
Functional Genomic Analysis of the Regulatory Network in
The let-7 microRNA (miRNA) regulates cellular differentiation across many animal species. Loss of let-7 activity causes abnormal development in Caenorhabditis elegans and unchecked cellular proliferation in human cells, which contributes to tumorigenesis. These defects are due to improper expression of protein-coding genes normally under let-7 regulation. While some direct targets of let-7 have been identified, the genome-wide effect of let-7 insufficiency in a developing animal has not been fully investigated. Here we report the results of molecular and genetic assays aimed at determining the global network of genes regulated by let-7 in C. elegans. By screening for mis-regulated genes that also contribute to let-7 mutant phenotypes, we derived a list of physiologically relevant potential targets of let-7 regulation. Twenty new suppressors of the rupturing vulva or extra seam cell division phenotypes characteristic of let-7 mutants emerged. Three of these genes, opt-2, prmt-1, and T27D12.1, were found to associate with Argonaute in a let-7–dependent manner and are likely novel direct targets of this miRNA. Overall, a complex network of genes with various activities is subject to let-7 regulation to coordinate developmental timing across tissues during worm development.
Published in the journal:
. PLoS Genet 9(3): e32767. doi:10.1371/journal.pgen.1003353
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1003353
Summary
The let-7 microRNA (miRNA) regulates cellular differentiation across many animal species. Loss of let-7 activity causes abnormal development in Caenorhabditis elegans and unchecked cellular proliferation in human cells, which contributes to tumorigenesis. These defects are due to improper expression of protein-coding genes normally under let-7 regulation. While some direct targets of let-7 have been identified, the genome-wide effect of let-7 insufficiency in a developing animal has not been fully investigated. Here we report the results of molecular and genetic assays aimed at determining the global network of genes regulated by let-7 in C. elegans. By screening for mis-regulated genes that also contribute to let-7 mutant phenotypes, we derived a list of physiologically relevant potential targets of let-7 regulation. Twenty new suppressors of the rupturing vulva or extra seam cell division phenotypes characteristic of let-7 mutants emerged. Three of these genes, opt-2, prmt-1, and T27D12.1, were found to associate with Argonaute in a let-7–dependent manner and are likely novel direct targets of this miRNA. Overall, a complex network of genes with various activities is subject to let-7 regulation to coordinate developmental timing across tissues during worm development.
Introduction
MicroRNAs (miRNAs) are an abundant class of regulatory genes that control many cellular and developmental processes [1]. The biogenesis of miRNAs requires multiple steps, beginning with transcription by RNA polymerase II to produce capped and polyadenylated primary transcripts [2], [3]. These transcripts are processed sequentially by the RNase III enzymes Drosha and Dicer, resulting in the ∼22 nucleotide (nt) single stranded mature miRNA. The mature miRNA is incorporated into the RNA induced silencing complex (RISC), which uses the miRNA as a sequence specific guide to find and mediate regulation of target mRNAs. The miRISC usually induces translational repression and destabilization of the target mRNA through mechanisms that are still being determined [4], [5].
let-7 was originally discovered as a miRNA controlling developmental timing in Caenorhabditis elegans [6], [7]. The lethality associated with mutations in this gene is at least partly due to vulval rupturing, where internal organs burst out of the egg-laying pore. Additionally, lateral hypodermal seam cells fail to terminally differentiate at the larval to adult transition in let-7 mutants. These phenotypes place let-7 in the heterochronic pathway, which includes genes that regulate the temporal identity of cell divisions and fates [6], [8]. let-7 regulates developmental timing, in part, through the direct target genes lin-41 and hbl-1 [6], [7], [9], [10]. These genes, in turn, regulate the transcription factor lin-29, which directly controls terminal differentiation in the hypodermis [6], [7], [9], [10]. Several transcription factors, such as the nuclear hormone receptor daf-12, the forkhead transcription factor pha-4 and the zinc finger protein die-1, genetically interact with let-7 and are also likely direct targets [11]. Genetic mutation or RNAi depletion of any one of these let-7 targets is sufficient to at least partially rescue the lethality of let-7 mutants.
The let-7 miRNA is a widely conserved animal miRNA and its role in regulating differentiation also appears to be conserved [12], [13], [14]. Typically, expression of let-7 family miRNAs is negligible in stem cells and in early embryonic tissues and is then up-regulated as cells take on more differentiated fates. In worms and mammalian cells, the LIN-28 RNA binding protein is largely responsible for keeping let-7 miRNA levels low during early development [15]. LIN-28 prevents the maturation of let-7 family miRNAs by blocking Drosha or Dicer processing or promoting destabilization of let-7 precursors [16], [17], [18], [19], [20], [21], [22], [23]. The abnormally low expression of let-7 detected in various types of tumors has been linked, in some cases, to aberrant up-regulation of LIN-28 [24]. Additionally, let-7 and LIN-28 have opposing effects on insulin sensitivity in mice [25], [26]. This is due at least in part to direct targeting of several metabolic genes by let-7 miRNA.
Consistent with its role in promoting differentiated states, decreased expression of let-7 miRNA has been associated with numerous types of cancer [14]. In fact, one of the first discovered targets of let-7 in humans is RAS, a notorious oncogene [27]. Since then, many genes that promote cell division or antagonize the differentiated state have been implicated as direct or indirect targets of let-7 regulation [28], [29], [30], [31], [32], [33]. Remarkably, the introduction of let-7 miRNA into lung or breast tumors in mouse models has been shown to halt tumor growth in vivo [31], [34], [35]. Thus, let-7 functions as a tumor suppressor in at least in some settings, where it represses the expression of genes needed for oncogenesis.
To understand how let-7 or any miRNA controls a cellular process, the genes it regulates must be identified. Many computational prediction approaches have been taken to match miRNAs to targets [11], [36], [37], [38], [39], [40], [41]. However, the limited overlap of predicted targets between programs suggests that a consensus regarding the rules for target recognition is yet to be reached. The best defined motif for target recognition is perfect pairing of miRNA nucleotides 2–7, called the “seed” region, with a target sequence [42]. Deviations from seed pairing can be compensated for by strong pairing of the 3′ end of the miRNA or “centered sites”, where the middle portion of the miRNA forms consecutive base pairs with the target [42], [43]. Several validated target sites fail to conform to any of these motifs [42], [44]. Furthermore, other features, such as location within an mRNA and RNA secondary structure surrounding the miRNA complementary sequence influence whether a target site will be recognized in vivo. Ultimately, the endogenous context of the target site and the cellular environment will determine which sequences will be recognized and regulated by miRISC.
Numerous experimental methods have complemented the in silico endeavors to match miRNAs with direct targets. Traditional genetic as well as RNAi-based suppressor screens have uncovered major targets of the first described miRNAs in C. elegans [6], [7], [11], [27], [45], [46]. More high-throughput methods have been based on the general role of miRNAs in down-regulating mRNA and protein levels of their targets [5], [47]. Microarray or RNA-seq analysis of transcripts up-regulated when a miRNA is absent can provide lists of potential direct targets [48], [49], [50], [51]. Likewise, large-scale proteomics analyses have been used to detect proteins sensitive to changes in expression of specific miRNAs [52], [53], [54]. More recently, ribosome profiling has been developed as an indirect method for assessing changes in the translation status of mRNAs, leading to the conclusion that regulation by miRISC generally results in target mRNA destabilization [55], [56]. A more direct approach for detecting targets of miRISC is to capture mRNAs associated with Argonaute complexes. RNA immunoprecipitation (RIP) or cross-linking followed by IP (CLIP) protocols have been used to identify entire transcripts or the actual mRNA sequences in contact with Argonaute, respectively [57], [58], [59], [60], [61], [62], [63], [64], [65], [66]. These types of experiments demonstrate that an mRNA is recognized by miRISC but do not necessarily reveal the identity of the miRNA involved or if the interaction is regulatory.
We combined several molecular and genetic methods to identify physiologically relevant targets of let-7 in C. elegans. Our approach for discovering new let-7 regulatory targets takes advantage of let-7 dependent expression differences of the known targets, including lin-41 [67], [68]. We postulated that other direct targets would also be mis-regulated in let-7 mutants. Therefore, in vivo expression changes were analyzed in wild-type (WT) and let-7 mutant animals using microarray analysis to identify a list of relevant candidate target genes. This list of genes was further refined by computational target predictions and expression analysis in the downstream heterochronic mutant, lin-29. The relevance of the up-regulated genes for let-7 phenotypes was tested through RNAi-based suppressor screens. These genetic analyses revealed twenty new downstream effectors of let-7 phenotypes, including multiple transcription factors and metabolic proteins. Several of these genes also affect let-7 dependent phenotypes seen in lin-28 mutants revealing a complex genetic interaction with let-7. By showing let-7 dependent association with Argonaute, we were able to confirm three new direct targets of let-7 with binding sites in the 3′ UTRs as well as in coding regions.
Results
While some direct targets of the let-7 miRNA are known, a full picture of the let-7 regulatory network remains largely uncharacterized. Although several groups have attempted to identify let-7 targets in C. elegans, the criteria and, consequently, the predicted targets from these approaches have minimal overlap [11], [36], [38], [39], [40], [41], [69]. We have undertaken a multi-step approach for the discovery and validation of let-7 targets in C. elegans, beginning with analysis of global, let-7-dependent gene expression changes, and followed by genetic interaction analysis of candidates. Final validation of direct targets was confirmed by let-7 dependent RISC association (Figure 1).

Widespread gene mis-regulation in worms deficient for let-7 activity
We have previously shown let-7-dependent mRNA destabilization of known direct targets [67], suggesting that in addition to giving a general picture of let-7 function, microarray analysis of gene-misregulation in let-7 mutants will provide a basis for the discovery of new direct targets. The let-7(n2853) mutation changes the fifth G to an A in the mature let-7 miRNA [6], which destabilizes target interactions and results in up-regulation of lin-41 mRNA, an established let-7 target [7], [67], [70]. To identify globally the genes regulated by let-7, six independent and paired wild-type and let-7(n2853) fourth larval stage (L4) RNA samples were labeled and hybridized to Affymetrix arrays. 2216 genes were up-regulated, and 1905 genes were down-regulated in the let-7(n2853) mutants compared to WT worms (FDR<0.05) (Table S1). By microarray analysis, most of the differentially expressed genes were only modestly mis-regulated, as only 42 genes were up-regulated >2-fold (Table 1) and 49 were down-regulated by >2-fold (Table 2). Illustrating the role of let-7 as a master regulator of development, the up-regulated genes were enriched for Biological Process Gene Ontology (GO) terms representing larval growth and development (Table S1). The up-regulated genes represent direct, including the known targets lin-41, daf-12, and hbl-1, and indirect targets of let-7 repression. To further investigate the regulatory relationships between let-7 and the up-regulated genes, a combination of computational and molecular-genetic criteria were used to enrich for direct target candidates among the up-regulated genes.


Enrichment of let-7 complementary sequences in the 3′ UTRs of genes up-regulated in let-7 mutants
Direct mRNA targets of miRNAs typically have partially complementary miRNA binding sites, making prediction of miRNA targets from genomic sequence difficult [42], and many groups have developed a variety of rules for target recognition [11], [36], [38], [39], [40], [41], [69]. To enrich for biologically relevant candidates and allow for non-canonical binding sites, we searched for enriched 6-mer sequences in the 3′ UTRs of the genes up-regulated in let-7 mutants. Two conserved 6-mers complementary to let-7 mature sequence were enriched in the 3′ UTRs in the up-regulated gene set (Table S1). As expected, the nucleotides TACCTC, which are complementary to the let-7 seed sequence (nucleotides 2–7 of a mature miRNA), were enriched, consistent with the prevailing model for miRNA target recognition [42]. Also enriched was AACCTA, complementary to nucleotides 9–14 of let-7, which overlaps with the newly described “centered sites” observed for some miRNA target interactions [43]. 158 genes that were up-regulated in let-7 mutants had at least one of these two 6-mers in their 3′ UTRs. The presence of strong seed enrichment in the up-regulated gene set led us to include an additional 8 and 5 up-regulated, predicted targets found by the seed based algorithms PicTar and TargetScan respectively, for further analysis. From the three prediction methods, there were 167 unique direct target candidates, including the known targets lin-41, daf-12, and hbl-1.
Elimination of likely indirect downstream targets of let-7 regulation
We also employed an alternative filter to select potential let-7 targets independent of preconceptions about base pairing requirements. let-7 is near the end of a genetic pathway controlling developmental timing in C. elegans [71]. Negative regulation of lin-41 by let-7 in late larval stages allows the transcription factor LIN-29 to accumulate and to directly control the terminal differentiation of multiple cell types [6], [7], [72], [73]. In let-7 mutants, lin-41 persists in late larval stages where it can continue to negatively regulate lin-29 [6], [7]. Thus, in let-7 mutants, larval genes turned off by lin-29 will be up-regulated in addition to direct targets of let-7. In lin-29 mutants, the same downstream larval genes should be up-regulated, yet the upstream direct targets of let-7 should be unaffected. By analyzing gene-expression in lin-29 versus let-7 mutants, novel targets can be found that may not have obvious binding sites.
Three lin-29(n333) mutant L4 RNA samples paired with wildtype and let-7(n2853) samples were collected, labeled and hybridized to Affymetrix microarrays. In lin-29(n333), 3030 genes were up-regulated and 1994 genes were down-regulated relative to WT samples (Table S2). Consistent with a role for lin-29 in directing terminal differentiation and adult fates, genes up-regulated in lin-29 mutants were enriched for GO terms for larval development (Table S2). In comparison to WT, 930 common genes were up-regulated in both let-7(n2853) and lin-29(n333) and 649 common genes were down-regulated in both. We selected the 192 genes that were up-regulated in both of the let-7(n2853) vs. WT and the let-7(n2853) vs. lin-29(n333) pairs, which included lin-41, and daf-12, as possible direct targets (Tables S1 and S3). Combining the candidates that emerged from the computational and mRNA expression analyses, there were 340 candidates to test for genetic interactions with let-7.
Several transcription factors suppress vulval rupture in let-7 mutants
To identify functionally important genes among the list of candidates, we used RNAi screens to find genetic interactions by suppression of let-7 mutant phenotypes. The let-7 mutant worms display an array of developmental timing defects at the larval to adult transition including rupturing (Rup) of the intestine and gonads through the vulva [6], [7]. The developmental defects observed in let-7 mutants are caused by the over-expression of direct regulatory targets such as lin-41 and hbl-1, and some of these defects can be suppressed by RNAi knockdown of these targets in let-7 mutants [6], [7], [9], [10]. RNAi mediated suppression of vulval rupturing in let-7 mutants has been used to find new genetic interactions in sets of computationally predicted targets and in genes on chromosome I [11], [38], [74]. However, many of the candidate genes from our global expression analyses have not been assayed for vulval rupture and, thus, we were able to discover novel genetic suppressors.
Using the Ahringer feeding RNAi library [75], the Vidal feeding RNAi library [76] and a few clones we generated, 308 genes out of the 340 candidates were tested for suppression of vulval rupturing in the let-7(mn112) null strain. Homozygous let-7(mn112) mutants die at the late larval stages and must be maintained by a wild-type copy of the let-7 gene coming from a balanced translocation or a rescuing transgene [6], [7]. To grow a population of let-7(mn112) mutants to be able to score suppression, we generated a transgenic strain in which the worms were maintained by the presence of an extrachromosomal array (Ex[let-7(+); myo-2::GFP]), which contains a let-7 rescue fragment, allowing the mutants to survive, and the myo-2 promoter driving expression of a GFP marker in the pharynx to indicate the presence of the array (Figure 2A). To identify new suppressors of vulval rupturing, worms were grown synchronously from the L1 stage on bacteria expressing dsRNA targeting candidate genes or empty vector, as a negative control, and populations of non-transgenic animals were scored for the rate of vulval rupturing at the late larval and young adult stages (Figure 2B–2C). Nine clones exhibited larval growth arrest and therefore could not be scored for suppression. Empty vector clones were scored eight independent times as a negative control and 86–97% of these non-rescued worms ruptured at the time of scoring. We considered clones in which less than 75% of the population exhibited rupturing as suppressors (Figure 2D), consistent with a previous screen [11]. From this, 22 suppressors were retested and 16 clones again met the suppression threshold, including known suppressors lin-41, daf-12, and hbl-1 (Table 3) (Figure 2C–2D). Transcription factors constitute approximately half of the rupturing suppressors (7 of 16), several of which are involved in development including fos-1, lin-11, and sox-2 [77], [78], [79], [80], [81]. Enrichment of a different set of transcription factors was also noted by the Slack lab as genetic suppressors of their computational let-7 predictions [11].


let-7–dependent seam cell cycle exit is controlled by a diverse set of downstream genes
To broaden the search for genes that interact with let-7 beyond those involved in vulval rupture, we reasoned that novel targets might control other phenotypes found in let-7 mutants. In addition to the rupturing phenotype, let-7 mutants also have defects in the terminal differentiation of their seam cells, a specialized type of hypodermal cell [6], [7], [82]. Seam cells undergo significant changes during the larval to adult transition, including fusion of the seam cells, cessation of division, and the secretion of the adult cuticular structure known as alae [83]. Exit of the seam cells from the cell cycle and secretion of alae have been shown to be retarded in let-7 mutants [6], [7], [82]. Interestingly, seam cell fusion was unaffected in let-7(mn112) null mutants, suggesting that some aspects of seam cell terminal differentiation are let-7 independent (Figure S1). We chose to focus on the cell cycle exit defect, in which the seam cells fail to stop dividing at the larval to adult transition [82], as this would be the first screen for suppression of this phenotype and likely to uncover novel genetic interactions. Candidate RNAi clones from the rupturing suppression screen were tested for suppression of the cell cycle exit defect in let-7(n2853) mutants also carrying the integrated transgene Int[scm::GFP], which expresses a nuclear localized GFP specifically expressed in seam cells. The number of GFP positive seam cell nuclei were counted in at least 20 young adult worms (Figure 3A). Candidates were considered suppressed if they had significantly less nuclei than empty vector grown at the same time, p<0.05 using a Mann-Whitney U test. The 23 suppressing clones yielded 10 reproducible suppressors upon retest (Figure 3B and Table 3).

Among the suppressors were lin-41 and daf-12, which suppress two other let-7 phenotypes, vulval rupture and alae formation [6], [7], [11]. Thus lin-41 and daf-12 RNAi are sufficient to suppress all previously described phenotypes of let-7 mutants. Though hbl-1 RNAi also suppresses rupturing and alae formation defects, it is not surprising that it does not suppress the extra seam cell nuclei defect because hbl-1 loss of function mutants also have an increase in the seam cell nuclei number [10]. Of the 306 clones screened, 7 clones caused larval arrest and could not be scored. Consistent with previous work by the Gilleard lab [84], elt-1 RNAi led to the loss of most of the seam cells during larval development, rendering it inconclusive for suppression. Suppressors of the supernumerary seam cell divisions in let-7(n2853) represent a diverse set of gene functions and there is only modest overlap with the rupturing suppressors, suggesting that the two phenotypes are likely under separate genetic control (Table 3).
Candidate let-7 targets differentially affect vulva formation
The twenty-three candidate let-7 targets were also tested for potential roles in a vulva formation abnormality due to precocious let-7 expression. The loss of function lin-28(n719) mutants exhibit a partially penetrant temperature-sensitive protruding multiple vulva (pmuv) phenotype that is dependent on let-7. At 25°C, this phenotype is expressed in ∼67% of the lin-28(n719) population with the remaining worms displaying a single protruding vulva (pvul) (Figure 4A). In the presence of the let-7(mn112) null allele, the pmuv phenotype is no longer observed in lin-28(n719) worms, and 100% of the double mutant population expresses the pvul phenotype (Figure 4A). Thus, the pmuv phenotype is dependent on let-7, and suggests that the precocious expression of let-7 in the lin-28 mutants might prematurely repress targets needed to regulate vulval cell patterning. We predicted that further suppression of such targets by RNAi would enhance the pmuv phenotype in lin-28(n719) worms. To identify potential targets that act in this pathway, the percent of the population exhibiting pmuv was scored for lin-28(n719) mutants subjected to RNAi of the 23 candidates. RNAi of three genes produced the expected enhancement of the pmuv phenotype (Figure 4B), suggesting that inappropriate down-regulation of these candidates in lin-28 mutants contributes to mis-specification of vulval cell fates. This enhanced phenotype is dependent on let-7 because the pmuv phenotype is almost entirely absent in lin-28 mutant worms that also lack let-7 activity (lin-28(n719);let-7(mn112)) (Figure 4C). Surprisingly, another set of genes significantly decreased the incidence of pmuv in lin-28(n719) (Figure 4B) and, in the case of nhr-25, the pvul phenotype was also suppressed in the lin-28(n719);let-7(mn112) double mutants (Figure 4C). These results suggest that some of the candidate genes may have a more complicated relationship with let-7, possibly affecting let-7 expression or activity in tissue-specific feedback loops.

Novel targets associated with ALG-1 in a let-7–dependent manner
miRNAs repress target mRNA expression through their association with Argonaute proteins allowing them to act as sequence-specific guides for the RISC complex [4], [5]. Taking advantage of the recent global map of Argonaute Like Gene 1 (ALG-1) binding sites in C. elegans [66], we searched for these sites in the twenty-three suppressors. Eight of the twenty-three suppressing genes had significant ALG-1 binding sites within their 3′ UTRs and coding regions. This group included the known let-7 targets, such as daf-12 and lin-41, as well as hbl-1, which is also a target of other let-7 miRNA family members (Table 3) [7], [9], [10], [11], [85], [86], [87].
To test if let-7 is responsible for the interaction of ALG-1 with these genes, we analyzed their association with ALG-1 using RNA immunoprecipitation (RIP) in wild-type and let-7(n2853) worms (Figure 5A). Genes regulated by let-7 are expected to be enriched in wild-type samples versus let-7 mutant samples, while genes targeted by other miRNAs should be amplified similarly in both strains. Four independent RIPs were analyzed, and targets enriched in the wild-type for at least 2 of the 4 replicates were considered to be dependent on let-7 for ALG-1 association. The known targets lin-41 and daf-12, served as positive controls with both showing let-7-dependent enrichment in the ALG-1 IP. fos-1 was used as a negative control as it did not have any significant CLIP reads nor did fos-1 sequences amplify from the RIPs in either worm strain. lin-14 was also used as a negative control because it is a known target of a different miRNA, lin-4, and as expected there was no significant change in ALG-1 binding in let-7 mutants compared to WT. daf-9 and adt-2 had significant CLIP reads but could not be verified as targets through the RIP analysis. adt-2 had similar levels in the WT and let-7(n2853) mutant strains suggesting it may be targeted by a different miRNA, which could mask any let-7 dependent RISC association.
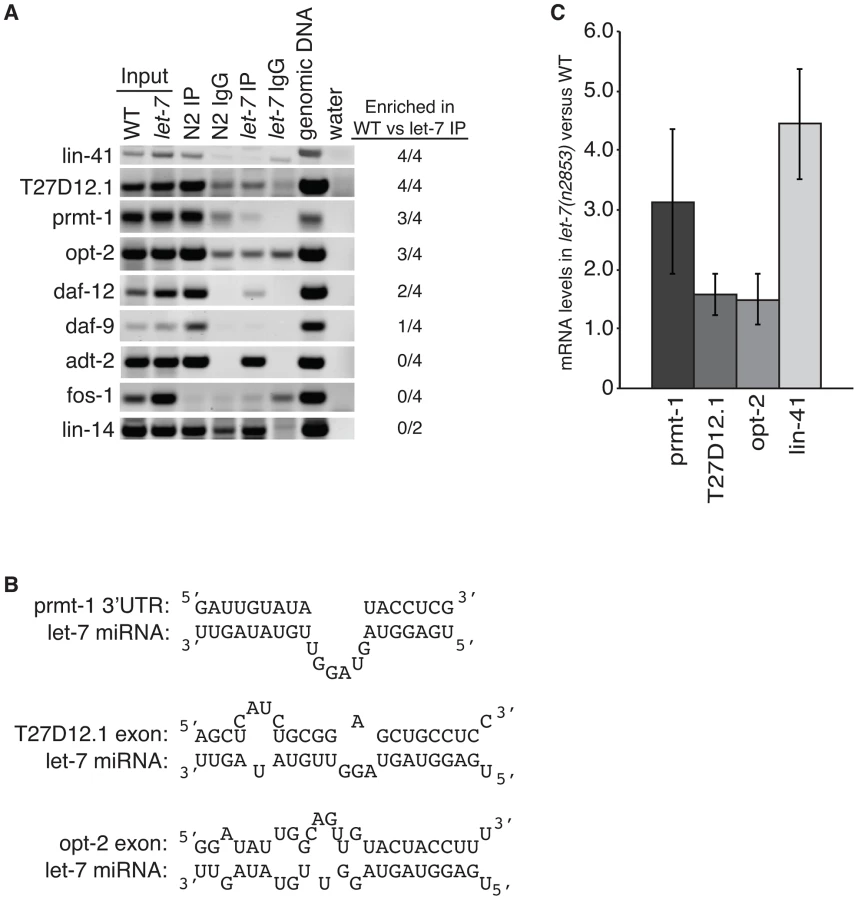
Three novel targets were identified: prmt-1, opt-2, and T27D12.1. They were all enriched in the WT compared to the let-7(n2853) RIP (Figure 5A) and are, therefore, associated with ALG-1 in a let-7-dependent manner. Furthermore, we found let-7 complementary sites (LCS) within the ALG-1 binding sites of these targets (Figure 5B), supporting these genes as new direct targets of let-7. Interestingly, T27D12.1 and opt-2, which contain predicted target sites in coding exon sequences, showed weak mis-regulation at the mRNA level in let-7(n2853) versus WT worms (Figure 5C). In contrast, prmt-1 and the positive control lin-41, which contain 3′UTR target sites, were up-regulated over three-fold at the mRNA level in the let-7 mutant worms. These data are consistent with the global correlation observed between changes in mRNA levels and ALG-1 binding to 3′UTR, but not coding exon sequences [88].
Discussion
The let-7 miRNA is exceptional in its conservation and essential role in cellular differentiation across species [13]. Loss of let-7 activity results in lethality in worms and contributes to oncogenesis in mammalian tissues [14], [89]. Since these effects are due to mis-regulation of let-7 targets, identification of the biologically relevant genes regulated by this miRNA has been a paramount research goal. Through a combination of genetic and molecular screens in C. elegans, we have uncovered twenty-three genes that are up-regulated in let-7 mutants and contribute to the developmental abnormalities characteristic of these mutants. Three of these genes, lin-41, daf-12 and hbl-1, are the best previously characterized let-7 targets in C. elegans, validating the sensitivity of our approach [6], [7], [9], [10], [11]. Unexpectedly, a subset of the genes that suppressed let-7 mutant phenotypes also suppressed a lin-28 phenotype that is due to up-regulation of let-7 expression, suggesting nonlinear pathways between these targets and let-7 in vulval precursor cells. Three genes, prmt-1, opt-2, and T27D12.1, were found to associate with the miRNA complex in a let-7 dependent manner and, thus, emerged as likely novel direct targets of let-7.
A large fraction of the transcriptome is mis-regulated in let-7(n2853) worms. Based on microarray analyses, most of these changes are less than two-fold. However, let-7(n2853) is a temperature sensitive loss of function strain that maintains some let-7 activity even at the non-permissive temperatures. Accordingly, the fold change in let-7 target mRNA expression for lin-41, for example, is less dramatic in let-7(n2853) compared to wild type at the L4 stage than it is in stages before (L2) and after (L4) let-7 expression in wild type worms [67]. By using reproducibility in the direction of change, instead of the absolute fold difference in mRNA levels, we identified twenty new genes in the let-7 pathway that exhibited only modest expression differences in let-7 mutants. In fact of our list of let-7 suppressors, only lin-41 and daf-12 were mis-regulated by more than two-fold by microarray analyses. The large number of down-regulated genes in let-7(n2853) mutants likely represents indirect targets, reflecting mis-regulation of direct targets that transcriptionally regulate some of these genes. Over one-third of the genes up and down-regulated in let-7(n2853) were changed in the same direction in lin-29 mutants, indicating that failure to trigger the lin-29-dependent transcriptional program also accounts for many of the mis-regulated genes in let-7 mutants.
Considering that the two well-established targets of let-7, lin-41, and daf-12, suppress both the rupturing vulva and extra seam cell phenotypes of let-7 mutants, it was surprising to find almost entirely distinct sets of new genes affecting one phenotype versus the other. The opt-2 gene was the only additional suppressor of both phenotypes, suggesting that different pathways largely control maturation of the vulva and seam cells. While it is not entirely understood why let-7 mutants rupture through the vulva, it has been postulated that improper cell fusions during vulva formation cause weakening and destabilization of this structure. Fourteen new genes were found to suppress the bursting vulva phenotype when subjected to RNAi conditions, none of which overlapped with the previously described suppressors of this let-7 phenotype [11], [74]. A distinction from these studies is that we screened for suppression in null let-7(mn112) worms as opposed to the weaker let-7(n2853) strain. Two of the let-7(n2853) suppressors identified in Grosshans et al., 2005, lin-59 and lss-18, were found to be up-regulated in let-7 mutants by our microarray analyses. However, these candidates failed to suppress the rupturing of let-7(mn112) worms, in agreement with the previous study [11]. Many of the genes we identified as suppressors of vulva rupturing encode transcription factors, a category also prominent on the list of potential let-7 targets described in Grosshans et al., 2005 [11]. Genes involved in translation make up another class of let-7(n2853) suppressors [74]. A combined approach, incorporating let-7 target predictions by PicTar, reporter assays and screens for suppression of rupturing in let-7(n2853), resulted in twelve potential new targets [38]. Of the genes that passed the genetic test, only fos-1 is in common with our list of bursting suppressors. Another group tested 181 genes with various criteria for being potential let-7 targets for changes in protein levels in WT versus let-7(n2853) worms [54]. Of the nineteen candidates up-regulated in let-7 mutants, nine also suppressed rupturing in let-7(n2853). Three of these suppressors, T19A6.2, Y47GA.10, and F46B6.7, were up-regulated in our microarray data. However, they failed suppress vulva rupturing in the null let-7(mn112) background and, thus, did not appear on our final list of candidates. An important consideration when screening for suppression of vulva rupturing is that in some cases the effect may be indirect due to slow or halted development or the absence of vulva formation. These caveats were avoided by using the let-7(mn112) strain containing the extrachromosomal let-7 rescue construct, as RNAi clones that affected development regardless of the presence of the let-7 transgene could be flagged. Nonetheless, the observation that RNAi of many different genes results in suppression of the rupturing phenotype in let-7 mutants points to the existence of cross-regulatory pathways that are sensitive to down-regulation of a single target.
Reiteration of seam cell nuclear divisions at the transition to adulthood is another characteristic of let-7 mutants [11], [82]. In C. elegans, the lateral seam cells undergo an asymmetric division in which one daughter cell differentiates while the other repeats this pattern at each larval stage [83]. In let-7 mutants, the seam cells inappropriately undergo the larval type division instead of differentiating to the adult fate, where the cells normally fuse and cease dividing [6]. The heterochronic gene lin-29 is downstream of let-7 and is a master regulator of seam cell differentiation [6], [73]. The failure of seam cells to properly differentiate in let-7 mutants seems to be largely due to a lack of lin-29 activity [6], [7]. How let-7 positively regulates the expression of LIN-29 protein is presently unknown. Our screen identified eight new genes that suppress the supernumerary seam cell divisions of let-7(n2853) mutants. Three of these suppressors, opt-2, prmt-1, T27D12.1, are likely direct targets of let-7 since their association with Argonaute is dependent on this miRNA. The group of extra seam cell suppressors includes factors with a variety of predicted functions that could potentially contribute to mis-regulation of lin-29.
In C. elegans, processing of the let-7 miRNA early in larval development is inhibited by LIN-28 protein [21], [23]. In lin-28(n719) mutants, let-7 miRNA is expressed precociously, resulting in premature repression of its targets. One effect of this mis-regulation is the development of protruding multiple vulvas in lin-28 mutants grown at 25°C. This partially penetrant pmuv phenotype is dependent on let-7 because lin-28(n719);let-7(mn112) strains only produce single protruding vulvas. Since early accumulation of let-7 miRNA is expected to cause premature down-regulation of targets, we anticipated that further silencing of potential targets by RNAi would enhance the pmuv phenotype in lin-28(n719) worms. Three candidates, fos-1, ZK1236.1 and T08B2.8, emerged as enhancers, pointing to roles for these genes in vulval fate specification. Surprisingly, there were also several candidates that decreased the percentage of pmuv in lin-28(n719) worms including, nhr-25, hbl-1, sox-1, prmt-1, and nduf-7. Since this effect is also observed when let-7 is removed from lin-28(n719), these suppressors potentially feedback to regulate the expression or function of let-7 in vulval precursor cells. Feedback loops between let-7 family members and targets, such as daf-12 and hbl-1, in other tissues have been previously demonstrated [9], [10], [85], [86], [87], [90].
Multiple lines of molecular and genetic evidence support opt-2, prmt-1 and T27D12.1 as new direct targets of let-7 regulation. One of the targets, opt-2, may be a general downstream effector in the let-7 pathway as down-regulation of opt-2 suppresses phenotypes in the vulva and seam cells. Before this study, opt-2 was not a predicted let-7 target because it lacks complementarity to the 5′ end of the miRNA (seed) in its 3′UTR. However, a single ALG-1 binding site is present in the second last exon of opt-2 and this region includes a predicted let-7 binding site. opt-2 (also known as pept-1) is a member of the peptide transporter family and facilitates uptake of di - and tri-peptides in the intestine [91], [92]. Loss of opt-2 activity slows development, alters fat accumulation and enhances stress resistance 91,93. Although opt-2 appears to be exclusively expressed in the intestine, loss of this factor causes global changes in gene expression [94], [95]. Reporters driven by the let-7 promoter also show intestinal expression, suggesting that let-7 miRNA is available for directly regulating opt-2 in this tissue [96], [97], [98]. The ability of opt-2 RNAi to suppress let-7 phenotypes in vulval and seam cells suggests that signaling from the intestine influences development of these tissues.
Another likely direct target, T27D12.1, also seems to be regulated by let-7 through sequences in its open reading frame. This gene lacks predicted target sites for let-7 in its 3′UTR but came through our screen as a modestly up-regulated gene in let-7(n2853) that was capable of suppressing the extra seam cell phenotype of these mutants. T27D12.1 contains one ALG-1 binding site in its 3′UTR and one in a coding exon, but only the exonic region includes an obvious LCS, which conforms to seed-pairing with the allowance of a G-U pair. T27D12.1 is predicted to encode a sodium/phosphate transporter protein but little else is known about this factor.
The more conventional miRNA target, prmt-1, has an LCS within its 3′UTR and was previously predicted by the mirWIP and PITA algorithms as a let-7 target [40], [41]. While prmt-1 has ALG-1 binding sites in its 3′UTR as well as coding exon sequences, only the 3′UTR site includes an obvious let-7 complementary site. Although there is not a canonical LCS in the 3′UTRs of mammalian homologs of prmt-1, there are several well conserved potential let-7 binding sites (Figure S2). prmt-1 encodes a protein arginine methyltransferase, and it has been shown in mammalian cells to be a major contributor to methylation of histone 4 at arg-3, leading to transcriptional activation [99], [100]. Additionally, PRMT-1 has been shown to methylate arginine residues on other types of proteins in mammalian cells and C. elegans [93], [101]. Recently, it was discovered that PRMT-1 methylates DAF-16, a key transcription factor in the insulin pathway [101]. This modification prevents phosphorylation of DAF-16 by AKT, thus, keeping it in an active state to promote the expression of longevity-related genes. prmt-1 has a broad expression pattern that is largely overlapping with let-7 transcriptional reporters [96], [97], [98], [101]. Down-regulation of prmt-1 by let-7 in late larval stages could influence the lifespan of worms by causing reduced methylation and, hence, activity of DAF-16.
Our combination of molecular and genetic screens revealed a complex network of genes that interact with let-7 in C. elegans. This approach was sensitive enough to detect the established let-7 targets, lin-41, daf-12 and hbl-1. While these genes are regulated at the mRNA level, other targets that are only subject to translational repression would be missed by focusing on transcripts up-regulated in let-7 mutants. However, the microarray data revealed that thousands of genes are mis-regulated when there is insufficient let-7 activity, supporting a widespread role for this miRNA in regulating, directly and indirectly, gene expression. A set of the up-regulated genes proved to be biologically relevant for the developmental abnormalities that arise in the absence of let-7 activity. At least three of these genes, which encode transport proteins and a modifying enzyme, appear to be new direct targets of let-7. In conclusion, let-7 appears to regulate a variety of direct targets, which in turn influences the expression of hundreds of other genes. Loss of this miRNA alone results in extensive changes in gene expression and abnormal development in multiple tissues, supporting the role of let-7 as a master gene regulator.
Materials and Methods
Nematode strains
The C. elegans strains were cultured at 15°C or 25°C under standard conditions [102]. Worms were synchronized by hypochlorite treatment and development was initiated by plating arrested L1 hatchlings on NGM plates seeded with OP50 bacteria or RNAi bacteria on RNAi plates. Strains used in this study include the following: wild type (WT) Bristol N2, MT7626 let-7(n2853), MT333 lin-29(n333), MT1524 lin-28(n719), PQ79 mnDp1(X/V)/+; unc-3(ed151) let-7(mn112); Ex[let-7(+); myo-2::GFP], PQ270 mnDp1(X/V)/+; unc-3(ed151) let-7(mn112); lin-28(n719), PQ293 let-7(n2853); Int[scm::GFP].
RNAi experiments
Seam cell nuclei were counted at 40 hr (25°C) in 20 adult PQ293 let-7(n2853); Int[scm::GFP] worms grown on vector control or gene specific RNAi plates for one generation. Suppression was determined by a Mann-Whitney U test comparing worms on each RNAi vector to those on the empty L4440 control vector grown on the same day. Bursting suppression was scored as more than 25% non-bursting, non-green (non-rescued) 40 hr adult PQ79 mnDp1(X/V)/+; unc-3(ed151) let-7(mn112); Ex[let-7(+); myo-2::GFP] worms grown at 25°C. All suppressing clones were retested using the same criteria for reproducibility. All clones suppressing at least one phenotype were verified by sequencing. Fifty to one hundred lin-28(n719) or lin-28(n719);let-7(mn112) worms were grown on RNAi until 48 hr (25°C) adults and then scored for the protruding multivulva (Pmuv) or protruding single vulva (Pvul) phenotypes. Suppression/enhancement was determined by a Ttest comparing worms on each RNAi clone to those on the empty L4440 control vector grown at the same time for 4 or 5 independent RNAi experiments.
Microarray analysis
Six paired replicates of L4 RNA from WT or let-7(n2853) worms were prepared and labeled as per manufacturer's instructions (Affymetrix, Santa Clara) and hybridized to Affymetrix C. elegans Gene microarrays. Three of the paired replicates of WT and let-7(n2853) were also paired with lin-29(n333) replicates for array analysis. To assess the significance of differential gene expression between the two groups, a paired t-statistic was computed. CEL files obtained after scanning were analyzed by using Affymetrix APT tools and Robust Multi-array Average (RMA)-sketch normalized [103]. Annotation files for the probe sets were obtained from Affymetrix. The paired t-test statistic was utilized to compute differences between groups for each probe set. Probe sets were mapped to custom gene structures generated from Refseq annotations obtained from ce2 at the UCSC genome browser. Gene ontology analyses were performed using the database for annotation, visualization and integrated discovery (DAVID) and the Functional Annotation Clustering Tool [104], [105]. Classifications were set to the highest stringency and the recommended enrichment score of ≥1.3 was applied. To search for enriched motifs in the gene lists, pair-wise alignments between C. briggsae (cb1) and C. elegans (ce2) were obtained from the UCSC genome browser. 3′UTR exons were spliced together to generate the sequence if necessary, and then extended to 2000 bases from the stop codon. 6-mer enrichment in genes up-regulated in let-7(n2853) versus non-regulated genes was computed using methods described in [106].
RNA immunoprecipitation (RIP)
RIP assays were preformed as previously described [23], [88]. Synchronized WT and let-7(n2853) worms were grown at 25°C for 29 hours before being cross-linked by UV treatment. Equal amounts of lysates were pre-cleared before immunoprecipitation with the anti-ALG-1 antibody (Thermo Fisher Scientific) or control IgG (Caltag Laboratories) and protein G Dynabeads (Invitrogen). Immunoprecipitated material was subjected to Proteinase K treatment and RNA extraction before reverse transcription using random oligo priming. The resulting cDNA was used in PCR with the primers listed in Table S4.
qPCR
RNA was isolated from WT and let-7(n2853) worms grown at 25°C for 28 hours. qPCR was performed on cDNA with SYBR green (Applied Biosystems) and 10 uM of each forward and reverse primer on an ABI Prism 7000 real time PCR machine. Primers are listed in Table S4.
Supporting Information
Zdroje
1. AaltoAP, PasquinelliAE (2012) Small non-coding RNAs mount a silent revolution in gene expression. Curr Opin in Cell Biol 24 (3)
333–40 doi:10.1016/j.ceb.2012.03.006.
2. KimVN, HanJ, SiomiMC (2009) Biogenesis of small RNAs in animals. Nature reviews Molecular cell biology 10 : 126–139.
3. WinterJ, JungS, KellerS, GregoryRI, DiederichsS (2009) Many roads to maturity: microRNA biogenesis pathways and their regulation. Nature cell biology 11 : 228–234.
4. HuntzingerE, IzaurraldeE (2011) Gene silencing by microRNAs: contributions of translational repression and mRNA decay. Nature reviews Genetics 12 : 99–110.
5. PasquinelliAE (2012) MicroRNAs and their targets: recognition, regulation and an emerging reciprocal relationship. Nature reviews Genetics 13 : 271–282.
6. ReinhartBJ, SlackFJ, BassonM, PasquinelliAE, BettingerJC, et al. (2000) The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature 403 : 901–906.
7. SlackFJ, BassonM, LiuZ, AmbrosV, HorvitzHR, et al. (2000) The lin-41 RBCC gene acts in the C. elegans heterochronic pathway between the let-7 regulatory RNA and the LIN-29 transcription factor. Molecular cell 5 : 659–669.
8. AmbrosV, HorvitzHR (1984) Heterochronic mutants of the nematode Caenorhabditis elegans. Science 226 : 409–416.
9. AbrahanteJE, DaulAL, LiM, VolkML, TennessenJM, et al. (2003) The Caenorhabditis elegans hunchback-like gene lin-57/hbl-1 controls developmental time and is regulated by microRNAs. Developmental cell 4 : 625–637.
10. LinSY, JohnsonSM, AbrahamM, VellaMC, PasquinelliA, et al. (2003) The C elegans hunchback homolog, hbl-1, controls temporal patterning and is a probable microRNA target. Developmental cell 4 : 639–650.
11. GrosshansH, JohnsonT, ReinertKL, GersteinM, SlackFJ (2005) The temporal patterning microRNA let-7 regulates several transcription factors at the larval to adult transition in C. elegans. Developmental cell 8 : 321–330.
12. PasquinelliAE, ReinhartBJ, SlackF, MartindaleMQ, KurodaMI, et al. (2000) Conservation of the sequence and temporal expression of let-7 heterochronic regulatory RNA. Nature 408 : 86–89.
13. MondolV, PasquinelliAE (2012) Let's make it happen: the role of let-7 microRNA in development. Current topics in developmental biology 99 : 1–30.
14. BoyerinasB, ParkSM, HauA, MurmannAE, PeterME (2010) The role of let-7 in cell differentiation and cancer. Endocrine-related cancer 17: F19–36.
15. ThorntonJE, GregoryRI (2012) How does Lin28 let-7 control development and disease? Trends Cell Biol 474–82 doi:10.1016/j.tcb.2012.06.001.
16. HeoI, JooC, ChoJ, HaM, HanJ, et al. (2008) Lin28 mediates the terminal uridylation of let-7 precursor MicroRNA. Molecular cell 32 : 276–284.
17. NewmanMA, ThomsonJM, HammondSM (2008) Lin-28 interaction with the Let-7 precursor loop mediates regulated microRNA processing. RNA 14 : 1539–1549.
18. RybakA, FuchsH, SmirnovaL, BrandtC, PohlEE, et al. (2008) A feedback loop comprising lin-28 and let-7 controls pre-let-7 maturation during neural stem-cell commitment. Nature cell biology 10 : 987–993.
19. ViswanathanSR, DaleyGQ, GregoryRI (2008) Selective blockade of microRNA processing by Lin28. Science 320 : 97–100.
20. HeoI, JooC, KimYK, HaM, YoonMJ, et al. (2009) TUT4 in concert with Lin28 suppresses microRNA biogenesis through pre-microRNA uridylation. Cell 138 : 696–708.
21. LehrbachNJ, ArmisenJ, LightfootHL, MurfittKJ, BugautA, et al. (2009) LIN-28 and the poly(U) polymerase PUP-2 regulate let-7 microRNA processing in Caenorhabditis elegans. Nature structural & molecular biology 16 : 1016–1020.
22. PiskounovaE, PolytarchouC, ThorntonJE, LaPierreRJ, PothoulakisC, et al. (2011) Lin28A and Lin28B inhibit let-7 microRNA biogenesis by distinct mechanisms. Cell 147 : 1066–1079.
23. Van WynsberghePM, KaiZS, MassirerKB, BurtonVH, YeoGW, et al. (2011) LIN-28 co-transcriptionally binds primary let-7 to regulate miRNA maturation in Caenorhabditis elegans. Nature structural & molecular biology 18 : 302–308.
24. ViswanathanSR, PowersJT, EinhornW, HoshidaY, NgTL, et al. (2009) Lin28 promotes transformation and is associated with advanced human malignancies. Nature genetics 41 : 843–848.
25. ZhuH, Shyh-ChangN, SegreAV, ShinodaG, ShahSP, et al. (2011) The Lin28/let-7 axis regulates glucose metabolism. Cell 147 : 81–94.
26. FrostRJ, OlsonEN (2011) Control of glucose homeostasis and insulin sensitivity by the Let-7 family of microRNAs. Proceedings of the National Academy of Sciences of the United States of America 108 : 21075–21080.
27. JohnsonSM, GrosshansH, ShingaraJ, ByromM, JarvisR, et al. (2005) RAS is regulated by the let-7 microRNA family. Cell 120 : 635–647.
28. JohnsonCD, Esquela-KerscherA, StefaniG, ByromM, KelnarK, et al. (2007) The let-7 microRNA represses cell proliferation pathways in human cells. Cancer research 67 : 7713–7722.
29. BoyerinasB, ParkSM, ShomronN, HedegaardMM, VintherJ, et al. (2008) Identification of let-7-regulated oncofetal genes. Cancer research 68 : 2587–2591.
30. ShellS, ParkSM, RadjabiAR, SchickelR, KistnerEO, et al. (2007) Let-7 expression defines two differentiation stages of cancer. Proceedings of the National Academy of Sciences of the United States of America 104 : 11400–11405.
31. YuF, YaoH, ZhuP, ZhangX, PanQ, et al. (2007) let-7 regulates self renewal and tumorigenicity of breast cancer cells. Cell 131 : 1109–1123.
32. LeeYS, DuttaA (2007) The tumor suppressor microRNA let-7 represses the HMGA2 oncogene. Genes & development 21 : 1025–1030.
33. MayrC, HemannMT, BartelDP (2007) Disrupting the pairing between let-7 and Hmga2 enhances oncogenic transformation. Science 315 : 1576–1579.
34. Esquela-KerscherA, TrangP, WigginsJF, PatrawalaL, ChengA, et al. (2008) The let-7 microRNA reduces tumor growth in mouse models of lung cancer. Cell cycle 7 : 759–764.
35. KumarMS, ErkelandSJ, PesterRE, ChenCY, EbertMS, et al. (2008) Suppression of non-small cell lung tumor development by the let-7 microRNA family. Proceedings of the National Academy of Sciences of the United States of America 105 : 3903–3908.
36. EnrightAJ, JohnB, GaulU, TuschlT, SanderC, et al. (2003) MicroRNA targets in Drosophila. Genome biology 5: R1.
37. LewisBP, ShihIH, Jones-RhoadesMW, BartelDP, BurgeCB (2003) Prediction of mammalian microRNA targets. Cell 115 : 787–798.
38. LallS, GrunD, KrekA, ChenK, WangYL, et al. (2006) A genome-wide map of conserved microRNA targets in C. elegans. Current biology : CB 16 : 460–471.
39. MirandaKC, HuynhT, TayY, AngYS, TamWL, et al. (2006) A pattern-based method for the identification of MicroRNA binding sites and their corresponding heteroduplexes. Cell 126 : 1203–1217.
40. KerteszM, IovinoN, UnnerstallU, GaulU, SegalE (2007) The role of site accessibility in microRNA target recognition. Nature genetics 39 : 1278–1284.
41. HammellM, LongD, ZhangL, LeeA, CarmackCS, et al. (2008) mirWIP: microRNA target prediction based on microRNA-containing ribonucleoprotein-enriched transcripts. Nature methods 5 : 813–819.
42. BartelDP (2009) MicroRNAs: target recognition and regulatory functions. Cell 136 : 215–233.
43. ShinC, NamJW, FarhKK, ChiangHR, ShkumatavaA, et al. (2010) Expanding the microRNA targeting code: functional sites with centered pairing. Molecular cell 38 : 789–802.
44. RigoutsosI (2009) New tricks for animal microRNAS: targeting of amino acid coding regions at conserved and nonconserved sites. Cancer research 69 : 3245–3248.
45. WightmanB, HaI, RuvkunG (1993) Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell 75 : 855–862.
46. LeeRC, FeinbaumRL, AmbrosV (1993) The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 75 : 843–854.
47. ThomsonDW, BrackenCP, GoodallGJ (2011) Experimental strategies for microRNA target identification. Nucleic acids research 39 : 6845–6853.
48. HuangJC, BabakT, CorsonTW, ChuaG, KhanS, et al. (2007) Using expression profiling data to identify human microRNA targets. Nature methods 4 : 1045–1049.
49. LimLP, LauNC, Garrett-EngeleP, GrimsonA, SchelterJM, et al. (2005) Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature 433 : 769–773.
50. SchmitterD, FilkowskiJ, SewerA, PillaiRS, OakeleyEJ, et al. (2006) Effects of Dicer and Argonaute down-regulation on mRNA levels in human HEK293 cells. Nucleic acids research 34 : 4801–4815.
51. SoodP, KrekA, ZavolanM, MacinoG, RajewskyN (2006) Cell-type-specific signatures of microRNAs on target mRNA expression. Proceedings of the National Academy of Sciences of the United States of America 103 : 2746–2751.
52. BaekD, VillenJ, ShinC, CamargoFD, GygiSP, et al. (2008) The impact of microRNAs on protein output. Nature 455 : 64–71.
53. SelbachM, SchwanhausserB, ThierfelderN, FangZ, KhaninR, et al. (2008) Widespread changes in protein synthesis induced by microRNAs. Nature 455 : 58–63.
54. JovanovicM, ReiterL, PicottiP, LangeV, BoganE, et al. (2010) A quantitative targeted proteomics approach to validate predicted microRNA targets in C. elegans. Nature methods 7 : 837–842.
55. GuoH, IngoliaNT, WeissmanJS, BartelDP (2010) Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 466 : 835–840.
56. StadlerM, ArtilesK, PakJ, FireA (2012) Contributions of mRNA abundance, ribosome loading, and post - or peri-translational effects to temporal repression of C. elegans heterochronic miRNA targets. Genome Res 22 (12)
2418–26 doi:10.1101/gr.136515.111.
57. BeitzingerM, PetersL, ZhuJY, KremmerE, MeisterG (2007) Identification of human microRNA targets from isolated argonaute protein complexes. RNA biology 4 : 76–84.
58. EasowG, TelemanAA, CohenSM (2007) Isolation of microRNA targets by miRNP immunopurification. RNA 13 : 1198–1204.
59. HendricksonDG, HoganDJ, HerschlagD, FerrellJE, BrownPO (2008) Systematic identification of mRNAs recruited to argonaute 2 by specific microRNAs and corresponding changes in transcript abundance. PLoS ONE 3: e2126 doi:10.1371/journal.pone.0002126.
60. KarginovFV, ConacoC, XuanZ, SchmidtBH, ParkerJS, et al. (2007) A biochemical approach to identifying microRNA targets. Proceedings of the National Academy of Sciences of the United States of America 104 : 19291–19296.
61. LandthalerM, GaidatzisD, RothballerA, ChenPY, SollSJ, et al. (2008) Molecular characterization of human Argonaute-containing ribonucleoprotein complexes and their bound target mRNAs. RNA 14 : 2580–2596.
62. ZhangL, DingL, CheungTH, DongMQ, ChenJ, et al. (2007) Systematic identification of C. elegans miRISC proteins, miRNAs, and mRNA targets by their interactions with GW182 proteins AIN-1 and AIN-2. Molecular cell 28 : 598–613.
63. ChiSW, ZangJB, MeleA, DarnellRB (2009) Argonaute HITS-CLIP decodes microRNA-mRNA interaction maps. Nature 460 : 479–486.
64. HafnerM, LandthalerM, BurgerL, KhorshidM, HausserJ, et al. (2010) Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell 141 : 129–141.
65. LeungAK, YoungAG, BhutkarA, ZhengGX, BossonAD, et al. (2011) Genome-wide identification of Ago2 binding sites from mouse embryonic stem cells with and without mature microRNAs. Nature structural & molecular biology 18 : 237–244.
66. ZisoulisDG, LovciMT, WilbertML, HuttKR, LiangTY, et al. (2010) Comprehensive discovery of endogenous Argonaute binding sites in Caenorhabditis elegans. Nature structural & molecular biology 17 : 173–179.
67. BaggaS, BrachtJ, HunterS, MassirerK, HoltzJ, et al. (2005) Regulation by let-7 and lin-4 miRNAs results in target mRNA degradation. Cell 122 : 553–563.
68. DingXC, GrosshansH (2009) Repression of C. elegans microRNA targets at the initiation level of translation requires GW182 proteins. The EMBO journal 28 : 213–222.
69. LewisBP, BurgeCB, BartelDP (2005) Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120 : 15–20.
70. VellaMC, ChoiEY, LinSY, ReinertK, SlackFJ (2004) The C. elegans microRNA let-7 binds to imperfect let-7 complementary sites from the lin-41 3′UTR. Genes & development 18 : 132–137.
71. NimmoRA, SlackFJ (2009) An elegant miRror: microRNAs in stem cells, developmental timing and cancer. Chromosoma 118 : 405–418.
72. BettingerJC, LeeK, RougvieAE (1996) Stage-specific accumulation of the terminal differentiation factor LIN-29 during Caenorhabditis elegans development. Development 122 : 2517–2527.
73. RougvieAE, AmbrosV (1995) The heterochronic gene lin-29 encodes a zinc finger protein that controls a terminal differentiation event in Caenorhabditis elegans. Development 121 : 2491–2500.
74. DingXC, SlackFJ, GrosshansH (2008) The let-7 microRNA interfaces extensively with the translation machinery to regulate cell differentiation. Cell cycle 7 : 3083–3090.
75. KamathRS, FraserAG, DongY, PoulinG, DurbinR, et al. (2003) Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature 421 : 231–237.
76. RualJF, CeronJ, KorethJ, HaoT, NicotAS, et al. (2004) Toward improving Caenorhabditis elegans phenome mapping with an ORFeome-based RNAi library. Genome research 14 : 2162–2168.
77. SternbergPW (2005) Vulval development. WormBook : the online review of C elegans biology 1–28.
78. SherwoodDR, ButlerJA, KramerJM, SternbergPW (2005) FOS-1 promotes basement-membrane removal during anchor-cell invasion in C. elegans. Cell 121 : 951–962.
79. MohamadnejadM, SwensonES (2008) Induced pluripotent cells mimicking human embryonic stem cells. Archives of Iranian medicine 11 : 125–128.
80. YuJ, VodyanikMA, Smuga-OttoK, Antosiewicz-BourgetJ, FraneJL, et al. (2007) Induced pluripotent stem cell lines derived from human somatic cells. Science 318 : 1917–1920.
81. TayY, ZhangJ, ThomsonAM, LimB, RigoutsosI (2008) MicroRNAs to Nanog, Oct4 and Sox2 coding regions modulate embryonic stem cell differentiation. Nature 455 : 1124–1128.
82. HayesGD, FrandAR, RuvkunG (2006) The mir-84 and let-7 paralogous microRNA genes of Caenorhabditis elegans direct the cessation of molting via the conserved nuclear hormone receptors NHR-23 and NHR-25. Development 133 : 4631–4641.
83. SulstonJE, HorvitzHR (1977) Post-embryonic cell lineages of the nematode, Caenorhabditis elegans. Developmental biology 56 : 110–156.
84. SmithJA, McGarrP, GilleardJS (2005) The Caenorhabditis elegans GATA factor elt-1 is essential for differentiation and maintenance of hypodermal seam cells and for normal locomotion. Journal of cell science 118 : 5709–5719.
85. AbbottAL, Alvarez-SaavedraE, MiskaEA, LauNC, BartelDP, et al. (2005) The let-7 MicroRNA family members mir-48, mir-84, and mir-241 function together to regulate developmental timing in Caenorhabditis elegans. Developmental cell 9 : 403–414.
86. BethkeA, FielenbachN, WangZ, MangelsdorfDJ, AntebiA (2009) Nuclear hormone receptor regulation of microRNAs controls developmental progression. Science 324 : 95–98.
87. HammellCM, KarpX, AmbrosV (2009) A feedback circuit involving let-7-family miRNAs and DAF-12 integrates environmental signals and developmental timing in Caenorhabditis elegans. Proceedings of the National Academy of Sciences of the United States of America 106 : 18668–18673.
88. ZisoulisDG, KaiZS, ChangRK, PasquinelliAE (2012) Autoregulation of microRNA biogenesis by let-7 and Argonaute. Nature 486 : 541–544.
89. BussingI, SlackFJ, GrosshansH (2008) let-7 microRNAs in development, stem cells and cancer. Trends in molecular medicine 14 : 400–409.
90. RoushSF, SlackFJ (2009) Transcription of the C. elegans let-7 microRNA is temporally regulated by one of its targets, hbl-1. Developmental biology 334 : 523–534.
91. MeissnerB, BollM, DanielH, BaumeisterR (2004) Deletion of the intestinal peptide transporter affects insulin and TOR signaling in Caenorhabditis elegans. The Journal of biological chemistry 279 : 36739–36745.
92. VeljkovicE, StasiukS, SkellyPJ, ShoemakerCB, VerreyF (2004) Functional characterization of Caenorhabditis elegans heteromeric amino acid transporters. The Journal of biological chemistry 279 : 7655–7662.
93. YamagataK, DaitokuH, TakahashiY, NamikiK, HisatakeK, et al. (2008) Arginine methylation of FOXO transcription factors inhibits their phosphorylation by Akt. Molecular cell 32 : 221–231.
94. NehrkeK (2003) A reduction in intestinal cell pHi due to loss of the Caenorhabditis elegans Na+/H+ exchanger NHX-2 increases life span. The Journal of biological chemistry 278 : 44657–44666.
95. SpanierB, LaschK, MarschS, BennerJ, LiaoW, et al. (2009) How the intestinal peptide transporter PEPT-1 contributes to an obesity phenotype in Caenorhabditits elegans. PLoS ONE 4: e6279 doi:10.1371/journal.pone.0006279.
96. Esquela-KerscherA, JohnsonSM, BaiL, SaitoK, PartridgeJ, et al. (2005) Post-embryonic expression of C. elegans microRNAs belonging to the lin-4 and let-7 families in the hypodermis and the reproductive system. Developmental dynamics : an official publication of the American Association of Anatomists 234 : 868–877.
97. JohnsonSM, LinSY, SlackFJ (2003) The time of appearance of the C. elegans let-7 microRNA is transcriptionally controlled utilizing a temporal regulatory element in its promoter. Developmental biology 259 : 364–379.
98. MartinezNJ, OwMC, Reece-HoyesJS, BarrasaMI, AmbrosVR, et al. (2008) Genome-scale spatiotemporal analysis of Caenorhabditis elegans microRNA promoter activity. Genome research 18 : 2005–2015.
99. StrahlBD, BriggsSD, BrameCJ, CaldwellJA, KohSS, et al. (2001) Methylation of histone H4 at arginine 3 occurs in vivo and is mediated by the nuclear receptor coactivator PRMT1. Current biology : CB 11 : 996–1000.
100. WangH, HuangZQ, XiaL, FengQ, Erdjument-BromageH, et al. (2001) Methylation of histone H4 at arginine 3 facilitating transcriptional activation by nuclear hormone receptor. Science 293 : 853–857.
101. TakahashiY, DaitokuH, HirotaK, TamiyaH, YokoyamaA, et al. (2011) Asymmetric arginine dimethylation determines life span in C. elegans by regulating forkhead transcription factor DAF-16. Cell metabolism 13 : 505–516.
102. BrennerS (1974) The genetics of Caenorhabditis elegans. Genetics 77 : 71–94.
103. IrizarryRA, HobbsB, CollinF, Beazer-BarclayYD, AntonellisKJ, et al. (2003) Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 4 : 249–264.
104. Huang daW, ShermanBT, LempickiRA (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature protocols 4 : 44–57.
105. Huang daW, ShermanBT, LempickiRA (2009) Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic acids research 37 : 1–13.
106. YeoGW, Van NostrandEL, LiangTY (2007) Discovery and analysis of evolutionarily conserved intronic splicing regulatory elements. PLoS Genet 3: e85 doi:10.1371/journal.pgen.0030085.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2013 Číslo 3
Nejčtenější v tomto čísle
- Fine Characterisation of a Recombination Hotspot at the Locus and Resolution of the Paradoxical Excess of Duplications over Deletions in the General Population
- Molecular Networks of Human Muscle Adaptation to Exercise and Age
- Recurrent Rearrangement during Adaptive Evolution in an Interspecific Yeast Hybrid Suggests a Model for Rapid Introgression
- Genome-Wide Association Study and Gene Expression Analysis Identifies as a Predictor of Response to Etanercept Therapy in Rheumatoid Arthritis