
RSR-2, the Ortholog of Human Spliceosomal Component SRm300/SRRM2, Regulates Development by Influencing the Transcriptional Machinery
Protein components of the spliceosome are highly conserved in eukaryotes and can influence several steps of the gene expression process. RSR-2, the Caenorhabditis elegans ortholog of the human spliceosomal protein SRm300/SRRM2, is essential for viability, in contrast to the yeast ortholog Cwc21p. We took advantage of mutants and RNA interference (RNAi) to study rsr-2 functions in C. elegans, and through genetic epistasis analysis found that rsr-2 is within the germline sex determination pathway. Intriguingly, transcriptome analyses of rsr-2(RNAi) animals did not reveal appreciable splicing defects but instead a slight global decrease in transcript levels. We further investigated this effect in transcription and observed that RSR-2 colocalizes with DNA in germline nuclei and coprecipitates with chromatin, displaying a ChIP-Seq profile similar to that obtained for the RNA Polymerase II (RNAPII). Consistent with a novel transcription function we demonstrate that the recruitment of RSR-2 to chromatin is splicing-independent and that RSR-2 interacts with RNAPII and affects RNAPII phosphorylation states. Proteomic analyses identified proteins associated with RSR-2 that are involved in different gene expression steps, including RNA metabolism and transcription with PRP-8 and PRP-19 being the strongest interacting partners. PRP-8 is a core component of the spliceosome and PRP-19 is the core component of the PRP19 complex, which interacts with RNAPII and is necessary for full transcriptional activity. Taken together, our study proposes that RSR-2 is a multifunctional protein whose role in transcription influences C. elegans development.
Published in the journal:
. PLoS Genet 9(6): e32767. doi:10.1371/journal.pgen.1003543
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1003543
Summary
Protein components of the spliceosome are highly conserved in eukaryotes and can influence several steps of the gene expression process. RSR-2, the Caenorhabditis elegans ortholog of the human spliceosomal protein SRm300/SRRM2, is essential for viability, in contrast to the yeast ortholog Cwc21p. We took advantage of mutants and RNA interference (RNAi) to study rsr-2 functions in C. elegans, and through genetic epistasis analysis found that rsr-2 is within the germline sex determination pathway. Intriguingly, transcriptome analyses of rsr-2(RNAi) animals did not reveal appreciable splicing defects but instead a slight global decrease in transcript levels. We further investigated this effect in transcription and observed that RSR-2 colocalizes with DNA in germline nuclei and coprecipitates with chromatin, displaying a ChIP-Seq profile similar to that obtained for the RNA Polymerase II (RNAPII). Consistent with a novel transcription function we demonstrate that the recruitment of RSR-2 to chromatin is splicing-independent and that RSR-2 interacts with RNAPII and affects RNAPII phosphorylation states. Proteomic analyses identified proteins associated with RSR-2 that are involved in different gene expression steps, including RNA metabolism and transcription with PRP-8 and PRP-19 being the strongest interacting partners. PRP-8 is a core component of the spliceosome and PRP-19 is the core component of the PRP19 complex, which interacts with RNAPII and is necessary for full transcriptional activity. Taken together, our study proposes that RSR-2 is a multifunctional protein whose role in transcription influences C. elegans development.
Introduction
RNA splicing is a highly conserved process in eukaryotes that transforms primary transcripts, or pre-mRNAs, into mature mRNAs through the removal of intronic sequences [1], [2]. This process is accomplished by the spliceosome, which is a large and dynamic RNA-protein complex. Components of the spliceosome include five small nuclear ribonucleoprotein particles (snRNPs) and a variable number of other protein factors (over 100 have been indentified) [3]. Genetic studies of these spliceosome factors have been hampered due to their essential functions. As a consequence, most of the functional information about these proteins comes from biochemical studies to the detriment of genetic approaches. The fact that spliceosome components are well conserved through evolution enables model organisms to be used to explore the functions of individual proteins of this macromolecular complex. The manageable genetics of C. elegans highlight this model organism as a powerful approach to the functional dissection of the elements of this sophisticated RNA-protein machine.
SR proteins are an evolutionarily conserved family characterized by an RNA recognition motif (RRM) and a region rich in arginine and serine dipeptides (RS domain) [4]. Human SRm300/SRRM2, yeast Cwc21p, and C. elegans RSR-2 are called “SR-related” proteins because they do not contain RRM motifs. RS domains are important for protein-protein interactions and are present in many splicing factors. However, the RS domain can also be found in other types of proteins such as chromatin modifiers and transcriptional regulators [5]. Different gene expression steps need to be interconnected for efficient performance, and SR proteins are important in such crosstalk because they have been associated with transcription-related activities, constitutive and alternative splicing, mRNA nuclear export, nonsense-mediated decay and mRNA translation [6], [7]. An increasing number of cis-acting and trans-acting splicing mutations affecting RNA processing have been implicated in human diseases [8]. In particular, modification of SR protein sequences or alteration of their target motifs may lead to several diseases including cancer [6].
Human SRm300/SRRM2 is a much larger protein than its yeast and worm orthologs (2296 vs. 135 and 425 amino acids, respectively) and contains a highly conserved N-terminal region that bears a cwf21 motif. In yeast and humans, this motif is known to be required for interaction with Prp8 (Figure 1A) [9]. The 150 N-terminal amino acids of SRm300/SRRM2 and RSR-2 share 50.6% similarity and 43.3% identity. The 159 N-terminal amino acids of human SRm300/SRRM2 can functionally substitute Cwc21 in yeast, supporting the functional conservation from yeast to humans [9]. SRm300/SRRM2 was initially identified by mass spectrometry as a protein associated with SRm160 in a complex that promotes pre-mRNA splicing [10]. However, specific immunodepletion of SRm300/SRRM2 does not prevent splicing of specific pre-mRNAs in HeLa cell splicing extracts, which have previously been shown to require the SRm160/300 complex [11]. In yeast, exhaustive functional analyses have shown that, although cwc21 mutants do not display pronounced defects in pre-mRNA processing, cwc21 may be involved in multiple splicing-related processes by acting redundantly with other genes such as Isy1, Ntc20 and Syf2 that encode components of the Prp19-associated complex (also known as NTC complex in yeast) [9], [12]. Recently, a novel function in transcription elongation, which is probably independent of its role in splicing, has been identified for the Prp19 complex [13].
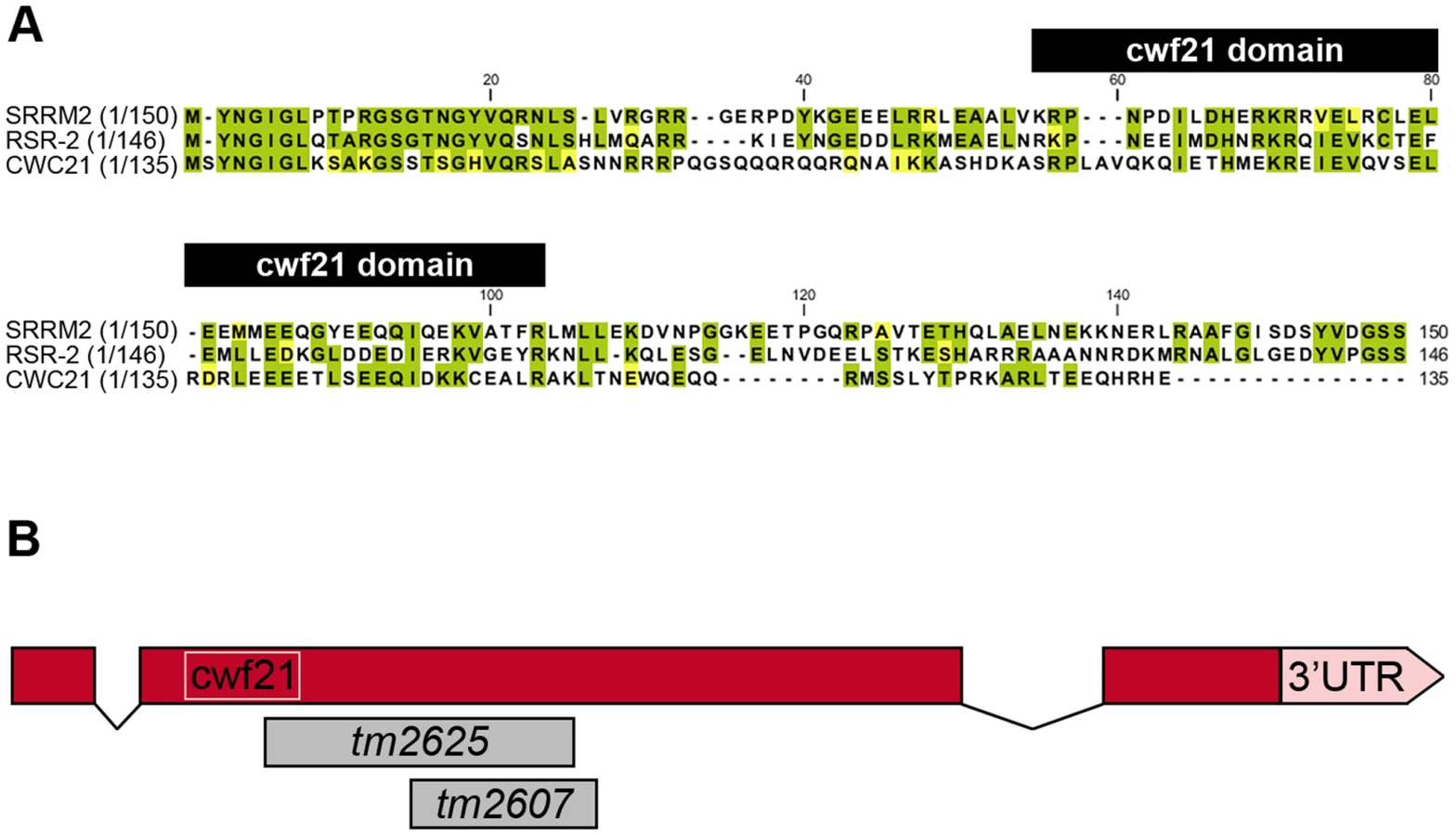
Although the SRm300/SRRM2 yeast ortholog cwc21 is not essential for viability, the inactivation of its C. elegans homolog rsr-2 by double-stranded RNA (dsRNA) microinjection produces larval-arrested animals [14]. Other specific phenotypes, including extra intestinal nuclei and synthetic genetic interactions, have been observed after inactivation of rsr-2 through RNAi by feeding [15]. Despite the widespread observation of rsr-2 RNAi phenotypes, a comprehensive approach to identifying rsr-2 functional mechanisms has not so far been made [14], [15].
The fact that splicing occurs cotranscriptionally expands the possibilities of components of the spliceosome influencing transcription and/or chromatin remodeling [16]. Several lines of evidence support the influence of splicing on transcription. For example: (i) there are splicing factors that associate with the RNA Polymerase II (RNAPII) C-terminal domain (CTD) to stimulate transcriptional elongation [17]–[19], (ii) RNAPII activity is higher in intron-containing than in intronless genes [20], (iii) 5′ splice sites stimulate transcription even in the absence of splicing [21], (iv) U1 snRNPs and the Prp19 complex are present in actively transcribed intronless genes [13], [16], [22], and (v) transcriptional regulators have been identified as components of the spliceosome [23].
In this study, we describe an rsr-2 mutation that, although informative, is disadvantageous for a genetic dissection of rsr-2 because it induces growth arrest at early larval stages. Therefore, we have devised an rsr-2 RNAi protocol to render a reliable and reproducible phenotype as a solid system in order to study rsr-2 functions taking genetic and genomic approaches, including epistasis analyses, tiling arrays, chromatin immunoprecipitation-sequencing (ChIP-Seq) and RNA-Seq. We complemented these studies with proteomic analyses to demonstrate that RSR-2, despite its interaction with spliceosome components, has an impact on the transcriptional machinery that is critical to the correct development of a multicellular organism.
Results
Investigating rsr-2 functions by mutations and RNAi
RNAi assays have shown that rsr-2 is essential for C. elegans development. rsr-2 RNAi approaches by microinjection or by feeding (which has a weaker effect) give rise to a variety of phenotypes, from larval and embryonic lethality to reduced brood size and sterility [14], [15], [24]. At our request, the Japanese National Bioresource Project (NBRP) generated two deletion alleles for rsr-2 (Figure 1B). After several backcrosses to remove other possible mutations, we found that alleles tm2607 (196 bp deletion+1 bp insertion) and tm2625 (337 bp deletion+2 bp insertion) produced viable animals and arrested larvae respectively. The allele tm2607 produced a truncated protein that was 65 amino acids shorter. Although several arginine and serine residues were eliminated, the truncation did not affect the cwf21 motif and the deleted region turned out not to be essential for RSR-2 functions (Figure S5B). In contrast to tm2607, the tm2625 mutation is a deleterious out-of-frame deletion/insertion affecting the cwf21 motif.
We characterized rsr-2(tm2625) mutants and determined that the larval arrest phenotype was not temperature-dependent. After five days of post-embryonic development, animals homozygous for the tm2625 mutation were approximately 0.3 mm long, while heterozygous rsr-2(tm2625) and homozygous rsr-2(tm2607) worms reached the standard adult size of 1 mm (Figure 2A, B). According to their size, tm2625 animals were arrested at the L1–L2 stage [25]. We next assessed whether cell differentiation occurred correctly in these mutants by using the MH33 antibody, a marker for intestinal cell differentiation. The rsr-2(tm2625) animals showed a dramatic defect in MH33 staining (Figure 2C), which was in agreement with the previous observation of a dysfunctional gut detected in the progeny of animals microinjected with rsr-2 dsRNA (Longman et al. 2001).

The acute larval arrest phenotype was difficult to track and dissect genetically. Therefore, we established an RNAi protocol to generate a population with a reproducible and milder phenotype. Synchronized L1 larvae fed at 25°C with bacteria producing rsr-2 dsRNA showed a partial depletion of rsr-2 mRNA levels in wild type and viable tm2607 animals, as revealed by semiquantitative reverse transcription PCR (RT-PCR) (Figure 2D). Wild type worms exposed to rsr-2 RNAi reached the adult stage, but these animals were sterile. Sterility was primarily caused by a defect in the sperm/oocyte switch producing worms without oocytes and with more sperm than present in wild type animals (Figure 2E). The same phenotype was observed in rsr-2(tm2607); rsr-2(RNAi) animals (data not shown). To exclude the possibility that this rsr-2 RNAi phenotype was caused by a general inhibition of splicing, we used the transgenic strain BL3466, which has an intron-containing GFP construct that is expressed in intestinal cells upon constitutive splicing [26]. While GFP levels were significantly reduced after RNAi-mediated knockdown of the heterogeneous nuclear ribonucleoprotein hrp-1 positive control, cat-1(RNAi) negative control and rsr-2(RNAi) did not affect GFP expression (Figure 2F). This experiment indicates that constitutive splicing is not severely altered in rsr-2(RNAi) worms.
rsr-2 regulates the germline sex determination pathway
To determine where rsr-2 functions within the sperm/oocyte decision pathway, we conducted epistasis experiments. We first used mutants for gld-3 and fog-1 that only generate oocytes and are positioned at the beginning and end of the germline sex determination pathway, respectively (Figure S1A) [27]. gld-3(q741); rsr-2(RNAi) worms only produced sperm displaying a masculinization of germline phenotype (Mog). In contrast, fog-1(q253); rsr-2(RNAi) worms developed only oocytes. Therefore, our RNAi assays indicated that rsr-2 is genetically downstream of gld-3 and upstream of fog-1, thereby confirming the presence of rsr-2 in the germline sex determination pathway. We next investigated the genetic interaction of rsr-2 with other components of the pathway (Table 1) and found that rsr-2 is mostly upstream of fem-3 and cooperates with fbf-1, fbf-2, nos-3 and puf-8 in the control of the sperm/oocyte switch.

The existence of genes such as fem-3, which regulates the germline and the somatic sex determination pathways (Figure S1B) [28], as well as the ubiquitous expression of rsr-2 in somatic cells, encouraged us to examine whether rsr-2 could also be involved in the somatic sex determination pathway. To address this question, we used the somatic expression of a lacZ reporter transgene controlled by the fem-3 3′UTR [29]. Whereas control animals carrying the lacZ::fem-3 3′ UTR did not express the reporter, rsr-2 RNAi induced strong lacZ expression (Figure S2A). However, rsr-2 RNAi did not cause lacZ expression in lacZ::tra-2 3′ UTR transgenic animals, indicating the specificity of RSR-2 in regulating 3′ UTRs (Figure S2B). Therefore, rsr-2 acts upstream of fem-3 in both somatic and germ cells.
During germline development, another cell fate decision takes place prior to the spermatogenesis/oogenesis switch: the transition of germ cells from a proliferating state into meiosis. If this switch fails, cells continue proliferating, resulting in a tumorous germline. To investigate whether rsr-2 regulates this process, we first injected rsr-2 dsRNA to induce a stronger depletion of RSR-2 during development, but rsr-2(RNAi) escapers that reached the adult stage were Mog sterile animals and did not have tumorous germlines. Moreover, using genetic analysis and mitotic markers, we found that rsr-2 RNAi did not seem to affect the mitosis/meiosis transition (Figure S3). Therefore, these findings suggest that rsr-2 is essential for some, but not all, developmental processes in the germline.
Tiling arrays indicate that rsr-2(RNAi) L4 worms have slightly lower global transcript levels while splicing remains unaltered
To shed light on the molecular functions of rsr-2 during C. elegans development, we used Affymetrix tiling arrays to examine not only transcript levels, but also intron retention events. We purified total RNA from synchronized L4 animals, when the sperm/oocyte switch takes place, grown on gfp and rsr-2 dsRNA-expressing bacteria. We used two biological replicates for each condition, and raw data (in CEL files) were analyzed using the Tiling Analysis Software (TAS) developed by Affymetrix. This analysis provided information about the levels of 30,430 transcripts. To estimate gene expression in gfp(RNAi) and rsr-2(RNAi) animals, we plotted mean signal intensities for transcripts, exons and introns by chromosomes (Figure 3A). These analyses indicated that the rsr-2 RNAi produced a slight overall reduction in transcript, exon and intron levels in all chromosomes, with the exception of the X chromosome. Mean signal intensities and normalized values for transcripts, exons and introns are shown in Table S1.

In order to obtain more information regarding the downregulated genes that cause the reduction of transcript intensity signals in rsr-2(RNAi) animals, we selected genes that had expression levels 20% higher or lower than the control. Using these criteria, we identified 1726 transcripts (from 1540 genes) that were upregulated and 2453 transcripts (from 2208 genes) that were downregulated. These two gene lists were uploaded to modMine (http://intermine.modencode.org), a web resource that assesses the intersection of genes of interest with gene classes generated by other studies [30]. We did not associate upregulated genes with any gene class, but we found that our list of downregulated genes was enriched in germline genes, particularly those involved in spermatogenesis (Figure S4). The decrease in transcripts required for spermatogenesis suggests that, although rsr-2(RNAi) animals produce an excess of sperm, these may not be properly differentiated at the developmental time when the experiment was performed. The fact that sperm-enriched genes are almost entirely absent from chromosome X [31] may explain why rsr-2(RNAi) worms had lower transcript levels in all chromosomes except the X chromosome.
Since our initial functional analysis of rsr-2 was focused on germline sex determination, we performed quantitative RT-PCR (qRT-PCR) for some components of this pathway. In accordance with our tiling arrays, where we found a trend towards downregulation among germline genes, all of the tested genes with the exception of fog-1 (enriched in sperm) [32] showed a reduction of mRNA levels in rsr-2(RNAi) compared with gfp(RNAi) control animals (Figure 3B). We next examined whether the lower level of expression of germline sex determination genes in rsr-2(RNAi) animals was caused by mechanisms other than splicing. Unspliced transcripts, which often contain premature termination codons (PTCs) in intron sequences, are the target of nonsense-mediated decay (NMD) degradation [33]. Consequently, if NMD is operating we would not be able to detect whether rsr-2 RNAi causes a defect in splicing. Thus, we performed semiquantitative RT-PCR of germline transcripts from smg-5(r860); rsr-2(RNAi) animals, in which smg-5 mutants have a defective NMD pathway [34]. We detected abnormal transcripts in our positive control, but did not observe aberrant transcripts for any of the germline-tested genes (Figure 3C).
In conclusion, our rsr-2 RNAi protocol partially inactivates rsr-2, blocking the developmental switch from spermatogenesis to oogenesis. Such rsr-2 inactivation produces a general decrease in transcript levels that cannot be explained by altered constitutive splicing, which suggests that rsr-2 may regulate the gene expression of processes other than splicing.
rsr-2 is ubiquitously expressed in somatic cells but presents a restricted pattern in the germline
We next generated transgenic strains that enabled for the cellular expression of rsr-2 and subcellular location of RSR-2 to be visualized [35], [36]. Given that extrachromosomal repetitive arrays are silenced in the germline, the transgene rsr-2 promoter::GFP::Histone2B::rsr-2 3′UTR was linearized and microinjected together with digested genomic DNA to make complex arrays, which gave rise to germline expression for several generations. Since most of the gene expression regulation in the germline occurs through post-transcriptional repression [37], we included the rsr-2 3′UTR region in this molecular construct. This transgene showed widespread expression of rsr-2 in somatic lineages but a restricted pattern in the germline in which the GFP signal was low or absent in the most distal part where mitosis takes place (Figure 4A). In addition, we corroborated the exclusion of rsr-2 expression from the distal end of the germline at mRNA level by in situ hybridization (Figure 4B).

In order to analyze the subcellular localization of RSR-2, we generated a transgenic strain carrying the construct cerEx04[rsr-2 promoter::GFP::rsr-2 genomic fragment (exons, introns and 3′UTR)] using gene bombardment. These transgenic animals showed clear nuclear localization of RSR-2 (Figure 4C). Similar to other splicing factors [38], GFP::RSR-2 forms nuclear speckles that vary in number and size depending on the cell type (Figure 4D).
Taken together, the expression data indicate that rsr-2 is expressed ubiquitously in somatic cells, and that RSR-2 is localized in the nucleus and concentrated in speckles. However, the lack of ubiquitous rsr-2 expression in the germline, where different cellular processes occur at different locations, is intriguing.
RSR-2 associates with chromatin and interacts with RNAPII affecting its distribution and phosphorylation states
We generated a rabbit polyclonal antibody against RSR-2 (Q5092) and confirmed its specificity by western blots and in immunofluorescence assays (Figure S5A and S5C). Interestingly, anti-RSR-2 staining overlapped with chromatin as indicated by confocal images of germ nuclei (Figure 4E). We next performed ChIP-Seq experiments to investigate the association of RSR-2 with chromatin at the L4 stage using RNAPII as a control. Surprisingly, the anti-RSR-2 antibody was capable of precipitating chromatin in a ChIP-Seq pattern that resembled that of anti-RNAPII (8WG16) (Figure 5A and S10). As expected, RSR-2 peaks disappeared in worms treated with rsr-2(RNAi) (Figure 5A).

Given the similarities between RSR-2 and RNAPII regarding chromatin occupancy, we next wondered whether reduced levels of RSR-2 could influence RNAPII distribution and activity. As a consequence of knocking down rsr-2 by RNAi, the shape of the RNAPII ChIP-Seq profile changed in many loci throughout the genome (Figure S10). We validated and quantified this change by ChIP-qPCR of distinct genomic fragments of the gene phy-2, which is downregulated in rsr-2(RNAi) L4 and L3 animals (Figure 5B, Table S1 and S3). Moreover, we analyzed the distribution of peaks along an averaged gene model using the SeqSolve program, employing the MACS method for peak calling [39]. This gene model was subdivided into five regions according to the peak position with respect to the closest transcription start site (TSS). The occupancy of RSR-2, compared with that of RNAPII, was greater at promoters and TSS zones and lower within genes (Figure 5C). After RNAi-mediated knock-down of rsr-2, RNAPII peaks were slightly enriched at the 5′ region (promoters and TSSs) and reduced at the 3′ end (peaks containing PolyA) (Figure 5C). Even after considering the noise generated from investigating different cell types in a population of L4 worms, our data suggest that, globally, rsr-2 RNAi modified the distribution of RNAPII along the genes. This modification of RNAPII distribution may affect the transcriptional cycle and result in reduced mRNA production.
To explore further the influence of RSR-2 on RNAPII efficiency we examined whether rsr-2 RNAi could alter the phosphorylation of RNAPII, which is informative of its activity. The C-terminal domain (CTD) of RNAPII consists of multiple repeats of a heptamer sequence that presents three serine residues (Ser-2, Ser-5 and Ser-7), which are subjected to reversible phosphorylation during transcription. In the canonical RNAPII cycle, the CTD is hypophosphorylated at the promoter and becomes phosphorylated at Ser-5 upon the initiation of transcription. As RNAPII proceeds in the 5′-to-3′ direction, phosphorylated Ser-5 (P-Ser-5) decreases and Ser-2 (P-Ser-2) increases, favoring the recruitment of 3′-end processing factors [40]. We used the anti-RNAPII antibody 8WG16, which binds to both hyperphosphorylated and hypophosphorylated isoforms [41], [42], and found that rsr-2(RNAi) extracts have more hyperphosphorylated RNAPII than control gfp(RNAi) extracts as assayed by western blot (Figure S6). We validated this observation by using a different RNAPII antibody (N-20), which also recognizes the two phosphorylation states (Figure 6A). This result could be contradictory since rsr-2 RNAi produces an increment of active RNAPII and, at the same time, a slight global decrease in the number of transcripts. However, as argued in the discussion, an increment of active RNAPII does not always correlate with more efficient transcription.

Finally, we investigated whether RSR-2 and RNAPII can interact by using a coimmunoprecipitation assay (CoIP). We performed CoIP and found that both Ser-2 and Ser-5 RNAPII phosphorylated forms are able to coimmunoprecipitate with RSR-2. It seems that the interaction between RSR-2 and P-Ser-5 has a stronger binding affinity than that between RSR-2 and P-Ser-2 (Figure 6B), which supports the observed biased abundance of RSR-2 towards the 5′ end of genes. Together, these observations suggest that RSR-2 may be involved in the regulation of transcription based on the findings that (i) RSR-2 physically interacts with RNAPII, (ii) RSR-2 associates with chromatin with a pattern similar to that of RNAPII, and (iii) the reduction of RSR-2 levels modifies RNAPII distribution and the balance of RNAPII phosphorylated forms.
Splicing-independent recruitment of RSR-2 to transcriptionally active genes
Our ChIP-Seq analyses identified 6889 RNAPII peaks in 4679 genes, and 5445 RSR-2 peaks in 4338 genes (p-value cutoff 0.00001) (Table S2). Most of these peaks were present in both RSR-2 and RNAPII ChIP-Seq profiles (Figure S7). Interestingly, 74 of the RSR-2 peaks were located in intronless genes (551 intronless genes identified in the WS220 version of the genome). This finding indicates that the recruitment of RSR-2 to chromatin can be independent of splicing. Since the C. elegans genome is quite compact, we scanned the Genome Browser for RSR-2 peaks located at single exon genes that did not have genes expressing multi-exonic transcripts in the immediate vicinity (Figure 7A). To further validate these peaks we performed ChIP-qPCR along several intronless genes and concluded that both proteins, RNAPII and RSR-2, bind to intronless genes at regulatory and coding sequences (Figure 7B).

RSR-2 interacts with proteins related to splicing and transcription
We next immunopurified RSR-2-containing complexes from two biological replicates of wild type mixed-stage worm populations. Most of the proteins identified as RSR-2 interactors were components of the spliceosome, but we also detected proteins related to stress response, transcription, chromatin regulation, translation and ubiquitination (Table 2 and S4). A large number of peptides of the core spliceosome protein PRP-8 were identified, which is consistent with the physical interaction found between Cwc21p and Prp8p in yeast [9], [43]. Moreover, we detected interactions with snRNPs, which have also been observed in its human counterpart SRm300/SRRM2 [44]. Interestingly, we identified three proteins that are functionally related to both splicing and transcription: SKP-1, KIN-3, and PRP-19. SKP-1 is the ortholog of PRP45 in yeast and SKIP/SWN1 in humans. It has previously been shown that SKIP/SWN1 plays independent roles in splicing and transcription elongation [45]. KIN-3 encodes the ortholog of the human catalytic subunit of CKIIα, which is a protein kinase that phosphorylates several substrates regulating transcription and physically interacts with the splicing factor PRPF3 [46]. Finally, PRP-19 is the ortholog of human PRPF19, and the conserved Prp19 complex (Prp19C) is involved in pre-mRNA processing and transcriptional efficiency [13].

We expanded our proteomic analysis by performing two-dimensional electrophoresis (2-DE) and mass spectrometry analysis of one differential spot between a sample immunoprecipitated with anti-RSR-2 and a control sample (non-specific rabbit immunoglobulin) (Figure 8). Interestingly, we found 22 unique peptides corresponding to PRP-19, which validated its interaction with RSR-2. Thus, our proteomic analyses of RSR-2-containing complexes support the hypothesis that RSR-2 is part of the spliceosome but has the capacity to influence the transcriptional machinery through several protein-protein interactions.

Convergences and divergences between rsr-2 and prp-8
Since PRP-8 interacts with RSR-2 and is a protein located at the catalytic core of the spliceosome, we were interested in comparing the transcriptomes of rsr-2(RNAi), prp-8(RNAi) and control gfp(RNAi). We conducted RNA-Seq analysis in L3 animals because prp-8(RNAi) animals exhibited a severe developmental delay after L3 in our RNAi feeding conditions. Interestingly, there was a substantial number of transcripts (much more than expected by chance, 131 vs. 7 respectively) that were significantly downregulated (q≤0.05) in both rsr-2 and prp-8 deficient animals (Figure 9A) (Table S3). However, the overlap among upregulated genes was meaningless (Figure 9A). These results suggest common functions for RSR-2 and PRP-8 in controlling gene expression. Therefore, we further analyzed the common list of transcripts downregulated in prp-8 and rsr-2(RNAi) animals using modMine [30]. We found gene ontology (GO) terms for “body morphogenesis” and “molting cycle”, which included genes with high expression levels during postembryonic development.

Sequencing analyses revealed that prp-8(RNAi) transcriptomes accumulated more reads in introns than rsr-2 and gfp(RNAi) samples (Figure 9B). Therefore, using semiquantitative RT-PCR, we investigated the retention of introns, in wild type N2 animals and non-mediated decay (NMD) mutants, for two of the genes downregulated in both rsr-2 and prp-8(RNAi) animals (Figure 9C). We observed intron retention events in prp-8(RNAi) but not in rsr-2(RNAi) samples. Thus, we concluded that although both PRP-8 and RSR-2 are part of the spliceosome, after partial inactivation through RNAi the contribution to splicing catalysis was only obvious for PRP-8. Nevertheless, we cannot rule out the possibility that the full depletion of RSR-2 activity could somehow affect pre-mRNA processing.
We next assessed the possible effect of rsr-2 RNAi in alternative splicing by evaluating the predominance of spliced forms from our RNA-Seq data. We studied ten genes that have been reported to present alternative splicing at the L3 stage [47] and did not observe drastic variation in the balance of these alternative spliced transcripts (Figure S9). However, we cannot rule out the possibility that a stronger depletion of RSR-2 levels could favor one particular transcript that influences worm development.
Taken together, we hypothesize that during development, mild rsr-2 and prp-8 deficiencies may primarily affect highly transcribed genes. Nevertheless, the involvement of defects in splicing or in transcription in the eventual reduction of gene expression requires further exploration.
Discussion
In recent decades, our functional knowledge of spliceosome components has largely been driven by biochemical studies, because genetic analyses have been hampered by the essential functions of these proteins. The discovery of RNAi and the possibility of modulating its efficacy allowed for the mimicking of phenotypes caused by hypomorphic alleles. This study shows that reduced levels of C. elegans RSR-2 blocks the switch from spermatogenesis to oogenesis. Since RSR-2 is a homolog of the human splicing coactivator SRm300/SRRM2 and the yeast cwc21 spliceosome component, it was expected that rsr-2 inactivation would primarily affect the splicing process. However, we did not find appreciable alterations in constitutive splicing in animals with reduced levels of RSR-2. Instead, we observed a slight global reduction in the abundance of transcripts at L4 when the sperm/oocyte switch takes place. To explain the rsr-2(RNAi) phenotype in germline sex determination we propose a model in which low RSR-2 levels cause a decrease in the expression of genes, both activators and repressors, within the complex gene network governing the sperm/oocyte switch. The outcome of such global downregulation is that the abundance of FEM-3 protein is not low enough to trigger the switch to oogenesis (Figure S8). Grades of rsr-2 inactivation and the consequent reduction of the transcriptional levels required to manifest a phenotype may vary depending on each developmental process. Our study suggests that an RSR-2 deficit may more strongly affect processes driven by the action of high levels of transcripts at certain developmental times, such as during germline sex determination at the L4 stage or cuticle formation at the L3 stage. A parallelism could be established with the effect of hypomorphic mutations of certain splicing factors that cause the rare autosomal dominant human disease Retinitis Pigmentosa (adRP). adRP patients suffer a pathology of the retina, which is a tissue with a high transcriptional activity in adults, whereas other tissues are unaffected.
Interestingly, mog genes are a group of splicing-related genes that are also required for the sperm-to-oocyte switch [2], [48]–[52]. There are similarities and differences between mog genes and rsr-2, the former of which includes: (i) RSR-2 and MOG proteins are nuclear, (ii) rsr-2 and mog are required for somatic repression of a lacZ::fem-3 3′UTR transgene [29], (iii) mog genes and rsr-2 are located upstream of fem-3 in the sex determination pathway [27], (iv) mog mutants, similar to rsr-2(RNAi) animals, do not show general splicing defects [50], [52]–[54], (v) we have detected a physical interaction between MOG-2 and RSR-2 (Table 2) and (vi) we have shown a link between RSR-2 and transcription, and mog genes interact with the zinc-finger protein MEP-1 [27], [55]. However there are differences that set rsr-2 apart from mog genes, including: (i) most of the MOG proteins are DEAD/H-box RNA helicases, whereas RSR-2 lacks motifs with the capacity to bind or rearrange RNA, (ii) in contrast to rsr-2, all mog genes have a role in germline proliferation and function synthetically with gld-3 [48], (iii) a restricted expression pattern in the germline is unique for rsr-2, and (iv) rsr-2(RNAi) at 20°C in the rrf-1(pk1417) background, which confers RNAi resistance in the soma, produced 40% of Mog animals (data not shown) while RNAi of mog and other splicing-related genes produce much lower proportion of Mog worms under the same conditions [56]. Therefore, the similarities and differences between rsr-2 and mog genes suggest that they may share some functions but also have independent roles.
RNA processing can occur before transcription is completed, so both molecular mechanisms need to be functionally coupled to ensure efficient gene expression [57], [58]. Proteomic analyses of complexes containing RSR-2 and its orthologs have identified not only pure splicing factors, but also proteins involved in chromatin remodeling and transcription [9], [12], [44], [59]. Due to the recently identified role of the PRP19 complex in transcriptional elongation, the physical interaction of RSR-2 and its yeast and human orthologs with components of PRP19 complexes [9], [12], [44], [60] are particularly helpful for explaining the participation of RSR-2 in transcription proposed in this study [13]. Such interactions would also be functional because the yeast PRP19 complex components Isy1 and Cwc21 are functionally redundant [9], [43]. Moreover, the PRP19 complex, similar to RSR-2, is recruited to intron-containing as well as intronless genes in a transcription-dependent manner [13]. In addition, the physical interaction between RSR-2 and the worm orthologs of human CKIIα and SKIP/SWN1, which are implicated in transcriptional elongation [18], [45], [61], further reinforces the nexus between the RSR-2 interactome and regulation of transcriptional efficiency.
Beyond these interactions of RSR-2 with proteins playing roles in transcription, our study reveals that RSR-2 coimmunoprecipitates with RNAPII. The interaction between RSR-2 and RNAPII is not rare since some splicing factors directly interact with the C-terminal tail (CTD) of the polymerase [62]. Moreover, reduced levels of RSR-2 have an impact on global RNAPII distribution and phosphorylation. The increase of the active hyperphosphorylated RNAPII form observed after rsr-2 RNAi does not necessarily result in more efficient transcriptional activity. During animal development RNAPII often stalls or pauses just after the initiation of transcription because the accumulation of transcriptionally engaged RNAPII close to the TSS is a strategy for regulating the expression of genes in stimulus-responsive pathways and of regulators in signaling pathways [41], [63], [64]. Moreover, induced DNA damage causes CTD hyperphosphorylation beyond normal levels, and this hyperphosphorylation is associated with slow transcription [65].
This study has shown that RSR-2 is committed to the two largest nuclear gene expression machineries, its subcellular location being compatible with this. RSR-2 distribution is mainly nuclear: it is present in nuclear speckles or clearly overlaps with DNA as in the case of the germ cells. Human SRm300/SRRM2 is also nuclear and localizes to nuclear speckles in a range of human cells types [11], [66], [67]. Although several RNA processing factors have been detected in speckles of this type, transcriptional factors are also detected within such structures [38]. In fact, although nuclear speckles are commonly known as RNA processing bodies, they are often in the close vicinity of transcriptionally active chromosome territories [68].
Although this report is more concerned with the evidence for a new role of RSR-2 in transcription, we cannot rule out the possibility that the total absence of RSR-2 might somehow affect pre-mRNA processing. We also cannot discount the possibility that RSR-2 could play a potential role in alternative splicing, since transcriptional elongation can affect the outcome of alternative splicing events [69]. However, rsr-2(RNAi) animals did not show any evident alteration in the expression of alternative splice forms.
Some of our results relate rsr-2 with prp-8, which is a gene encoding a protein located at the catalytic core of the spliceosome. First, our proteomic studies revealed an interaction between RSR-2 and PRP-8. Second, using permissive RNAi conditions (feeding from L2–L3 stage) to inactivate prp-8 and F53B7.3 (Isy1 yeast homolog) genes, we observed the Mog phenotype (data not shown). Third, RNA-Seq revealed that PRP-8 and RSR-2 have target genes in common, which are downregulated in prp-8 and rsr-2(RNAi) animals. However, an excess of intron retention was only observed in prp-8(RNAi) worms. There are two hypotheses to explain why prp-8 and rsr-2 regulate the expression of a similar subset of genes. First, rsr-2 RNAi may affect splicing by producing immature transcripts that are undetectable in our experimental conditions (because RNA-Seq coverage is insufficient or semiquantitative RT-PCR is unable to detect low levels of immature transcripts). The second possibility is that partial prp-8 inactivation may cause a defect in transcription as a consequence of the functional coupling between the splicing and transcriptional machineries. In other words, a defect in prp-8 function might compromise splicing efficacy and transcription may respond to such a drawback by downregulating its activity. Thus, slowing down the active RNAPII would provide sufficient time for the spliceosome to rectify the problem and avoid the accumulation of immature transcripts.
Many splicing factors are essential genes that do not have functional redundancies that can cover their loss of function. It is possible that, as demonstrated in cells, during animal development splicing presents a buffering capacity due to the coupling with transcription to deal with partial loss of function of some spliceosome components.
Our data fit a model in which RSR-2 is a component of both the spliceosome and the RNAPII complex, but where a partial reduction of RSR-2 levels has a greater impact on transcriptional efficiency than on pre-mRNA processing. Importantly, the effect on transcription seems to be critical for C. elegans development and viability. The use of nuclear run-on assays and ChIP experiments with antibodies specific to various RNAPII forms in C. elegans will aid our understanding of how RSR-2 regulates transcription of genes [70], [71]. Since rsr-2 was identified as a genetic interactor of lin-35 Retinoblastoma [15], another aspect that needs to be explored is the impact of rsr-2 inactivation on chromosome modifications and nucleosome positioning due to the functional connection between chromatin, splicing and transcription [72].
Materials and Methods
Strains
Standard methods were used to culture and manipulate worms [73]. We used the wild type strain Bristol N2 and the following alleles and balancers: fog-1(q253) I, smg-5(r860) I, smg-1(r861) I, rrf-1(pk1417) I; glp-1(oz264) III, rrf-1(pk1417) I; gld-3(q730) II, rsr-2(tm2625)/mIn1[(dpy-10(e128)mIs14(myo-2::GFP)] II, rsr-2(tm2607) II, gld-3(q741)/mIn1[dpy-10(e128)mIs14(myo-2::GFP)] I, fbf-1(ok91) II, fbf-2(q738) II, nos-3(q650) II, puf-8(ok302) II, puf-8(q725) II, pha-1(e2123) III, fem-3(e2006) IV, and fog-2(q71) V. We also used the following transgenes: qIs43[lacZ::fem-3(+)], qIs15[lacZ::fem-3(q96gf)], wt8Di[lacZ::tra-2(+)], inIs173[pNvitgfp], cerEx01[rsr-2 promoter::gfp::Histone2B::rsr-2 3′UTR], and cerEx04[rsr-2 promoter::gfp::rsr-2 genomic fragment+rsr-2 3′UTR].
RNA-mediated interference (RNAi)
To induce RNA-mediated interference (RNAi) by feeding, nematode growth medium (NGM) plates were supplemented with 50 µg/ml ampicillin, 12.5 µg/ml tetracycline, and 1 mM IPTG. Plates seeded with the corresponding RNAi clone, validated by PCR and/or sequencing, from either ORFeome library [24] or Ahringer library [43] were used to feed wild type synchronized L1 animals. To interfere rsr-2 expression by microinjection, dsRNA was synthesized by using MEGAscript T7 kit (Ambion Cat. No. AM1333). Wild type young adults were injected with 1 ng/µl of rsr-2 dsRNA and F1 progeny was grown at 25°C.
Semiquantitative RT-PCR
Synchronized L4 worms were harvested [25], washed and frozen in TRIReagent (MRC Inc, Cat. No. TR-118) to perform total RNA isolation. cDNA was synthesized with oligo(dT) primers using the RevertAid H Minus First Strand cDNA synthesis kit (Fermentas Cat.No. K1632) following the manufacturer's instructions.
In situ hybridization of mRNA
In situ hybridization was performed mainly following the protocol described by Lee & Schedl [74], although gonad dissection, washes and incubations were performed on a multi-well Pyrex plate (Electron Microscopy Sciences). A sense probe was used as a negative control.
Immunostaining
Dissected gonads were fixed with 4% paraformaldehyde (Electron Microscopy Sciences Cat. No. 15710) and processed following standard immunostaining protocols. We used the freezing-cracking protocol for larval immunostaining [75]. The RSR-2 antibody was raised in rabbits against the immunogen sequence of the RSR-2 protein, which comprises aminoacids 39 to 138, and was affinity purified (Sdix, Strategic Diagnostics Inc. USA). Antibodies and dilutions used for immunofluorescence in this study were: anti-RSR-2 (Q56092) 1∶600; anti-GFP (A11120) (Molecular Probes) 1∶100; anti-intermediate filament (MH33) (DSHB) 1∶40; Alexa Fluor 488 IgG (Molecular Probes) 1∶400; and Alexa Fluor 568 IgG (Molecular Probes) 1∶400.
Generation of GFP reporters and transgenic animals
Molecular constructs were made by the Gateway recombinational cloning system (Invitrogen). Transgenic animals were generated by microinjection or bombardment with gold particles (Biolistic Helium Gun, Caenotec). Bombardment: N2 young adults were shot with 16 ng/µl of the reporter cerEx04. Transgenic animals from the progeny were selected through the puromycin/neomycin selection system [76]. Microinjection: 4 ng/µl of the linearized molecular construct cerEx01 were microinjected together with digested bacterial genomic DNA (80 ng/µl) and linearized pRF4 (2 ng/µl) (roller marker) to make complex arrays.
LacZ reporter assay
Animals carrying different lacZ::3′UTR reporter transgenes were synchronized at the L1 stage and fed with RNAi clones at 25°C. Adults were heat shocked for 2 hours at 30°C, recovered for 2 hours at 25°C, and processed to score for lacZ expression. Processing of samples and scoring was performed as described by Gallegos et al. [29].
Tiling arrays
Wild type N2 hermaphrodites were synchronized at the L1 stage [25] and total RNA from rsr-2 and gfp(RNAi) worms grown at 25°C was isolated 36 hours after L1 arrest. cDNA was synthesized and hybridized to Affymetrix C. elegans tiling array 1.0 (ref 900935). Affymetrix Tiling Analysis Software (TAS) V.1.1.02 was used to analyze the data. Briefly, the array signals were extracted comparing the mismatch probe values (MM) with their perfect match probes (PM). Then, the biological replicates were quantile-normalized and smoothed by a sliding window procedure (window size of 110 bp), and signals were scaled to a median intensity of 100. To detect regions of active transcription, interval analyses were made to obtain the corresponding transfrags, using in the algorithm a maxgap of 30 bp and a minrun of 50 bp for all data sets. Finally, using the tools implemented in the Galaxy platform [77], [78], and based on the information contained in the worm genome assembly WS180, the array signal values were used to infer the mean signal intensity for each gene in control and rsr-2(RNAi) worms, as an indirect estimate of whole gene expression. These estimates were used to recognize up - and down-regulated genes in the knocked-down population, and also to characterize the transcription patterns of each chromosome.
Real-time PCR assays
For RNA analysis, total RNA was isolated and cDNA synthesized with oligo(dT) primers. For DNA analysis, chromatin was immunoprecipitated and purified as detailed later in this section. In both cases, a LightCycler 480 SYBR Green I Master kit (Roche Cat. No. 04707516001) was used to determine gene expression or the amount of immunoprecipitated DNA respectively.
Protein analysis
Western blots were performed according to standard protocols (the detailed protocol can be found in the supporting Text S1). Antibodies and dilutions used were: anti-RSR-2 Q5092 (Sdix) (1∶500), anti-RNAPII N-20 (Santa Cruz Biotech) (1∶1000), phospho Ser-2 RNAPII A300-654A (Bethyl Laboratories, Inc) (1∶5000), phospho Ser-5 RNAPII A300-655A (Bethyl Laboratories, Inc) (1∶5000) and anti-ACTIN C4 (MP Biomedicals) (1∶500).
Chromatin immunoprecipitation - sequencing (ChIP-Seq)
ChIP assays were carried out as described previously by Zhong et al., with minor modifications [42]. Worms were collected at the L4 stage and 0.5 ml of packed larvae (≈2.2 mg of total protein) were crosslinked, sonicated and immunoprecipitated using anti-RSR-2 (Q5092) and anti-RNAPII (8WG16) antibodies (10 µg of each antibody). The immunoprecipitated DNA fragments and the input DNA were used to prepare libraries for sequencing using the Illumina GA platform, through the modENCODE sequencing facility set up in the Snyder laboratory (Stanford University). In order to run four samples in one flow cell, sequencing libraries were bar-coded and multiplexed as described in Lefrançois et al. [79]. ChIP-Seq fastq files were processed in Galaxy and mapped against the C. elegans WS220 genome version to generate SAM files. SAM files were converted to BAM files also in Galaxy [77]. Binding peaks from ChIP-Seq data (BAM files) were called using SeqSolve software, which uses the MACS algorithm to identify ChIP peaks [39].
RNA sequencing
Wild type synchronized L1 worms [25] were fed on rsr-2, prp-8 and gfp dsRNA-expressing bacteria at 25°C. After 26 hours, the three populations were harvested and frozen with TRIReagent. Total RNA was purified, including small RNAs, using the mirVana miRNA isolation kit (Ambion). Ribosomal RNA was depleted with the RiboMinus Eukaryote Kit (Invitrogen). RNA quality and integrity were evaluated with the Experion Bioanalyzer (Bio-Rad). Libraries for sequencing were made by using the Illumina TruSeq RNA Sample Preparation Kit. These libraries were run through a Genome Analyzer IIx Ultrasequencer (Illumina). Each sample yields more than 10 millions reads. The resulting fastq files were trimmed and mapped to the version WS225 of the C. elegans genome by using TopHat to generate BAM files that were analyzed in SeqSolve under the default settings (false discovery rate of 0.05). Reads displaying multiple mapping were filtered out. SeqSolve was used to perform a differential transcript expression analysis between rsr-2(RNAi) and gfp(RNAi) samples, and between prp-8(RNAi) and gfp(RNAi) samples. Transcripts covered by more than 5 reads were tested. This analysis uses Cufflinks/Cuffdiff [80] to quantify and identify transcripts with a significant level of expression between different conditions. In this analysis, expression values were normalized in FPKM (fragments per kilobase of exon per million fragments mapped). To analyze intron retention, a file containing intron sequences (excluding those introns with internal genes or ncRNAs) was used as a reference genome.
Immunoprecipitation
N2 worms were recovered from plates and washed three times with M9 buffer and twice with a buffer containing 50 mM HEPES-KOH pH 7.6, 1 mM EDTA, 140 mM KCL, 0.5% NP40 and 1% glycerol. Worms resuspended in the same buffer (proteases and phosphatases inhibitors added) were added dropwise into liquid nitrogen and worm-pearls were ground to a fine powder in liquid nitrogen using a pestle and mortar. Worm powder was thawed on ice and lysate passed through a 20-gauge syringe prior to a 2-minute centrifugation at 16,000 g and 4°C. Protein was quantified and immunoprecipitated with the Invitrogen immunoprecipitation kit (143-21D) applying minor modifications (see supporting Text S1 for details). For western blot, samples were resuspended in Laemli buffer and boiled for 5 minutes at 95°C. For proteomic assays, complexes were eluted and precipitated overnight with cold acetone. Antibodies used were: unspecific rabbit IgG (Santa Cruz sc-2027), anti-RSR-2 Q5092 (Sdix), phospho Ser-2 RNAPII A300-654A and phospho Ser-5 RNAPII A300-655A (Bethyl laboratories Inc).
Two-dimensional electrophoresis, silver staining and peptide digestion
Protein pellet samples were resuspended in rehydration buffer (7 M urea, 2 M thiourea, 2% CHAPS, DTT 80 mM, 0.5% IPG pH 3–10 buffer (GE)), brought up to a final volume of 130 µl, and loaded onto Immobiline DryStrip pH 3–10 7 cm (GE). Second-dimension electrophoresis SDS-PAGE was performed with a 7.5% polyacrylamide gel for 75 minutes at constant voltage (100 V; Mini Protean Hoefer system, GE). Gels were stained with a modified short silver nitrate stain (2), scanned with a GS 8000 Calibrated Densitometer (Bio-Rad), and the images obtained were processed and analyzed with Quantity One (Bio-Rad) image analysis software. Spots of interest were selected, manually excised from gels and subjected to in-gel digestion.
MudPIT
Precipitated proteins were dissolved in 50 µl of resuspension buffer (6 M urea, 200 mM ammonium bicarbonate), reduced with dithiothreitol (0.2 µM, 1 hour, 37°C) and alkylated in the dark with iodoacetamide (0.4 µM, 30 minutes, 25°C). The protein mixture was then diluted six-fold with 200 mM ammonium bicarbonate and digested with 6 µg of trypsin (Promega, cat # V5113) overnight at 37°C. Samples were acidified with formic acid and cleaned up on a home-made Empore C18 column (3M, St. Paul, MN, USA) [81].
LC-MS/MS
Samples were subjected to liquid chromatography-tandem mass spectrometry and data were acquired with Xcalibur software. Data were processed using Proteome Discoverer beta version 1.3.0.339 (see supporting Text S1 for details).
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. WahlMC, WillCL, LuhrmannR (2009) The spliceosome: design principles of a dynamic RNP machine. Cell 136 : 701–718.
2. ZahlerAM (2012) Pre-mRNA splicing and its regulation in Caenorhabditis elegans. Worm Book 1–21.
3. JuricaMS, MooreMJ (2003) Pre-mRNA splicing: awash in a sea of proteins. Mol Cell 12 : 5–14.
4. ShepardPJ, HertelKJ (2009) The SR protein family. Genome Biol 10 : 242.
5. BoucherL, OuzounisCA, EnrightAJ, BlencoweBJ (2001) A genome-wide survey of RS domain proteins. Rna 7 : 1693–1701.
6. LongJC, CaceresJF (2009) The SR protein family of splicing factors: master regulators of gene expression. Biochem J 417 : 15–27.
7. ZhongXY, WangP, HanJ, RosenfeldMG, FuXD (2009) SR proteins in vertical integration of gene expression from transcription to RNA processing to translation. Mol Cell 35 : 1–10.
8. WangGS, CooperTA (2007) Splicing in disease: disruption of the splicing code and the decoding machinery. Nat Rev Genet 8 : 749–761.
9. GraingerRJ, BarrassJD, JacquierA, RainJC, BeggsJD (2009) Physical and genetic interactions of yeast Cwc21p, an ortholog of human SRm300/SRRM2, suggest a role at the catalytic center of the spliceosome. Rna 15 : 2161–2173.
10. BlencoweBJ, IssnerR, NickersonJA, SharpPA (1998) A coactivator of pre-mRNA splicing. Genes Dev 12 : 996–1009.
11. BlencoweBJ, BaurenG, EldridgeAG, IssnerR, NickersonJA, et al. (2000) The SRm160/300 splicing coactivator subunits. Rna 6 : 111–120.
12. KhannaM, Van BakelH, TangX, CalarcoJA, BabakT, et al. (2009) A systematic characterization of Cwc21, the yeast ortholog of the human spliceosomal protein SRm300. Rna 15 : 2174–2185.
13. ChanaratS, SeizlM, StrasserK (2011) The Prp19 complex is a novel transcription elongation factor required for TREX occupancy at transcribed genes. Genes Dev 25 : 1147–1158.
14. LongmanD, McGarveyT, McCrackenS, JohnstoneIL, BlencoweBJ, et al. (2001) Multiple interactions between SRm160 and SR family proteins in enhancer-dependent splicing and development of C. elegans. Curr Biol 11 : 1923–1933.
15. CeronJ, RualJF, ChandraA, DupuyD, VidalM, et al. (2007) Large-scale RNAi screens identify novel genes that interact with the C. elegans retinoblastoma pathway as well as splicing-related components with synMuv B activity. BMC Dev Biol 7 : 30.
16. BrodyY, Shav-TalY (2011) Transcription and splicing: when the twain meet. Transcription 2 : 216–220.
17. DasR, YuJ, ZhangZ, GygiMP, KrainerAR, et al. (2007) SR proteins function in coupling RNAP II transcription to pre-mRNA splicing. Mol Cell 26 : 867–881.
18. DermodyJL, DreyfussJM, VillenJ, OgundipeB, GygiSP, et al. (2008) Unphosphorylated SR-like protein Npl3 stimulates RNA polymerase II elongation. PLoS One 3: e3273.
19. LinS, Coutinho-MansfieldG, WangD, PanditS, FuXD (2008) The splicing factor SC35 has an active role in transcriptional elongation. Nat Struct Mol Biol 15 : 819–826.
20. FurgerA, O'SullivanJM, BinnieA, LeeBA, ProudfootNJ (2002) Promoter proximal splice sites enhance transcription. Genes Dev 16 : 2792–2799.
21. DamgaardCK, KahnsS, Lykke-AndersenS, NielsenAL, JensenTH, et al. (2008) A 5′ splice site enhances the recruitment of basal transcription initiation factors in vivo. Mol Cell 29 : 271–278.
22. SpiluttiniB, GuB, BelagalP, SmirnovaAS, NguyenVT, et al. (2010) Splicing-independent recruitment of U1 snRNP to a transcription unit in living cells. J Cell Sci 123 : 2085–2093.
23. KornblihttAR, de la MataM, FededaJP, MunozMJ, NoguesG (2004) Multiple links between transcription and splicing. Rna 10 : 1489–1498.
24. RualJF, CeronJ, KorethJ, HaoT, NicotAS, et al. (2004) Toward improving Caenorhabditis elegans phenome mapping with an ORFeome-based RNAi library. Genome Res 14 : 2162–2168.
25. Porta-de-la-RivaM, FontrodonaL, VillanuevaA, CeronJ (2012) Basic Caenorhabditis elegans Methods: Synchronization and Observation. J Vis Exp (64)L e4019. doi:10.379/4019
26. MacMorrisM, BrockerC, BlumenthalT (2003) UAP56 levels affect viability and mRNA export in Caenorhabditis elegans. Rna 9 : 847–857.
27. EllisR, SchedlT (2007) Sex determination in the germ line. Worm Book 1–13.
28. ZarkowerD (2006) Somatic sex determination. Worm Book 1–12.
29. GallegosM, AhringerJ, CrittendenS, KimbleJ (1998) Repression by the 3′ UTR of fem-3, a sex-determining gene, relies on a ubiquitous mog-dependent control in Caenorhabditis elegans. Embo J 17 : 6337–6347.
30. ContrinoS, SmithRN, ButanoD, CarrA, HuF, et al. (2012) modMine: flexible access to modENCODE data. Nucleic Acids Res 40: D1082–1088.
31. ReinkeV, GilIS, WardS, KazmerK (2004) Genome-wide germline-enriched and sex-biased expression profiles in Caenorhabditis elegans. Development 131 : 311–323.
32. LamontLB, KimbleJ (2007) Developmental expression of FOG-1/CPEB protein and its control in the Caenorhabditis elegans hermaphrodite germ line. Dev Dyn 236 : 871–879.
33. RamaniAK, NelsonAC, KapranovP, BellI, GingerasTR, et al. (2009) High resolution transcriptome maps for wild-type and nonsense-mediated decay-defective Caenorhabditis elegans. Genome Biol 10: R101.
34. AndersKR, GrimsonA, AndersonP (2003) SMG-5, required for C.elegans nonsense-mediated mRNA decay, associates with SMG-2 and protein phosphatase 2A. Embo J 22 : 641–650.
35. EvansTC (2006) Transformation and microinjection. Worm Book 1–15.
36. MerrittC, SeydouxG (2010) Transgenic solutions for the germline. Worm Book 1–21.
37. MerrittC, RasolosonD, KoD, SeydouxG (2008) 3′ UTRs are the primary regulators of gene expression in the C. elegans germline. Curr Biol 18 : 1476–1482.
38. LamondAI, SpectorDL (2003) Nuclear speckles: a model for nuclear organelles. Nat Rev Mol Cell Biol 4 : 605–612.
39. ZhangY, LiuT, MeyerCA, EeckhouteJ, JohnsonDS, et al. (2008) Model-based analysis of ChIP-Seq (MACS). Genome Biol 9: R137.
40. BuratowskiS (2009) Progression through the RNA polymerase II CTD cycle. Mol Cell 36 : 541–546.
41. BaughLR, DemodenaJ, SternbergPW (2009) RNA Pol II accumulates at promoters of growth genes during developmental arrest. Science 324 : 92–94.
42. ZhongM, NiuW, LuZJ, SarovM, MurrayJI, et al. (2010) Genome-wide identification of binding sites defines distinct functions for Caenorhabditis elegans PHA-4/FOXA in development and environmental response. PLoS Genet 6: e1000848.
43. KamathRS, FraserAG, DongY, PoulinG, DurbinR, et al. (2003) Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature 421 : 231–237.
44. McCrackenS, LongmanD, MarconE, MoensP, DowneyM, et al. (2005) Proteomic analysis of SRm160-containing complexes reveals a conserved association with cohesin. J Biol Chem 280 : 42227–42236.
45. BresV, GomesN, PickleL, JonesKA (2005) A human splicing factor, SKIP, associates with P-TEFb and enhances transcription elongation by HIV-1 Tat. Genes Dev 19 : 1211–1226.
46. LehnertS, GotzC, KartariusS, SchaferB, MontenarhM (2008) Protein kinase CK2 interacts with the splicing factor hPrp3p. Oncogene 27 : 2390–2400.
47. RamaniAK, CalarcoJA, PanQ, MavandadiS, WangY, et al. (2011) Genome-wide analysis of alternative splicing in Caenorhabditis elegans. Genome Res 21 : 342–348.
48. BelfioreM, PugnaleP, SaudanZ, PuotiA (2004) Roles of the C. elegans cyclophilin-like protein MOG-6 in MEP-1 binding and germline fates. Development 131 : 2935–2945.
49. GrahamPL, SchedlT, KimbleJ (1993) More mog genes that influence the switch from spermatogenesis to oogenesis in the hermaphrodite germ line of Caenorhabditis elegans. Dev Genet 14 : 471–484.
50. KasturiP, ZanettiS, PassannanteM, SaudanZ, MullerF, et al. (2010) The C. elegans sex determination protein MOG-3 functions in meiosis and binds to the CSL co-repressor CIR-1. Dev Biol 344 : 593–602.
51. KonishiT, UodomeN, SugimotoA (2008) The Caenorhabditis elegans DDX-23, a homolog of yeast splicing factor PRP28, is required for the sperm-oocyte switch and differentiation of various cell types. Dev Dyn 237 : 2367–2377.
52. ZanettiS, MeolaM, BochudA, PuotiA (2011) Role of the C. elegans U2 snRNP protein MOG-2 in sex determination, meiosis, and splice site selection. Dev Biol 354 : 232–241.
53. PuotiA, KimbleJ (1999) The Caenorhabditis elegans sex determination gene mog-1 encodes a member of the DEAH-Box protein family. Mol Cell Biol 19 : 2189–2197.
54. PuotiA, KimbleJ (2000) The hermaphrodite sperm/oocyte switch requires the Caenorhabditis elegans homologs of PRP2 and PRP22. Proc Natl Acad Sci U S A 97 : 3276–3281.
55. BelfioreM, MathiesLD, PugnaleP, MoulderG, BarsteadR, et al. (2002) The MEP-1 zinc-finger protein acts with MOG DEAH box proteins to control gene expression via the fem-3 3′ untranslated region in Caenorhabditis elegans. Rna 8 : 725–739.
56. KerinsJA, HanazawaM, DorsettM, SchedlT (2010) PRP-17 and the pre-mRNA splicing pathway are preferentially required for the proliferation versus meiotic development decision and germline sex determination in Caenorhabditis elegans. Dev Dyn 239 : 1555–1572.
57. de AlmeidaSF, Carmo-FonsecaM (2012) Design principles of interconnections between chromatin and pre-mRNA splicing. Trends Biochem Sci 37 : 248–253.
58. PanditS, WangD, FuXD (2008) Functional integration of transcriptional and RNA processing machineries. Curr Opin Cell Biol 20 : 260–265.
59. MieleA, MedinaR, van WijnenAJ, SteinGS, SteinJL (2007) The interactome of the histone gene regulatory factor HiNF-P suggests novel cell cycle related roles in transcriptional control and RNA processing. J Cell Biochem 102 : 136–148.
60. RenL, McLeanJR, HazbunTR, FieldsS, Vander KooiC, et al. (2011) Systematic two-hybrid and comparative proteomic analyses reveal novel yeast pre-mRNA splicing factors connected to Prp19. PLoS One 6: e16719.
61. KroganNJ, KimM, AhnSH, ZhongG, KoborMS, et al. (2002) RNA polymerase II elongation factors of Saccharomyces cerevisiae: a targeted proteomics approach. Mol Cell Biol 22 : 6979–6992.
62. de AlmeidaSF, Carmo-FonsecaM (2008) The CTD role in cotranscriptional RNA processing and surveillance. FEBS Lett 582 : 1971–1976.
63. GilchristDA, FrommG, dos SantosG, PhamLN, McDanielIE, et al. (2012) Regulating the regulators: the pervasive effects of Pol II pausing on stimulus-responsive gene networks. Genes Dev 26 : 933–944.
64. ZeitlingerJ, StarkA, KellisM, HongJW, NechaevS, et al. (2007) RNA polymerase stalling at developmental control genes in the Drosophila melanogaster embryo. Nat Genet 39 : 1512–1516.
65. MunozMJ, Perez SantangeloMS, ParonettoMP, de la MataM, PelischF, et al. (2009) DNA damage regulates alternative splicing through inhibition of RNA polymerase II elongation. Cell 137 : 708–720.
66. LinCL, LeuS, LuMC, OuyangP (2004) Over-expression of SR-cyclophilin, an interaction partner of nuclear pinin, releases SR family splicing factors from nuclear speckles. Biochem Biophys Res Commun 321 : 638–647.
67. ZimowskaG, ShiJ, MungubaG, JacksonMR, AlpatovR, et al. (2003) Pinin/DRS/memA interacts with SRp75, SRm300 and SRrp130 in corneal epithelial cells. Invest Ophthalmol Vis Sci 44 : 4715–4723.
68. SpectorDL, LamondAI (2010) Nuclear Speckles. Cold Spring Harb Perspect Biol
69. DujardinG, LafailleC, PetrilloE, BuggianoV, Gomez AcunaLI, et al. (2013) Transcriptional elongation and alternative splicing. Biochim Biophys Acta 1829 : 134–140.
70. Garrido-LeccaA, BlumenthalT (2010) RNA polymerase II C-terminal domain phosphorylation patterns in Caenorhabditis elegans operons, polycistronic gene clusters with only one promoter. Mol Cell Biol 30 : 3887–3893.
71. GuangS, BochnerAF, BurkhartKB, BurtonN, PavelecDM, et al. (2010) Small regulatory RNAs inhibit RNA polymerase II during the elongation phase of transcription. Nature 465 : 1097–1101.
72. AllemandE, BatscheE, MuchardtC (2008) Splicing, transcription, and chromatin: a menage a trois. Curr Opin Genet Dev 18 : 145–151.
73. StiernagleT (2006) Maintenance of C. elegans. Worm Book 1–11.
74. LeeMH, SchedlT (2006) RNA in situ hybridization of dissected gonads. Worm Book 1–7.
75. DuerrJS (2006) Immunohistochemistry. Worm Book 1–61.
76. SempleJI, Garcia-VerdugoR, LehnerB (2010) Rapid selection of transgenic C. elegans using antibiotic resistance. Nat Methods 7 : 725–727.
77. BlankenbergD, Von KusterG, CoraorN, AnandaG, LazarusR, et al. (2010) Galaxy: a web-based genome analysis tool for experimentalists. Curr Protoc Mol Biol Chapter 19: Unit 19 10 11–21.
78. GoecksJ, NekrutenkoA, TaylorJ (2010) Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol 11: R86.
79. LefrancoisP, EuskirchenGM, AuerbachRK, RozowskyJ, GibsonT, et al. (2009) Efficient yeast ChIP-Seq using multiplex short-read DNA sequencing. BMC Genomics 10 : 37.
80. TrapnellC, RobertsA, GoffL, PerteaG, KimD, et al. (2010) Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7 : 562–578.
81. RappsilberJ, MannM, IshihamaY (2007) Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat Protoc 2 : 1896–1906.
82. GersteinMB, LuZJ, Van NostrandEL, ChengC, ArshinoffBI, et al. (2010) Integrative analysis of the Caenorhabditis elegans genome by the modENCODE project. Science 330 : 1775–1787.
83. BaughLR, HillAA, SlonimDK, BrownEL, HunterCP (2003) Composition and dynamics of the Caenorhabditis elegans early embryonic transcriptome. Development 130 : 889–900.
84. MeissnerB, WarnerA, WongK, DubeN, LorchA, et al. (2009) An integrated strategy to study muscle development and myofilament structure in Caenorhabditis elegans. PLoS Genet 5: e1000537.
85. WangX, ZhaoY, WongK, EhlersP, KoharaY, et al. (2009) Identification of genes expressed in the hermaphrodite germ line of C. elegans using SAGE. BMC Genomics 10 : 213.
86. AllenMA, HillierLW, WaterstonRH, BlumenthalT (2011) A global analysis of C. elegans trans-splicing. Genome Res 21 : 255–264.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2013 Číslo 6
Nejčtenější v tomto čísle
- BMS1 Is Mutated in Aplasia Cutis Congenita
- Sex-stratified Genome-wide Association Studies Including 270,000 Individuals Show Sexual Dimorphism in Genetic Loci for Anthropometric Traits
- Distinctive Expansion of Potential Virulence Genes in the Genome of the Oomycete Fish Pathogen
- Distinct Neuroblastoma-associated Alterations of Impair Sympathetic Neuronal Differentiation in Zebrafish Models