levels when O2 levels vary [16]. Fine tuning of ompF and ompC expression by OmpR has been observed in response to medium osmolarity due to the presence of multiple upstream OmpR boxes with different affinities [80]. Conversely, the highly cooperative mode of occupancy at the astC and acs promoters would likely render the expression of these operons exquisitely sensitive to changes in ArcA-P levels; thus, expression may more closely resemble an on-off switch. Ultimately, such flexibility in transcriptional regulatory outputs may be an important means for linking the redox sensing properties of the ArcAB two component system with the global optimization of carbon oxidation pathway levels. Further studies are underway to examine the contribution of different binding site architectures to both DNA binding and transcriptional regulation.
Materials and Methods
Growth conditions
All strains were grown in MOPS minimal medium [87] with 0.2% glucose at 37°C and sparged with a gas mix of 95% N2 and 5% CO2 (anaerobic) or 70% N2, 5% CO2, and 25% O2 (aerobic). Cells were harvested during mid-log growth (OD600 of ∼0.3 on a Perkin Elmer Lambda 25 UV/Vis Spectrophotometer).
Construction of promoter-lacZ fusions and β-galactosidase assays
A paaA promoter-lacZ fusion was constructed as described previously [88] by amplifying the region from +15 to −194 relative to the translation start using primers flanked by XhoI or BamHI restriction sites. A TAA stop codon was incorporated after codon 5 to terminate translation from the Shine-Dalgarno sequence present in this region. The resulting PCR fragment was digested with XhoI and BamHI and directionally cloned into plasmid pPK7035. This lacZ promoter construct was then recombined into the chromosomal lac operon as previously described [88] to create the paaA promoter-lacZ fusion and then transduced using P1 vir into MG1655 and PK9416 (ΔarcA) to creating PK9959 and PK9960 (Table S10). For assays with paaA, 1 mM phenylacetic acid (Sigma Aldrich) was added to the minimal glucose media. To terminate cell growth and any further protein synthesis chloramphenicol (final concentration, 20 µg/ml) was added, and cells were placed on ice until assayed for β-galactosidase activity [89]. β-galactosidase values represent the average of at least three replicates.
Cloning, overexpression and purification of His6-ArcA
arcA was amplified with primers which incorporated a NheI restriction site, a His6-tag and a Tev protease cleavage site (order listed in 5′-3′ direction) on the 5′ end of the gene and a XhoI site at the 3′ end. The NheI and XhoI digested fragments were cloned into plasmid pET 21-d to generate plasmid PK9431 for protein production. E. coli BL21(DE3), containing PK9431 was grown at 37°C until an OD600 of 0.5–0.6 was reached then 1 mM isopropyl-1-thio-β-D-galactopyranoside (IPTG) was added. After seven hours at 30°C, cells were harvested, suspended in 5 mM imidazole, 50 mM Tris-Cl, pH 8.3 and 0.3 M NaCl and lysed by sonication. His6-ArcA was isolated from cell lysates by passage over a Ni-NTA column pre-equilibrated with 5 mM imidazole, washing extensively with the same buffer followed by 50 mM imidazole, and then eluting with a linear gradient of 50–500 mM imidazole. Fractions containing the overexpressed His6-ArcA, determined by electrophoresis, were dialyzed against 50 mM Tris-Cl, pH 8.0 and 0.1 M NaCl and concentrated. Antibodies to ArcA were obtained from Harlan (Indianapolis, In), affinity purified prior to use and determined to be specific to ArcA by Western blot (data not shown). For DNase I footprinting, the His6 tag was removed from ArcA by overnight incubation with tobacco etch virus (TEV) protease at 4°C and passage over a Ni2+-agarose column (Qiagen). The protein concentration of ArcA (reported here as monomers) was determined with the Coomassie Plus protein assay reagent (Pierce), using bovine serum albumin as a standard.
Chromatin immunoprecipitation followed by hybridization to a microarray chip or high-throughput sequencing
ChIP was performed as previously described [90] using the affinity purified ArcA polyclonal antibodies. ChIP DNA along with corresponding input DNA were amplified by linker-mediated PCR and labeled with Cy3 or Cy5-random 9-mers then hybridized as previously described [49] to custom-made E. coli K-12 MG1655 tiled genome microarrays (Roche NimbleGen, Inc, Madison, WI). The hybridized microarrays were scanned using NimbleGen Hybridization System 4 and the PMT was adjusted as previously described [49]. Quantile normalization (“normalize.quantiles” in the R package VSN) [91] was used to obtain the same empirical distribution across the Cy3 and Cy5 channels and across biological replicate arrays to correct for dye intensity bias and to minimize microarray-to-microarray absolute intensity variations as previously described [92]. The log2 of the ratio of experimental signals (Cy5) to control signals (Cy3) was calculated. Regions of the genome enriched for occupancy by ArcA were identified using TAMALPAIS [93] L2 and L3 stringency levels (95th percentile/p<0.0001 and 98th percentile/p<0.05 of the log2 ratio for each chip, respectively) with the anaerobic fermentative ArcA data. Only enriched regions that were significant in both biological replicates were considered, resulting in the identification of 194 binding regions. Four false positives were eliminated from the data set by analyzing technical replicate ChIP-chip results from a strain lacking arcA (PK9416; Table S11). Fifty-three false positives were eliminated because we found that they resulted from ArcA co-immunoprecipitating with RNA polymerase at highly transcribed regions (Figure S4; Table S12; Text S1; Table S12) leaving 137 regions. The phosphorylation dependence of ArcA DNA binding at these sites was determined by performing a single biological replicate ArcA ChIP-chip experiment under aerobic conditions. For visualization, the anaerobic ArcA biological replicates were averaged then median smoothed using a 300 bp window using MochiView [94].
For ChIP-seq, enriched ChIP DNA from two additional biological replicates from anaerobic ArcA samples were submitted to the University of Wisconsin-Madison DNA Sequencing Facility for library construction and Illumina sequencing performed as previously described [49]. A total of 1,364,908 and 12,074,358 reads were obtained for the ChIP replicates. Greater than 90% and 80% of these reads, respectively, mapped uniquely to the K12 MG1655 genome (version U00096.2) using the software package SOAP release 2.20, allowing no more than two mismatches [95]. The CSDeconv algorithm [41] was then used to determine significantly enriched regions in high resolution using both ChIP-seq replicates and two anaerobic input samples [49] from the same sequencing run as the ArcA ChIP samples. Reads that mapped uniquely within the seven rRNA operon regions were eliminated to allow the algorithm to run more efficiently. CSDeconv was run with Matlab v7.11.0 (R2010b) using the following parameters: LLR = 21.75 and alpha = 800 for replicate one and LLR = 22 and alpha = 550 for replicate two. The find_enriched function was modified to account for differences in sequencing depth between the IP and Input samples. Correction factors of 2.98 (replicate 1) and 0.6579 (replicate 2), calculated by dividing the number of unique reads in the Input sample by the number of reads in the ChIP sample for replicates one and two, respectively, were multiplied by nip and the forward and reverse kernel density calculations for both the forward and reverse strands of the ChIP sample. FDRs of 0.0154 and 0.0156 for replicates one and two, respectively, were calculated by a sample swap (the number of peaks in the Input over the ChIP sample divided by the number of detections in the ChIP over the control sample). From 222 enriched regions generated from two independent ChIP-seq replicates, 146 ArcA-P binding regions (Table S1) were obtained using the same filtering criteria described for ChIP-chip (Table S12; Text S1). For visualization of the ChIP-seq data, the raw tag density at each position was calculated using QuEST version 2.0 [96] and normalized as tag density per million uniquely mapped reads.
The final list of 176 binding regions was obtained by searching binding regions that were found in only one ChIP-seq replicate (48) or were unique to ChIP-chip (28) with the ArcA box PWM (see below) using a cutoff of 10 bits as 99% of ArcA boxes in the alignment have an individual information content of 10 bits or greater. An ArcA binding site was identified in 30 of these binding regions (15 from ChIP-chip and 15 from ChIP-seq) which were, therefore, combined with the 146 regions found in both ChIP-seq replicates to produce the final list of 176 ArcA chromosomal binding regions (Table S1).
ArcA PWM construction and identification of predicted ArcA binding sites
Based on the improved resolution of ChIP-seq, sequence corresponding to a 200 bp window around each of the 146 CSDeconv binding regions (averages of the two replicates) was searched for a common motif using MEME [42] with the parameters -mod zoops -nmotifs 1 -minw 18 -maxw 25. Using the alignment from MEME, a sequence logo was built using the Delila software package with the delila, encode, rseq, dalvec, and makelogo programs [97]. A PWM generated from this alignment was used to search the 146 binding regions with a cutoff of 9 bits as this represents the lowest scoring ArcA box included in the MEME alignment. Using the program localbest, only the best scoring ArcA box within a 200 bp region was retained due to several instances of overlapping ArcA-P boxes being identified (sites with three and four DR elements). The resulting 128 ArcA-P boxes were used to make the final sequence logo (Figure 3A). The delila program ri [97] was used to calculate the information content of individual sequences within the positions −3 and 14, which ranged from 9.1 to 21 bits (Table S3). A PWM derived from the conservation of bases between positions −3 and 14 in these 128 ArcA-P boxes, is referred to throughout the paper as the ArcA box PWM. No unique motif was identified within the 18 binding regions without a match to the ArcA box. The scan program [97] was used to search DNA sequences upstream of differentially expressed operons that were not enriched in ChIP using the ArcA box PWM. The E. coli K12 genome sequence [98] was obtained from GenBank (v. U00096.2) and a bit score cutoff of 15 bp bits was used as this represents the average information content of the ArcA box PWM. The localbest program was used to select the best scoring ArcA box within a 200 bp region in cases where two sites were predicted in close proximity.
To construct the 10 bp PWM corresponding to a single direct repeat element, positions −3 to 6 and 8 to 17 from the 128 sequences used to make the ArcA box sequence logo were aligned as they correspond to the nucleotides contacted by each PhoB monomer in the crystal structure of the C-terminus of PhoB bound to its PhoB box [99]. Due to the identical spacing between DR elements and the highly similar nucleotide compositions of the PhoB and ArcA boxes, this structure likely serves as a good model for the nucleotides contacted by each ArcA monomer. A bit score cutoff of 0, which represents the theoretical lowest limit of binding [97], was used to search a 100 bp region surrounding each identified ArcA box with the scan program to identify sites with additional repeat elements. Where displayed, sequence walkers were used to visualize matches to the ArcA-P binding site using the lister program [100].
Gene expression profiling with a microarray
An in-frame ΔarcA deletion strain was constructed by replacing the coding region of arcA (codons 2–238) with a CmR resistance cassette flanked by FLP recognition target (FRT) sites from plasmid pKD32 in strain BW25993/pKD46, as described previously [101] to generate PK7510. Transduction with P1 vir was used to move the arcA::cat allele into MG1655 to produce PK7514. The CmR cassette of PK7514 was removed by transforming this strain with pCP20-encoding FLP recombinase [101] then screening for loss of Cm, generating PK9416 (Table S10). The deletion was confirmed by sequencing.
RNA was isolated from triplicate MG1655 and ΔarcA (PK9416) strains using a hot-phenol method [102]. The RNA was reverse transcribed to cDNA, labeled with Cy3-random 9-mers and hybridized onto the Roche NimbleGen E. coli 4plex Expression Array Platform (4×72,000 probes, Catalog Number A6697-00-01) as previously described [49]. The expression data was normalized using Robust Multi-Array (RMA) [103] and statistical analysis was performed with Arraystar III software (DNASTAR). Transcripts exhibiting a statistically significant (moderated t-test p-value<0.05) change in expression greater than 2-fold were considered differentially expressed and grouped into operons using operon definitions in EcoCyc [47] if at least two of the genes in a particular operon exhibited differential expression.
End product analysis
Samples (2 ml) for end product analysis were collected during log phase, the transition to stationary phase and in stationary phase (Figure 7A). Cells were removed by passage through a 0.2 µm filter and the supernatant was stored at −80°C prior to analysis. For each sample, glucose, pyruvic acid, succinic acid, lactic acid, formic acid, acetic acid, and ethanol were separated by high-performance liquid chromatography (HPLC) and subsequently quantified as previously described [104].
DNase I footprinting
Plasmids containing predicted ArcA-P binding sites were generated by PCR amplification of chromosomal DNA with primers flanked by XhoI or BamHI restriction sites and cloned into pPK7179 or pPK7035 (for the icdA promoter)(Table S10). The positions of the promoter fragments relative to the previously identified transcription start sites are as follows: for icdA [23], −216 to +65; for acs (P2) [105], −172 to +44; for phoH (P2) [86], −161 to +20; for paaA [106], −132 to +55; for astC [107], −166 to +62; for putP (P1) [108], −120 to +56; for trxC [45], −118 to +50 ; for dctA [109], −185 to +32. The icdA fragment contains two promoters: one whose expression is dependent on ArcA (P1) and a second promoter whose expression is dependent on FruR (P2) [23], [110]. To examine icdA expression from only P1 in future expression analyses, transcription from P2 was eliminated using the site-directed mutagenesis protocol described in [111] to mutate the −10 site from cattat to cggtga. DNA fragments were isolated from pPK7179 or pPK9476 (icdA) after digestion with XhoI and BamHI, radiolabelled at the 3′ BamHI end with [α-32P]-dGTP (PerkinElmer) and Sequenase Version 2.0 (USB Scientific), isolated from a non-denaturing 5% acrylamide gel and subsequently purified with elutip-d columns (Schleicher and Schuell). ArcA was phosphorylated by incubating with 50 mM disodium carbamyl phosphate (Sigma Aldrich) in 50 mM Tris, pH 7.9, 150 mM NaCl, and 10 mM MgCl2 for 1 h at 30°C [24] and immediately used in the binding assays. Footprinting assays were performed by incubating phosphorylated ArcA with labeled DNA (∼5 nM) for 10 min at 30°C in 40 mM Tris (pH 7.9), 30 mM KCl, 100 µg/ml BSA and 1 mM DTT followed by the addition of 2 µg/ml DNase I (Worthington) for 30 s. The DNase I reaction was terminated by the addition of sodium acetate and EDTA to final concentrations of 300 mM and 20 mM, respectively. The reaction mix was ethanol precipitated, resuspended in urea loading dye, heated for 60 s at 90°C, and loaded onto a 7 M urea, 8% polyacrylamide gel in 0.5× TBE buffer. An A+G ladder was made by formic acid modification of the radiolabeled DNA, followed by piperidine cleavage [112]. The reaction products were visualized by phosphorimaging.
Data deposition
All genome-wide data from this publication have been deposited in NCBI's Gene Expression Omnibus (GSE46415.
Supporting Information
Zdroje
1. PackerL, CadenasE (2011) Lipoic acid: energy metabolism and redox regulation of transcription and cell signaling. J Clin Biochem Nutr 48 : 26–32.
2. van HoekMJ, MerksRM (2012) Redox balance is key to explaining full vs. partial switching to low-yield metabolism. BMC Syst Biol 6 : 22.
3. KornasA, KuzniakE, SlesakI, MiszalskiZ (2010) The key role of the redox status in regulation of metabolism in photosynthesizing organisms. Acta Biochim Pol 57 : 143–151.
4. TrachoothamD, LuW, OgasawaraMA, NilsaRD, HuangP (2008) Redox regulation of cell survival. Antioxid Redox Signal 10 : 1343–1374.
5. BergerF, Ramirez-HernandezMH, ZieglerM (2004) The new life of a centenarian: signalling functions of NAD(P). Trends Biochem Sci 29 : 111–118.
6. BrekasisD, PagetMS (2003) A novel sensor of NADH/NAD+ redox poise in Streptomyces coelicolor A3(2). EMBO J 22 : 4856–4865.
7. AbateC, PatelL, RauscherFJ (1990) Redox regulation of fos and jun DNA-binding activity in vitro. Science 249 : 1157–1161.
8. DanonA, MayfieldSP (1994) Light-regulated translation of chloroplast messenger RNAs through redox potential. Science 266 : 1717–1719.
9. HaddadJJ (2004) Oxygen sensing and oxidant/redox-related pathways. Biochem Biophys Res Commun 316 : 969–977.
10. SchaferFQ, BuettnerGR (2001) Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic Biol Med 30 : 1191–1212.
11. ZhangQ, PistonDW, GoodmanRH (2002) Regulation of corepressor function by nuclear NADH. Science 295 : 1895–1897.
12. RutterJ, ReickM, WuLC, McKnightSL (2001) Regulation of clock and NPAS2 DNA binding by the redox state of NAD cofactors. Science 293 : 510–514.
13. SnoepJL, de GraefMR, WestphalAH, de KokA, Teixeira de MattosMJ, et al. (1993) Differences in sensitivity to NADH of purified pyruvate dehydrogenase complexes of Enterococcus faecalis, Lactococcus lactis, Azotobacter vinelandii and Escherichia coli: implications for their activity in vivo. FEMS Microbiol Lett 114 : 279–283.
14. LeonardoMR, DaillyY, ClarkDP (1996) Role of NAD in regulating the adhE gene of Escherichia coli. J Bacteriol 178 : 6013–6018.
15. AlvarezAF, GeorgellisD (2010) In vitro and in vivo analysis of the ArcB/A redox signaling pathway. Methods Enzymol 471 : 205–228.
16. RolfeMD, Ter BeekA, GrahamAI, TrotterEW, AsifHM, et al. (2011) Transcript profiling and inference of Escherichia coli K-12 ArcA activity across the range of physiologically relevant oxygen concentrations. J Biol Chem 286 : 10147–10154.
17. IuchiS, LinEC (1992) Purification and phosphorylation of the Arc regulatory components of Escherichia coli. J Bacteriol 174 : 5617–5623.
18. GeorgellisD, KwonO, LinEC (2001) Quinones as the redox signal for the Arc two-component system of bacteria. Science 292 : 2314–2316.
19. MalpicaR, FrancoB, RodriguezC, KwonO, GeorgellisD (2004) Identification of a quinone-sensitive redox switch in the ArcB sensor kinase. Proc Natl Acad Sci U S A 101 : 13318–13323.
20. BekkerM, AlexeevaS, LaanW, SawersG, Teixeira de MattosJ, et al. (2010) The ArcBA two-component system of Escherichia coli is regulated by the redox state of both the ubiquinone and the menaquinone pool. J Bacteriol 192 : 746–754.
21. GeorgellisD, KwonO, LinEC (1999) Amplification of signaling activity of the Arc two-component system of Escherichia coli by anaerobic metabolites. An in vitro study with different protein modules. J Biol Chem 274 : 35950–35954.
22. HolmAK, BlankLM, OldigesM, SchmidA, SolemC, et al. (2010) Metabolic and transcriptional response to cofactor perturbations in Escherichia coli. J Biol Chem 285 : 17498–17506.
23. ChaoG, ShenJ, TsengCP, ParkSJ, GunsalusRP (1997) Aerobic regulation of isocitrate dehydrogenase gene (icd) expression in Escherichia coli by the arcA and fnr gene products. J Bacteriol 179 : 4299–4304.
24. LynchAS, LinEC (1996) Transcriptional control mediated by the ArcA two-component response regulator protein of Escherichia coli: characterization of DNA binding at target promoters. J Bacteriol 178 : 6238–6249.
25. ChoBK, KnightEM, PalssonBO (2006) Transcriptional regulation of the fad regulon genes of Escherichia coli by ArcA. Microbiology 152 : 2207–2219.
26. CunninghamL, GeorgellisD, GreenJ, GuestJR (1998) Co-regulation of lipoamide dehydrogenase and 2-oxoglutarate dehydrogenase synthesis in Escherichia coli: characterization of an ArcA binding site in the lpd promoter. FEMS Microbiol Lett 169 : 403–408.
27. ShenJ, GunsalusRP (1997) Role of multiple ArcA recognition sites in anaerobic regulation of succinate dehydrogenase (sdhCDAB) gene expression in Escherichia coli. Mol Microbiol 26 : 223–236.
28. PellicerMT, LynchAS, De WulfP, BoydD, AguilarJ, et al. (1999) A mutational study of the ArcA-P binding sequences in the aldA promoter of Escherichia coli. Mol Gen Genet 261 : 170–176.
29. PellicerMT, FernandezC, BadiaJ, AguilarJ, LinEC, et al. (1999) Cross-induction of glc and ace operons of Escherichia coli attributable to pathway intersection. Characterization of the glc promoter. J Biol Chem 274 : 1745–1752.
30. DrapalN, SawersG (1995) Purification of ArcA and analysis of its specific interaction with the pfl promoter-regulatory region. Mol Microbiol 16 : 597–607.
31. NesbitAD, FleischhackerAS, TeterSJ, KileyPJ (2012) ArcA and AppY antagonize IscR repression of hydrogenase-1 expression under anaerobic conditions, revealing a novel mode of O2 regulation of gene expression in Escherichia coli. J Bacteriol 194 : 6892–6899.
32. LiuX, De WulfP (2004) Probing the ArcA-P modulon of Escherichia coli by whole genome transcriptional analysis and sequence recognition profiling. J Biol Chem 279 : 12588–12597.
33. SalmonKA, HungSP, SteffenNR, KruppR, BaldiP, et al. (2005) Global gene expression profiling in Escherichia coli K12: effects of oxygen availability and ArcA. J Biol Chem 280 : 15084–15096.
34. GerasimovaAVGM, MakeevVY, MironovAAaFA (2003) ArcA regulator of Gamma-Proteobacteria - Identification of the Binding Signal and Description of the Regulon. Biophysics 48: S21–S25.
35. WangX, GaoH, ShenY, WeinstockGM, ZhouJ, et al. (2008) A high-throughput percentage-of-binding strategy to measure binding energies in DNA-protein interactions: application to genome-scale site discovery. Nucleic Acids Res 36 : 4863–4871.
36. OgasawaraH, TeramotoJ, YamamotoS, HiraoK, YamamotoK, et al. (2005) Negative regulation of DNA repair gene (uvrA) expression by ArcA/ArcB two-component system in Escherichia coli. FEMS Microbiol Lett 251 : 243–249.
37. LeeYS, HanJS, JeonY, HwangDS (2001) The Arc two-component signal transduction system inhibits in vitro Escherichia coli chromosomal initiation. J Biol Chem 276 : 9917–9923.
38. JeongJY, KimYJ, ChoN, ShinD, NamTW, et al. (2004) Expression of ptsG encoding the major glucose transporter is regulated by ArcA in Escherichia coli. J Biol Chem 279 : 38513–38518.
39. MikaF, HenggeR (2005) A two-component phosphotransfer network involving ArcB, ArcA, and RssB coordinates synthesis and proteolysis of sigmaS (RpoS) in E. coli. Genes Dev 19 : 2770–2781.
40. TardatB, TouatiD (1993) Iron and oxygen regulation of Escherichia coli MnSOD expression: competition between the global regulators Fur and ArcA for binding to DNA. Mol Microbiol 9 : 53–63.
41. LunDS, SherridA, WeinerB, ShermanDR, GalaganJE (2009) A blind deconvolution approach to high-resolution mapping of transcription factor binding sites from ChIP-seq data. Genome Biol 10: R142.
42. BaileyTL, ElkanC (1994) Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol Biol 2 : 28–36.
43. GaoR, StockAM (2009) Biological insights from structures of two-component proteins. Annu Rev Microbiol 63 : 133–154.
44. GrimaudR, EzratyB, MitchellJK, LafitteD, BriandC, et al. (2001) Repair of oxidized proteins. Identification of a new methionine sulfoxide reductase. J Biol Chem 276 : 48915–48920.
45. RitzD, PatelH, DoanB, ZhengM, AslundF, et al. (2000) Thioredoxin 2 is involved in the oxidative stress response in Escherichia coli. J Biol Chem 275 : 2505–2512.
46. O'HandleySF, FrickDN, DunnCA, BessmanMJ (1998) Orf186 represents a new member of the Nudix hydrolases, active on adenosine(5′)triphospho(5′)adenosine, ADP-ribose, and NADH. J Biol Chem 273 : 3192–3197.
47. KeselerIM, Collado-VidesJ, Santos-ZavaletaA, Peralta-GilM, Gama-CastroS, et al. (2011) EcoCyc: a comprehensive database of Escherichia coli biology. Nucleic Acids Res 39: D583–590.
48. MuirM, WilliamsL, FerenciT (1985) Influence of transport energization on the growth yield of Escherichia coli. J Bacteriol 163 : 1237–1242.
49. MyersKS, YanH, OngIM, ChungD, LiangK, et al. (2013) Genome-scale Analysis of Escherichia coli FNR Reveals Complex Features of Transcription Factor Binding. PLoS Genet 9: e1003565.
50. KimD, HongJS, QiuY, NagarajanH, SeoJH, et al. (2012) Comparative analysis of regulatory elements between Escherichia coli and Klebsiella pneumoniae by genome-wide transcription start site profiling. PLoS Genet 8: e1002867.
51. VolbedaA, DarnaultC, ParkinA, SargentF, ArmstrongFA, et al. (2013) Crystal structure of the O(2)-tolerant membrane-bound hydrogenase 1 from Escherichia coli in complex with its cognate cytochrome b. Structure 21 : 184–190.
52. AtlungT, BrondstedL (1994) Role of the transcriptional activator AppY in regulation of the cyx appA operon of Escherichia coli by anaerobiosis, phosphate starvation, and growth phase. J Bacteriol 176 : 5414–5422.
53. MaZ, GongS, RichardH, TuckerDL, ConwayT, et al. (2003) GadE (YhiE) activates glutamate decarboxylase-dependent acid resistance in Escherichia coli K-12. Mol Microbiol 49 : 1309–1320.
54. TramontiA, ViscaP, De CanioM, FalconiM, De BiaseD (2002) Functional characterization and regulation of gadX, a gene encoding an AraC/XylS-like transcriptional activator of the Escherichia coli glutamic acid decarboxylase system. J Bacteriol 184 : 2603–2613.
55. GongS, RichardH, FosterJW (2003) YjdE (AdiC) is the arginine:agmatine antiporter essential for arginine-dependent acid resistance in Escherichia coli. J Bacteriol 185 : 4402–4409.
56. MatesAK, SayedAK, FosterJW (2007) Products of the Escherichia coli acid fitness island attenuate metabolite stress at extremely low pH and mediate a cell density-dependent acid resistance. J Bacteriol 189 : 2759–2768.
57. DurandS, StorzG (2010) Reprogramming of anaerobic metabolism by the FnrS small RNA. Mol Microbiol 75 : 1215–1231.
58. BoysenA, Moller-JensenJ, KallipolitisB, Valentin-HansenP, OvergaardM (2010) Translational regulation of gene expression by an anaerobically induced small non-coding RNA in Escherichia coli. J Biol Chem 285 : 10690–10702.
59. MakinoK, AmemuraM, KawamotoT, KimuraS, ShinagawaH, et al. (1996) DNA binding of PhoB and its interaction with RNA polymerase. J Mol Biol 259 : 15–26.
60. PrattLA, SilhavyTJ (1994) OmpR mutants specifically defective for transcriptional activation. J Mol Biol 243 : 579–594.
61. SlauchJM, RussoFD, SilhavyTJ (1991) Suppressor mutations in rpoA suggest that OmpR controls transcription by direct interaction with the alpha subunit of RNA polymerase. J Bacteriol 173 : 7501–7510.
62. CotterPA, MelvilleSB, AlbrechtJA, GunsalusRP (1997) Aerobic regulation of cytochrome d oxidase (cydAB) operon expression in Escherichia coli: roles of Fnr and ArcA in repression and activation. Mol Microbiol 25 : 605–615.
63. FerrandezA, GarciaJL, DiazE (2000) Transcriptional regulation of the divergent paa catabolic operons for phenylacetic acid degradation in Escherichia coli. J Biol Chem 275 : 12214–12222.
64. PapenfortK, SaidN, WelsinkT, LucchiniS, HintonJC, et al. (2009) Specific and pleiotropic patterns of mRNA regulation by ArcZ, a conserved, Hfq-dependent small RNA. Mol Microbiol 74 : 139–158.
65. MandinP, GottesmanS (2010) Integrating anaerobic/aerobic sensing and the general stress response through the ArcZ small RNA. EMBO J 29 : 3094–3107.
66. AlexeevaS, HellingwerfKJ, Teixeira de MattosMJ (2003) Requirement of ArcA for redox regulation in Escherichia coli under microaerobic but not anaerobic or aerobic conditions. J Bacteriol 185 : 204–209.
67. LevanonSS, SanKY, BennettGN (2005) Effect of oxygen on the Escherichia coli ArcA and FNR regulation systems and metabolic responses. Biotechnol Bioeng 89 : 556–564.
68. EvansMR, FinkRC, Vazquez-TorresA, PorwollikS, Jones-CarsonJ, et al. (2011) Analysis of the ArcA regulon in anaerobically grown Salmonella entericasv. Typhimurium. BMC Microbiol 11 : 58.
69. IuchiS, AristarkhovA, DongJM, TaylorJS, LinEC (1994) Effects of nitrate respiration on expression of the Arc-controlled operons encoding succinate dehydrogenase and flavin-linked L-lactate dehydrogenase. J Bacteriol 176 : 1695–1701.
70. BidartGN, RuizJA, de AlmeidaA, MendezBS, NikelPI (2012) Manipulation of the anoxic metabolism in Escherichia coli by ArcB deletion variants in the ArcBA two-component system. Appl Environ Microbiol 78 : 8784–8794.
71. MatsubaraM, KitaokaSI, TakedaSI, MizunoT (2000) Tuning of the porin expression under anaerobic growth conditions by his-to-Asp cross-phosphorelay through both the EnvZ-osmosensor and ArcB-anaerosensor in Escherichia coli. Genes Cells 5 : 555–569.
72. NystromT, LarssonC, GustafssonL (1996) Bacterial defense against aging: role of the Escherichia coli ArcA regulator in gene expression, readjusted energy flux and survival during stasis. EMBO J 15 : 3219–3228.
73. JonesSA, ChowdhuryFZ, FabichAJ, AndersonA, SchreinerDM, et al. (2007) Respiration of Escherichia coli in the mouse intestine. Infect Immun 75 : 4891–4899.
74. GovantesF, OrjaloAV, GunsalusRP (2000) Interplay between three global regulatory proteins mediates oxygen regulation of the Escherichia coli cytochrome d oxidase (cydAB) operon. Mol Microbiol 38 : 1061–1073.
75. AtlungT, SundS, OlesenK, BrondstedL (1996) The histone-like protein H-NS acts as a transcriptional repressor for expression of the anaerobic and growth phase activator AppY of Escherichia coli. J Bacteriol 178 : 3418–3425.
76. WybornNR, MessengerSL, HendersonRA, SawersG, RobertsRE, et al. (2002) Expression of the Escherichia coli yfiD gene responds to intracellular pH and reduces the accumulation of acidic metabolic end products. Microbiology 148 : 1015–1026.
77. SirkoA, ZeheleinE, FreundlichM, SawersG (1993) Integration host factor is required for anaerobic pyruvate induction of pfl operon expression in Escherichia coli. J Bacteriol 175 : 5769–5777.
78. StrohmaierH, NoigesR, KotschanS, SawersG, HogenauerG, et al. (1998) Signal transduction and bacterial conjugation: characterization of the role of ArcA in regulating conjugative transfer of the resistance plasmid R1. J Mol Biol 277 : 309–316.
79. RolfeMD, OconeA, StapletonMR, HallS, TrotterEW, et al. (2012) Systems analysis of transcription factor activities in environments with stable and dynamic oxygen concentrations. Open Biol 2 : 120091.
80. YoshidaT, QinL, EggerLA, InouyeM (2006) Transcription regulation of ompF and ompC by a single transcription factor, OmpR. J Biol Chem 281 : 17114–17123.
81. KimSK, KimuraS, ShinagawaH, NakataA, LeeKS, et al. (2000) Dual transcriptional regulation of the Escherichia coli phosphate-starvation-inducible psiE gene of the phosphate regulon by PhoB and the cyclic AMP (cAMP)-cAMP receptor protein complex. J Bacteriol 182 : 5596–5599.
82. KasaharaM, MakinoK, AmemuraM, NakataA, ShinagawaH (1991) Dual regulation of the ugp operon by phosphate and carbon starvation at two interspaced promoters. J Bacteriol 173 : 549–558.
83. YangC, HuangTW, WenSY, ChangCY, TsaiSF, et al. (2012) Genome-wide PhoB binding and gene expression profiles reveal the hierarchical gene regulatory network of phosphate starvation in Escherichia coli. PLoS One 7: e47314.
84. PerkinsTT, DaviesMR, KlemmEJ, RowleyG, WilemanT, et al. (2013) ChIP-seq and transcriptome analysis of the OmpR regulon of Salmonella enterica serovars Typhi and Typhimurium reveals accessory genes implicated in host colonization. Mol Microbiol 87 : 526–538.
85. GriffithKL, GrossmanAD (2008) A degenerate tripartite DNA-binding site required for activation of ComA-dependent quorum response gene expression in Bacillus subtilis. J Mol Biol 381 : 261–275.
86. KimSK, MakinoK, AmemuraM, ShinagawaH, NakataA (1993) Molecular analysis of the phoH gene, belonging to the phosphate regulon in Escherichia coli. J Bacteriol 175 : 1316–1324.
87. NeidhardtFC, BlochPL, SmithDF (1974) Culture medium for enterobacteria. J Bacteriol 119 : 736–747.
88. KangY, WeberKD, QiuY, KileyPJ, BlattnerFR (2005) Genome-wide expression analysis indicates that FNR of Escherichia coli K-12 regulates a large number of genes of unknown function. J Bacteriol 187 : 1135–1160.
89. Miller JH (1972) Experiments in molecular genetics. [Cold Spring Harbor, N.Y.]: Cold Spring Harbor Laboratory.
90. DavisSE, MooneyRA, KaninEI, GrassJ, LandickR, et al. (2011) Mapping E. coli RNA polymerase and associated transcription factors and identifying promoters genome-wide. Methods Enzymol 498 : 449–471.
91. HuberW, von HeydebreckA, SultmannH, PoustkaA, VingronM (2002) Variance stabilization applied to microarray data calibration and to the quantification of differential expression. Bioinformatics 18 Suppl 1: S96–104.
92. DufourYS, LandickR, DonohueTJ (2008) Organization and evolution of the biological response to singlet oxygen stress. J Mol Biol 383 : 713–730.
93. BiedaM, XuX, SingerMA, GreenR, FarnhamPJ (2006) Unbiased location analysis of E2F1-binding sites suggests a widespread role for E2F1 in the human genome. Genome Res 16 : 595–605.
94. HomannOR, JohnsonAD (2010) MochiView: versatile software for genome browsing and DNA motif analysis. BMC Biol 8 : 49.
95. LiR, YuC, LiY, LamTW, YiuSM, et al. (2009) SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics 25 : 1966–1967.
96. ValouevA, JohnsonDS, SundquistA, MedinaC, AntonE, et al. (2008) Genome-wide analysis of transcription factor binding sites based on ChIP-Seq data. Nat Methods 5 : 829–834.
97. SchneiderTD (1997) Information content of individual genetic sequences. J Theor Biol 189 : 427–441.
98. BlattnerFR, PlunkettG (1997) The complete genome sequence of Escherichia coli K-12. Science 277 : 1453–1462.
99. BlancoAG, SolaM, Gomis-RuthFX, CollM (2002) Tandem DNA recognition by PhoB, a two-component signal transduction transcriptional activator. Structure 10 : 701–713.
100. SchneiderTD (1997) Sequence walkers: a graphical method to display how binding proteins interact with DNA or RNA sequences. Nucleic Acids Res 25 : 4408–4415.
101. DatsenkoKA, WannerBL (2000) One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97 : 6640–6645.
102. KhodurskyAB, BernsteinJA, PeterBJ, RhodiusV, WendischVF, et al. (2003) Escherichia coli spotted double-strand DNA microarrays: RNA extraction, labeling, hybridization, quality control, and data management. Methods Mol Biol 224 : 61–78.
103. BolstadBM, IrizarryRA, AstrandM, SpeedTP (2003) A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 19 : 185–193.
104. SchwalbachMS, KeatingDH, TremaineM, MarnerWD, ZhangY, et al. (2012) Complex physiology and compound stress responses during fermentation of alkali-pretreated corn stover hydrolysate by an Escherichia coli ethanologen. Appl Environ Microbiol 78 : 3442–3457.
105. BeattyCM, BrowningDF, BusbySJ, WolfeAJ (2003) Cyclic AMP receptor protein-dependent activation of the Escherichia coli acs P2 promoter by a synergistic class III mechanism. J Bacteriol 185 : 5148–5157.
106. FerrandezA, MinambresB, GarciaB, OliveraER, LuengoJM, et al. (1998) Catabolism of phenylacetic acid in Escherichia coli. Characterization of a new aerobic hybrid pathway. J Biol Chem 273 : 25974–25986.
107. FraleyCD, KimJH, McCannMP, MatinA (1998) The Escherichia coli starvation gene cstC is involved in amino acid catabolism. J Bacteriol 180 : 4287–4290.
108. NakaoT, YamatoI, AnrakuY (1987) Nucleotide sequence of putC, the regulatory region for the put regulon of Escherichia coli K12. Mol Gen Genet 210 : 364–368.
109. DaviesSJ, GolbyP, OmraniD, BroadSA, HarringtonVL, et al. (1999) Inactivation and regulation of the aerobic C(4)-dicarboxylate transport (dctA) gene of Escherichia coli. J Bacteriol 181 : 5624–5635.
110. ProstJF, NegreD, OudotC, MurakamiK, IshihamaA, et al. (1999) Cra-dependent transcriptional activation of the icd gene of Escherichia coli. J Bacteriol 181 : 893–898.
111. NesbitAD, GielJL, RoseJC, KileyPJ (2009) Sequence-specific binding to a subset of IscR-regulated promoters does not require IscR Fe-S cluster ligation. J Mol Biol 387 : 28–41.
112. MaxamAM, GilbertW (1980) Sequencing end-labeled DNA with base-specific chemical cleavages. Methods Enzymol 65 : 499–560.
113. SchneiderTD, StephensRM (1990) Sequence logos: a new way to display consensus sequences. Nucleic Acids Res 18 : 6097–6100.
114. NeuwegerH, PersickeM, AlbaumSP, BekelT, DondrupM, et al. (2009) Visualizing post genomics data-sets on customized pathway maps by ProMeTra-aeration-dependent gene expression and metabolism of Corynebacterium glutamicum as an example. BMC Syst Biol 3 : 82.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2013 Číslo 10
Nejčtenější v tomto čísle
- A GDF5 Point Mutation Strikes Twice - Causing BDA1 and SYNS2
- Dominant Mutations in Identify the Mlh1-Pms1 Endonuclease Active Site and an Exonuclease 1-Independent Mismatch Repair Pathway
- Eleven Candidate Susceptibility Genes for Common Familial Colorectal Cancer
- The Histone H3 K27 Methyltransferase KMT6 Regulates Development and Expression of Secondary Metabolite Gene Clusters