
Evidence of Gene–Environment Interactions between Common Breast Cancer Susceptibility Loci and Established Environmental Risk Factors
Various common genetic susceptibility loci have been identified for breast cancer; however, it is unclear how they combine with lifestyle/environmental risk factors to influence risk. We undertook an international collaborative study to assess gene-environment interaction for risk of breast cancer. Data from 24 studies of the Breast Cancer Association Consortium were pooled. Using up to 34,793 invasive breast cancers and 41,099 controls, we examined whether the relative risks associated with 23 single nucleotide polymorphisms were modified by 10 established environmental risk factors (age at menarche, parity, breastfeeding, body mass index, height, oral contraceptive use, menopausal hormone therapy use, alcohol consumption, cigarette smoking, physical activity) in women of European ancestry. We used logistic regression models stratified by study and adjusted for age and performed likelihood ratio tests to assess gene–environment interactions. All statistical tests were two-sided. We replicated previously reported potential interactions between LSP1-rs3817198 and parity (Pinteraction = 2.4×10−6) and between CASP8-rs17468277 and alcohol consumption (Pinteraction = 3.1×10−4). Overall, the per-allele odds ratio (95% confidence interval) for LSP1-rs3817198 was 1.08 (1.01–1.16) in nulliparous women and ranged from 1.03 (0.96–1.10) in parous women with one birth to 1.26 (1.16–1.37) in women with at least four births. For CASP8-rs17468277, the per-allele OR was 0.91 (0.85–0.98) in those with an alcohol intake of <20 g/day and 1.45 (1.14–1.85) in those who drank ≥20 g/day. Additionally, interaction was found between 1p11.2-rs11249433 and ever being parous (Pinteraction = 5.3×10−5), with a per-allele OR of 1.14 (1.11–1.17) in parous women and 0.98 (0.92–1.05) in nulliparous women. These data provide first strong evidence that the risk of breast cancer associated with some common genetic variants may vary with environmental risk factors.
Published in the journal:
. PLoS Genet 9(3): e32767. doi:10.1371/journal.pgen.1003284
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1003284
Summary
Various common genetic susceptibility loci have been identified for breast cancer; however, it is unclear how they combine with lifestyle/environmental risk factors to influence risk. We undertook an international collaborative study to assess gene-environment interaction for risk of breast cancer. Data from 24 studies of the Breast Cancer Association Consortium were pooled. Using up to 34,793 invasive breast cancers and 41,099 controls, we examined whether the relative risks associated with 23 single nucleotide polymorphisms were modified by 10 established environmental risk factors (age at menarche, parity, breastfeeding, body mass index, height, oral contraceptive use, menopausal hormone therapy use, alcohol consumption, cigarette smoking, physical activity) in women of European ancestry. We used logistic regression models stratified by study and adjusted for age and performed likelihood ratio tests to assess gene–environment interactions. All statistical tests were two-sided. We replicated previously reported potential interactions between LSP1-rs3817198 and parity (Pinteraction = 2.4×10−6) and between CASP8-rs17468277 and alcohol consumption (Pinteraction = 3.1×10−4). Overall, the per-allele odds ratio (95% confidence interval) for LSP1-rs3817198 was 1.08 (1.01–1.16) in nulliparous women and ranged from 1.03 (0.96–1.10) in parous women with one birth to 1.26 (1.16–1.37) in women with at least four births. For CASP8-rs17468277, the per-allele OR was 0.91 (0.85–0.98) in those with an alcohol intake of <20 g/day and 1.45 (1.14–1.85) in those who drank ≥20 g/day. Additionally, interaction was found between 1p11.2-rs11249433 and ever being parous (Pinteraction = 5.3×10−5), with a per-allele OR of 1.14 (1.11–1.17) in parous women and 0.98 (0.92–1.05) in nulliparous women. These data provide first strong evidence that the risk of breast cancer associated with some common genetic variants may vary with environmental risk factors.
Introduction
Both genetic and non-genetic factors are involved in the etiology of breast cancer. Known susceptibility variants include rare high-risk mutations, principally in BRCA1 and BRCA2, more moderate susceptibility variants in genes such as PALB2, CHEK2 and ATM, and more than 20 common genetic susceptibility variants conferring modest increased risks, principally identified through genome-wide association studies. Taken together, the known susceptibility variants have been estimated to explain about 20–25% of the observed familial breast cancer risk [1]. There is still limited knowledge about how the relative risks of common susceptibility loci might be modified by the established reproductive and lifestyle risk factors (referred to as environmental risk factors) for breast cancer. Such knowledge could provide insights into common biological pathways for cancer development and further our understanding of breast cancer etiology for specific tumor subtypes. Previous reports of a possible interaction between variants in FGFR2 and use of menopausal hormone therapy (MHT) were not confirmed [2]–[6]. All recent large studies found no statistically significant evidence of multiplicative gene-environment interaction between several common susceptibility loci and established risk factors for breast cancer after allowing for multiple comparisons [2], [6], [7]. The strongest previously reported findings were for an interaction between LSP1-rs3817198 and number of births (P-value = 0.002), between CASP8-rs104585 and alcohol consumption (P-value = 0.003), and between 5p12-rs10941679 and use of estrogen-only MHT (P-value = 0.007) [2], [6], [7]. This lack of statistical evidence of interaction beyond that expected by chance may be partly due to limited power to detect weak gene-environment interactions and not having considered specific subtypes of breast cancer. We used pooled data from 24 studies participating in the Breast Cancer Association Consortium (BCAC) to evaluate whether the relative risks of single nucleotide polymorphisms (SNPs) at 23 published loci vary according to levels of 10 established environmental risk factors [8]. Since there is etiologic heterogeneity by subtypes of breast cancer, we also carried out these assessments for breast cancer with positive and negative estrogen receptor (ER) status [9].
Results
Up to 34,793 invasive cases and 41,099 controls of self-reported European ancestry were included in these analyses (Table 1). Based on 18,532 cases and 25,341 controls from 16 population-based studies, we found the expected associations between the environmental risk factors and breast cancer risk (Table 2). As expected, significant effect heterogeneity by age (as a surrogate for menopausal status) was observed only for body mass index (BMI) (P-value = 0.007).


Except for TGFB1-rs1982073, all SNPs showed highly significant associations with breast cancer overall (Table 3). Eleven SNPs showed evidence of heterogeneity in the OR by ER status at p<0.01. The per-allele OR overall and for subsets of women with information available for the risk factors considered were very similar to those previously published and provided no evidence of bias in OR estimates related to data availability (data not shown).

The strongest evidence was found for modification of the association with LSP1-rs3817198 by number of births in parous women (Pinteraction per birth increase in parous women = 2.4×10−6) (Table 4; Figure 1 showing individual study results). Since this interaction was previously assessed in BCAC, we reassessed the interaction in 6266 cases and 3899 controls not included in the previous report [7]. The SNP association still varied significantly with number of births in parous women (Pinteraction = 1.6×10−3), thus independently replicating the previous finding. The results were consistent across studies (Pheterogeneity = 0.37) (Figure 1B). In the overall dataset, the per-allele OR (95% confidence interval) for rs3817198 ranged from 1.03 (0.96–1.10) in parous women with one birth to 1.26 (1.16–1.37) in women with four or more births (Figure 2) and in comparison was 1.08 (1.01–1.16) in nulliparous women (Table S4).
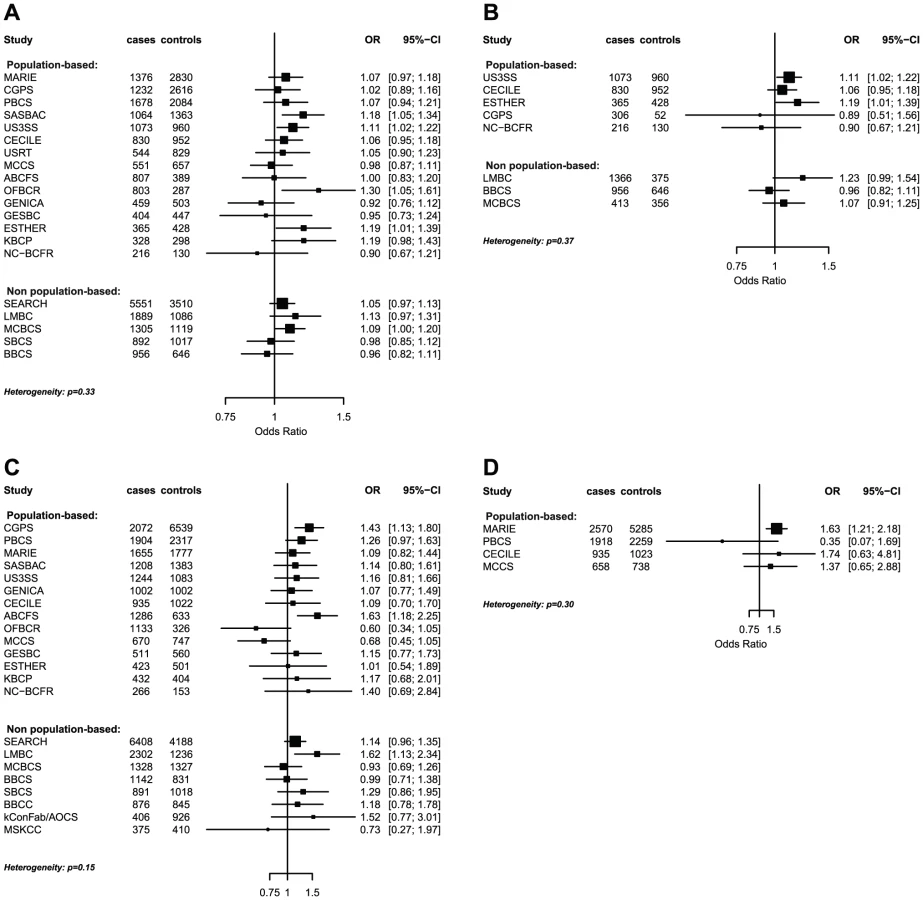


The polymorphism 1p11.2-rs11249433 was associated with breast cancer in parous (1.14, 1.11–1.17) but not nulliparous women (0.98, 0.92–1.05) (Pinteraction = 5.3×10−5). The interaction was non-significantly stronger for risk of ER-positive than ER-negative tumours (Pheterogeneity = 0.13, Table S5, Table S6), corresponding to this SNP being more strongly associated with ER-positive disease (Table 3). When restricted to ER-positive breast cancer, the per-allele OR for rs11249433 was 1.16 (1.13–1.20) in parous women and 0.97 (0.90–1.04) in nulliparous women (Pinteraction = 1.6×10−5) (Table 4). There was no significant heterogeneity in the interaction ORs by study (Figure 1C).
For the previously reported potential interaction between CASP8-rs1045485 (in complete LD with rs17468277) and alcohol consumption (<1 versus ≥1 drink/day) [6], we found moderate evidence when assessing effect modification by alcohol intake per 10 g/day increase (Pinteraction per 10 g/day = 3.0×10−3) (Table S4). However, when alcohol intake was dichotomized at 20 g/day (approximately 2 drinks/day), the estimated per-allele OR for CASP8-rs17468277 was 0.91 (0.84–0.98) in those who drank <20 g/day and 1.45 (1.14–1.85) in those who drank ≥20 g/day (Pinteraction = 3.1×10−4) (Table 4, Figure 1D).
We observed weaker evidence of differences in the associations with breast cancer for three further SNPs according to use of MHT and for one SNP according to age at first birth. These included rs13387042 and current use of combined estrogen/progestagen MHT (yes/no) (Pinteraction = 2.4×10−3), rs2823093 and current use of estrogen only MHT (yes/no) (Pinteraction = 6.6×10−3), rs999737 and duration of estrogen only MHT among current users (Pinteraction = 4.0×10−3), and rs614367 and age at first birth among parous women (Pinteraction = 9.1×10−3) (Table S4).
The observed SNP-environmental interaction ORs were not altered substantially (less than 8% change in the interaction ORs) when adjusting for additional covariates. These additional covariates included (when not the potentially effect-modifying variable of interest) ever parous (yes/no), number of births, BMI, age surrogate for postmenopausal status (≥54 years), interaction of BMI and postmenopausal status (≥54 years), current use of MHT, past use of MHT, duration of oral contraceptives (OC) use, lifetime alcohol intake, smoking (pack-years) (Table S7). Subjects with missing covariable information were excluded from these analyses, leading to considerably reduced sample sizes. Restricting the analyses to only 16 population-based studies did not change the results substantially (i.e., less than 3%) (Table S8).
The false-positive report probability (FPRP) was below 0.2 at a prior probability greater than 0.001 for the replicated effect modification of LSP1-rs3817198 by number of births and 1p11.2-rs11249433 and being ever parous. For the effect modification of CASP8-rs17468277 by alcohol intake ≥20 g/day, the FPRP was below 0.2 at a prior probability greater than 0.01. For the four potential interactions reported above, the FPRP was only below 0.2 at a prior probability greater than 0.05. (Table S9).
Discussion
We carried out a comprehensive evaluation of potential gene-environment interactions between 23 established common susceptibility variants for breast cancer and 10 established environmental risk factors, using 18 variables. Compared to the previous analysis, the present dataset from BCAC included 5 new population-based studies as well as additional study participants from some studies [7]. We examined additional environmental risk factors (14 variables), and 11 additional recently identified common susceptibility loci.
In our previous report, the strongest evidence of effect modification (P-value = 0.002) was observed for LSP1-rs3817198 by number of births [7]. The highly consistent and significant finding based on the present analysis of only additional cases and controls provided clear independent replication. We also show that the interaction holds for both ER-positive and ER-negative disease. This lack of heterogeneity is biologically plausible since neither the SNP nor number of births show heterogeneity by ER status in association with breast cancer risk [9], [10]. Only ever parous versus nulliparous but not the number of births in parous women was assessed for gene-environment interaction in two previous studies [2], [6]. Consistent with our data indicating no differential effects by ever parous compared to never parous, they did not find evidence of interaction between LSP1-rs3817198 and ever being parous. The rs3817198 SNP is located on the short arm of chromosome 11 and lies within LSP1, encoding lymphocyte-specific protein 1, an intracellular F-actin binding protein, although the gene underlying the association has not been definitively identified. The SNP lies close to the H19/IGF2 imprinted region, and the association of breast cancer with rs3817198 has been reported to be restricted to the paternally inherited allele [11]. The effect heterogeneity of LSP1-rs3817198 by number of births appears to be partly due to a significant negative correlation between number of rs3817198 C alleles and number of births in parous women (P-value = 0.002), which was found both in the data of our previous report as well as the additional data for the present analysis. Although not statistically significant, the mean number of children was also reported to be lower in women carrying the CC genotype in the Million Women Study [6]. Also of interest is that LSP1-rs3817198 has been associated with mammographic density, consistent with the direction of the breast cancer association [12]. Mammographic density has also been found to be reduced after a full-term pregnancy, particularly with greater number of births [13], [14].
We also replicated the strongest finding reported in the Million Women Study based on 7,610 cases and 10,196 controls [6]. In that study, the per-allele OR of CASP8-rs1045485 (or rs17468277 in our dataset) was 0.99 (0.92–1.07) in those who reported <1 drink/day and 1.23 (1.09–1.38) in those who reported ≥1 drink/day (P-value = 0.003). Our observation of an increased per-allele OR of 1.45 (1.14–1.85) for those who reported high alcohol intake ≥20 g/day and 0.91 (0.84–0.98) for those who consume less provides independent replication of this SNP-environmental interaction. Although one drink corresponds to an intake of approximately 10 g alcohol, the Million Women study reported the strongest risk increase in breast cancer for women consuming at least 15 drinks per week (RR 1.29 (1.23–1.35)) [15], corresponding to approximately to 2 drinks per day (20 g alcohol). There is no known functional effect of CASP8-rs1045485, however, it is associated with a risk haplotype in CASP8, which is more strongly associated with breast cancer risk [16], [17]. Caspase 8 is an important initiator of apoptosis and is activated in response to DNA damage that can be caused by alcohol consumption through ethanol-related oxidative stress [18].
Ever being parous, but not number of births, was found to modify the effect of a different SNP, 1p11.2-rs11249433, in particular for ER-positive breast cancer. This SNP shows significantly stronger association with risk of ER-positive tumors than of ER-negative tumors [19]. In nulliparous women, rs11249433 was not associated with risk of ER-positive disease, whereas in parous women, the per-allele OR of 1.14 was slightly greater than the overall OR of 1.12. The Breast and Prostate Cancer Cohort Consortium evaluated interactions between 13 of the 23 genetic loci and 9 risk factors, including 1p11.2-rs11249433 and ever parous. They found no evidence for this interaction (P-value = 0.79), with per-allele OR of 1.09 (1.04–1.14) in parous and 1.11 (0.99–1.24) in nulliparous women [2]. These ORs are not in the same relative direction as our finding with respect to ever being parous. This may be in part due to misclassification of parity if information on parity for participants of the cohort studies was only available at time of recruitment and therefore incomplete with reference to the diagnosis of breast cancer. Their analysis was based on 8,576 cases and 11,892 controls, which had considerably lower statistical power than the present study. The SNP rs11249433 is located on the short arm of chromosome 1 close to the centromere, which makes it hard to map. The nearest known genes are FCGR1B (low-affinity Fc gamma receptor family) and NOTCH2 (coding a transmembrane receptor protein). Recently, a study reported a positive association of NOTCH2 mRNA expression with the breast cancer risk allele of rs11249433 [20]. This association was strongest with the subgroup of ER-positive breast tumors without TP53 mutation, providing some evidence that the increased risk of ER-positive breast cancer might be due to differences in NOTCH2 expression [20].
The evidence for the other four potential interactions mentioned in the results was considerably weaker and confirmation of these findings in further studies is therefore required. Three of these involved effect modification by use of MHT. The effect modification of RAD51L1-rs999737 by duration of estrogen only MHT in current users is particularly interesting because this polymorphism has been associated with mammographic density in the same direction as the breast cancer association [12]. Mammographic density has also been found to be increased in postmenopausal women among users of MHT [21].
RAD51L1 is a member of the Rad51-like proteins that play a crucial role in homologous recombinational repair [22]. Rare deleterious mutations in other genes of this pathway, including BRCA1 and BRCA2, confer a high risk of breast cancer [1], [23]. Menopausal hormone therapy has been suggested to alter breast cancer risk in BRCA1 mutation carriers although the evidence is still limited [24]. It is thus plausible that estrogen only MHT modifies the relative risk for genetic variants in RAD51L1 on breast cancer risk.
NRIP1 (nuclear receptor–interacting protein 1), also called RIP140 (receptor-interacting protein 140), is known to interact with ERα, repress ER signaling and inhibit its mitogenic effects [25]. Exposure to exogenous estrogens through MHT, which stimulate ER signalling, could therefore alter the association of NRIP1 rs2823093 with breast cancer.
It is less clear how 2q35-rs13387042 might be modified by current combined estrogen/progestagen MHT use since the gene involved at this locus is still unknown. The SNP is located on the short arm of chromosome 2 and lies in a linkage disequilibrium (LD) block containing no known gene(s) or non-coding RNAs. The closest known genes are TNP1 (transition protein 1), IGFBP5 (insulin-like growth factor binding protein 5), IGFBP2 (insulin-like growth factor binding protein 2) and TNS1 (tensin 1/matrix-remodelling-associated protein 6) [26]. The observed effect modification would suggest that the gene involved may be responsive to steroid hormones.
Both Campa et al. and the Million Women Study investigated potential interactions with MHT (overall use) [2], [6]. Neither study reported evidence for interaction between 2q35-rs13387042 or RAD51L1-rs999737 with MHT and breast cancer risk. However, neither study considered current use of MHT even though elevated risks for breast cancer have been clearly established for current use and not for past use [6], [27], [28]. Yet Campa et al. found differences in OR estimates for 2q35-rs13387042 by ever use of combined estrogen/progestagen MHT in the same direction as our results for current combined estrogen/progestagen MHT use, with a per-allele OR of 0.83 (0.78–0.89) in non-users and 0.77 (0.69–0.86) in ever combined estrogen/progestagen MHT users (P-value = 0.26) (in their Supplementary Table 5). We were not able to confirm the previously suggested possible interaction of 5p12-rs10941679 or FGFR2 variants with MHT and other factors [2]–[5]. Our data suggest that age at first birth in parous women may modify the effect of 11q13-rs614367, which is located in a region containing no known genes [29]. This newly identified SNP has not been previously assessed for interaction with environmental risk factors.
One of the strengths of our study is the large sample size, required for assessing weak to moderate gene-environment interactions, particularly when marker SNPs instead of causal variants are used [30]. We assessed gene-environment interaction separately for ER-positive and ER-negative disease, thereby accounting for heterogeneity by ER status in risk associated with both genetic and environmental factors. However, statistical power was still limited to detect interactions in susceptibility to ER-negative disease. Although selection bias is likely to affect estimates of environmental main effects, under reasonable assumptions, it should not influence the assessment of multiplicative gene-environment interactions or estimates of SNP relative risks [31]. Furthermore, both non-differential and differential misclassification of exposure tend to underestimate the multiplicative interaction parameter rather than yield spurious evidence of interaction [32]. To reduce potential bias due to population stratification, we restricted our analyses to subjects of European ancestry and stratified by study in all analyses. The robustness of our findings to differences in study design was supported by sensitivity analyses considering only data from population-based studies. The interaction estimates also did not change substantially when adjusting for further covariates: the p-values were however higher due to the considerably reduced sample sizes. The absence of study heterogeneity in the estimates of gene-environment interactions provides further reassurance of the robustness of the findings.
The effect modifications identified in our study are relatively weak and should result in small differences in risk estimates of joint effects compared to those based on models assuming multiplicative effects. However, most of the SNPs investigated are only markers of the underlying causal variants and underestimate the effects of the causal variants if linkage disequilibrium is incomplete [33]. Thus, gene-environment interactions with the underlying causal variant could have a greater modifying effect on the relative risk [30]. These findings also underline the importance of investigating interactions separately for causally distinct subtypes of breast cancer in future assessments of gene-environment interaction.
In summary, we provide strong evidence of effect modification of LSP1-rs3817198 by number of births and of CASP8-rs1045485 by alcohol consumption. For some additional common genetic variants, the associations with breast cancer risk may vary with environmental factors. However, there is little evidence for multiplicative gene-environment interactions for most susceptibility loci and environmental risk factors. Understanding the biological implications of the observed interactions could provide further insight into the etiology of breast cancer. The potential impact of these results on risk prediction for breast cancer needs to be considered in future studies.
Methods
Study participants and risk factor data
We used primary data from the studies in BCAC. All studies had approval from the relevant ethics committees and all participants gave informed consent. A centralized BCAC database of information about common risk factors and tumor characteristics was constructed to facilitate studies of potential modifications of SNP associations by other risk factors. A multi-step data harmonization procedure was used to reconcile differences in individual study questionnaires. The reference age for cohort studies was calculated at time of enrollment and for case-control studies at date of diagnosis for cases and at date of interview for controls. All time-dependent variables were assessed at reference age. This analysis included only subjects of European ancestry that had genotype data for at least 3 SNPs and provided information on at least one of the established risk factors. Relevant data were available from 24 studies, including 16 population-based studies (14 case-control and 2 prospective cohort studies) and 8 non-population-based studies (Table 1, Table S1, Table S2). Subsets of data from 19 studies (with 11 population-based) were included in a previous report that assessed interactions between 12 susceptibility variants, reproductive history, BMI and breast cancer risk [7].
SNP selection and genotyping
We included 21 SNPs found to be associated with overall breast cancer risk at genome-wide statistical significance (p<5×10−7) [10], [25], [34] and SNPs for TGFB1 and CASP8 from candidate gene studies [17] (Table S3). For three loci, 14q24.1/RAD51L1, 12p11, CASP8, a surrogate SNP in high linkage disequilibrium (LD) (r2 = 1 in HapMap CEU) was genotyped in a subset (Table 3 footnote) [19], [25], [35].
Genotyping was performed in the framework of BCAC by Taqman and iPlex assays and underwent quality control as described previously [10], [19], [25], [34], [36], [37]. Genotype data were excluded from analysis on a study-by-study basis according to the following BCAC quality control (QC) guidelines: 1) any sample that consistently failed for >20% of the SNPs within a genotyping round, 2) all samples on any one plate that had a call rate <90% for any one SNP, 3) all genotype data for any SNP where overall call rate was <95%, 4) all genotype data for any SNP where duplicate concordance was <98%. In addition, for any SNP where the P-value for departure from Hardy-Weinberg proportions for controls was <0.005, clustering of the intensity plots was reviewed manually and the data excluded if clustering was judged to be poor.
Statistical methods
We used logistic regression to assess the main effects of the SNP and environmental risk factors on invasive breast cancer risk. Analyses were adjusted for study as a categorical variable and reference age as a continuous variable. Odds ratios (OR) and their 95% confidence intervals (CI) were calculated for the SNP associations assuming a log-additive model and tested for association with a one degree of freedom trend test. All statistical tests were two-sided.
The assessment of associations with the environmental risk factors was based on data only from the 16 population-based studies to ensure unbiased estimates for comparison with established effect sizes. The variables considered were analyzed as continuous (age at menarche, number of births in parous women, age at first birth, usual BMI, height, duration of oral contraceptive use, duration of current use of estrogen-progestagen combined therapy, duration of current use of estrogen-only therapy, pack-years of cigarette smoking, mean lifetime daily grams of alcohol intake, recent physical activity in hours per week), or as dichotomous (ever parous, ever breastfed, ever OC use, ever smoked, current EPT use, current ET use) (Table 2). Analyses were performed for all women as well as separately for women aged <54 years and ≥54 years, considering the age groups as surrogates of pre - and postmenopausal status, Differential effects by menopausal status were assessed by adding an interaction term. For all categorical variables, the lowest level of exposure (or no use) was used as the reference. For evaluating current use of MHT by type, we used never use of MHT as the reference category and additionally adjusted for former use of MHT and other MHT type, as appropriate.
To test for interactions between SNPs and environmental risk factors, we fitted for each SNP two logistic models, a model with terms for the SNP and the risk factor of interest and another model with additionally an interaction term for the product between the SNP (number of risk alleles) and the risk factor variable. We modeled the interaction based on the risk factor variable definitions employed for the main effects. All analyses were stratified by study and adjusted for age as a continuous variable. The likelihood ratio test was used to compare the difference between the two models and departure from independent multiplicative effects of the SNP and the risk factor. BMI was the only variable found to show differential effects by menopausal status, which is consistent with the literature [38]. Therefore, interaction between SNPs and BMI was assessed separately for pre - and postmenopausal women whereas all other risk factors were evaluated regardless of menopausal status. To assess study heterogeneity, we calculated odds ratios for interaction for each individual study, adjusting for age, and reported P-values for heterogeneity using a Q-test. Subjects with missing data for a particular SNP or environmental factor were excluded from the respective analysis. We also calculated stratum specific per-allele ORs for each SNP: age at menarche (≤11, 12–13, ≥14 years), number of births (1,2,3, ≥4), age at first birth (<20, 20–24, 25–29, ≥30 years), usual BMI (<25, 25–29, ≥30), height (<160, 160–164, 165–169, ≥170 cm), duration of oral contraceptive use and of menopausal hormone use (0, >0–<5, 5–<10, ≥10 years), mean lifetime alcohol intake (0, 0–<10, 10–<20, ≥20 g/day), pack-years of smoking (0, 1–<10, 10–<20, ≥20), and physical activity (0, >0–<3.5, ≥3.5–<7, ≥7 h/week).
For SNP-environment interactions with associated P-value<10−3, we also compared results after adjusting for additional covariates. We performed a total of 414 (23 SNPs x 18 risk variables) tests. To account for chance findings due to multiple comparisons, we calculated the false positive report probability (FPRP) for SNP-environment interactions with associated P-value<10−3 [39]. The FPRP depends on the prior probability that the association between the SNP and breast cancer is modified by the environmental risk factor, the power of the present study, and the observed P-value. Since the prior probability of the assessed multiplicative interactions varies depending on subjective evaluation of existing evidence, we calculated the FPRPs for prior probabilities ranging from 0.05 to 0.0001. We considered findings with FPRP below 0.2 to be noteworthy results, as previously proposed [39].
In secondary analyses, we examined associations and effect modifications separately for women with ER-positive tumors and ER-negative tumors, each compared to all controls. Effect heterogeneity by ER status was tested using case-case analysis.
Data harmonization was performed using an ACCESS database and transformation of the data elements was performed using SAS (Release 9.2). All other data analyses were conducted using SAS (Release 9.2) and the R programming language [40].
Supporting Information
Zdroje
1. MavaddatN, AntoniouAC, EastonDF, Garcia-ClosasM (2010) Genetic susceptibility to breast cancer. Mol Oncol 4 : 174–191.
2. CampaD, KaaksR, Le MarchandL, HaimanCA, TravisRC, et al. (2011) Interactions between genetic variants and breast cancer risk factors in the breast and prostate cancer cohort consortium. J Natl Cancer Inst 103 : 1252–1263.
3. KawaseT, MatsuoK, SuzukiT, HirakiA, WatanabeM, et al. (2009) FGFR2 intronic polymorphisms interact with reproductive risk factors of breast cancer: results of a case control study in Japan. Int J Cancer 125 : 1946–1952.
4. PrenticeRL, HuangY, HindsDA, PetersU, PettingerM, et al. (2009) Variation in the FGFR2 gene and the effects of postmenopausal hormone therapy on invasive breast cancer. Cancer Epidemiol Biomarkers Prev 18 : 3079–3085.
5. RebbeckTR, DeMicheleA, TranTV, PanossianS, BuninGR, et al. (2009) Hormone-dependent effects of FGFR2 and MAP3K1 in breast cancer susceptibility in a population-based sample of post-menopausal African-American and European-American women. Carcinogenesis 30 : 269–274.
6. TravisRC, ReevesGK, GreenJ, BullD, TipperSJ, et al. (2010) Gene-environment interactions in 7610 women with breast cancer: prospective evidence from the Million Women Study. Lancet %19 375 : 2143–2151.
7. MilneRL, GaudetMM, SpurdleAB, FaschingPA, CouchFJ, et al. (2010) Assessing interactions between the associations of common genetic susceptibility variants, reproductive history and body mass index with breast cancer risk in the breast cancer association consortium: a combined case-control study. Breast Cancer Res 12: R110.
8. Breast Cancer Association Consortium (2006) Commonly studied single-nucleotide polymorphisms and breast cancer: results from the Breast Cancer Association Consortium. J Natl Cancer Inst 98 : 1382–1396.
9. YangXR, Chang-ClaudeJ, GoodeEL, CouchFJ, NevanlinnaH, et al. (2011) Associations of breast cancer risk factors with tumor subtypes: a pooled analysis from the Breast Cancer Association Consortium studies. J Natl Cancer Inst 103 : 250–263.
10. BroeksA, SchmidtMK, ShermanME, CouchFJ, HopperJL, et al. (2011) Low penetrance breast cancer susceptibility loci are associated with specific breast tumor subtypes: findings from the Breast Cancer Association Consortium. Hum Mol Genet 20 : 3289–3303.
11. KongA, SteinthorsdottirV, MassonG, ThorleifssonG, SulemP, et al. (2009) Parental origin of sequence variants associated with complex diseases. Nature 462 : 868–874.
12. VachonCM, ScottCG, FaschingPA, HallP, TamimiRM, et al. (2012) Common Breast Cancer Susceptibility Variants in LSP1 and RAD51L1 Are Associated with Mammographic Density Measures that Predict Breast Cancer Risk. Cancer Epidemiol Biomarkers Prev
13. ButlerLM, GoldEB, GreendaleGA, CrandallCJ, ModugnoF, et al. (2008) Menstrual and reproductive factors in relation to mammographic density: the Study of Women's Health Across the Nation (SWAN). Breast Cancer Res Treat 112 : 165–174.
14. LoehbergCR, HeusingerK, JudSM, HaeberleL, HeinA, et al. (2010) Assessment of mammographic density before and after first full-term pregnancy. Eur J Cancer Prev 19 : 405–412.
15. AllenNE, BeralV, CasabonneD, KanSW, ReevesGK, et al. (2009) Moderate alcohol intake and cancer incidence in women. J Natl Cancer Inst 101 : 296–305.
16. CampNJ, ParryM, KnightS, AboR, ElliottG, et al. (2012) Fine-mapping CASP8 risk variants in breast cancer. Cancer Epidemiol Biomarkers Prev 21 : 176–181.
17. CoxA, DunningAM, Garcia-ClosasM, BalasubramanianS, ReedMW, et al. (2007) A common coding variant in CASP8 is associated with breast cancer risk. Nat Genet 39 : 352–358.
18. DumitrescuRG, ShieldsPG (2005) The etiology of alcohol-induced breast cancer. Alcohol 35 : 213–225.
19. FigueroaJD, Garcia-ClosasM, HumphreysM, PlatteR, HopperJL, et al. (2011) Associations of common variants at 1p11.2 and 14q24.1 (RAD51L1) with breast cancer risk and heterogeneity by tumor subtype: findings from the Breast Cancer Association Consortium. Hum Mol Genet 20 : 4693–4706.
20. FuYP, EdvardsenH, KaushivaA, ArhancetJP, HoweTM, et al. (2010) NOTCH2 in breast cancer: association of SNP rs11249433 with gene expression in ER-positive breast tumors without TP53 mutations. Mol Cancer 9 : 113.
21. BoydNF, MelnichoukO, MartinLJ, HislopG, ChiarelliAM, et al. (2011) Mammographic density, response to hormones, and breast cancer risk. J Clin Oncol 29 : 2985–2992.
22. LioYC, MazinAV, KowalczykowskiSC, ChenDJ (2003) Complex formation by the human Rad51B and Rad51C DNA repair proteins and their activities in vitro. J Biol Chem 278 : 2469–2478.
23. MeindlA, HellebrandH, WiekC, ErvenV, WappenschmidtB, et al. (2010) Germline mutations in breast and ovarian cancer pedigrees establish RAD51C as a human cancer susceptibility gene. Nat Genet 42 : 410–414.
24. ChlebowskiRT, PrenticeRL (2008) Menopausal hormone therapy in BRCA1 mutation carriers: uncertainty and caution. J Natl Cancer Inst 100 : 1341–1343.
25. GhoussainiM, FletcherO, MichailidouK, TurnbullC, SchmidtMK, et al. (2012) Genome-wide association analysis identifies three new breast cancer susceptibility loci. Nat Genet 44 : 312–318.
26. StaceySN, ManolescuA, SulemP, RafnarT, GudmundssonJ, et al. (2007) Common variants on chromosomes 2q35 and 16q12 confer susceptibility to estrogen receptor-positive breast cancer. Nat Genet 39 : 865–869.
27. BeralV (2003) Breast cancer and hormone-replacement therapy in the Million Women Study. Lancet 362 : 419–427.
28. Flesch-JanysD, SlangerT, MutschelknaussE, KroppS, ObiN, et al. (2008) Risk of different histological types of postmenopausal breast cancer by type and regimen of menopausal hormone therapy. Int J Cancer 123 : 933–941.
29. TurnbullC, AhmedS, MorrisonJ, PernetD, RenwickA, et al. (2010) Genome-wide association study identifies five new breast cancer susceptibility loci. Nat Genet 42 : 504–507.
30. HeinR, BeckmannL, Chang-ClaudeJ (2008) Sample size requirements for indirect association studies of gene-environment interactions (G x E). Genet Epidemiol 32 : 235–245.
31. MorimotoLM, WhiteE, NewcombPA (2003) Selection bias in the assessment of gene-environment interaction in case-control studies. Am J Epidemiol 158 : 259–263.
32. Garcia-ClosasM, ThompsonWD, RobinsJM (1998) Differential misclassification and the assessment of gene-environment interactions in case-control studies. Am J Epidemiol 147 : 426–433.
33. ZondervanKT, CardonLR (2004) The complex interplay among factors that influence allelic association. Nat Rev Genet 5 : 89–100.
34. FletcherO, JohnsonN, OrrN, HoskingFJ, GibsonLJ, et al. (2011) Novel breast cancer susceptibility locus at 9q31.2: results of a genome-wide association study. J Natl Cancer Inst 103 : 425–435.
35. ThomasG, JacobsKB, KraftP, YeagerM, WacholderS, et al. (2009) A multistage genome-wide association study in breast cancer identifies two new risk alleles at 1p11.2 and 14q24.1 (RAD51L1). Nat Genet 41 : 579–584.
36. MilneRL, BenitezJ, NevanlinnaH, HeikkinenT, AittomakiK, et al. (2009) Risk of estrogen receptor-positive and -negative breast cancer and single-nucleotide polymorphism 2q35-rs13387042. J Natl Cancer Inst 101 : 1012–1018.
37. MilneRL, GoodeEL, Garcia-ClosasM, CouchFJ, SeveriG, et al. (2011) Confirmation of 5p12 as a susceptibility locus for progesterone-receptor-positive, lower grade breast cancer. Cancer Epidemiol Biomarkers Prev 20 : 2222–2231.
38. van den BrandtPA, SpiegelmanD, YaunSS, AdamiHO, BeesonL, et al. (2000) Pooled analysis of prospective cohort studies on height, weight, and breast cancer risk. Am J Epidemiol 152 : 514–527.
39. WacholderS, ChanockS, Garcia-ClosasM, El GhormliL, RothmanN (2004) Assessing the probability that a positive report is false: an approach for molecular epidemiology studies. J Natl Cancer Inst 96 : 434–442.
40. R Development Core Team (2011) R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2013 Číslo 3
Nejčtenější v tomto čísle
- Fine Characterisation of a Recombination Hotspot at the Locus and Resolution of the Paradoxical Excess of Duplications over Deletions in the General Population
- Molecular Networks of Human Muscle Adaptation to Exercise and Age
- Recurrent Rearrangement during Adaptive Evolution in an Interspecific Yeast Hybrid Suggests a Model for Rapid Introgression
- Genome-Wide Association Study and Gene Expression Analysis Identifies as a Predictor of Response to Etanercept Therapy in Rheumatoid Arthritis