
Distinct Neuroblastoma-associated Alterations of Impair Sympathetic Neuronal Differentiation in Zebrafish Models
Heterozygous germline mutations and deletions in PHOX2B, a key regulator of autonomic neuron development, predispose to neuroblastoma, a tumor of the peripheral sympathetic nervous system. To gain insight into the oncogenic mechanisms engaged by these changes, we used zebrafish models to study the functional consequences of aberrant PHOX2B expression in the cells of the developing sympathetic nervous system. Allelic deficiency, modeled by phox2b morpholino knockdown, led to a decrease in the terminal differentiation markers th and dbh in sympathetic ganglion cells. The same effect was seen on overexpression of two distinct neuroblastoma-associated frameshift mutations, 676delG and K155X - but not the R100L missense mutation - in the presence of endogenous Phox2b, pointing to their dominant-negative effects. We demonstrate that Phox2b is capable of regulating itself as well as ascl1, and that phox2b deficiency uncouples this autoregulatory mechanism, leading to inhibition of sympathetic neuron differentiation. This effect on terminal differentiation is associated with an increased number of phox2b+, ascl1+, elavl3− cells that respond poorly to retinoic acid. These findings suggest that a reduced dosage of PHOX2B during development, through either a heterozygous deletion or dominant-negative mutation, imposes a block in the differentiation of sympathetic neuronal precursors, resulting in a cell population that is likely to be susceptible to secondary transforming events.
Published in the journal:
. PLoS Genet 9(6): e32767. doi:10.1371/journal.pgen.1003533
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1003533
Summary
Heterozygous germline mutations and deletions in PHOX2B, a key regulator of autonomic neuron development, predispose to neuroblastoma, a tumor of the peripheral sympathetic nervous system. To gain insight into the oncogenic mechanisms engaged by these changes, we used zebrafish models to study the functional consequences of aberrant PHOX2B expression in the cells of the developing sympathetic nervous system. Allelic deficiency, modeled by phox2b morpholino knockdown, led to a decrease in the terminal differentiation markers th and dbh in sympathetic ganglion cells. The same effect was seen on overexpression of two distinct neuroblastoma-associated frameshift mutations, 676delG and K155X - but not the R100L missense mutation - in the presence of endogenous Phox2b, pointing to their dominant-negative effects. We demonstrate that Phox2b is capable of regulating itself as well as ascl1, and that phox2b deficiency uncouples this autoregulatory mechanism, leading to inhibition of sympathetic neuron differentiation. This effect on terminal differentiation is associated with an increased number of phox2b+, ascl1+, elavl3− cells that respond poorly to retinoic acid. These findings suggest that a reduced dosage of PHOX2B during development, through either a heterozygous deletion or dominant-negative mutation, imposes a block in the differentiation of sympathetic neuronal precursors, resulting in a cell population that is likely to be susceptible to secondary transforming events.
Introduction
Neuroblastoma is an embryonal malignancy of the peripheral sympathetic nervous system (PSNS) that arises from the developing neural crest and manifests as neoplasms in sympathetic ganglia or adrenal medulla. The oncogenic events culminating in neuroblastoma are thought to occur very early in development, consistent with the status of this tumor as the most common cancer of infants [1]. A defining feature of neuroblastic tumors is their broad spectrum of cellular differentiation, ranging from undifferentiated cells that indicate a poor prognosis to those showing greater differentiation and predicting a generally favorable outcome [2]. This heterogeneity suggests that dysregulated differentiation of sympathetic progenitor cells plays a key role in neuroblastoma pathogenesis. Direct evidence for this model comes from the identification of heterozygous germline mutations in the homeodomain transcription factor PHOX2B, a regulator of sympathetic neuronal differentiation [3]–[8]. Such mutations are also found in patients with other neural crest-derived disorders, including congenital central hypoventilation syndrome (CCHS) and Hirschprung's disease, characterized by absent or abnormal development of the noradrenergic neurons in the brain stem and colon, respectively [3], [5], [7], [8].
The vast majority of embryonal tumors like neuroblastoma arise from aberrant genetic and epigenetic changes that control the survival, proliferation and differentiation of specific tissues. Hence, one way to decipher the tumorigenic contribution of germline changes in highly conserved genes such as PHOX2B is to understand how they perturb normal development. The sympathetic nervous system is derived from the neural crest, a multipotent embryonic structure consisting of a transient population of cells that migrate from the neural tube to the region of the dorsal aorta during development [9]–[11]. At the dorsal aorta, prespecified SOX10-positive sympathetic progenitors, under the influence of bone morphogenetic proteins (BMPs), start the process of differentiation into noradrenergic neurons [12]. Phox2b and the basic helix-loop-helix (bHLH) factor Ascl1 are the first transcription factors that appear upon initiation of differentiation of sympathoadrenal precursors [13]–[15]. Phox2a is expressed downstream of both Phoxb and Ascl1 [13], [16], [17] but its role in the initial stages of the sympathetic lineage remains undefined. Further neuronal differentiation occurs under the influence of the transcription factors Hand2 [18], [19], Gata2/3 [20] and Tfap2a [21], [22], which interact in a complex regulatory network to ultimately induce the expression of tyrosine hydroxylase (Th) and dopamine beta hydroxylase (Dbh) - two enzymes required for catecholamine production that serve as markers of terminal sympathetic neuronal differentiation [23]–[25]
Extensive work in murine and avian models has established Phox2b as a key regulator of autonomic neuron development, as its complete absence leads to embryonic lethality in mice due to the failure of sympathetic nervous system formation [15], [26]. Phox2b is first expressed in the murine peripheral sympathetic primordial at the dorsal aorta at E10.5 [13], [27]. By E13.5, it is expressed in all sympathetic progenitor cells including those of the superior cervical ganglia, paravertebral and pelvic ganglia and the adrenal medulla [27], and expression continues into the early postnatal period [28]. As the ganglia/adrenal medullae mature and the sympathoblasts differentiate, TH - and DBH-expression increases while PHOX2B expression decreases, so that by P28, only 12% of TH-positive sympathetic neurons have PHOX2B expression (compared to approximately 60% at P4) [28]. Hence, Phox2b is downregulated during terminal neuronal differentiation [28]. Phox2b also has growth inhibitory effects, as its overexpression promotes cell cycle exit and inhibits the proliferation of cultured sympathetic neurons [29], [30]. Most of the activity of the Phox2b promoter depends on an autoregulatory loop enabling the gene to regulate its own activity and that of other genes [31].
The human PHOX2B gene maps to chromosome 4p13 and consists of three exons encoding a highly conserved 314–amino-acid protein with two polyalanine repeats of 9 and 20 residues C-terminal to the homeodomain. Unlike the case in CCHS, which is defined largely by polyalanine repeat expansion mutations that lead to expansion of the second polyalanine tract [8], [32], [33], germline PHOX2B mutations associated with neuroblastoma tend to be (i) missense alterations in highly conserved regions [4], [7], [33] or (ii) mutations that result in a frameshift, giving rise to an altered or truncated protein lacking the second polyalanine motif [5], [8], [33], [34]. More recently, whole-allele deletions resulting in the reduction of the protein have been reported [35]. Still, the mechanisms by which these different classes of alterations predispose individuals to tumor development in the PSNS remain somewhat unclear. Partial loss of function with the preserved ability to suppress cellular proliferation but not to promote differentiation [36], complete loss of function due to functional haploinsufficiency [37] and both dominant-negative and gain-of-function effects [30], [34], [38] have all been proposed as contributors to neuroblastoma predisposition. In the single in vivo study to date, heterozygous insertion of two frameshift variants, 931del5 and 693del8, in the mouse Phox2b locus resulted in impaired proliferation of sympathetic ganglion progenitors and biased differentiation towards the glial lineage, leading the authors to conclude that these mutants exhibited both dominant-negative and gain-of-function effects [38].
In this study we sought to analyze the effects of aberrant PHOX2B expression on sympathetic neuron development for each of the major classes of neuroblastoma-associated mutants. We selected the zebrafish model for this purpose because its development occurs ex utero, and the embryos survive considerably longer than mouse embryos, permitting analysis of the PSNS at later stages of sympathetic neuron differentiation and maintenance [39]. We show here that allelic PHOX2B deletion, modeled by morpholino (MO) knockdown, leads to a decrease in sympathetic neuronal differentiation in the PSNS. A similar loss of differentiation was observed upon overexpression of a neuroblastoma-linked truncation mutation (K155X) [8] and a frameshift mutation (676delG) [5] in the presence of endogenous phox2b, indicating that these variants function in a dominant-negative manner. By contrast, the R100L missense mutation [7] lacked any discernible effect on sympathetic ganglion development in the zebrafish embryo. We demonstrate further that the decrease in terminal differentiation was associated with an increased number of undifferentiated sympathetic neuronal precursors that were resistant to the effects of retinoic acid (RA), and generated a pool of developmentally arrested cells that could serve as targets for future transforming events.
Results
phox2b is expressed in cells of the PSNS in the zebrafish
To establish the zebrafish as a model for studying PHOX2B function in PSNS development, we analyzed expression of the phox2b gene in the superior cervical ganglion (SCG). The SCG is the earliest sympathetic ganglion to develop, starting at about 36 hours postfertilization (hpf) and progressing in size until about 4 days postfertilization (dpf) (Figure 1A–1D) [18], [21], [39], [40]. SCG cells are located ventral to the notochord between somites 1 and 4 and are easily visualized as several smaller cell aggregates that progressively increase in size, ultimately forming two separate ganglia in an hourglass shape. phox2b expression is first seen in the SCG cells at 36 hpf extending caudally in two irregular parallel rows to form the primary sympathetic ganglion chain to somite 11 (Figure 1A–1D). It remains robust in the SCG until 72 hpf (Figure 1C) when it gradually begins to be replaced by increasing numbers of cells expressing dbh and th, markers of terminal differentiation (Figures 1E–1F), whose expression is first seen at about 48hpf. This expression pattern, especially in the peripheral sympathetic ganglia, and the close sequence homology with human PHOX2B [41] (Figure S1) support the use of the zebrafish to model PHOX2B function in the PSNS.

phox2b deficiency leads to decreased PSNS differentiation
To study the consequences of allelic PHOX2B deletion in patients with neuroblastoma [35], we performed morpholino (MO) knockdown of zebrafish phox2b, using two non-overlapping antisense oligonucleotide sequences targeted to the phox2b gene: a translation-blocking MO (MOATG) and a splice-blocking MO (MOsplice) directed to the second exon/intron splice junction (Figure S2A). phox2b knockdown was confirmed by immunoblotting in the case of the ATG MO (which showed ∼70–80% knockdown) and RT-PCR for the splice MO (Figure S2B). Experiments were performed in both wild-type and p53 mutant embryos (tp53M214K/M214K) [42] to account for potential nonspecific effects associated with some MO injections, with similar results obtained in both backgrounds (Figure S2C). MO knockdown of phox2b led to a marked reduction in th and dbh expression in the SCG at 3 dpf, as compared with mismatched control MO-injected (MOMM) or uninjected wild-type (WT) siblings (Figures S3A, S3B, S3D, S3E, S3G, S3H). This phenotype was consistent with both the ATG and the splice MOs. To ensure that the decrease in th and dbh was not secondary to a general delay in development due to MO injection, we examined the SCG at 4 dpf (Figures 2A, 2B, 2D, 2E, 2G, 2H) and later (5 dpf; data not shown), again noting a decrease in the expression of these genes. To confirm that the phenotype was specific to phox2b, we rescued the th- and dbh-expressing cells by coexpressing human PHOX2B mRNA with both MOs, which led to an increase in th and dbh expression in the SCG (Figures 2C, 2E; 2G, 2H; Figures S3C, S3F; 3G, 3H). These results indicate that PHOX2B is necessary and sufficient for the terminal differentiation of sympathetic neuronal precursors.

The 676delG and K155X variants cause a decrease in terminal differentiation of sympathetic progenitors
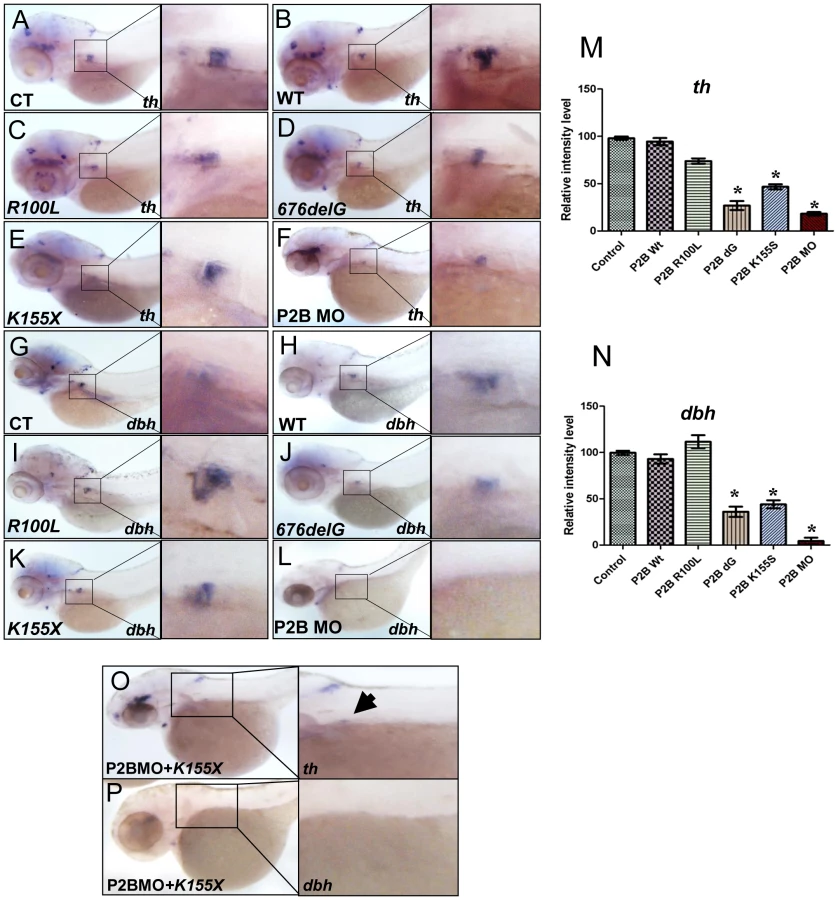
We next examined the effects of three distinct neuroblastoma-associated PHOX2B mutations on PSNS development (Figure S4, Figure 3). Overexpression of the 676delG frameshift [5] and K155X truncation [8] variants led to a significant decrease in the expression of th (Figure 3D, 3E, 3M) and dbh in the SCG (Figure 3J, 3K, 3N) as compared to that in control (Figure 3A, 3G) and WT (Figure 3B, 3H) phox2b RNA-injected animals, but had a similar effect to MO knockdown (Figure 3F, 3L). By contrast, overexpression of the R100L homeodomain missense mutation [7] did not lead to a discernible change in the expression of either th or dbh in the SCG (Figure 3C, 3I). To mimic the heterozygous situation seen in patients with PHOX2B mutations, we repeated these experiments in the setting of phox2b MO knockdown. In this context, overexpression of K155X and 676delG led to an even more striking reduction and, in some embryos, to an almost complete absence of th and dbh expression in the SCG (Figure 3O, 3P). These results suggest that the block in differentiation imposed by the 676delG and K155X mutations cannot be rescued by the expression of endogenous wild-type phox2b; rather, these variants appear to function dominant-negatively.
phox2b deficiency prevents retinoic acid-induced sympathetic progenitor cell differentiation
PHOX2B complements exogenous differentiating agents such as retinoic acid (RA) by promoting cellular differentiation in vitro [36]. To determine whether phox2b loss might obviate the induction of differentiation by RA, we treated control and phox2b MO embryos with various concentrations of 13-cis-retinoic acid, which is commonly used in the treatment of patients with neuroblastoma (Figure 4A–4F). Control-injected embryos showed an increase in th expression in the SCG after RA treatment (Figure 4A–4C, 4G), which was not apparent in the SCG of the phox2b morphant embryos (Figure 4D–4F, 4G). A similar impairment in RA-induced differentiation was observed after expression of the 676delG variant and, to a lesser extent, the K155X variant (Figure 4H–4P). Similar effects in dbh expression were also seen (data not shown). Together, these findings reinforce the strict requirement for Phox2b in the differentiation of sympathetic neuronal progenitors and suggest that loss of this transcription factor cannot be overcome with RA treatment.

phox2b deficiency induces distinct effects on genes involved in PSNS differentiation
To assess the impact of decreased phox2b function on the transcription factors that mediate noradrenergic differentiation in the zebrafish, we analyzed their expression following MO knockdown. We observed that mRNA expression of phox2b itself was markedly increased despite the decrease in Phox2b protein induced by MO knockdown (Figure 5A, 5B; Figure S5A, S5B). During normal development in the zebrafish, phox2b expression is first seen in sympathetic ganglia precursors at 36 hpf and, as the cells undergo differentiation, decreases to lower levels by 4 dpf (this study), corresponding to E10.5 to E13 in mice [6]. This increase in phox2b RNA expression on abrogation of the protein is consistent with studies that Phox2b regulates its own expression [31]. We also noted that expression of the zebrafish ortholog of ASCL1, ascl1, was strikingly increased in the SCG in phox2b morphants (Figure 5C, 5D; Figure S5C, S5D). ascl1 is expressed only transiently in the SCG with maximal expression at 48 hpf, decreasing to less than 10% in 3-dpf embryos [18]. The increased expression of ascl1 transcripts in the SCG of phox2b morphants suggests that Phox2b negatively regulates ascl1 transcription as well. Similar to the effect seen with phox2b MO knockdown, overexpression of the neuroblastoma-associated variants also led to increased ascl1 expression in the SCG, with the 676delG and K155X mutants inducing higher ascl1 expression than did R100L (Figure 5E–5I). Phox2a, a homologue of Phox2b, that is expressed downstream of both Ascl1 and Phoxb during murine sympathetic neural development [13], [17] was unchanged in phox2b morphant embryos (Figure 5J, 5K). Finally, expression of hand2 [19], [43], gata3 [20], [44] and tfap2a [21], [22], three other transcriptional regulators implicated in the control of sympathetic neuronal differentiation [10], [18] was decreased on phox2b knockdown (Figure S5E–J). Overexpression of the 676delG and K155X variants also led to a decrease in gata3 and tfap2a expression (Figure S5K–P). Together, these results suggest that these genes are regulated by phox2b during sympathetic neuronal differentiation and are affected by perturbations in its function.

phox2b deficiency leads to an increase in progenitor cells in the SCG
We surmised that reduced expression of the th and dbh noradrenergic differentiation markers in the SCG of phox2b morphants might reflect either apoptosis of terminally differentiated sympathetic neurons or the failure of precursors to differentiate. There was no evidence of an increase in apoptosis in the SCG as determined by acridine orange staining of phox2b MO-injected embryos (data not shown). However, simultaneous analysis of th and phox2b expression by double labeling of the phox2b morphants showed an increase in phox2b expression with a concomitant decrease in th expression in the SCG compared to stage-matched control MO-injected embryos (Figures 6A, 6B). Analysis of these embryos using qRT-PCR confirmed the presence of significantly increased phox2b expression in the face of decreased th (Figure 6E). The same results were obtained when double labeling was performed with dbh and phox2b, the decrease in dbh-expressing cells being accompanied by increased expression of phox2b in morphants compared to controls (Figures 6C, 6D). We were unable to test whether this was also true in embryos in which PHOX2B variants were overexpressed because the high level of sequence homology between human and zebrafish phox2b made it difficult to interpret the results. To confirm that these phox2b- and ascl1-expressing SCG cells were arrested early in differentiation, we analyzed the expression of elavl3 (HuC), the well-characterized, evolutionarily conserved neural differentiation marker that is normally only detected in mature postmitotic neurons and has been used to demonstrate neuronal differentiation in the zebrafish [41], [45]–[47] (Figures 6F–6K). We observed a decrease in elavl3 expression in the phox2b morphants compared to controls, with the extent of reduction comparable to that of th and dbh, suggesting that the SCG is populated mainly by undifferentiated cells. Overexpression of the 676delG and K155X mutants, but not R100L, also led to a decrease in elavl3 expression in the SCG of these embryos (Figures 6H–K). Together, these data suggest that a decrease in Phox2b levels, either through MO knockdown or overexpression of dominant-negative mutations, blocks sympathetic cells at a progenitor stage marked by high phox2b and ascl1 expression. Such cells cannot proceed to the next developmental stage, involving hand2, gata3 and tfap2a, and therefore cannot undergo terminal PSNS differentiation characterized by the expression of th and dbh.

ascl1 is not essential for differentiation of SCG cells in zebrafish
During noradrenergic development in the chick embryo, Phox2b and Ascl1 are expressed together in response to BMP signaling [44], [48]; in fact, elimination of Ascl1 has also been reported to cause impaired sympathetic differentiation [14], [16]. To determine whether ascl1 is as critical as phox2b to PSNS differentiation in the zebrafish, we studied the effects of ascl1 knockdown using a translation-blocking MO (Figure S6A; Figure 7A, 7B, 7C, 7E). Abrogation of ascl1 alone led to only a marginal decrease in th and dbh expression in the SCG (Figure 7B, 7E, 7G), while a striking decrease in both th and dbh expression occurred with simultaneous knockdown of both ascl1 and phox2b, similar to the result with phox2b knockdown alone (Figure 7C, 7F, 7G). Moreover, in contrast to the outcome of phox2b knockdown, expression of hand2, gata3 and tfap2a was not affected in the ascl1 morphants (Figure S6B–G). These findings identify Phox2b as the central driver of terminal differentiation of sympathetic neurons in the zebrafish model.

ascl1 does not regulate phox2b in zebrafish
Observations in murine models have revealed that PHOX2B and ASCL1 cross regulate each other [6], [14], [16], [49]. The fact that ascl1 expression was significantly increased in the phox2b morphants suggested a regulatory effect of phox2b on ascl1 (Figure 5C,5D). To determine if Ascl1 had the same effect on phox2b in the zebrafish model, we studied the effects of ascl1 knockdown on phox2b expression. While causing a decrease in its own expression at both the RNA and protein levels (Figure 7H, 7I, Figure S6A), knockdown of ascl1 had no effect on phox2b expression (Figure 7K, 7L). Similarly, overexpression of ascl RNA also did not affect phox2b expression (Figure S6H, S6I). However, knockdown of both genes simultaneously led to a striking increase in ascl1 (Figure 7J) and phox2b (Figure 7M) mRNA expression in the SCG. These results suggest that although Phox2b is capable of regulating ascl1 in the zebrafish model, the latter does not have any appreciable impact on phox2b. Rather, Ascl1 appears to primarily regulate phox2a. Unlike knockdown of phox2b, which did not affect phox2a expression, we observed a significant reduction in phox2a expression in the SCG following ascl1 knockdown (Figure 7N, 7O). These results are consistent with the finding that Ascl1 is required for the expression of Phox2a in sympathetic and parasympathetic murine ganglionic anlagen [14], [16], with Phox2b assuming a less important role.
Discussion
In this study, we relied on a zebrafish model of PSNS development to demonstrate that a reduction in phox2b expression due to MO knockdown or overexpression of the neuroblastoma-associated 676delG frameshift and K155X truncation variants of PHOX2B inhibits the terminal differentiation of sympathetic neuron progenitors (Figure 8). Our findings indicate that intact Phox2b function is essential for the normal development of cells in the sympathetic ganglia, and that in the context of phox2b deficiency, differentiation cannot be induced by exogenous agents.

In contrast to the remainder of the nervous system, where the onset of neuronal differentiation is coupled with cell cycle exit, an increase in the number of sympathetic neurons during development reflects the proliferation of differentiated sympathetic neurons rather than the proliferation and subsequent differentiation of neuronal progenitors [50], [51]. Indeed, studies in the chick embryo have revealed that the vast majority of postmitotic sympathetic neurons are generated through the proliferation of already differentiated neurons [50]–[53]. An alteration in Phox2b function in the zebrafish, by either MO knockdown or overexpression of certain neuroblastoma-associated variants, led to a block in the differentiation of sympathetic progenitor cells. First, there was a large increase in phox2b transcript-expressing cells at the expense of th- and dbh-expressing cells in the SCG (Figure 6). Second, these phox2b-expressing cells were unable to proceed to subsequent differentiation steps, even when stimulated with RA, whose signaling can induce noradrenergic differentiation in the zebrafish (Figure 4) [21]. Third, in addition to decreased th and dbh expression, the SCG cells also showed reduced expression of the mature neuronal marker elavl3 (Figure 6).
In our study, specific types of PHOX2B variants exhibited distinct effects on sympathetic neuronal differentiation, with the R100L missense mutation lacking an effect on differentiation, while the 676delG frameshift and K155X truncation mutations exhibited impaired differentiation, an effect that was all the more prominent with simultaneous MO knockdown. Findings similar to ours were reported by Reiff et al (2010) on overexpression of the K155X mutation in primary chick sympathetic neuron cultures, although in this system the 676delG variant lacked any apparent effect on the expression of terminal differentiation markers [30]. In agreement with our data, another group reported resistance to differentiation with RA in human neuroblastoma cells engineered to express the 676delG variant (in the presence of endogenous PHOX2B) [36]. The R100L homeodomain variant, on the other hand, did not appear to affect sympathetic neuronal differentiation in the zebrafish, similar to findings in the above mentioned study in avian sympathetic neurons, where this mutant elicited increased cell proliferation only [30].
The basis for the varied effects of distinct mutations on sympathetic neuronal differentiation is not entirely clear. To some extent, they reflect intrinsic differences between the in vitro and in vivo models in which the variants were tested, as well as different degrees of forced or blocked expression in target cells. Ultimately, however, the impact of PHOX2B mutants on the differentiation of immature sympathetic neurons depends on the protein structures that are modified. The R100L missense variant, for example, is defined by a minimal change in its homeodomain, whereas both 676delG and K155X possess modifications due to an altered reading frame or deletion of critical coding sequences within the C-terminus, respectively, either of which can lead to the disruption of essential protein-protein interactions or, in some instances, to novel interactions not seen with the WT protein. Indeed, misfolding and oligomerization have been demonstrated with frameshift mutations of PHOX2B and cellular mislocalization and cytoplasmic aggregation with truncated variants [54]. The 693del8 frameshift mutant, for example, is thought to interact with proteins that do not bind to the WT protein [38]. Similarly, we have noted that the 676delG and K155X variants do not bind to certain proteins recognized by WT PHOX2B, resulting in impaired neuroblastoma cell differentiation (George et al, unpublished observations). Thus, it is not surprising that the K155X truncation mutation, unlike the R100L missense change, generates a protein that not only stimulates sympathetic neuron proliferation [30], but also has a dominant–negative inhibitory effect on cell differentiation.
Furthermore, since both frameshift and truncation changes in the PHOX2B gene appear to generate stable proteins that lack the transactivation potential of WT PHOX2B, [30], [36], [37], [55] their contribution to neuroblastoma predisposition could be twofold. By interacting with different modifier proteins, these mutants could preserve the ability of PHOX2B to suppress cellular proliferation while abolishing its pro-differentiation regulatory effects. At the same time, as has been shown previously [30] these transactivation–impaired variants likely compete with intact PHOX2B for critical promoter sequences on neuronal differentiation-linked target genes, resulting in a dominant-negative repressive effect on their expression. This hypothesis is supported by the failure of the WT phox2b to rescue the arrested differentiation of sympathetic neuronal progenitors expressing either the 676delG or K155X variants.
The gene dosage of PHOX2B also appears to have played a role in determining the disease phenotype in our study. Indeed, fish with MO knockdown of phox2b but no mutations or deletions consistently showed arrested differentiation of sympathetic neuronal progenitors. This result would account for a neuroblastoma case recently reported by Jennings et al [35] in which a heterozygous deletion of PHOX2B was associated with both CCHS and a neural crest tumor. Thus, PHOX2B deficiency due to whole-allele deletion should be considered another mechanism whereby individuals might acquire a predisposition to neuroblastoma. The precise oncogenic mechanism of PHOX2B deficiency and its associated phenotypes will require additional study, but it seems likely that dosage reduction beyond 50% (sub-haploinsufficiency) may be required, as mice that were haploinsufficient for Phox2b did not develop tumors [56].
Neuroblastoma development in TH-MYCN transgenic mice begins with hyperplastic lesions in early postnatal sympathetic ganglia that are composed predominantly of Phox2b-positive but Th-negative neuronal progenitors [28]. This indicates a central role for aberrant PHOX2B regulation of immature sympathetic neurons in neuroblastoma predisposition. Our studies suggest a model (Figure 8) in which aberrant PHOX2B function through either allelic deletion or dominant-negative mutations promote the same endpoint: impaired differentiation of sympathetic neuronal progenitor cells while promoting the expansion of a population of undifferentiated cells likely to be susceptible to so-called second “hits” such as MYCN amplification.
Materials and Methods
Ethics statement
All experiments involving zebrafish conformed to the regulatory standards and guidelines of the Dana-Farber Cancer Institute (DFCI) and the Brigham and Women's Hospital (BWH) Institutional Animal Care and Use Committee.
Fish husbandry and strains
Zebrafish were maintained at the DFCI and BWH zebrafish facilities under standard conditions [57]. Embryos were raised in E3 medium supplemented with 0.003% 1-phenyl-2-thiourea (PTU) at 28.5°C to allow visualization of internal structures. The p53−/− mutant allele tp53M214K/M214K was kindly provided by A. Thomas Look [42]. tp53M214K/M214K and wild-type embryos of the AB strain were obtained by natural group mating. The embryos were staged according to established morphological criteria [57].
Morpholino injections
Morpholinos (MOs) were obtained from GeneTools, LLC (Philomath, OR) based on the published GenBank sequences for phox2b and ascl1 (phox2b P2BT2 splice donor: 5′-AAGTAAGCGGAGAATGTCCCACCTG; mismatched phox2b P2BT2 splice donor: AcTAAcCGGAcAATcTCCgACCTG; phox2b ATG: 5′-TATACATTGAAAAGGCTCAGTGGAG; mismatched phox2b ATG: TATAgATTcAAAAccCTCAcTGGAG; ascl1 ATG: 5′ - CCATCTTGGCGGTGATGTCCATTTC; mismatched ascl1 ATG: CCATgTTGGCcGTcATcTCgATTTC). For knockdown of phox2b, two nonoverlapping MOs were used (ATGK1, designed to target the start site; and P2BT2, designed to block splicing at the phox2b exon 2-intron 2 boundary). Embryos at the one to two-cell stages were injected with up to 4 ng of MO diluted with 1× Danieau's solution with Phenol Red. Injected embryos were raised to 12, 24, 36, 72, 96 and 120 hpf and processed for in situ hybdridization. Generalized nonspecific necrosis in MO-injected and mismatched control MO-injected AB embryos was alleviated using the tp53M214K/M214K strain [42]. MO concentrations were optimized so that the lowest amount necessary to retain the normal phenotype while demonstrating effects of absent phox2b expression was used. The observed phenotypes were consistent between the ATG and splice MO. Care was taken to stage-match the embryos as much as possible with mismatched MO-injected controls so as to eliminate phenotypes due to developmental delay.
Capped RNA overexpression
Human PHOX2B (a generous gift from K-S Kim) and the PHOX2B mutant 676delG (kindly provided by J. Maris) were subcloned into the pCS2+ vector. Site directed mutagenesis was used to generate PHOX2B mutant constructs R100L and K155X using the Quickchange II Site-directed mutagenesis kit (Stratagene). ascl1 was amplified from a whole embryo cDNA library and cloned into pCS2+. The SP6 Message machine kit (Ambion) was used to transcribe synthetic capped RNA. Embryos were injected with 50–100 pg of mRNA at the one - to two-cell stages. Injected embryos were raised at different time points ranging from 12 hpf to 4 dpf and fixed with 4% paraformaldehyde for in situ hybridization.
Antisense probes
The following antisense RNA probes were generous gifts: phox2b and phox2a (S. Guo), dbh and elavl3 (J-S Lee). Other probes (th, ascl1, tfap2a, hand2, gata3) were generated by amplifying the Danio rerio ORFs from a whole embryo cDNA library using PCR primers based on published GeneBank sequences. The PCR products were subcloned into the EcoRI/XhoI sites of pCS2+ vector. Following sequence verification, antisense riboprobes were generated by in vitro transcription with DIG RNA labeling kit Sp6/T7 (Roche).
Whole-mount in situ hybridization and immunofluorescence
Embryos at different developmental stages were collected and fixed in 4% (w/v) paraformaldehyde (Sigma). Whole-mount in situ hybridization was performed as described [58]. Images of zebrafish embryos were taken using an Olympus SZX12 microscope and a digital camera.
Intensity measurements
Intensity measurements were performed using Image-Pro Plus (IPP) software (MediaCybernetics, PA). Identical regions were selected using the rectangle tool set to a constant area. Mean optical density (MOD) and total per area (TPA) in boxed areas were used for pixel intensity. Differences between two groups were analyzed using Student's t-test.
Retinoic acid treatment
At 2 or 3 dpf embryos injected with a control MO and phox2b MO, were subjected to 13-cis retinoic acid treatment for 24 hrs at three concentrations 1 nM, 10 nM, and 100 nM diluted in PTU egg water. DMSO was used as a control. Embryos were then fixed and gene expression was analyzed using in-situ hybridization.
RT-PCR
Embryos were homogenized in TRIzol reagent by passing through a 22G needle. Total RNA was extracted as described in the manufacturer's instructions (Invitrogen). Primer pairs encompassing the full-length phox2b gene were designed and used to amplify phox2b in the MO-injected embryos with the Superscript First strand synthesis kit (Invitrogen). 50 pg of 1st strand cDNA was used to amplify phox2b using the Expand High Fidelity Plus PCR system (Roche).
Western blotting
Immunoblotting was performed according to standard methods. The following antibodies were used: anti-PHOX2B (Santa Cruz, N14 cat# sc-48627), anti-ASCL1 (Santa Cruz, sc-28688 ASCL1-H56). Anti-beta tubulin antibody (Abcam 6046-100) was used as a loading control.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. BagatellR, Beck-PopovicM, LondonWB, ZhangY, PearsonAD, et al. (2009) Significance of MYCN amplification in international neuroblastoma staging system stage 1 and 2 neuroblastoma: a report from the International Neuroblastoma Risk Group database. J Clin Oncol 27 : 365–370.
2. ParkJR, EggertA, CaronH (2008) Neuroblastoma: biology, prognosis, and treatment. Pediatr Clin North Am 55 : 97–120, x.
3. AmielJ, LaudierB, Attie-BitachT, TrangH, de PontualL, et al. (2003) Polyalanine expansion and frameshift mutations of the paired-like homeobox gene PHOX2B in congenital central hypoventilation syndrome. Nat Genet 33 : 459–461.
4. McConvilleC, ReidS, BaskcombL, DouglasJ, RahmanN (2006) PHOX2B analysis in non-syndromic neuroblastoma cases shows novel mutations and genotype-phenotype associations. Am J Med Genet A 140 : 1297–1301.
5. MosseYP, LaudenslagerM, KhaziD, CarlisleAJ, WinterCL, et al. (2004) Germline PHOX2B mutation in hereditary neuroblastoma. Am J Hum Genet 75 : 727–730.
6. PattynA, MorinX, CremerH, GoridisC, BrunetJF (1999) The homeobox gene Phox2b is essential for the development of autonomic neural crest derivatives. Nature 399 : 366–370.
7. TrochetD, BourdeautF, Janoueix-LeroseyI, DevilleA, de PontualL, et al. (2004) Germline mutations of the paired-like homeobox 2B (PHOX2B) gene in neuroblastoma. Am J Hum Genet 74 : 761–764.
8. Weese-MayerDE, Berry-KravisEM, ZhouL, MaherBS, SilvestriJM, et al. (2003) Idiopathic congenital central hypoventilation syndrome: analysis of genes pertinent to early autonomic nervous system embryologic development and identification of mutations in PHOX2b. Am J Med Genet A 123A: 267–278.
9. Bronner-FraserM (2003) Hierarchy of events regulating neural crest induction. Harvey Lect 99 : 129–144.
10. RohrerH (2011) Transcriptional control of differentiation and neurogenesis in autonomic ganglia. Eur J Neurosci 34 : 1563–1573.
11. Le DouarinNM, CreuzetS, CoulyG, DupinE (2004) Neural crest cell plasticity and its limits. Development 131 : 4637–4650.
12. MorikawaY, ZehirA, MaskaE, DengC, SchneiderMD, et al. (2009) BMP signaling regulates sympathetic nervous system development through Smad4-dependent and -independent pathways. Development 136 : 3575–3584.
13. PattynA, MorinX, CremerH, GoridisC, BrunetJF (1997) Expression and interactions of the two closely related homeobox genes Phox2a and Phox2b during neurogenesis. Development 124 : 4065–4075.
14. GuillemotF, LoLC, JohnsonJE, AuerbachA, AndersonDJ, et al. (1993) Mammalian achaete-scute homolog 1 is required for the early development of olfactory and autonomic neurons. Cell 75 : 463–476.
15. StankeM, JunghansD, GeissenM, GoridisC, ErnsbergerU, et al. (1999) The Phox2 homeodomain proteins are sufficient to promote the development of sympathetic neurons. Development 126 : 4087–4094.
16. HirschMR, TiveronMC, GuillemotF, BrunetJF, GoridisC (1998) Control of noradrenergic differentiation and Phox2a expression by MASH1 in the central and peripheral nervous system. Development 125 : 599–608.
17. LoL, TiveronMC, AndersonDJ (1998) MASH1 activates expression of the paired homeodomain transcription factor Phox2a, and couples pan-neuronal and subtype-specific components of autonomic neuronal identity. Development 125 : 609–620.
18. LucasME, MullerF, RudigerR, HenionPD, RohrerH (2006) The bHLH transcription factor hand2 is essential for noradrenergic differentiation of sympathetic neurons. Development 133 : 4015–4024.
19. HowardM, FosterDN, CserjesiP (1999) Expression of HAND gene products may be sufficient for the differentiation of avian neural crest-derived cells into catecholaminergic neurons in culture. Dev Biol 215 : 62–77.
20. LimKC, LakshmananG, CrawfordSE, GuY, GrosveldF, et al. (2000) Gata3 loss leads to embryonic lethality due to noradrenaline deficiency of the sympathetic nervous system. Nat Genet 25 : 209–212.
21. HolzschuhJ, Barrallo-GimenoA, EttlAK, DurrK, KnapikEW, et al. (2003) Noradrenergic neurons in the zebrafish hindbrain are induced by retinoic acid and require tfap2a for expression of the neurotransmitter phenotype. Development 130 : 5741–5754.
22. Barrallo-GimenoA, HolzschuhJ, DrieverW, KnapikEW (2004) Neural crest survival and differentiation in zebrafish depends on mont blanc/tfap2a gene function. Development 131 : 1463–1477.
23. FrancisNJ, LandisSC (1999) Cellular and molecular determinants of sympathetic neuron development. Annu Rev Neurosci 22 : 541–566.
24. YangC, KimHS, SeoH, KimCH, BrunetJF, et al. (1998) Paired-like homeodomain proteins, Phox2a and Phox2b, are responsible for noradrenergic cell-specific transcription of the dopamine beta-hydroxylase gene. J Neurochem 71 : 1813–1826.
25. GoridisC, RohrerH (2002) Specification of catecholaminergic and serotonergic neurons. Nat Rev Neurosci 3 : 531–541.
26. PattynA, MorinX, CremerH, GoridisC, BrunetJF (1999) The homeobox gene Phox2b is essential for the development of autonomic neural crest derivatives. Nature 399 : 366–370.
27. TiveronMC, HirschMR, BrunetJF (1996) The expression pattern of the transcription factor Phox2 delineates synaptic pathways of the autonomic nervous system. J Neurosci 16 : 7649–7660.
28. AlamG, CuiH, ShiH, YangL, DingJ, et al. (2009) MYCN promotes the expansion of Phox2B-positive neuronal progenitors to drive neuroblastoma development. Am J Pathol 175 : 856–866.
29. DubreuilV, HirschMR, PattynA, BrunetJF, GoridisC (2000) The Phox2b transcription factor coordinately regulates neuronal cell cycle exit and identity. Development 127 : 5191–5201.
30. ReiffT, TsarovinaK, MajdazariA, SchmidtM, del PinoI, et al. (2010) Neuroblastoma phox2b variants stimulate proliferation and dedifferentiation of immature sympathetic neurons. J Neurosci 30 : 905–915.
31. CargninF, FloraA, Di LascioS, BattaglioliE, LonghiR, et al. (2005) PHOX2B regulates its own expression by a transcriptional auto-regulatory mechanism. J Biol Chem 280 : 37439–37448.
32. MateraI, BachettiT, PuppoF, Di DucaM, MorandiF, et al. (2004) PHOX2B mutations and polyalanine expansions correlate with the severity of the respiratory phenotype and associated symptoms in both congenital and late onset Central Hypoventilation syndrome. J Med Genet 41 : 373–380.
33. TrochetD, O'BrienLM, GozalD, TrangH, NordenskjoldA, et al. (2005) PHOX2B genotype allows for prediction of tumor risk in congenital central hypoventilation syndrome. Am J Hum Genet 76 : 421–426.
34. van LimptV, SchrammA, van LakemanA, SluisP, ChanA, et al. (2004) The Phox2B homeobox gene is mutated in sporadic neuroblastomas. Oncogene 23 : 9280–9288.
35. JenningsLJ, YuM, RandCM, KravisN, Berry-KravisEM, et al. (2012) Variable human phenotype associated with novel deletions of the PHOX2B gene. Pediatr Pulmonol 47 : 153–161.
36. RaabeEH, LaudenslagerM, WinterC, WassermanN, ColeK, et al. (2008) Prevalence and functional consequence of PHOX2B mutations in neuroblastoma. Oncogene 27 : 469–476.
37. BachettiT, MateraI, BorghiniS, Di DucaM, RavazzoloR, et al. (2005) Distinct pathogenetic mechanisms for PHOX2B associated polyalanine expansions and frameshift mutations in congenital central hypoventilation syndrome. Hum Mol Genet 14 : 1815–1824.
38. NagashimadaM, OhtaH, LiC, NakaoK, UesakaT, et al. (2012) Autonomic neurocristopathy-associated mutations in PHOX2B dysregulate Sox10 expression. J Clin Invest 122 : 3145–3158.
39. StewartRA, LeeJS, LachnitM, LookAT, KankiJP, et al. (2010) Studying peripheral sympathetic nervous system development and neuroblastoma in zebrafish. Methods Cell Biol 100 : 127–152.
40. GuoS, BrushJ, TeraokaH, GoddardA, WilsonSW, et al. (1999) Development of noradrenergic neurons in the zebrafish hindbrain requires BMP, FGF8, and the homeodomain protein soulless/Phox2a. Neuron 24 : 555–566.
41. ElworthyS, PintoJP, PettiferA, CancelaML, KelshRN (2005) Phox2b function in the enteric nervous system is conserved in zebrafish and is sox10-dependent. Mech Dev 122 : 659–669.
42. BerghmansS, MurpheyRD, WienholdsE, NeubergD, KutokJL, et al. (2005) tp53 mutant zebrafish develop malignant peripheral nerve sheath tumors. Proc Natl Acad Sci U S A 102 : 407–412.
43. HendershotTJ, LiuH, ClouthierDE, ShepherdIT, CoppolaE, et al. (2008) Conditional deletion of Hand2 reveals critical functions in neurogenesis and cell type-specific gene expression for development of neural crest-derived noradrenergic sympathetic ganglion neurons. Dev Biol 319 : 179–191.
44. TsarovinaK, PattynA, StubbuschJ, MullerF, van der WeesJ, et al. (2004) Essential role of Gata transcription factors in sympathetic neuron development. Development 131 : 4775–4786.
45. KelshRN, EisenJS (2000) The zebrafish colourless gene regulates development of non-ectomesenchymal neural crest derivatives. Development 127 : 515–525.
46. DuttonKA, PaulinyA, LopesSS, ElworthyS, CarneyTJ, et al. (2001) Zebrafish colourless encodes sox10 and specifies non-ectomesenchymal neural crest fates. Development 128 : 4113–4125.
47. MarusichMF, FurneauxHM, HenionPD, WestonJA (1994) Hu neuronal proteins are expressed in proliferating neurogenic cells. J Neurobiol 25 : 143–155.
48. HowardMJ, StankeM, SchneiderC, WuX, RohrerH (2000) The transcription factor dHAND is a downstream effector of BMPs in sympathetic neuron specification. Development 127 : 4073–4081.
49. StankeM, StubbuschJ, RohrerH (2004) Interaction of Mash1 and Phox2b in sympathetic neuron development. Mol Cell Neurosci 25 : 374–382.
50. RohrerH, ThoenenH (1987) Relationship between differentiation and terminal mitosis: chick sensory and ciliary neurons differentiate after terminal mitosis of precursor cells, whereas sympathetic neurons continue to divide after differentiation. J Neurosci 7 : 3739–3748.
51. TsarovinaK, SchellenbergerJ, SchneiderC, RohrerH (2008) Progenitor cell maintenance and neurogenesis in sympathetic ganglia involves Notch signaling. Mol Cell Neurosci 37 : 20–31.
52. DiCicco-BloomE, Townes-AndersonE, BlackIB (1990) Neuroblast mitosis in dissociated culture: regulation and relationship to differentiation. J Cell Biol 110 : 2073–2086.
53. PotznerMR, TsarovinaK, BinderE, Penzo-MendezA, LefebvreV, et al. (2010) Sequential requirement of Sox4 and Sox11 during development of the sympathetic nervous system. Development 137 : 775–784.
54. TrochetD, HongSJ, LimJK, BrunetJF, MunnichA, et al. (2005) Molecular consequences of PHOX2B missense, frameshift and alanine expansion mutations leading to autonomic dysfunction. Hum Mol Genet 14 : 3697–3708.
55. TrochetD, de PontualL, StrausC, GozalD, TrangH, et al. (2008) PHOX2B germline and somatic mutations in late-onset central hypoventilation syndrome. Am J Respir Crit Care Med 177 : 906–911.
56. CrossSH, MorganJE, PattynA, WestK, McKieL, et al. (2004) Haploinsufficiency for Phox2b in mice causes dilated pupils and atrophy of the ciliary ganglion: mechanistic insights into human congenital central hypoventilation syndrome. Hum Mol Genet 13 : 1433–1439.
57. Westerfield M (1995) The zebrafish book, A guide for the laboratory use of zebrafish (Danio rerio). Eugene, Oregon: University of Oregon Press.
58. ThisseC, ThisseB, SchillingTF, PostlethwaitJH (1993) Structure of the zebrafish snail1 gene and its expression in wild-type, spadetail and no tail mutant embryos. Development 119 : 1203–1215.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2013 Číslo 6
Nejčtenější v tomto čísle
- BMS1 Is Mutated in Aplasia Cutis Congenita
- Sex-stratified Genome-wide Association Studies Including 270,000 Individuals Show Sexual Dimorphism in Genetic Loci for Anthropometric Traits
- Distinctive Expansion of Potential Virulence Genes in the Genome of the Oomycete Fish Pathogen
- Distinct Neuroblastoma-associated Alterations of Impair Sympathetic Neuronal Differentiation in Zebrafish Models