
Budding Yeast ATM/ATR Control Meiotic Double-Strand Break (DSB) Levels by Down-Regulating Rec114, an Essential Component of the DSB-machinery
An essential feature of meiosis is Spo11 catalysis of programmed DNA double strand breaks (DSBs). Evidence suggests that the number of DSBs generated per meiosis is genetically determined and that this ability to maintain a pre-determined DSB level, or “DSB homeostasis”, might be a property of the meiotic program. Here, we present direct evidence that Rec114, an evolutionarily conserved essential component of the meiotic DSB-machinery, interacts with DSB hotspot DNA, and that Tel1 and Mec1, the budding yeast ATM and ATR, respectively, down-regulate Rec114 upon meiotic DSB formation through phosphorylation. Mimicking constitutive phosphorylation reduces the interaction between Rec114 and DSB hotspot DNA, resulting in a reduction and/or delay in DSB formation. Conversely, a non-phosphorylatable rec114 allele confers a genome-wide increase in both DSB levels and in the interaction between Rec114 and the DSB hotspot DNA. These observations strongly suggest that Tel1 and/or Mec1 phosphorylation of Rec114 following Spo11 catalysis down-regulates DSB formation by limiting the interaction between Rec114 and DSB hotspots. We also present evidence that Ndt80, a meiosis specific transcription factor, contributes to Rec114 degradation, consistent with its requirement for complete cessation of DSB formation. Loss of Rec114 foci from chromatin is associated with homolog synapsis but independent of Ndt80 or Tel1/Mec1 phosphorylation. Taken together, we present evidence for three independent ways of regulating Rec114 activity, which likely contribute to meiotic DSBs-homeostasis in maintaining genetically determined levels of breaks.
Published in the journal:
. PLoS Genet 9(6): e32767. doi:10.1371/journal.pgen.1003545
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1003545
Summary
An essential feature of meiosis is Spo11 catalysis of programmed DNA double strand breaks (DSBs). Evidence suggests that the number of DSBs generated per meiosis is genetically determined and that this ability to maintain a pre-determined DSB level, or “DSB homeostasis”, might be a property of the meiotic program. Here, we present direct evidence that Rec114, an evolutionarily conserved essential component of the meiotic DSB-machinery, interacts with DSB hotspot DNA, and that Tel1 and Mec1, the budding yeast ATM and ATR, respectively, down-regulate Rec114 upon meiotic DSB formation through phosphorylation. Mimicking constitutive phosphorylation reduces the interaction between Rec114 and DSB hotspot DNA, resulting in a reduction and/or delay in DSB formation. Conversely, a non-phosphorylatable rec114 allele confers a genome-wide increase in both DSB levels and in the interaction between Rec114 and the DSB hotspot DNA. These observations strongly suggest that Tel1 and/or Mec1 phosphorylation of Rec114 following Spo11 catalysis down-regulates DSB formation by limiting the interaction between Rec114 and DSB hotspots. We also present evidence that Ndt80, a meiosis specific transcription factor, contributes to Rec114 degradation, consistent with its requirement for complete cessation of DSB formation. Loss of Rec114 foci from chromatin is associated with homolog synapsis but independent of Ndt80 or Tel1/Mec1 phosphorylation. Taken together, we present evidence for three independent ways of regulating Rec114 activity, which likely contribute to meiotic DSBs-homeostasis in maintaining genetically determined levels of breaks.
Introduction
In most sexually reproducing organisms, meiotic recombination is initiated by programmed catalysis of DNA double strand breaks (DSBs) by Spo11, an evolutionarily conserved type II topoisomerase-like transesterase [1]. In Saccharomyces cerevisiae, where the process is best understood, Spo11 activity requires nine additional proteins, five of which are meiosis specific (Rec102, Rec104, Rec114, Mei4, and Mer2), and four that are expressed during both meiosis and vegetative growth (Rad50, Mre11, Xrs2, and Ski8) [2]. These proteins interact with each other and/or with Spo11 to form a complex referred to as the Spo11 - or DSB-complex, or DSB-machinery, and participate in the Spo11 transesterase reaction that leads to the formation of a DSB (reviewed in [2]).
Meiotic DSBs are essential for meiosis; nevertheless, each break represents a potentially lethal or mutagenic DNA lesion that must be repaired before the first meiotic division (MI). As such, Spo11 catalysis is tightly regulated at the temporal, spatial, and quantitative levels. For instance, the catalysis does not normally take place until the locus has undergone replication [3], [4]. When it occurs, DSB-catalysis takes place preferentially at loci referred to as DSB hotspots rather than randomly throughout the genome [5]–[7]. The number of breaks catalyzed per meiosis is also developmentally programmed; in yeast or mammals, the number is approximately 150–250 per meiosis, whereas in Drosophila, it is about 25 [6]–[10].
Maintaining the number of meiotic DSBs at the developmentally programmed level would require both positive and negative means of regulating break formation. Although much is known about the genetic requirements for DSB formation [2], factors and mechanisms involved in monitoring the extent of breakage and/or limiting the number of breaks remain largely elusive. Recent studies suggested a role for the mammalian ATM kinase and its Drosophila and budding yeast homologs, tefu+ and TEL1, respectively, in down-regulating meiotic DSB formation [8], [9], [11]. These proteins are members of the ATM/ATR family of conserved signal transduction kinases involved in fundamental DNA/chromosomal processes such as DNA replication, DNA damage repair, recombination, and checkpoint regulation [12], [13]. They also play a key role(s) in many essential meiotic processes including interhomolog bias in DSB repair [14], meiotic recombination checkpoint regulation [15], and sex chromosome inactivation in mammals [16].
Here we present evidence that Rec114, an evolutionarily conserved Spo11-accessory protein and an essential component of the meiotic DSB-machinery [2], is a direct target of Tel1/Mec1, the budding yeast ATM/ATR homologues. Several Spo11-accessory proteins are proposed to be anchored at the chromosome axes and interact transiently with DSB hotspots at chromatin loops to promote cleavage [17]–[21]. Tel1/Mec1 phosphorylation of Rec114 upon DSB formation down-regulates its interaction with DSB hotspots and leads to reduced levels of Spo11 catalysis. Further analyses showed two additional means of down-regulating Rec114: synapsis associated removal at the onset of pachytene, as previously observed [17], [22], and Ndt80-dependent turnover. We propose a model whereby multiple means of regulating Rec114 activity contribute to meiotic DSB homeostasis in maintaining the number of breaks at the developmentally programmed level.
Results
Rec114 is a Tel1/Mec1 target
Budding yeast Tel1 and Mec1, like their mammalian counterparts, ATM and ATR, are serine/threonine kinases [23]. These kinases preferentially phosphorylate their substrates on serine (S) or threonine (T) residues that precede glutamine (Q) residues, so called SQ/TQ or [S/T]Q motifs. Many known targets of the ATM/ATR family proteins contain [S/T]Q cluster domains (SCDs), defined as a region where three or more SQ or TQ motifs are found within a tract of 100 residues or less [24].
As a means to investigate a role of Tel1/Mec1 in regulating DSB formation, we explored the possibility that they might directly phosphorylate one or more of the nine Spo11-accesssory proteins mentioned above. Rec114, an evolutionarily conserved meiosis specific chromosomal protein, was the most likely target with eight SQ/TQ consensus phosphorylation sites, seven of which are found in two clusters, referred to as SCD1 and SCD2 (Figure 1A). Western blot analysis using polyclonal α-Rec114 antibodies [17] revealed the appearance of slower migrating Rec114 species (Figure 1A). The putative phosphorylated isoform(s) of Rec114 was more prominent in a strain expressing a tagged version of REC114, REC114-13xMYC (Figure 1B “WT”). The tagged version also persisted for longer, showing that despite conferring full spore viability the tag changed some of Rec114's characteristics (see below). In both REC114 and REC114-13xMYC strains, the slower migrating species became prominent by 4 hours, corresponding to meiotic prophase in the current experimental condition [14].
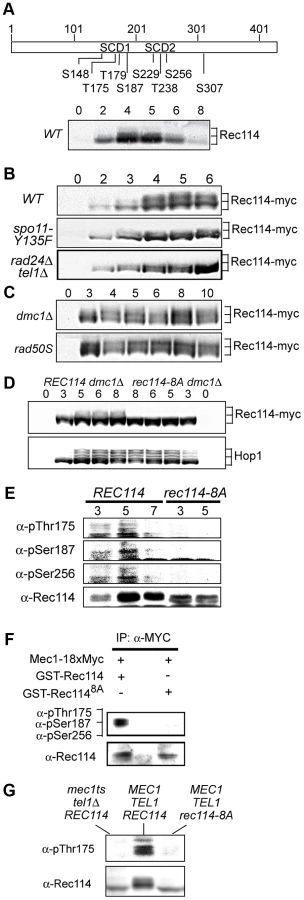
DSBs formed by Spo11 activates Tel1/Mec1, which in turn, directly phosphorylate a number of targets including H2AX, Sae2/Com1, (the ortholog of human CtIP), Hop1, and Zip1 [14], [25]–[27]. To test whether the Rec114 phosphorylation was also dependent on meiotic DSBs, we assessed the effect of spo11-Y135F, a catalytically inactive allele of SPO11 [1]. The gel shift was not detected in protein from spo11-Y135F strains, indicting it is dependent on DSB formation (Figure 1B).
Next, we tested the dependence of the Rec114 mobility shift on TEL1/MEC1. To this end, we assessed Rec114 migration patterns in a rad24Δ tel1Δ strain. In a rad24Δ tel1Δ strain, the Tel1/Mec1 signaling is down-regulated to a level comparable to that in mec1Δ tel1Δ cells kept viable by a suppressor mutation, sml1Δ; however, rad24Δ tel1Δ cells do not exhibit the severe meiotic progression defect observed in the latter [14]. We found that Rec114 mobility shift was reduced in a rad24Δ tel1Δ background (Figure 1B). The reduction was also observed at the restrictive temperature in a tel1Δ strain carrying the temperature sensitive mec1-4 allele [28] (Figure 1G).
Defects in meiotic recombination or synapsis activate Tel1 - or Mec1 - checkpoint response [12], [14], [15], [26], [27], [29]. In rad50S, mre11S (“S” for separation of function), or com1Δ/sae2Δ backgrounds, Spo11 remains covalently bound to the break ends, preventing their further processing. Accumulation of unprocessed meiotic DSBs in these mutants triggers a TEL1-dependent checkpoint response [30]–[32]. Elimination of the meiotic recombinase Dmc1, on the other hand, leads to accumulation of hyper-resected break ends that are loaded with single strand DNA (ssDNA) binding proteins and activates a MEC1-mediated checkpoint response [15], [33]. During Tel1 - or Mec1-checkpoint response, a number of targets, including Hop1 and Com1/Sae2, remain hyper-phosphorylated, reflecting the increased kinase activity of Tel1/Mec1. We found that both the extent and duration of Rec114 mobility shift seemed also enhanced in a rad50S or dmc1Δ background (Figure 1C), consistent with the possibility that Rec114 might be a target of Tel1/Mec1.
To further address the role(s) of Tel1/Mec1 in Rec114 mobility shift, we examined its migration pattern in a strain expressing a rec114 allele, rec114-8A, where all of the S or T residues of the eight Tel1/Mec1 consensus sites were replaced by a non-phosphorylatable alanine (A). We found that Rec114 mobility shift was abolished in a rec114-8A dmc1Δ strain (Figure 1D), indicating that the observed shift is due to a modification(s) at one or more of the eight Tel1/Mec1 consensus sites.
To confirm in vivo phosphorylation of Rec114 at a specific residue(s) during normal meiosis, we generated phospho-specific antibodies against three of the eight ATM/ATR consensus sites in Rec114. T175 and S187 were chosen based on their biological relevance (Table 1; see analysis below); S265 was selected using a software tool that predicts kinase-specific phosphorylation sites (GPS 2.1; Supporting Online Material). Using these phospho-specific antibodies, we performed Western blot analyses on samples taken from strains expressing either WT or the non-phosphorylatable allele, rec114-8A. The results showed that each of the three phospho-specific antibodies generated signals in the WT samples but not the rec114-8A, confirming in vivo phosphorylation of Rec114 at these three sites (Figure 1E). Finally, we demonstrated that purified Mec1 could directly phosphorylate one or more of the three confirmed in vivo Rec114 phosphorylation sites in vitro (Figure 1F). Taken together, we conclude that Rec114 is a DSB dependent target of Tel1/Mec1 during normal meiosis.

Synthetic interaction between rec114-phosphomimetic and spo11-hypomorphic alleles
To investigate function(s) of Tel1/Mec1 phosphorylation of Rec114, the effect of mutating the S or T residues of the eight Tel1/Mec1 consensus sites was examined. We began the analysis with two rec114 alleles, rec114-8A or rec114-8D, where the eight S or T were mutated to either a non-phosphorylatable alanine (A) or to a phospho-mimetic aspartic acid (D) residue, respectively. Spore viability of rec114-8A diploids was comparable to that of REC114 in all genetic backgrounds tested (Table 1). rec114-8D, in contrast, conferred haploinsufficiency and synthetic interactions with mutations that confer either a reduction in DSB-catalysis (e.g. spo11-HA and spo11-DA) [34] or sensitivity to such reduction (e.g. pch2Δ) [35] (Table 1). Thus, constitutively mimicking Tel1/Mec1 phosphorylation might be deleterious to meiosis. Alternatively, the effect might be due to protein misfolding caused by the introduction of eight closely spaced negative charges, which might have led to its degradation. Although we cannot rigorously rule out the latter, it appears unlikely, given that chromatin bound Rec1148D is more abundant than Rec114 (see analysis below), and also because replacing as few as two (T175 and S187) of the eight consensus sites with a phosphomimetic residue confers a rec114-8D like phenotype with respect to haploinsufficiency and synthetic interaction with spo11-hypomorphic alleles (Table 1). Notably, T175 and S187 of Rec114 are confirmed in vivo phosphorylation sites (Figure 1E).
Rec114 phosphorylation down-regulates Spo11 catalysis
The synthetic spore lethality interaction between rec114-phosphomimetic and spo11-hypomorphic alleles, which are known to confer sublethal reductions in crossover (CO) levels [34] (Table 1), suggested that the combined effects of the mutations may result in a lethal deficit in CO-formation. To test this, we assessed the effect of rec114-8D on CO-levels at the well characterized HIS4-LEU2 artificial meiotic recombination hotspot (Figure 2A) [36]. rec114-8D conferred a delay in the accumulation of COs, and about 25% reduction in the final level of COs; in rec114-8A, the level of COs was comparable to WT but they appeared earlier (Figure 2BC).

A reduction in CO-levels can result from either insufficient DSB levels and/or a defect(s) in CO homeostasis [34]. CO homeostasis refers to the notion that CO-levels tend to be maintained at the expense of noncrossovers (NCOs), and is, in part, based on the observation that strains expressing spo11-hypomorphic alleles exhibited only a modest reduction in the levels of COs despite the fact that their DSB levels, assessed in a rad50S background, were significantly lower than WT [34]. To determine whether the reduction in CO-levels in a rec114-8D strain was due to a defect in break formation and/or CO homeostasis, we measured DSB levels in a rec114-8D com1Δsae2Δ or rec114-8D rad50S strain using pulsed field gel electrophoresis (PFGE)/Southern analysis (Figure 2D; data not shown). The results showed that rec114-8D confers a dramatic reduction in the levels of DSBs on three different chromosomes examined, ChrIII, V, and VIII (Figure 2E; Figure S1 ABC; data not shown). We conclude that the modest reduction in CO-levels in a rec114-8D strain is likely due to a reduction in DSB levels, and that the observed synthetic interaction between rec114-phosphomimetic and spo11-hypomorphic alleles (Table 1) may result from additive impact of the two mutations on insufficient DSB-catalysis.
The above observations suggest that Tel1/Mec1 phosphorylation of Rec114, mimicked in rec114-8D, down-regulates DSB formation. If so, the absence of the phosphorylation in rec114-8A should lead to an increase in DSB levels, assuming that no other mechanism was acting redundantly. Indeed, a substantial increase could be observed for break sites near YCL064C or YCR048W on ChrIII (Figure 2EF). The extent of the increase was comparable to that observed in tel1Δ, a mutant reported to cause an increase in DSB levels [11]. Since Rec114 is a target of Tel1 and/or Mec1 (above), the latter suggests that Rec114 is likely to be a key target in mediating Tel1 negative regulation in DSB levels. Unlike rec114-8D, whose negative effect on break levels was obvious at all break sites analyzed on ChrIII, V, and VIII, we were only able to document the much subtler positive effect of rec114-8A or tel1Δ on ChrIII with this technology (Figure 2EF; Figure S1D–E and data not shown).
The dramatic effect of rec114-8D suggests that phosphorylation of some or all of the sites mutated is sufficient to strongly reduce Spo11 catalysis. The comparably modest increase in rec114-8A mutants, where Rec1148A is insensitive to Tel1/Mec1 negative control via phosphorylation at these sites, suggests that Rec1148A might mainly cause repeated cleavage by the same activated DSB machine near the break on the same chromatid, which would hardly increase the DSB signals measured by Southern; alternatively, it may point to the existence of additional mechanism(s) limiting break formation, and that it/they is/are yet to be discovered.
Unexpectedly, we found that the negative effect of rec114-8D on break level was notably attenuated in a dmc1Δ background compared to rad50S or com1Δ/sae2Δ (Figure 2G; data not shown). In a rec114-8D dmc1Δ strain, DSB levels reached about 75% of a REC114 dmc1Δ. In a RAD50 DMC1 background, the effect of rec114-8D was intermediate, between rad50S/com1Δ/sae2Δ and dmc1Δ (Figure S2). These observations show that the control of DSB formation is likely multi-layered and that feedbacks in addition to that by Rec114 phosphorylation exist.
Rec114 phosphorylation leads to a genome-wide reduction in DSB levels
As an independent means of assessing the effect of Rec114 phosphorylation on DSB levels, we performed a genome-wide Spo11-chromatin immunoprecipitation (ChIP) on CHIP assay (here on referred to as ChIP-chip), which confers greater resolution and offers easier normalization than a Southern blot based analysis (e.g. [7], [37]). In constructing the required strains for the analysis, we took into account the potential genetic interaction between various epitope tags of Spo11 and rec114 alleles as suggested by reduced spore viability of strains expressing tagged versions of either protein (Table 1; data not shown). We introduced the untagged versions of REC114, rec114-8A, or rec114-8D alleles into a rad50S strain expressing SPO11-18xMYC. Unlike spo11-6xHIS-3xHA, the SPO11-18xMYC did not affect spore viability of rec114-8D strains (data not shown). Spo11-myc ChIP was performed without the use of formaldehyde (FA) cross-linking to enrich for Spo11 proteins that had remained covalently bound to the break ends upon DNA-cleavage. To ensure the highest degree of comparability between the three REC114/rec114 allele backgrounds, the experiments were performed strictly in parallel for all steps from culturing to the final analysis. The resulting profiles of covalently bound Spo11 in the three backgrounds reproduced the published DSB hotspot profiles [7] with great precision (Figure 3A). A small fraction of signals, typically near telomeres and within pericentric regions, however, are not DSB specific, but identical among the three profiles (Figure 3A, areas denoted by *); these were used to superimpose the profiles (decile normalization, [17], Materials and Methods). Importantly, the three aligned profiles differ in the amplitude of hundreds of sharply defined positions in an almost invariable pattern: Spo11 signal in rec114-8A is higher than in wild type, while Spo11 in rec114-8D is strongly reduced (Figure 3A; Figure S3).

The results of statistical evaluation of the differences in these peaks is presented in Figure 3C. The following prediction was tested in this analysis: If DSB formation was indeed reduced in rec114-8D relative to rec114-8A, then the ratio of the Spo11 profiles of rec114-8A over rec114-8D, (hereon referred to as 8A/8D), should define DSB sites. In fact, the correlation between DSB hotspots and the 8A/8D peaks should be greater than that of not-normalized profiles. Indeed, profiles of these ratios identify near 100% of the published DSB hotspots (eg. Figure S3 A,D). When peaks of the ratio of these profiles were compared to the mapped hotspots at a resolution of 600 bp, >97% of the 1200 strongest Spo11 8A/8D peaks matched one of the 3600 DSB sites [7], (p<10−40, Figure S4A). The same was true for smaller selections; 62% of 500 strongest 8A/8D sites matched one of the 500 strongest DSB sites (p<10−40, Figure 3C), while 76% of 100 8A/8D matched 100 DSBs (p<10−20, Figure S4B). More detailed results showing the cumulative curves of distances compared to a null hypothesis (random) are provided in Figures 3Ci and Figure S4A,B. Although there are some peaks in the Spo11 profiles, where 8D>8A, less than 1% of the 500 strongest 8D/8A match the 500 DSBs, a strong anti-correlation (p<10−6) that excludes that there is significant 8D>8A at DSB sites (data not shown). Even for the smaller difference between WT and 8A, WT/8A produces a clear anti-correlation (Figure 3Ci). Being independent of decile or any other normalization, this analysis indicates that Spo11 catalysis at nearly all known hotspots is attenuated in the phospho-mimicking rec114-8D background. Furthermore, the degree of attenuation is roughly proportional to the hotspot strength in that the 100 strongest DSB peaks correspond to the 100 strongest Spo11 8A/8D peaks, whereas the 500 strongest DSB peaks to the 500 strongest Spo11 8A/8D peaks.
Analysis of the smaller differences between Spo11 profiles in rec114-8A and in REC114 by 500×500 comparison (500 hottest DSB hotspots against 500 strongest 8A/WT peaks) also produced a significant, although somewhat weaker, correlation (p<10−40, Figure 3Ci). We thus confirm with high significance, that Spo11 signals in the non-phosphorylatable rec114-8A are more abundant than in the wild type background, at least for the 500 strongest hotspots genome wide. The effect of rec114 mutations on the extent of Spo11 catalysis was confirmed further by qPCR analysis at a strong DSB site (YCR047C, Figure 3Aii). Taken together, these results strongly suggest genome-wide down-regulation of Spo11 catalysis by phosphorylation of Rec114, at least in the rad50S background.
In addition to axis-site binding, Rec114 also shows phosphorylation-sensitive interactions with DSB hotspots
Rec114 is a meiotic chromosome axis protein whose recruitment to the chromosomes is essential for Spo11 catalysis [17], [20], [22]. To test whether Tel1/Mec1 phosphorylation might down-regulate Spo11 catalysis by affecting Rec114's association with certain chromosomal positions, we performed genome wide Rec114 ChIP-chip analysis in strains expressing untagged versions of Rec114, Rec1148A or Rec1148D using a polyclonal antibody raised against Rec114 [17].
The analysis of Rec114 ChIP-chip after 4 hours in SPM showed enrichment of Rec114 at chromosome axes located nearby strong DSB hotspots (Figure 3Bi) as shown previously [17], [21]. Similar to the Spo11 profiles, the three Rec114 profiles became perfectly superimposed after decile normalization for many DSB-unspecific low signal peaks (Figure 3Bi). Within DSB-rich domains of ChrIII, signals at axis sites were strongest for Rec1148D, followed by Rec114 and then Rec1148A at axis sites. This relationship was confirmed by qChIP at one axis site over a meiotic time course (Figure 3Bii). Thus, Rec114-axis association appears to correlate negatively with DSB levels. We conclude that the reduction in DSB levels in a rec114-8D strain is not due to defects in Rec1148D -axes interaction.
RMM and other Spo11 accessory proteins are proposed to be anchored at the chromosome axes and interact transiently with DSB hotspots at chromatin loops to promote cleavage [17]–[21]. Given the apparent excess of Spo11-accessory proteins relative to the number of breaks catalyzed (e.g. [20]), such transient interaction is expected to manifest as small peaks near hotspots interspersed in a landscape of prominent axis signals. Indeed, for the hyperactive Rec1148A protein, nearly all of the strong DSB hotspots show small peaks overlapping the hotspots (Figure 3Bi, at 211.7kb; Figure 3Biii, v, Figure S5). These DSB associated peaks are stronger in Rec1148A than in wild type and are typically absent in Rec1148D. At strong hotspots, the profiles reversed their order noted above and become Rec1148A>Rec114>Rec1148D, although Rec1148D strongly dominates at the immediately adjacent axis sites (Figure 3Biii, v, Figure S5). Among the 35 strongest hotspots (as defined in [7]), 33 of them presented Rec1148A>Rec1148D (p<1.6×10−17), and all but one overlapped with local Rec1148A maximum in the DSB cluster (e.g. Figure 3Biii, iv, v). Comparing Rec114 association with a DSB site (YCR047C) and its neighboring axis site as a function of time, we observed that the extent of increase at the DSB site (Figure 3Bvi) is greater than the increase at the axis site (Figure 3Bii). Furthermore, the time dependent increase in the hotspot associated Rec114 exhibited Rec1148A>Rec114>Rec1148D (Figure 3Bvi).
Similar to arguments of the previous section, the following prediction was tested: If more Rec1148A bound to DSB sites than Rec1148D, peaks of the ratio of the profiles Rec1148A/Rec1148D (8A/8D) should map to DSB sites. Analysis shows that the majority of DSB-sites coincide with 8A/8D peaks (Figures S3 B, E). Indeed, comparison of the 500 strongest peaks and 500 hottest hotspots revealed a highly significant correlation (Figure 3C, p<10−37). Interestingly, 8A/WT and WT/8D peaks also exhibit significant correlations with DSB sites (p<10−19, 98% confidence interval of a random model plotted) suggesting the relation: 8A>WT>8D at DSB sites. Inversion of the DSB anti-correlated 8D profile also lead to the observed positive correlation of WT/8D (Figure 3Cii, ‘1/8D’ red circles), albeit with a weaker correlation than the 8A/8D (p<10−7) and WT/8D ratios (p<.04), lending solid statistical support to the interpretation Rec1148A>Rec114>Rec1148D at the 500 strongest DSB hotspots. Selecting just 100 strongest sites produced similar significances, while selecting more hotspots (3600) results in loss of significance, as the effect of 8A becomes insignificant compared to the effect of 1/8D for weak hotspots (Figure S4).
The parallel analysis of mutations with opposite effects on DSB hotspot binding provided an opportunity to unequivocally demonstrate genome-wide associations of Rec114 with DSB sites. In addition, these mutants reveal that interaction between Rec114 and DSB sites are negatively regulated by Tel1/Mec1 phosphorylation of Rec114.
Rec114 phosphorylation delays the onset of its NDT80-dependent turnover
The effects of Rec114 phosphorylation on its steady state protein levels were assessed by Western blot analysis (Figure 4) using the α-Rec114 antibody [17]. In a rec114-8A culture, a reduction in the protein levels, most notable at 5 and 6 hours, was observed (Figure 4A). In rec114-8D, protein persists longer, until the 6 and 8 hour time points. Thus, phosphorylation of Rec114 appears to increase not only its axis-association but also its steady state levels.

Ndt80 is a meiosis specific transcription factor required for pachytene exit and resolution of joint molecules (JMs). Some meiotic DSBs persist in an ndt80Δ background, suggesting its involvement in curtailment of break formation [38], [39], or a failure to repair some DSBs. To determine whether Ndt80 affected the stability of Rec114, we repeated the same Western blot analysis in an ndt80Δ background. Results revealed that Rec114 becomes stabilized in a REC114 ndt80Δ strain for at least 12 hours after transfer to SPM (Figure 4B), while it rapidly declined in NDT80 after 5 hours (Figure 4A). Thus, timely Rec114 degradation requires Ndt80. ndt80Δ also prevented the degradation of Rec1148A and Rec1148D (Figure 4B), suggesting that the observed differences in steady state protein levels in the mutants (Figure 4A) might be caused by differential timing of Ndt80 activation.
Rec114 phosphorylation delays its synapsis-associated removal from chromosomes
All Spo11-accessory proteins examined to date, including Rec114, are recruited to the chromosomes before the initiation of meiotic recombination, and remain chromosome-associated until Zip1 dependent homolog synapsis takes place [17], [20], [22], [40]. Zip1 is an evolutionarily conserved component of the central region of the synaptonemal complex (SC), and is required for homolog synapsis and meiotic recombination [41]–[43].
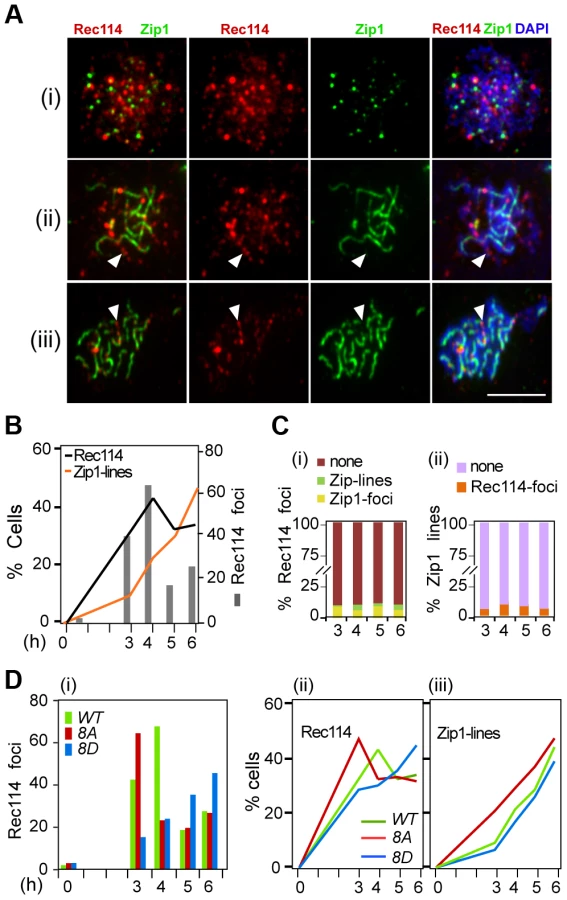
In early meiotic prophase, there is little overlap between Rec114 and Zip1; at later stages, Rec114 foci become less abundant and dimmer in synapsed chromosome regions but remain bright in unsynapsed regions of the same nucleus [17], [22]. These observations suggest that synapsis might promote the removal of Rec114 and its associated proteins Mei4 and Mer2. Combining this with the current observation that the extent of Rec114-axis association is affected by its phosphorylation status (Figure 3B) raised the possibility that Rec114 phosphorylation might affect the timing of synapsis. To address this, we performed co-immunostaining analyses of Rec114 and Zip1 using polyclonal antibodies raised against each protein (Supplementary Online Information). The experiment was conducted in an ndt80Δ background to exclude any influence by the NDT80 dependent Rec114 degradation (above).
Rec114 in the ndt80Δ background behaved as reported [17], [22], with Rec114 foci peaking at mid prophase just before the onset of synapsis, with little or no overlap between Rec114 and Zip1 staining (Figure 5A,C). The fraction of nuclei containing Rec1148A -foci decline more rapidly than Rec114, while that of Rec1148D containing nuclei remain abundant until at least 6 hours in SPM (Figure 5Di, ii), consistent with synapsis being affected by the status of Rec114 phosphorylation (Figure 5D iii). These observations show that synapsis-associated dissociation of Rec114 is Ndt80 independent. Depending on the Rec114 allele and the associated DSB frequency, synapsis occurs earlier or later, entailing earlier or later Ndt80 independent Rec114 removal. In ∼30% of ndt80Δ cells, some strong Rec114 foci persisted up to 6 hours into meiosis (Figure S6), consistent with the stabilization of the protein in ndt80Δ (Figure 4). Most of the Rec114 positive ndt80Δ cells exhibited dimming and/or disappearance of signal along SCs and a persistent polycomplex (PC) (Figure S6i, ii), in agreement with ‘SC-decay’ in ndt80 mutants [38]. All prominent Rec114 foci were on Zip1-free areas or in PC (Figure S6i, iii), suggesting that they might be aggregates of stripped Rec114 that cannot be degraded in an ndt80 background. The abundance of residual Rec114 present at these late time points is consistent with the aberrantly late DSBs observed in ndt80Δ [39].
Discussion
Rec114 is an evolutionarily conserved essential component of the meiotic DSB-machinery [17], [20], [22], [44]–[48]. Here we present evidence that phosphorylation of Rec114 reduces both its interaction with DSB-hotspots and DSB formation. Furthermore, it prolongs Rec114-axis association and delays the onset of NDT80 dependent turnover, suggesting the existence of a feedback system that couples the steady state DSB levels to post-leptotene regulation of Rec114 activity.
DSB-dependent Tel1/Mec1 phosphorylation of Rec114
Three independent studies have implicated a role of the ATM kinases in down-regulating Spo11 catalysis [8], [9], [11]. The evidence presented here implicates Rec114 as a physiologically relevant Mec1 and/or Tel1 target in this regulation. Results show a robust reduction of function for phospho-mimicking Rec1148D and a subtler increase in function in the rad50S background for Rec1148A. Why are these effects not symmetric? One trivial explanation could be that introducing eight aspartic acid residues into Rec1148D may render the protein partially non-functional, aside from mimicking phosphorylation. However, Rec1148D is a stable protein that binds well to chromatin, excluding general protein stability-, nuclear import - or chromatin-binding defect for Rec1148D. Furthermore, since Rec1148D behaves similar to Rec1142D (T175D, S187D) in terms of reduction of DSBs, inferred based on reduced spore viability in a spo11-HA background (Table 1), and two aspartic acid exchanges are less likely to strongly damage the protein, we favor an interpretation involving constitutive phospho-mimicking as explanation. If so, phosphorylation is sufficient to tune down DSB formation (e.g. Rec1148D or Rec1142D), while other effects might prevent the observation of a strong increase in break levels under constitutive “on” conditions (e.g. Rec1148A).
Several models (e.g. [14], [17], [49]) propose that a first negative feedback may be locally restricted to the activated DSB-machine and its surrounding chromatin loops. Phosphorylation of Rec114 would be ideally suited to mediate such a control. However, repeated cleavage of the already broken chromatid is not expected to lead to an increase of the DSB signal. Cleavage of hotspots on the intact sister chromatid could be responsible for the 20–30% increase observed by the ChIP-chip analysis in the rad50S background. Increased DSB formation in Rec1148A, even if only moderate, identifies Rec114 as a rate limiting target of negative feedback at least in the com1Δ/sae2Δ or rad50s background. On the other hand DSB formation is strongly impeded in Rec1148D (or Rec1142D), suggesting that phosphorylation affects a critical function of Rec114. Importantly, phosphomimicking Rec1148D shows a reduced interaction with DSB-hotspots suggesting a plausible mechanism explaining its reduced activity.
Synapsis dependent removal of Rec114
Budding yeast Rec114 physically interacts with Mei4 and Mer2, two other components of the DSB-machinery, to form a complex referred to as RMM (Rec114-Mei4-Mer2) [20], [22]. RMM foci become abundant in early meiotic prophase and the proteins accumulate on DNA sequences, which organize the chromosome axis upon condensation; when chromosomes synapse, RMM foci become dimmer and eventually disappear [17], [22], an observation confirmed here in a strain expressing untagged Rec114 (Figure 5). The strong correlation between the appearance of Zip1-lines and the disappearance of Rec114 foci from the synapsed regions of the chromosomes suggests that the process of synapsis itself could be removing RMM foci. Synapsis dependent removal of RMM occurs independently of Tel1/Mec1 phosphorylation and Ndt80 (and thus protein degradation), consistent with being an independent mechanism of down regulating DSB levels.
NDT80 dependent Rec114-turnover
During normal meiosis, steady state levels of meiotic DSBs decrease to the background level as cells proceed beyond pachytene with complete cessation of DSBs depending on Ndt80 [39]. The Ndt80 dependent Rec114 degradation reported here (Figure 4) presents a plausible mechanistic explanation for the latter.
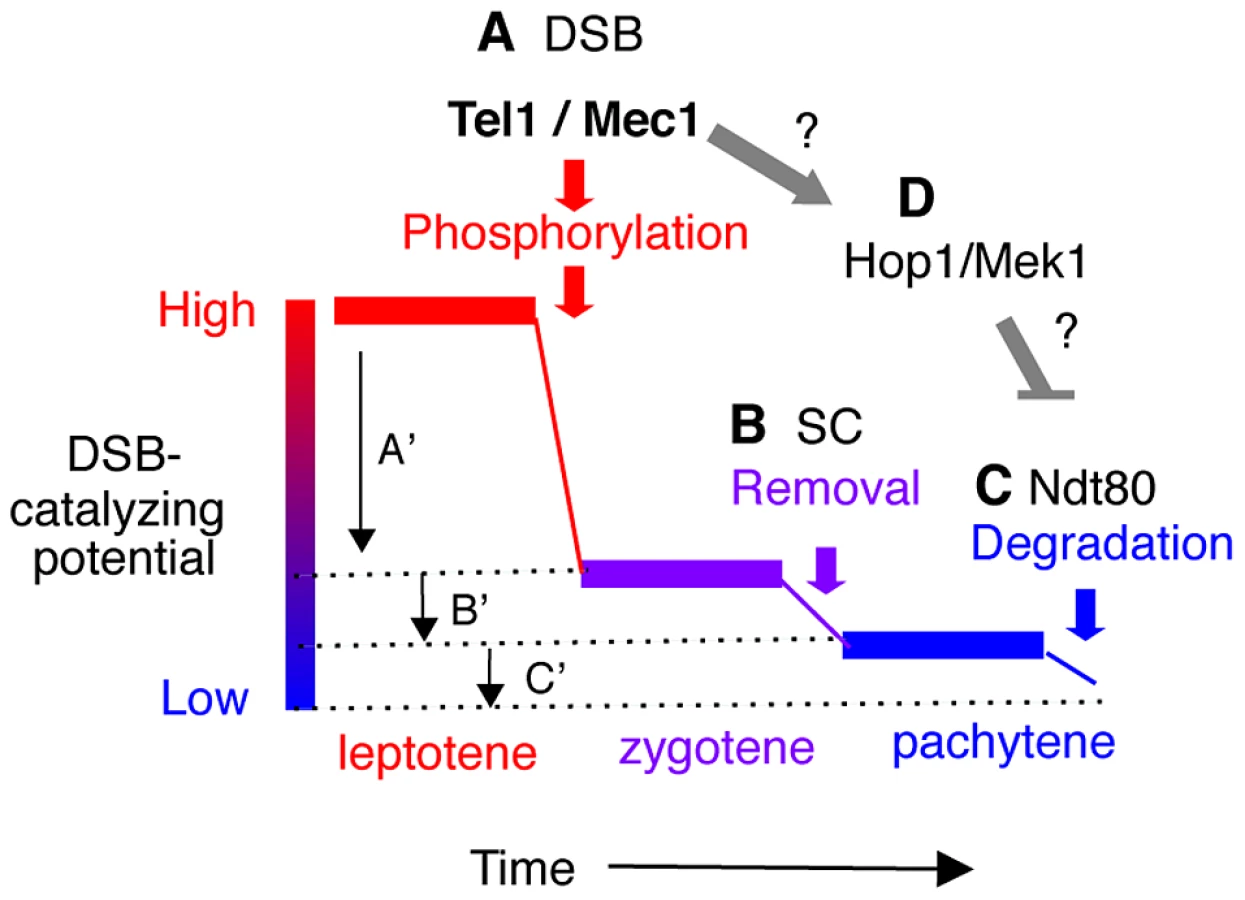
Normally, Ndt80 activation is coupled to meiotic DSB-repair and synapsis. In response to defects in either process (e.g. in a rad50S, dmc1Δ or zip1Δ background), Tel1/Mec1 prevent Ndt80 activation by hyper-phosphorylating Hop1/Mek1 [50]. Hop1 is an evolutionarily conserved meiotic chromosome axis protein that functions as a meiotic paralog of Rad9 in the Tel1/Mec1 signaling cascade [14]. Hyper-phosphorylation of Hop1, in turn, activates the checkpoint function of Mek1, a meiotic chromosome axis associated serine/threonine kinase, and a meiotic paralog of Rad53/Chk2 [14]. Importantly, the Tel1/Mec1-Hop1/Mek1-Ndt80 signaling pathway appears to also regulate normal meiotic progression [12], [38], [51]–[53], raising the possibility that the observed earlier or delayed onset of NDT80 dependent Rec114 turnover in rec114 phospho-mutants might be under Tel1/Mec1 regulation (Figure 6).
Meiotic DSB homeostasis, the tendency to maintain similar DSB levels under different circumstances
The term “meiotic DSB homeostasis” was originally introduced to refer to the phenomenon, whereby the accumulated DSB frequency in a chromosomal region appeared to be maintained at a constant level [54]. Here, we expand the meaning to include that the break frequency might be regulated not only at the regional, but also at genome wide level. A sophisticated system controlling chromosome synapsis and recombination is expected to operate, at least, at two levels:
First, the DSB machinery needs to be “informed” about the success of a particular DSB catalysis. This local negative feedback should be limited to the immediate environment and should prevent repeated cleavage of the already broken chromatid near the break. One manifestation of this local down regulation would be DSB interference, or “competitive inactivation” of weaker hotspots, by a nearby strong hotspot [54]–[57]. We show evidence that phosphorylation of Rec114 is a key step in communicating DNA breakage to the DSB machinery via Mec1 and/or Tel1.
Second, nucleus wide (global) signaling of successful completion of homolog synapsis and meiotic DSB repair should precede irreversible global inactivation of the DSB machinery. We present evidence for two feedback based mechanisms of such regulation: synapsis dependent removal and Ndt80 dependent degradation of Rec114. Linking the progression of synapsis to down regulation of DSB formation conveniently ensures that enough DSBs have been formed to guarantee successful homology search. However, synapsis alone does not appear to lead to irreversible inactivation of the DSB machinery. This ultimate decision is instead linked to the exit from pachytene, when the activation of Ndt80 provides some guarantee that early prophase events have successfully completed.
Evidence here and elsewhere indicates that cells have the means to prevent excessive DSB formation via negative feedback [8], [9], [11]. But what happens when the break level is too low? A key feature of these nested feedback loops (Figure 6) is that a reduced Spo11 activity, independent of its cause, will delay the completion of synapsis and the onset of Ndt80 activation, and thus provide more time to accumulate breaks, even at a low rate. For instance, delayed synapsis and Ndt80 activation in the DSB poor rec114-8D mutant, would delay RMM inactivation, likely prolonging its active lifespan and raising the level of DSBs eventually produced. Indeed, Rec1148D remained longer and in greater abundance at chromosome axes than Rec114 or Rec1148A. DSB homeostasis could also account for the apparent ‘catching up’ of break levels in a rec114-8D dmc1Δ strain (Figure 2G). For instance, defective recombination and synapsis in the mutant would severely compromise Zip1 dependent RMM removal, while dmc1Δ activation of Mec1-Hop1/Mek1 checkpoint response would prevent Ndt80 dependent Rec114 degradation, thus allowing Rec1148D to remain active. The fact that break levels in rec114-8D rad50S remain low, apparently unable to catch up, suggests that DSB repair beyond rad50S arrest point (e.g. endonucleolytic removal of Spo11 followed by break resection) might be required to activate the synapsis and/or Ndt80 based feedback loops (Figure 6).
DSB homeostasis may contribute significantly to the relatively mild effect on spore viability of mutants (e.g. spo11-hypomorphs) with a low rate of DSB formation that was up to now solely attributed to CO homeostasis. But clearly, postponing the inactivation of the DSB machinery in response to problems in synapsis and break repair helps to provide more DSBs, on which CO homeostasis can act to ensure correct chromosome segregation.
Materials and Methods
Standard yeast manipulation procedures and growth media are utilized. All strains were of the SK1 background (Table S1). Specific [S/T]Q to AQ and DQ mutations were introduced and sequenced to ensure that no additional mutations were created during the mutagenesis. Standard Western blot, Southern blot, and spread surface immunofluorescence techniques were used. Chip on CHIP and qPCR were performed as described in [17].
Yeast strains and media
Standard yeast manipulation procedures and growth media were utilized. All strains are of the SK1 background; relevant genotypes of the strains are listed in Table S1.
Construction of rec114 strains
The myc13 tag from a REC114-MYC13-HYGRO plasmid (pNS2) was removed to generate pJC15, an integration plasmid without an epitope tag. Specific [S/T]Q to AQ or DQ mutations were introduced into either pNS2 or pJC15 utilizing the QuickChange Multi Site-Directed Mutagenesis kit (Stratagene). The entire open reading frame (ORF) of each allele was sequenced to ensure that the allele did not contain any incidental mutation(s). Each rec114 allele was introduced into a rec114Δ::KanMX4 haploid strain (RCY336/337), where the endogenous REC114 gene was replaced by a kanamycin resistant gene. Transformants were identified based on their ability to grow on hygromycin plates but not on kanamycin. Southern blot and PCR analyses were performed on candidate colonies to confirm integration of a single copy of a specific rec114-HygroMX4 allele at the endogenous locus, replacing the rec114Δ::KanMX4 allele. Correct rec114 haploid transformants of each allele were taken through standard yeast genetics manipulation to generate corresponding rec114 homozygous diploid strains suitable for meiotic analyses.
Generation of phospho-specific Rec114 antibodies
Three of the eight S/T[Q] consensus sites in Rec114, T175, S187 and S256, were selected for generation of phospho-specific antibodies. T175 and S187 were chosen based on the fact that replacing these residues with a non-phosphorylatable alanine (A) confers haploinsufficiency and synthetic interaction with spo11 hypomorphic alleles (Table 1). S256 was chosen because it was one of the six residues within Rec114 that were predicted to be the most likely ATM/ATR phosphorylation sites (GPS2.1 software [58]). Specificity of each phospho-specific antibody was confirmed by Western blot analysis of rec114 strains, each expressing a rec114 allele missing a specific phosphorylation site(s).
Synchronous meiotic time course
Induction of synchronous meiosis is carried out according to the established protocols [17], [59]. All pre-growth and meiotic time courses were carried out at 30°C except for mec1-4ts tel1Δ sml1Δ meiosis, where the culture was kept at 23°C and shifted to 30°C 2 hours after transferring into sporulation medium (SPM).
Protein purification and manipulation methods
GST-REC114 and GST-rec114-8A plasmid-construction and protein expression were carried out as described [60]. To purify Mec1-myc18 from yeast cells, 500 ml of logarithmically growing cell cultures were subjected to 1 hour incubation with 0.1% methyl methanesulfonate (MMS) followed by Immunoprecipitation using Goat anti-myc-agarose antibodies (AbCam). Mec1-myc immunoprecipitates were mixed with reaction cocktail containing kinase buffer, cold ATP, and either GST-Rec114 or GST-Rec1148A. The mixtures were incubated at 30°C for 25 minutes and subjected to electrophoresis on SDS gels. Gels were transferred onto a nitrocellulose membrane and subjected to Western blot analyses using anti-Rec114 or phospho-specific antibodies.
Western blot analysis
Whole-cell extracts (WCE) were prepared from cell suspensions in 20% trichloroacetic acid (TCA) by agitation with glass beads. Precipitated proteins were solubilized in SDS-PAGE sample buffer, and appropriate dilutions were analyzed by SDS-PAGE and Western blotting. Antibodies for Western blotting were mouse monoclonal anti-myc (1∶1000, AbCam), rabbit polyclonal anti-Rec114 (1∶1000), anti-Phospho-Rec114-S187, anti-Phospho-Rec114 - T175, anti-Phospho-Rec114 - S265 (1∶1000, Cambridge Research Biomedicals), goat anti-mouse IgG conjugated to horseradish peroxidase (1∶10,000; Sigma-Aldrich), and donkey anti-rat IgG conjugated to horseradish peroxidase (1∶10,000; Sigma-Aldrich).
Southern blot analysis
Southern blot analysis following Pulse Field Gel electrophoresis (PFGE) using DNA prepared in agarose plugs or standard agarose gel electrophoresis were performed as described [61]. Exception was that the PFGE gels shown in Figure 2G and Figure S1A were run with the following modifications: initial switch time; 15 sec – final switch time; 32.5 sec, in order to better separate large chromosomes. For quantifying the level of DSBs, only the signals associated with breaks proximal to the probe was utilized to maximize the detection of chromosomes that acquired more than one break (see [3] for discussion).
Chromatin Immunoprecipitation on CHIP (ChIPchip) and quantitative PCR (qPCR)
Rec114 and Spo11-myc chromatin immunoprecipitation (ChIP), quantitative PCR (qPCR), and microarrays hybridization/analysis were performed as described [17].
Cytological methods
Surface spread meiotic chromosomes were prepared as described [14]. Staining was performed as described [14] with the following primary antibodies: rabbit polyclonal anti-Rec1141 (1 : 100, F. Klein, MFPL), mouse monoclonal anti-HA (12CAS, 1∶100, S. Ley, NIMR), mouse monoclonal anti-MYC (9E10, 1∶100, S. Ley, NIMR goat polyclonal anti-Zip1 (1∶50, SantaCruz Biotechnology). Secondary antibodies (Invitrogen) were used at a 1∶500 dilution: chicken anti-mouse Alexa-488, anti-goat Alexa-488, chicken anti-rabbit Alexa-594. Chromosomal DNA was stained with 1 ug/ml 4,6-diamino-2-phenylimide (DAPI). Images were recorded and analyzed using a Deltavision (DV3) workstation from Applied Precision Inc. with a Photometrics CoolSnap HQ (10–20 MHz) air cooled CCD camera and controlled by Softworx image acquisition and deconvolution software.
Significance
For comparing proportions (e.g. matching versus non-matching peaks) significance values were computed using Fisher's exact test (http://www.langsrud.com/fisher.htm).
Statistical analysis of the Chip on CHIP
data was done as described [17]. Briefly, CEL files were converted using Affymetrix's “tiling array software” (TAS) and expressed as ChIP relative to WCE (whole cell extract) or as ChIP relative to another ChIP. The output of TAS (intensities per chromosome position) was smoothed using bandwidths between 250 and 1000 bp (ksmooth, statistic package R) and plotted for all chromosomes. For microarrays “decile normalization” was used. It is often a good choice for automatic background correction [17]. For the profiles to be compared in this work, a single correction factor was determined per profile F = 1/(0.1 percentile). These factors were usually very close to 1. After multiplying all profile intensities with their correction factors, they were precisely superimposing on all background peaks (compare Fig. 3A 0–30 kb, and 115–155 kb, see brackets with asterisk).
Peak calling and peak matching
Peaks were called automatically as described previously [17]. To quantify the overlap between peaks of one profile and the meiotic hotspot map published by [7], we used their simplified list of hotspots, organized in 3600 blocks, listing start and end of each hotspot array and the number of mapped 5′-ends detected. We defined the distance of a peak to its nearest hotspot as the distance to the nearest edge of a hotspot block (even when the peak mapped inside the block). Most of these arrays have a very narrow width, (median 180 bp, mean 252 bp, 0.9 quantile 507 bp). In order to simulate random distributions, we generated random positions corresponding to the numbers of peaks to be tested and mapped them relative to the hotspot-blocks, the same way as the experimental data. (Simulations were repeated 100 times to obtain the 2% and 98% percentiles plotted). For example, for the comparison of the 500 highest peaks with the 500 strongest hotspots, the hotspots were sorted according to strength, the peaks were sorted according to strength and then the top 500 of each list were compared. For each of the 500 highest peaks, the distance to the nearest hotspot-block was determined and the distances accumulated and plotted.
Supporting Information
Zdroje
1. KeeneyS, GirouxCN, KlecknerN (1997) Meiosis-specific DNA double-strand breaks are catalyzed by Spo11, a member of a widely conserved protein family. Cell 88 : 375–384.
2. MurakamiH, KeeneyS (2008) Regulating the formation of DNA double-strand breaks in meiosis. Genes Dev 22 : 286–292.
3. BordeV, GoldmanAS, LichtenM (2000) Direct coupling between meiotic DNA replication and recombination initiation. Science 290 : 806–809.
4. HochwagenA, ThamWH, BrarGA, AmonA (2005) The FK506 binding protein Fpr3 counteracts protein phosphatase 1 to maintain meiotic recombination checkpoint activity. Cell 122 : 861–873.
5. BaudatF, NicolasA (1997) Clustering of meiotic double-strand breaks on yeast chromosome III. Proc Natl Acad Sci U S A 94 : 5213–5218.
6. BuhlerC, BordeV, LichtenM (2007) Mapping meiotic single-strand DNA reveals a new landscape of DNA double-strand breaks in Saccharomyces cerevisiae. PLoS Biol 5: e324.
7. PanJ, SasakiM, KniewelR, MurakamiH, BlitzblauHG, et al. (2011) A hierarchical combination of factors shapes the genome-wide topography of yeast meiotic recombination initiation. Cell 144 : 719–731.
8. JoyceEF, PedersenM, TiongS, White-BrownSK, PaulA, et al. (2011) Drosophila ATM and ATR have distinct activities in the regulation of meiotic DNA damage and repair. J Cell Biol 195 : 359–367.
9. LangeJ, PanJ, ColeF, ThelenMP, JasinM, et al. (2011) ATM controls meiotic double-strand-break formation. Nature 479 : 237–240.
10. ManceraE, BourgonR, BrozziA, HuberW, SteinmetzLM (2008) High-resolution mapping of meiotic crossovers and non-crossovers in yeast. Nature 454 : 479–485.
11. ZhangL, KimKP, KlecknerNE, StorlazziA (2011) Meiotic double-strand breaks occur once per pair of (sister) chromatids and, via Mec1/ATR and Tel1/ATM, once per quartet of chromatids. Proc Natl Acad Sci U S A 108 : 20036–20041.
12. CarballoJA, ChaRS (2007) Meiotic roles of Mec1, a budding yeast homolog of mammalian ATR/ATM. Chromosome Res 15 : 539–550.
13. HarperJW, ElledgeSJ (2007) The DNA damage response: ten years after. Mol Cell 28 : 739–745.
14. CarballoJA, JohnsonAL, SedgwickSG, ChaRS (2008) Phosphorylation of the axial element protein Hop1 by Mec1/Tel1 ensures meiotic interhomolog recombination. Cell 132 : 758–770.
15. LydallD, NikolskyY, BishopDK, WeinertT (1996) A meiotic recombination checkpoint controlled by mitotic checkpoint genes. Nature 383 : 840–843.
16. TurnerJM, AprelikovaO, XuX, WangR, KimS, et al. (2004) BRCA1, histone H2AX phosphorylation, and male meiotic sex chromosome inactivation. Curr Biol 14 : 2135–2142.
17. PanizzaS, MendozaMA, BerlingerM, HuangL, NicolasA, et al. (2011) Spo11-accessory proteins link double-strand break sites to the chromosome axis in early meiotic recombination. Cell 146 : 372–383.
18. AcquavivaL, SzekvolgyiL, DichtlB, DichtlBS, de La Roche Saint AndreC, et al. (2013) The COMPASS subunit Spp1 links histone methylation to initiation of meiotic recombination. Science 339 : 215–218.
19. SommermeyerV, BeneutC, ChaplaisE, SerrentinoME, BordeV (2013) Spp1, a Member of the Set1 Complex, Promotes Meiotic DSB Formation in Promoters by Tethering Histone H3K4 Methylation Sites to Chromosome Axes. Mol Cell 49 : 43–54.
20. MalekiS, NealeMJ, AroraC, HendersonKA, KeeneyS (2007) Interactions between Mei4, Rec114, and other proteins required for meiotic DNA double-strand break formation in Saccharomyces cerevisiae. Chromosoma 116 : 471–486.
21. MiyoshiT, ItoM, KugouK, YamadaS, FuruichiM, et al. (2012) A central coupler for recombination initiation linking chromosome architecture to S phase checkpoint. Mol Cell 47 : 722–733.
22. LiJ, HookerGW, RoederGS (2006) Saccharomyces cerevisiae Mer2, Mei4 and Rec114 form a complex required for meiotic double-strand break formation. Genetics 173 : 1969–1981.
23. MalloryJC, PetesTD (2000) Protein kinase activity of Tel1p and Mec1p, two Saccharomyces cerevisiae proteins related to the human ATM protein kinase. Proc Natl Acad Sci U S A 97 : 13749–13754.
24. TravenA, HeierhorstJ (2005) SQ/TQ cluster domains: concentrated ATM/ATR kinase phosphorylation site regions in DNA-damage-response proteins. Bioessays 27 : 397–407.
25. MahadevaiahSK, TurnerJM, BaudatF, RogakouEP, de BoerP, et al. (2001) Recombinational DNA double-strand breaks in mice precede synapsis. Nat Genet 27 : 271–276.
26. Cartagena-LirolaH, GueriniI, ViscardiV, LucchiniG, LongheseMP (2006) Budding Yeast Sae2 is an In Vivo Target of the Mec1 and Tel1 Checkpoint Kinases During Meiosis. Cell Cycle 5 : 1549–1559.
27. FalkJE, ChanAC, HoffmannE, HochwagenA (2010) A Mec1 - and PP4-dependent checkpoint couples centromere pairing to meiotic recombination. Dev Cell 19 : 599–611.
28. ChaRS, KlecknerN (2002) ATR homolog Mec1 promotes fork progression, thus averting breaks in replication slow zones. Science 297 : 602–606.
29. WuHY, BurgessSM (2006) Two distinct surveillance mechanisms monitor meiotic chromosome metabolism in budding yeast. Curr Biol 16 : 2473–2479.
30. AlaniE, PadmoreR, KlecknerN (1990) Analysis of wild-type and rad50 mutants of yeast suggests an intimate relationship between meiotic chromosome synapsis and recombination. Cell 61 : 419–436.
31. NairzK, KleinF (1997) mre11S–a yeast mutation that blocks double-strand-break processing and permits nonhomologous synapsis in meiosis. Genes Dev 11 : 2272–2290.
32. UsuiT, OgawaH, PetriniJH (2001) A DNA damage response pathway controlled by Tel1 and the Mre11 complex. Mol Cell 7 : 1255–1266.
33. BishopDK, ParkD, XuL, KlecknerN (1992) DMC1: a meiosis-specific yeast homolog of E. coli recA required for recombination, synaptonemal complex formation, and cell cycle progression. Cell 69 : 439–456.
34. MartiniE, DiazRL, HunterN, KeeneyS (2006) Crossover homeostasis in yeast meiosis. Cell 126 : 285–295.
35. ZandersS, Sonntag BrownM, ChenC, AlaniE (2011) Pch2 modulates chromatid partner choice during meiotic double-strand break repair in Saccharomyces cerevisiae. Genetics 188 : 511–521.
36. HunterN, KlecknerN (2001) The single-end invasion: an asymmetric intermediate at the double-strand break to double-holliday junction transition of meiotic recombination. Cell 106 : 59–70.
37. KugouK, OhtaK (2009) Genome-wide high-resolution chromatin immunoprecipitation of meiotic chromosomal proteins in Saccharomyces cerevisiae. Methods Mol Biol 557 : 285–304.
38. XuL, AjimuraM, PadmoreR, KleinC, KlecknerN (1995) NDT80, a meiosis-specific gene required for exit from pachytene in Saccharomyces cerevisiae. Mol Cell Biol 15 : 6572–6581.
39. AllersT, LichtenM (2001) Differential timing and control of noncrossover and crossover recombination during meiosis. Cell 106 : 47–57.
40. PrielerS, PenknerA, BordeV, KleinF (2005) The control of Spo11's interaction with meiotic recombination hotspots. Genes Dev 19 : 255–269.
41. SymM, EngebrechtJA, RoederGS (1993) ZIP1 is a synaptonemal complex protein required for meiotic chromosome synapsis. Cell 72 : 365–378.
42. StorlazziA, XuL, SchwachaA, KlecknerN (1996) Synaptonemal complex (SC) component Zip1 plays a role in meiotic recombination independent of SC polymerization along the chromosomes. Proc Natl Acad Sci U S A 93 : 9043–9048.
43. BornerGV, KlecknerN, HunterN (2004) Crossover/noncrossover differentiation, synaptonemal complex formation, and regulatory surveillance at the leptotene/zygotene transition of meiosis. Cell 117 : 29–45.
44. MolnarM, ParisiS, KakiharaY, NojimaH, YamamotoA, et al. (2001) Characterization of rec7, an early meiotic recombination gene in Schizosaccharomyces pombe. Genetics 157 : 519–532.
45. MaloneRE, BullardS, HermistonM, RiegerR, CoolM, et al. (1991) Isolation of mutants defective in early steps of meiotic recombination in the yeast Saccharomyces cerevisiae. Genetics 128 : 79–88.
46. AjimuraM, LeemSH, OgawaH (1993) Identification of new genes required for meiotic recombination in Saccharomyces cerevisiae. Genetics 133 : 51–66.
47. SasanumaH, HirotaK, FukudaT, KakushoN, KugouK, et al. (2008) Cdc7-dependent phosphorylation of Mer2 facilitates initiation of yeast meiotic recombination. Genes Dev 22 : 398–410.
48. KumarR, BourbonHM, de MassyB (2010) Functional conservation of Mei4 for meiotic DNA double-strand break formation from yeasts to mice. Genes Dev 24 : 1266–1280.
49. GoldfarbT, LichtenM (2010) Frequent and efficient use of the sister chromatid for DNA double-strand break repair during budding yeast meiosis. PLoS Biol 8: e1000520.
50. AcostaI, OntosoD, San-SegundoPA (2011) The budding yeast polo-like kinase Cdc5 regulates the Ndt80 branch of the meiotic recombination checkpoint pathway. Mol Biol Cell 22 : 3478–3490.
51. KatoR, OgawaH (1994) An essential gene, ESR1, is required for mitotic cell growth, DNA repair and meiotic recombination in Saccharomyces cerevisiae. Nucleic Acids Res 22 : 3104–3112.
52. HepworthSR, FriesenH, SegallJ (1998) NDT80 and the meiotic recombination checkpoint regulate expression of middle sporulation-specific genes in Saccharomyces cerevisiae. Mol Cell Biol 18 : 5750–5761.
53. PakJ, SegallJ (2002) Role of Ndt80, Sum1, and Swe1 as targets of the meiotic recombination checkpoint that control exit from pachytene and spore formation in Saccharomyces cerevisiae. Mol Cell Biol 22 : 6430–6440.
54. RobineN, UematsuN, AmiotF, GidrolX, BarillotE, et al. (2007) Genome-wide redistribution of meiotic double-strand breaks in Saccharomyces cerevisiae. Mol Cell Biol 27 : 1868–1880.
55. XuL, KlecknerN (1995) Sequence non-specific double-strand breaks and interhomolog interactions prior to double-strand break formation at a meiotic recombination hot spot in yeast. EMBO J 14 : 5115–5128.
56. WuTC, LichtenM (1995) Factors that affect the location and frequency of meiosis-induced double-strand breaks in Saccharomyces cerevisiae. Genetics 140 : 55–66.
57. de MassyB, NicolasA (1993) The control in cis of the position and the amount of the ARG4 meiotic double-strand break of Saccharomyces cerevisiae. EMBO J 12 : 1459–1466.
58. XueY, RenJ, GaoX, JinC, WenL, et al. (2008) GPS 2.0, a tool to predict kinase-specific phosphorylation sites in hierarchy. Mol Cell Proteomics 7 : 1598–1608.
59. PadmoreR, CaoL, KlecknerN (1991) Temporal comparison of recombination and synaptonemal complex formation during meiosis in S. cerevisiae. Cell 66 : 1239–1256.
60. GeymonatM, SpanosA, JensenS, SedgwickSG (2010) Phosphorylation of Lte1 by Cdk prevents polarized growth during mitotic arrest in S. cerevisiae. J Cell Biol 191 : 1097–1112.
61. MurakamiH, BordeV, NicolasA, KeeneyS (2009) Gel electrophoresis assays for analyzing DNA double-strand breaks in Saccharomyces cerevisiae at various spatial resolutions. Methods Mol Biol 557 : 117–142.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2013 Číslo 6
Nejčtenější v tomto čísle
- BMS1 Is Mutated in Aplasia Cutis Congenita
- Sex-stratified Genome-wide Association Studies Including 270,000 Individuals Show Sexual Dimorphism in Genetic Loci for Anthropometric Traits
- Distinctive Expansion of Potential Virulence Genes in the Genome of the Oomycete Fish Pathogen
- Distinct Neuroblastoma-associated Alterations of Impair Sympathetic Neuronal Differentiation in Zebrafish Models