
The Regulatory T Cell Lineage Factor Foxp3 Regulates Gene Expression through Several Distinct Mechanisms Mostly Independent of Direct DNA Binding
The suppressive activity of regulatory T cells provides the immune system with a mechanism to prevent detrimental immune responses, such as autoimmunity, attack of the beneficial commensal microbiota and rejection of the fetus. Intriguingly, expression of a single lineage factor Foxp3 is sufficient to completely reprogram T cells from a pro-inflammatory to a suppressive phenotype. Here, we show that Foxp3 alters the expression of thousands of genes through several independent mechanisms. In many cases, its own ability to bind to DNA appears to be dispensable, but rather it binds indirectly to the DNA by interaction with other transcription factors. Foxp3 then in turn recruits other proteins that affect gene expression through chromatin modification. For example, Foxp3 indirectly binds to the IL-2 promoter via interaction with the transcriptional activators c-Rel, AML-1 and NFAT. This leads to the Foxp3 mediated recruitment of class I histone deacetylases HDAC1, 2 and 3, which in turn counteracts the activation-induced hyper-acetylation of the promoter, thereby switching the gene off. In a way, Foxp3 hijacks pre-existing regulatory mechanism to reverse the transcriptional expression status of the target gene. By dissecting Foxp3 on a molecular level, we also show that this is only one of several independent mechanism utilised by Foxp3.
Published in the journal:
. PLoS Genet 11(6): e32767. doi:10.1371/journal.pgen.1005251
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1005251
Summary
The suppressive activity of regulatory T cells provides the immune system with a mechanism to prevent detrimental immune responses, such as autoimmunity, attack of the beneficial commensal microbiota and rejection of the fetus. Intriguingly, expression of a single lineage factor Foxp3 is sufficient to completely reprogram T cells from a pro-inflammatory to a suppressive phenotype. Here, we show that Foxp3 alters the expression of thousands of genes through several independent mechanisms. In many cases, its own ability to bind to DNA appears to be dispensable, but rather it binds indirectly to the DNA by interaction with other transcription factors. Foxp3 then in turn recruits other proteins that affect gene expression through chromatin modification. For example, Foxp3 indirectly binds to the IL-2 promoter via interaction with the transcriptional activators c-Rel, AML-1 and NFAT. This leads to the Foxp3 mediated recruitment of class I histone deacetylases HDAC1, 2 and 3, which in turn counteracts the activation-induced hyper-acetylation of the promoter, thereby switching the gene off. In a way, Foxp3 hijacks pre-existing regulatory mechanism to reverse the transcriptional expression status of the target gene. By dissecting Foxp3 on a molecular level, we also show that this is only one of several independent mechanism utilised by Foxp3.
Introduction
TReg cells, a subtype of CD4+ T cells, provide the immune system with a checkpoint preventing detrimental immune responses [1]. They not only avert the development of autoimmunity and preclude the rejection of the fetus by the maternal immune system [2–4], but also modulate the immune system to counteract excessive inflammation in response to pathogens and commensal microorganisms [5]. The lineage factor Foxp3, a member of the forkhead winged helix family of DNA binding factors, plays a key role in both TReg cell lineage commitment and TReg cell maintenance [6, 7]. Expression of Foxp3 is essential for TReg cell differentiation irrespective of whether the T cells commit to the TReg lineage during their development in the thymus or if they are induced in the periphery at a later stage [8]. Foxp3 acts in conjunction with specific changes in DNA-methylation to drive full commitment to the TReg identity [9] Lack or loss of functional Foxp3 leads to widespread autoimmunity caused by the absence of TReg cells [2, 10]. In the absence of Foxp3 it is possible for ‘would-be’ TReg cells to develop but these lack suppressive activity and have a more Th2-like phenotype [11].
Despite its importance in the development and maintenance of TReg cells, the molecular mechanisms involved in Foxp3-mediated changes of the transcriptional program governing TReg cell lineage commitment are poorly understood. Foxp3 has been shown to form macromolecular complexes, yet their exact composition is unknown [12]. A study using an unbiased proteomics approach suggests that Foxp3 interacts with in excess of 300 proteins [13]. It is unlikely that all of them bind Foxp3 at the same time and the composition, dynamics and function of complex formation remain to be resolved. For now, studies addressing individual interaction partners point to the complexity of Foxp3-mediated regulation of gene expression.
The forkhead domain (FKH) of Foxp3 has been shown to be important in its homo-dimerization and DNA binding [14]. The consensus sequence for Foxp3 DNA-binding appears to be rather degenerate and, as such, it has been proposed that Foxp3’s FKH has only low intrinsic affinity for DNA and therefore relies on hetero-dimerization with other transcription factors [15–17]. Indeed, Foxp3 interacts with nuclear factor of activated T cells (NFAT) through its FKH and forms heterodimers with Foxp1 through interactions with its leucine dimerization domain [16, 18]. It also interacts with Ikaros family member Eos and AML1 [19, 20], as well as the transcription factors GATA3, c-Rel and RORγT [13, 21, 22]. Further complexity is introduced by Foxp3's reported association with chromatin modifiers such as the class II histone deacetylases (HDAC) 7 and 9 as well as the histone acetyltransferase TIP-60 [23]. In most cases, the modes of interaction and their functional roles remain to be resolved.
Ectopic expression of Foxp3 in TH cells (TH::Foxp3) is sufficient to induce a TReg cell-like phenotype [24]. These TH::Foxp3 cells alter their transcriptional program to up-regulate many genes characteristic of TReg cells such as Cd25, Ctla4, Tnfrsf18 and Lag3 and to suppress pro-inflammatory genes such as Il2 [6, 24, 25]. Notably, TH::Foxp3 cells not only have suppressive activity ex vivo, but can also compensate for the absence of TReg cells in vivo [26]. Thus, ectopic expression of Foxp3 in TH cells provides us with a system to study the transcriptional changes mediated by this protein in the lineage commitment of TReg cells.
Here we explored how Foxp3 regulates gene expression on a molecular level. We analysed the transcriptomes of TH cells ectopically expressing either wild type or mutant Foxp3 and showed that Foxp3 is involved in the activation and suppression of genes equally. We find that Foxp3 lacking the DNA binding FKH still retains the capability to regulate a large number of its target genes while, in contrast, deletion of a proline-rich interaction domain (ProR) leads to a loss of most Foxp3-mediated gene regulation. Due to its extremely high proline content, this region is disordered, making it impossible to deduce any hints as to potential functional domains based on structural predictions. Instead, we used a variety of comparative genomics approaches to determine subdomains or motifs that are evolutionarily conserved due to their functional importance. Our analysis of the effects of deletion of these motifs on Foxp3-mediated transcriptional regulation suggests that subdomains within the ProR of Foxp3 are involved in several independent mechanisms of transcriptional control, regulating distinct sets of genes; one such mechanism is the recruitment of class I HDACs to Foxp3 target genes such as Il2 and Ifng. In summary, our data suggest that Foxp3 acts as a scaffold for the recruitment of interaction partners with diverse functions. Some of them target the complex to the relevant control elements of the genes to be regulated, while others provide the complex with effector functions such as the ability to modify chromatin to modulate gene expression.
Results
Identification of TReg relevant, Foxp3-regulated genes
To study the role of the functional domains of Foxp3 in gene regulation, we transduced naive CD4+CD25- TH cells with retroviral expression vectors encoding wild-type Foxp3, Foxp3 mutants or a GFP control. We ensured equivalent protein expression levels of all Foxp3 constructs used here (data shown in the relevant sections). The expression of many Foxp3-regulated genes is known to be influenced by cellular activation [15], so we rested the cells for 48h post transduction before purifying the mRNA for transcriptome analysis by RNA-seq. We obtained, on average, 1.0x108 reads per library with 7.7x107 properly paired reads mapping unambiguously to the mouse genome (GRCm38v75; S1 Table).
We determined the number of reads that mapped to each gene within the mouse genome and used these counts to identify genes that were significantly differentially expressed between samples. We then filtered the lists for biologically relevant changes. The expression level of each gene within a cell population falls within a bimodal distribution (S1A Fig) [27]. One mode contains genes without any expression (not expressed, NE) and those which are only expressed at a very low level (lowly expressed, LE, < 1 transcript per cell). The other mode contains highly expressed genes (HE) and it is this population that has been proposed to be functionally relevant [27]. We classified the gene expression of our samples according to this bimodal distribution (S1B Fig) and used this to filter the lists of differentially expressed genes. For each pairwise comparison, genes that were classified as NE or LE in both conditions were ignored, as they are likely to be representative of transcriptional noise rather than biologically relevant changes [27].
Foxp3 transduced (TH::Foxp3) cells are sufficient to compensate for the absence of TReg cells both in vitro and in vivo [6, 28] and this provides a tractable system in which to study Foxp3’s mechanisms of gene regulation. However, the transcriptional program of Foxp3-transduced TH cells is different from that of ex vivo TReg cells. We found that 8,128 genes were differentially expressed between naive TH and TReg cells using existing RNA-seq data [29], whereas 5,136 genes were differentially expressed between TH::control and TH::Foxp3 cells (S2A Fig). To ensure that we focussed in this study on Foxp3-regulated genes involved in TReg function we took the intersection of these sets, which represents 2,407 Foxp3-regulated, TReg-relevant genes. Expression of Foxp3 led to upregulation of 1,255 and downregulation of 1,152 TReg cell relevant genes (S2 Table) indicating a slight preference for Foxp3-upregulated genes within this list (P = 0.037, binomial test). The transcriptomic analyses presented here are robust to the choice of the TReg-relevant, Foxp3-regulated gene set; we also performed the same analysis of a gene set defined as above using 3,092 differentially expressed genes between TReg and naive TH cells from previously described microarray analyses [30], leading to 1,092 TReg-relevant Foxp3-regulated genes. The conclusions drawn from these analogous analyses were unchanged. The overlap between our set of 2,407 genes and this set of 1,092 genes was highly significant (p<1x1016, Fisher’s exact test, S2B Fig) and, since RNA-seq is typically more sensitive than microarray analysis [31], we proceeded with the larger gene set.
We analysed the list of 2,407 Foxp3-regulated, TReg-relevant genes to determine the gene ontology (GO) terms that are enriched within it (S3 Table). These included multiple immunology-related terms. Twenty-five percent (609) of the genes are classified as immunology-related by the ImmPort project [32]. This is a significant enrichment of immunology-related genes (P = 2.11×10–69, Fisher’s exact test).
Separation of Foxp3’s DNA binding function and its role in complex formation
Previously, much emphasis has been put on the importance of the FKH of Foxp3 since mutations that lead to immunodysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome were identified within the FKH [14] and mutations disrupting FKH function caused dysregulation of key TReg functional genes [18]. Indeed, the FKH has been shown to be involved in key functions of Foxp3 such as DNA-binding, asymmetrical homo-dimerization and hetero-dimerization with NFAT [14, 16, 18].
It is possible that point mutations within the FKH [18] are more widely disruptive to the structure of Foxp3, and so to separate FKH-mediated DNA-binding of Foxp3 from its role in ProR-mediated complex formation we generated three mutants. In Foxp3ΔProR we deleted exons 1 to 3, which encode the ProR (Fig 1A). Since the FKH contains nuclear localisation sequences (NLS) [33], we made two separate FKH-deletion mutants, one with the entire domain deleted (Foxp3ΔFKH) and one in which we added an unrelated SV40-derived NLS to the C-terminus of the protein (Foxp3∆FKHnls). Analyses of the transcriptional profiles of Foxp3-regulated genes by hierarchical clustering (Fig 1B) and principal component analysis (PCA) (Fig 1C) illustrate the differences in the transcriptional changes caused by mutation of Foxp3.
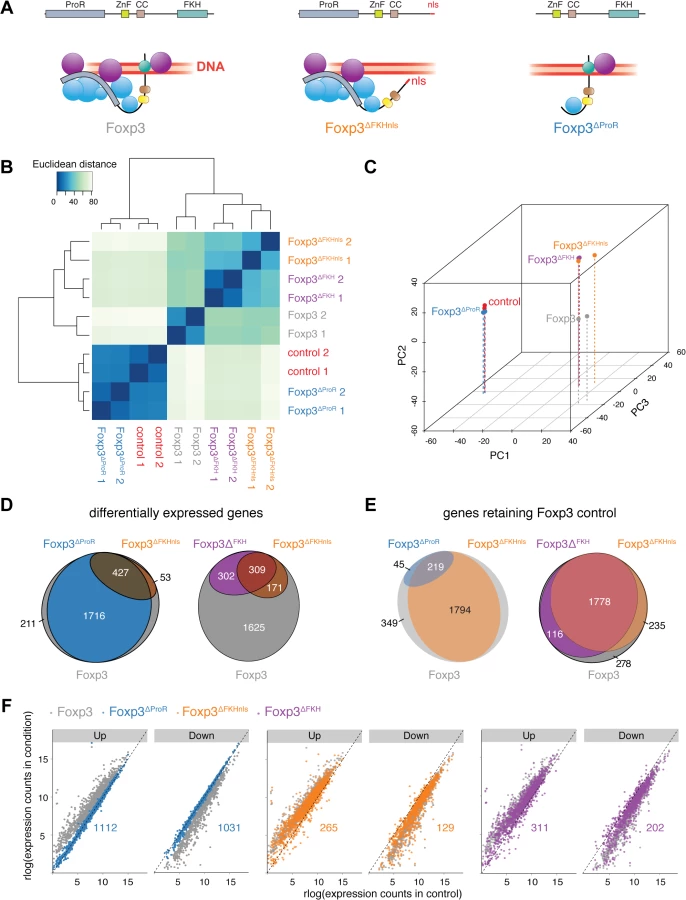
When assessing transcriptional changes caused by Foxp3 mutants we considered that enhanced upregulation of Foxp3-induced genes or downregulation of Foxp3-suppressed genes by Foxp3 mutants is unlikely to reflect a failure in Foxp3-mediated regulation. Thus, all those genes that exhibited such changes compared to their expression in TH::Foxp3 cells were classed as retaining Foxp3 control (S2C Fig). Conversely, genes expressed at levels closer to those seen in TH::control cells rather than in TH::Foxp3 cells were classed as “dysregulated”.
Transcriptomic analysis highlights key role for Foxp3 ProR
The ProR plays a pivotal role in Foxp3-mediated gene expression control. The transcriptomes of TH::Foxp3∆ProR cells clustered with those of the TH::control cells, suggesting a dramatic loss in Foxp3-mediated regulation in the absence of the ProR (Fig 1B and 1C) despite having equivalent Foxp3 protein levels as wild-type (S3A Fig). Differential gene expression analysis showed that the majority of relevant, Foxp3-regulated genes (2,143 out of 2,407 genes) were significantly differentially expressed in TH::Foxp3∆ProR cells compared with wild-type TH::Foxp3 cells (Fig 1D). In contrast, deletion of the FKH domain (Foxp3∆FKHnls) led to differential expression of only 480 genes. Analysis of the direction of differential expression (S2C Fig) revealed that only 264/2,407 genes (11%) retained Foxp3 control in TH::Foxp3∆ProR cells (Fig 1E), while 2,013 genes (84%) retained Foxp3 control in TH::Foxp3∆FKHnls cells. We compared the numbers of Foxp3-upregulated and Foxp3-downregulated genes with the distribution seen within the whole set of Foxp3-regulated, relevant genes (Fig 1F and Table 1). Both Foxp3∆FKHnls and Foxp3∆FKH preferentially dysregulated Foxp3-upregulated genes (P = 1.5×10–9 and 1.2×10–4 respectively, binomial tests). Collectively, these data indicate that the ProR plays a crucial role in Foxp3-dependent transcription, whereas the FKH is required for the regulation of fewer genes and appears dispensable in many cases.

The role of the FKH and Foxp3-mediated nuclear localisation
Previous work that disrupted the FKH domain of Foxp3 along with the C-terminal NLS found that this altered the nuclear localisation of the protein and prevented TReg suppressor function [34]. We used the Foxp3∆FKHnls and TH::Foxp3∆FKH mutants to explore the effects of presence or absence of an NLS alongside deletion of the FKH (Fig 2A).

Surprisingly, the transcriptomes of TH::Foxp3∆FKHnls and TH::Foxp3∆FKH cells clustered with those of the TH::Foxp3 cells (Fig 1B and 1C). We found that the majority of genes (1,778 out of 2,407) retained Foxp3-control in both TH::Foxp3∆FKH and TH::Foxp3∆FKHnls cells (Fig 1D and 1E). Flow cytometry analyses showed that the range of Foxp3 expression in cell populations transduced with each construct was comparable in both primary T cells (Fig 2B) and transfected HEK293T cells (Fig 2C). Furthermore, expression of Foxp3 in TH::Foxp3 cells was only slightly higher than that found in primary regulatory T cells (S4 Fig). Intracellular staining of the Foxp3 mutants ectopically expressed in HEK293T cells revealed that the absence of the FKH and NLS did not completely abrogate Foxp3 nuclear localisation, with Foxp3 in both cytoplasm and nucleus (Figs 2D and S5). The sets of genes dysregulated by the two FKH mutants do not overlap exactly (Fig 1D and 1E). This may be due to subtle differences in subcellular localisation of these mutants and suggests that the correct nuclear concentration of Foxp3 is important for gene regulation.
Although the majority of genes are not dysregulated upon deletion of the FKH, this does not mean that this domain is unimportant for Foxp3’s functions. Indeed, we find that genes encoding proteins known to be essential for TReg function (CTLA4, CD25, GITR, LAG3) are affected by the absence of the FKH in both TH::Foxp3∆FKH and TH::Foxp3∆FKHnls cells (Fig 2E), albeit the downregulation of CD62L is maintained in both TH::Foxp3∆FKH and TH::Foxp3∆FKHnls cells.
Comparative genomics suggests subdomains in ProR
We next sought to identify subregions of the Foxp3 ProR that are likely to be important in its control of gene expression. As the ProR is structurally disordered one cannot discern functional domains based on structural cues and so we developed an alternative strategy using comparative genomics. Evolution tends to conserve functionally important residues [28], clusters of which are indicative of functional subdomains. A comparison of multiple-sequence alignments of the amino-acid sequence of Foxp3 revealed that the sequence homology of orthologs across all vertebrates was restricted to the C-terminal half of the protein which contains the FKH, coiled coil domain (CC) and zinc finger domain (ZnF). The sequence-space occupied by the ProR in mammals was also present in non-mammalian vertebrates although in these species this region is not conserved (Fig 3A, top) and has low proline content (~5% vs ~20%). Stretches of proline-rich content first appeared in non-placental mammals (Fig 3A, top).
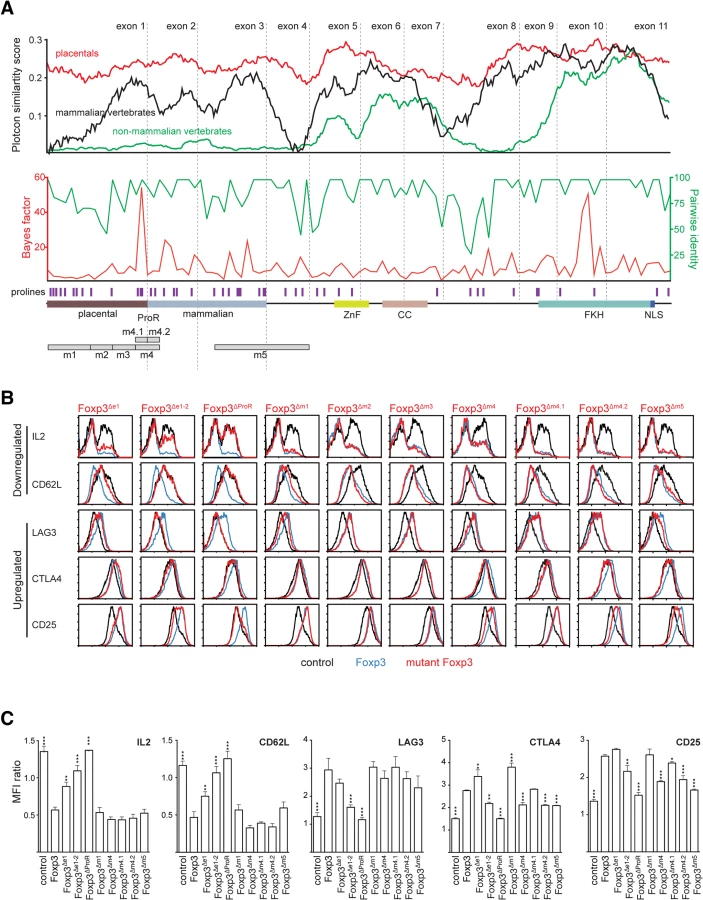
A codon alignment of Foxp3 identified stretches of residues where the number of synonymous (silent) mutations significantly exceeded that of non-synonymous (replacement) mutations (Bayes factor). We calculated the moving average of the Bayes factor for the event that regions within Foxp3 are under purifying selection [28], as well as the moving average of pairwise identity. Several regions within Foxp3 had high values for both, indicating that they are under purifying selection (Fig 3A, middle). Notably, apart from a peak in the FKH, the strongest signals of selection were found in the ProR, suggesting that once this region was acquired, it remained under strong evolutionary constraints.
Based on our analyses, the part of the ProR unique to placental mammals can be divided into 4 distinct regions (m1-m4), each of which is framed by prolines (Figs 3A, bottom, and S3B). Regions m1 and m3 have relatively high pairwise identity, but low average Bayes factor. In region m2, both the average pairwise identity and Bayes factor are low, indicating that this region is not under selective pressure. In contrast, region m4 is highly conserved and has a high average Bayes factor, suggesting that it is under strong purifying selection and thus likely to be of functional importance. An additional region, m5 which partially overlaps the ProR, was identified as highly conserved in all mammals.
Candidate protein analysis suggests distinct roles for ProR subdomains
Based on these findings we generated Foxp3 mutants in which ProR subdomains were either deleted or replaced with short stretches of alanines, ectopically expressed them in TH cells and used flow cytometry to assess their importance in the expression of proteins known to be important in TReg cell function (Fig 3B and 3C). We also assessed the effect of the removal of exon 1, exons 1 to 2 or the entire ProR. We ensured that the Foxp3 mutants were expressed at similar levels and localized to the nucleus of the transduced cells (S3A Fig). These results are summarized in Fig 4A.

Removal of the ProR led to a complete loss of Foxp3-mediated regulation of all TReg relevant proteins analysed (Foxp3ΔProR). The deletion of the first two exons had a similar effect, although the dysregulation of CTLA4 and CD25 was less marked (Foxp3Δe1-2). When we deleted exon 1, which is only conserved in placental mammals, Foxp3-mediated regulation of IL-2, CD62L and LAG-3 was impaired (Foxp3Δe1). The regulation of CTLA4 and CD25 remained unaffected by this mutation and ∆m1 (Foxp3Δe1, Foxp3Δm1). In contrast, replacement of region m4 with a stretch of five alanine residues severely affected Foxp3’s ability to regulate CTLA4 and CD25 (Foxp3Δm4), but did not affect the regulation of IL-2, CD62L and LAG3. Replacement of regions m2 and m3 with stretches of alanine residues had no effect on the regulation of any of these proteins. When we replaced region m5, we found that it only affected the expression of candidate proteins upregulated by Foxp3 (Foxp3Δm5).
Region m4 spans the junction between exons 1 and 2 but deletion of exon 1 did not dysregulate the proteins affected by removal of region m4. This implies functional importance of the second half of region m4, contained in exon 2 (Fig 4A). Thus we generated mutants with each half of m4 replaced separately by alanines (Foxp3Δm4.1 and Foxp3Δm4.2). The loss of function observed (Fig 3B and 3C) could be attributed to the four replaced amino acids (LVMV) in the N-terminal region of exon 2. This motif is fully conserved in all placentals (S6A Fig). qPCR analysis of target gene expression (S6B Fig) mirrored protein expression. The lack of overlap between the target genes affected by deletions of exon 1 and subdomain m4.2 suggests that they are regulated by two independent mechanisms (Fig 4A). Combined, these analyses suggest that the ProR mediates TReg cell-specific gene expression through more than one mechanism.
ProR subdomains confer distinct TReg functions
Foxp3∆ProR is unable to confer a TReg phenotype in both ex vivo suppression assays (Fig 4B) and in an in vivo model of autoimmune colitis induction (Fig 4C). However, only in vivo assessment revealed a functional role for the ProR subdomains (m1 to m5). Alanine replacement of region m4 or subdomain m4.2 led to the most severe impairment of the transduced cells in preventing weight loss (Fig 4C), confirming the importance of the LVMV motif for Foxp3 function. Deletion of region m1 led to a significant impairment of the transduced cells in preventing weight loss, albeit slightly less marked than that observed upon deletion of the entire exon 1. Alanine replacement of region m5 had a milder effect, not reaching statistical significance in our experiments (Fig 4C). While these assays confirm the importance of the subdomains for overall TReg function, they give no clues about the molecular mechanisms affected. The discrepancy between ex vivo and in vivo results is likely due to Foxp3 affecting TReg cell function via multiple mechanisms such as cell-cell interactions, homeostasis, tissue homing and effector function, which might not all be required in ex vivo assays.
Subdomains within the ProR link into distinct transcriptional regulatory mechanisms
A transcriptomic analysis of the effects of the subdomain deletions confirmed that the ProR links into several distinct mechanisms of gene regulation. Hierarchical clustering (Fig 4D) and PCA (Fig 4E) revealed that the transcriptomes of TH::Foxp3Δe1, TH::Foxp3Δm4.2 and TH::Foxp3Δm5 cells were much closer to that of TH::Foxp3 than to TH::Foxp3ΔProR cells. We examined the distribution of Foxp3-upregulated and Foxp3-downregulated genes within the lists of dysregulated genes for each mutant and compared these distributions with that within all 2,407 Foxp3-regulated genes (Table 1). Foxp3Δm4.2 appears to preferentially dysregulate Foxp3-downregulated genes. Deletion of region m5 affects the most genes (224/2407, Fig 4G; Foxp3∆m5), yet has the mildest phenotype in the in vivo model of autoimmune colitis induction (Fig 4C). In contrast, deletion of exon 1 which affects the least Foxp3 regulated genes results in a more marked phenotype in vivo (Fig 4C; Foxp3Δe1). Clearly, the number of genes that become dysregulated does not directly correlate with the severity of the in vivo phenotype and would require a much more detailed analysis to elucidate the role of the Foxp3 subdomains in the function of TReg cells. However, our approach allows us to draw firm conclusions regarding the control of sets of genes regulated by Foxp3. Alanine replacement of m4.2 dysregulated 210 genes. The effect of deletion of exon 1 was even smaller with only 21 genes becoming dysregulated in TH::Foxp3Δe1 cells (Fig 4F; Foxp3Δe1). Only one gene dysregulated by Foxp3∆m4.2 was also affected by deletion of exon 1 (Fig 4F) and all affected genes were strict subsets of the set of genes dysregulated in TH::Foxp3ΔProR. This strongly suggests that Foxp3 ProR region utilizes two independent mechanisms to regulate these gene sets.
Gene ontology enrichment analysis of lists of dysregulated genes
We analysed the lists of genes dysregulated by each Foxp3 mutant to determine the number of immunology-related genes as well as the GO-terms enriched within the lists (Tables 2 and S3). In each case there was a significant enrichment of immunology-related genes. We classified enriched GO terms as immunology-related or non-immunology-related and found that 12.7% of enriched terms were immunology related for the set of all Foxp3-regulated genes. The GO terms enriched within the lists of genes dysregulated by the Foxp3ΔFKH and Foxp3ΔFKHnls mutants contained a significantly higher proportion of immunology-related terms.

ProR-dependent, FKH-independent regulation of IL2
Many genes known to be important for TReg cell function are also upregulated by activation. For example, Ctla4 and Cd25 are upregulated in all activated TH cells [35, 36] but TReg cells constitutively express these genes irrespective of activation status [1, 6]. To avoid the confounding effects of activation we performed our analyses on resting cells. The expression of activation-induced IL-2 is known to be suppressed by Foxp3 in TReg cells [11, 37]. While in our transcriptomic data of resting cells Il2 transcript levels were low in all conditions, activation-induced IL2 expression was suppressed by Foxp3 at the protein level in re-activated cells (Figs 3B and 3C and 4A). We found that the ProR was indispensable for Foxp3-mediated IL2 suppression upon activation (Fig 5A; TH::Foxp3∆ProR), but contrary to results published previously [18], this suppressive effect was independent of the FKH (Fig 5A; TH::Foxp3∆FKHnls). We also observed downregulation of Il2 transcripts by Foxp3 when transduced cells were re-stimulated (Fig 5B). Il2 downregulation appeared to be very sensitive to changes in nuclear localisation efficiency since it is only dysregulated if the NLS is not present (Fig 5; TH::Foxp3ΔFKH). Deletion of exon 1 and exon1 to 2 had a partial effect (Fig 5; TH::Foxp3Δe1), while deletion of another part of the ProR, region m4.2 which had a marked effect on the behaviour of TH::Foxp3Δm4.2 cells in vivo (Fig 4C), had no effect on IL-2 expression (Fig 5; TH::Foxp3Δm4.2). These results, in combination with the fact that IL-2 plays a central role in the function and behaviour of TReg cells [37], led us to further investigate the regulation of IL-2 expression as an example of FKH-independent Foxp3-mediated control of gene expression involving the ProR. Foxp3-mediated histone-deacetylation of the Il2 and Ifng promoters has been proposed to downregulate the expression of these genes [24] and Foxp3 appears to interact with various chromatin modifiers. The class I histone-deacetylases (HDAC1, 2, 3), which are ubiquitously expressed in T cells, all appear to associate with the Foxp3 complex [13, 38]. However, little is known about the mode of interaction between Class I HDACs and Foxp3 and the mechanism by which this affects gene expression. The apparent importance of the ProR in the regulation of IL-2 led us to determine whether and how this domain might be involved in the interaction of Foxp3 with class I HDACs.

Foxp3 ProR directly interacts with class I HDACs
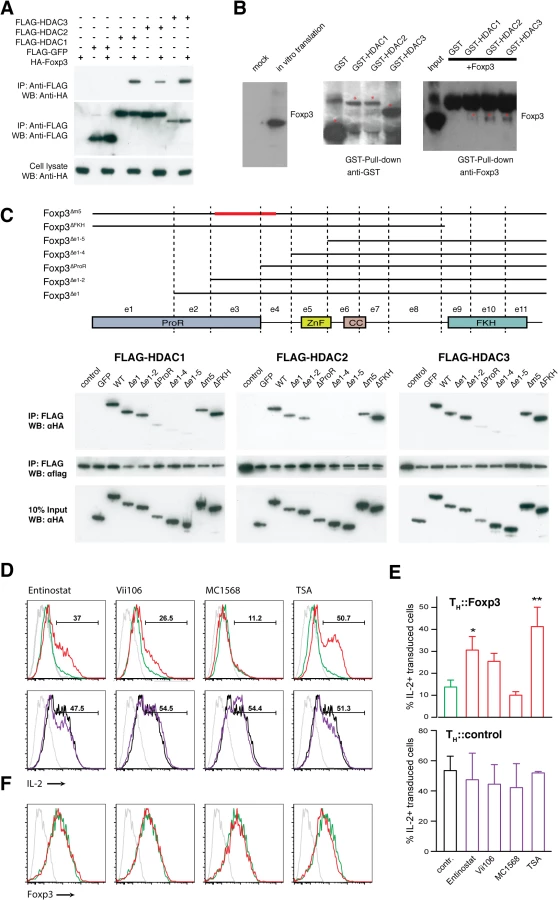
We found that the class I HDACs directly interact with the ProR of Foxp3. We confirmed the interaction of these proteins by co-expression of FLAG-tagged HDAC1, 2 or 3 and HA-tagged Foxp3 in HEK293T cells (Fig 6A) and in primary T cells (S7A Fig). Foxp3 could be co-precipitated with all three class I HDACs. To determine whether this interaction was direct we in vitro translated Foxp3 protein and incubated it with GST-tagged class I HDACs purified from E. coli. We found that all three HDACs can directly interact with Foxp3 (Fig 6B).
To map the interaction region, HA-tagged Foxp3 deletion mutants and FLAG-tagged class I HDACs were co-expressed in HEK293T cells (Fig 6C). Deletion of the FKH domain had no effect on the interaction nor did deletion of m5, which has been previously implicated in the interaction between Foxp3 and chromatin modifiers such as HDAC7 and Tip60 [23]. Deletion of ProR (exon 1–3) severely impaired the interaction with class I HDACs while deletion of exons 1–2 had only a partial effect. This might lead one to speculate that exon 3 is crucial for the interaction (Fig 6C). However, we found that deletion of Foxp3 exon 3 alone had little, if any, effect upon the interaction (S7B Fig). Together, our data suggest an altogether more complex mode of interaction between the ProR of Foxp3 and the class I HDACs. It is possible that the ProR contains multiple HDAC binding sites or that the binding of HDACs is strengthened by other molecules in the complex thus masking the effect of the deletion of their primary interaction site.
Foxp3’s suppression of IL2 is dependent on activity of class I HDACs
Inhibition of class I HDACs impairs Foxp3-mediated suppression of IL-2 expression. We treated TH::Control or TH::Foxp3 cells with HDAC inhibitors (Fig 6D and 6E). The class I HDAC inhibitors Entinostat and Vii106 impaired Foxp3-mediated suppression of IL-2. The pan-HDAC inhibitor TSA exhibited an even more marked effect while an inhibitor of class IIa HDACs, MC1568, had no effect on IL-2 regulation (Fig 6D and 6E). These effects were not due to downregulation of Foxp3 as there was no difference in Foxp3 levels between HDAC inhibitor treated and untreated cells (Fig 6F). None of the HDAC inhibitor treatments had an obvious effect on IL-2 in TH::control cells (Fig 6D and 6E). Our data shows that inhibition of class I HDACs activity impairs the suppression of IL-2 expression by Foxp3. We therefore decided to investigate whether the interaction between Foxp3 and class I HDACs facilitates their recruitment to the Il2 promoter. This in turn might explain how Foxp3 binding to the Il2 promoter leads to its de-acetylation thereby preventing activation-induced upregulation of Il2 [24]. As we could not exclude off target effects of the HDAC inhibitors, we also knocked down the class I HDACs using a cocktail of HDAC1, 2 and 3 siRNAs. Similar to our findings using the inhibitors we found a marked effect on IL-2 expression (S8 Fig).
Foxp3 recruitment to the Il2 promoter is FKH-independent
We found that Foxp3 is recruited effectively to the Il2 promoter in the absence of the FKH (Fig 7A–7C). This is contrary to the previous suggestion that Il2 regulation requires cooperative DNA binding via the heterodimerization of the Foxp3 FKH with NFAT [18]. We arbitrarily selected three sites upstream of the Il2 transcription start site for analysis by ChIP-qPCR (Fig 7A). Site A comprises the proximal part of the Il2 promoter and contains the binding sites of a multitude of transcription factors while site B is more distal with two AML1 binding sites [19]. Site C is intended to be a control just outside the Il2 promoter, and contains no obvious transcription factor binding sites. Consistent with previous work [24], we found that the recruitment of Foxp3 to Il2 promoter sites A and B is activation dependent (S7C–S7E Fig). ChIP-qPCR revealed that upon activation HA-Foxp3∆ProR and HA-Foxp3∆FKHnls were enriched at the Il2 promoter A and B sites to the same extent as wild type Foxp3, but we found no enrichment at the C site or at the Cd3ε promoter which is not regulated by Foxp3 (Fig 7B) [39]. The expression of tagged proteins was confirmed by Western blotting (Fig 7C). While these data do not preclude a role for the FKH in modulating Foxp3 binding to the Il2 promoter per se, our results cast doubt on this being the main mechanism of Foxp3 recruitment. The interaction of Foxp3 with other Il2-regulating transcription factors including AP-1, C-Rel and AML1 may be responsible for FKH-independent recruitment [19, 21, 40].

Foxp3 mediated deacetylation of the Il2 promoter requires ProR
The ProR is necessary for the recruitment of HDACs and subsequent deacetylation of the Il2 promoter during activation. HDAC1, 2 and 3 were equally enriched at Il2 promoter site A in TH::HA-Foxp3 and TH::HA-Foxp3∆FKHnls cells compared with TH::HA-GFP cells, but their enrichment was completely impaired in the absence of the ProR (Fig 7D). A similar result was observed at site B for HDAC2 (Fig 7E). As expected, we did not observe a significant enrichment of HDACs at site C nor at the Cd3ε promoter (Fig 7F and 7G). Expression levels of HDAC1, 2, 3 were similar in TH::HA-Foxp3 and TH::HA-GFP control cells (S7F and S7G Fig), so these effects were not due to the upregulation of HDACs by Foxp3.
Consistent with previous findings [41, 42], the Il2 promoter became hyperacetylated upon activation (Fig 7H). This was especially pronounced at the most proximal site A, but appeared to extend beyond the promoter itself to site C. The constitutively expressing gene Cd3ε promoter was highly acetylated regardless of activation state. The presence of HA-Foxp3 or HA-Foxp3∆FKHnls counteracted activation-induced acetylation of the Il2 promoter, but had no effect on the status of the Cd3ε promoter. In contrast, HA-Foxp3∆ProR was unable to abrogate activation-induced acetylation at the Il2 promoter (Fig 7I).
Importantly, this mechanism of transcriptional control by Foxp3 is not unique to Il2; we also observed analogous results at the promoter of the gene encoding IFN-γ. As was the case for Il2, Foxp3-mediated suppression of Ifng in activated T cells was lost upon deletion of the FKH domain but recovered upon addition of an NLS (S9A Fig). Both HA-Foxp3∆ProR and HA-Foxp3∆FKHnls were equally recruited to the Ifng promoter though at slightly lower levels than HA-Foxp3 (S9B Fig). HDAC2 and 3 were found to be enriched at the Ifng promoter in the presence of HA-Foxp3 or HA-Foxp3∆FKHnls while the enrichment was lost upon deletion of the ProR (S9C Fig) and therefore deacetylation did not occur for this mutant (S9D Fig).
Taken together, our data demonstrate that Foxp3 can be recruited to promoters in an FKH-independent fashion. The binding of Foxp3 leads to a ProR-dependent recruitment of class I HDACs, which in turn counteracts activation-induced hyperacetylation, thus inhibiting induction of gene expression.
Discussion
Arguably the most important finding of our study is that the various domains and subdomains of Foxp3 fulfil distinct, discernible functions in gene regulation. The vast majority of Foxp3-regulated genes are affected by loss of the ProR. It has been previously suggested that structural flexibility caused by the high proline content of the ProR might be key to its role in complex formation with a multitude of interaction partners [43, 44]. Indeed, we find clusters of residues under purifying selection, which we believe to represent conserved binding sites for a variety of interaction partners while the proline residues at cluster boundaries provide flexibility for dynamic complex formation. Our finding that selective deletion of these putative subdomains affects different, largely non-overlapping sets of genes suggests that Foxp3 utilizes different mechanistic pathways to regulate gene expression. Our data imply that the ProR links Foxp3 to at least three independent mechanism of regulation: one involves the subdomain encoded by exon 1, another requires a four amino-acid motif within exon 2 (m4.2) and a third is a more complex association of class I HDACs with the entire ProR.
Surprisingly, the FKH appeared to be dispensable for the transcriptional control of most Foxp3 regulated TReg relevant genes. However, one has to be very cautious when extending our findings to draw conclusions regarding the relative importance to TReg cell function of individual functional domains of Foxp3. It would be naive to conclude that the FKH is less functionally important than any other domain, because its deletion ‘only’ dysregulates the expression of 16% (395) of Foxp3 regulated genes. Within this FKH-dependent set are genes with known TReg function such as Ctla4. A conditional knockout of this single gene within TReg cells results in a complete loss of regulatory function and an IPEX-like phenotype [45]. Furthermore, the greater proportion of immunology-related GO terms enriched within the lists of genes dysregulated by deletion of the FKH may reflect the fact that the role of the FKH is more focused on regulating a greater breadth of immune-related functions than the ProR. The first IPEX mutations described were located in the FKH domain of human Foxp3 [46] and we have demonstrated previously that TH::Foxp3ΔFKH cells completely fail to compensate for the lack of TReg cells in mice [28]. Our findings here are consistent with IPEX mutations within the FKH domain causing pathological impairment of TReg function. IPEX mutations have now been found throughout Foxp3 with many of them occurring within the ProR [46, 47]. It is noteworthy that those IPEX mutations that destabilize Foxp3 on the mRNA or protein level have the most severe phenotypes as they lead to complete loss of Foxp3 function [46]. This is in agreement with our proposal that Foxp3 acts through multiple different mechanisms to exert its control upon distinct sets of genes.
We worried that deletion of the FKH may lead to an overestimation of the importance of this domain due to interference with nuclear localization of Foxp3. This was not the case. Despite lacking an NLS, Foxp3ΔFKH was not completely excluded from the nucleus and retained the majority of its regulatory capacity. This is likely due to it being shuttled to the nucleus by other transcription factors as has been demonstrated for its interaction with c-Rel [21]. However, the regulation of some genes appears to be more sensitive to a lower concentration of Foxp3 in the nucleus than others. We also found that reintroduction of an SV40-derived NLS can have detrimental effects on Foxp3 function leading to a loss of the regulation of another set of genes. This suggests that the correct nuclear concentration of Foxp3 is important due to its participation in the dynamic formation of different complexes. Therefore, modifications altering the stability of Foxp3 such as its acetylation, phosphorylation and ubiquitylation play an important role in regulating gene expression and TReg function [48–50].
Contrary to published data [18], we demonstrate that the FKH, which interacts with both NFAT and DNA, is dispensable for the suppression of Il2 expression. It cannot be ruled out that FKH point mutations or introduction of bulky residues described in this previous analysis [18] led to more widespread structural disturbances of Foxp3 and so affected more than just its ability to associate with NFAT through the FKH domain. Furthermore, the authors show that deletion of the Foxp3 N-terminal region (encompassing the ProR) dysregulates expression of IL-2, CD25 and CTLA-4 although a mechanism to explain this is not provided. We show that as long as sufficient Foxp3 is in the nucleus, a complex containing transcription factors and class I HDACs is formed. This does not preclude a role for FKH-mediated NFAT/Foxp3 interaction in Il2 regulation, but demonstrates that it is not essential in the context of modulation of histone acetylation at the Il2 promoter. Foxp3ΔFKHnls is efficiently recruited to the Il2 promoter. We propose that the FKH-independent interaction of Foxp3 with AML1 and/or c-Rel and/or AP-1 [19, 21, 40], possibly other factors mediates its recruitment to their binding sites in the IL2 promoter. Foxp3 can thus reverse the normally pro-transcriptional effects of DNA-bound transcription factors to ensure proper commitment to the TReg lineage.
It was previously shown that Foxp3 binding at the Il2 and Ifng promoters coincides with their deacetylation [24]. Here we provide the full molecular mechanism for HDAC-mediated suppression of Il2 and Ifng by Foxp3. We show that Foxp3 recruits class I HDACs to the promoters of Il2 and Ifng in a ProR-dependent, FKH-independent manner. Furthermore, we demonstrate that this Foxp3-mediated suppression of Il2 and Ifng in TH::Foxp3 cells is sensitive to inhibitors of class I HDACs or knockdown of class I HDACs. The fact that we were not able to define a specific interaction domain using co-immunoprecipitation of HDACs with Foxp3 mutants overexpressed in HEK293T cells implies a complex mode of interaction. Yet, we show using in vitro translated Foxp3 and bacterially-produced HDACs that class I HDACs interact directly with Foxp3. We propose that Foxp3 acts as a scaffold connecting DNA binding by its interaction partners to HDAC recruitment at promoters such as those of Il2 and Ifng. This in turn leads to suppression of gene expression by counteracting activation-induced histone hyperacetylation. This mechanism should not be confused with the proposed function of class II HDAC in the regulation of Foxp3 expression and TReg cell function [23, 51]. Whilst class I HDACs are known to deacetylate histones, class IIa HDACs are enzymatically inactive and act primarily through recruiting other proteins [52]. Whilst it has been proposed that class II HDACs in combination with the histone acetylase TIP-60 are involved in IL-2 suppression [23], their role in this process remains unclear. It appears that TIP-60 acetylates Foxp3 itself [23], which suggests an altogether more indirect effect on IL-2 expression.
Unlike conventional CD4 helper T cells, TReg cells fail to remodel the Il2 proximal promoter to become accessible chromatin upon stimulation [53]. This suggests that Foxp3-induced deacetylation of the Il2 promoter is involved in establishing silent chromatin during TReg fate commitment. This establishment of epigenetic state is mimicked in TH::Foxp3 cells. We believe it is very likely that this mechanism of suppression is involved in the Foxp3-mediated suppression of many genes whose promoters would otherwise become hyperacetylated.
More broadly, Foxp3 may act as a bridge between any of its DNA-bound interaction partners (including AML1, c-Rel and AP-1) and other factors that modulate gene expression via chromatin modifications (eg HMTs, HATs, DNA demethylases). Foxp3 could thus affect the expression of a large number of genes. The fact that the promoters and enhancers of many Foxp3-regulated genes are not directly bound by Foxp3 [15, 17] suggests that, in many cases, this might be independent of Foxp3’s own DNA-binding abilities as we observe in the case of Il2 and Ifng.
Since many of the genes that are functionally important in TReg cells are also transiently induced upon activation of TH cells [15], we analysed the transcriptomes of cells rested after transduction with the Foxp3 constructs. Yet, it is clear that activation of TReg cells also changes their transcriptional profiles. Indeed, we observe this in the case of Il2 and Ifng where Foxp3 counteracts activation-induced hyperacetylation. Further transcriptomic studies will be required to fully elucidate the additional biologically important layer of complexity that is introduced by signalling through the TCR in vivo [54].
We have devised a generalizable strategy to dissect Foxp3 on a molecular level and to analyse the function of individual domains and subdomains. We foresee that this approach will be useful in future studies by firstly suggesting a clear road map to further dissect Foxp3, secondly providing us with Foxp3 mutants only affecting a small number of candidate genes and thirdly allowing us to correlate Foxp3 subdomains with Foxp3 complex formation and function. This provides a pliable system in which to study lineage commitment of TReg cells as well as the functional roles of gene expression modules.
Materials and Methods
Mice and cell culture
BALB/c and B6.Foxp3(hCD2) mice[55] were maintained under specific pathogen-free conditions at the MRC-ARES animal facility (Cambridge, UK). CD4+CD25- T cells were isolated from BALB/c mice between 2–4 months of age by negatively depleting splenocytes stained with FITC-conjugated depletion cocktail antibodies anti-CD8, anti-CD19, anti-CD11b, anti-CD11c, anti-Ly6G, anti-TER119 and anti-CD25 using an AutoMACS separator (Miltenyi Biotec). Isolated mouse primary T cells were cultured in RPMI-1640 medium supplemented with 10% FCS, 50 μg/mL gentamicin and 50 μM 2-mercaptoethanol. For in vivo experiments, no animals were excluded from the analyses and no statistical method was used to predetermine sample size. Age and sex-matched animals were randomly assigned to groups and technicians performing the work and assessing outcomes were blinded to the treatments. All experimental procedures received local ethics committee approval and were conducted in accordance with the Home Office Animals (Scientific Procedures) Act of 1986. Human embryonic kidney 293T cells (ATCC, USA) were used as the packaging cell line for producing viral particles and as the host for co-expressing proteins for co-immunoprecipitation experiments. These were cultured in IMDM supplemented with 10% FCS and 50 μg/mL gentamicin. All cells were cultured in a humidified incubator at 37°C, with 5% CO2.
Antibodies
The antibodies used in this study are described in S4 Table.
Expression vectors
For retroviral transduction of cells, cDNA encoding full-length Foxp3 (with HA or FLAG tag when necessary), Foxp3 deletion mutants, negative control GFP or HDACs 1, 2, 3 and 7 were cloned into the bicistronic MLV-based retroviral plasmid m6p co-expressing a GPI-linked extracellular part of ratCD8a (rCD8a) [56]. For in vitro translation, cDNA encoding Foxp3 was cloned into the pCDNA3.1(+) vector (Invitrogen). For expression in E. coli, cDNA encoding HDAC1, 2 and 3 was cloned into pGEX-6P-1 (Amersham).
Virus production and T cell transduction
Virus supernatant was produced by transfecting 293T cells with equal amounts of retroviral vector and pCL-Eco packaging plasmid (Novus Biologicals) using the Phosphate Transfection Kit (Invitrogen). Supernatant of transfected cells was collected at 48 h and 72 h post-transfection and was snap-frozen in liquid nitrogen and stored at -20°C. For retroviral transduction, primary CD4+CD25 - T cells were activated with plate-coated anti-CD3 (1 μg/ml) and soluble anti-CD28 (1 μg/ml) for 16 h. Reactivated cells were then resuspended in fresh cRPMI medium with an equal volume of viral supernatant and 5ng/ml murine IL-2 (Peprotech) and 8 μg/ml polybrene. After 12 h incubation at 37 Co, with 5% CO2, cells were harvested by centrifugation at 1260xg for 1.5 h at 32°C and transferred to fresh cRPMI supplemented with IL-2.
RNA-seq library preparation and sequencing
Transduced CD4+CD25- primary T cells were rested for 48h in RPMI medium supplemented with IL-2 (5 ng/ml). Rested cells expressing the transduction marker rCD8a were sorted by FACS to >95% purity. mRNA isolation and cDNA synthesis were performed using the uMACS mRNA isolation kit and uMACS cDNA synthesis module (Miltenyi Biotec). 5x105 cells were used for each sample. Second strand synthesis was performed using E. coli DNA polymerase I (NEB), E. coli DNA ligase (NEB) and E. coli RNaseH (NEB) in second strand synthesis buffer (Invitrogen) at 16 °C for 2.5 h. Double-stranded DNA was fragmented by sonication using the Bioruptor UCD-400 (Diagenode), with 20 cycles of 30 s on, 30 s off. Fragmented samples were end-repaired using Klenow large fragment (NEB), T4 DNA polymerase (NEB) and T4 polynucleotide kinase (NEB) at 20°C for 30 min. DNA was purified using Zymo DNA Clean and Concentrator kits (Zymo). Purified blunt-ended DNA was 3’ adenylated using Klenow exo - fragment (NEB) at 37°C for 30 min. DNA was purified as before and adapters containing 6 bp barcodes from the sample prep oligonucleotide kit (Illumina) were ligated to 3’ A-tailed DNA fragment at 20°C for 30 min using T4 DNA ligase (NEB). Each DNA sample was amplified via 18-cycles of PCR using universal primers from the sample prep oligonucleotide kit, The PCR conditions are 30 sec at 98°C followed by 18 cycles of 10 sec at 98°C, 30 sec at 65°C, and 30 sec at 72°C, then a final step of 5mins at 72°C.The library was separated by agarose gel electrophoresis using a 2% gel and fragments between 200–400 bp were excised and purified using Minelute gel extraction kit (Qiagen). The concentration and size distribution of fragments was then determined with a Bioanalyzer High-Sensitivity DNA Chip (Agilent). RNA-seq libraries from different samples were multiplexed in equimolar concentrations for sequencing using a Hiseq 2000 (Illumina) at the Cancer Research Institute (Cambridge, UK).
RNA-seq data analysis
Reads were aligned against GRCm38 mouse genome release 75 using the spliced-read mapper GSNAP [57]. GSNAP command line parameters were “-A sam-B 5-t 38-n 1-Q—-nofails” so that only uniquely-mapped reads were reported. The htseq-count Python script [58] was used to determine the number of reads from each sample that mapped to each feature within the GRCm38v75 GTF file. The script was run in “union” mode so that reads overlapping more than one feature were ignored. Transcript lengths for each gene were calculated from the sum of all annotated non-overlapping exonic regions. Robust FPKM estimates were calculated using the DESeq2 R package [59]. DESeq2 was also used to calculate regularized log-transformed expression counts. RNA-seq data for naive TH and splenic TReg cells [29] were processed as for other data.
Differential expression analysis
Genes were assigned to high (HE), low (LE), intermediate (INT) and non-expressing (NE) categories by performing expectation-maximization curve fitting of a Gaussian mixture model on log2-transformed FPKMs using the R package ‘MClust’ as described previously [27]. Kernel density estimates of read count distributions were performed with the density function available in R using the default settings apart from setting the bandwidth to be twice the default. HE and LE class boundaries were set for a false discovery rate of 0.05 as described for the EpiChIP software [60] although the classification procedure was, here, reimplemented in R. Genes that fell between the expression boundaries were classified as ‘intermediate’. DESeq2 was used to calculate moderated fold changes and associated adjusted P values (using Wald tests with Benjamini Hochberg correction for multiple testing) for all genes in every pairwise comparison between samples. A gene was defined as differentially expressed between two samples if: (i) It was classified as INT or HE in at least one sequencing replicate and (ii) it had an adjusted p value of less than 0.05. Genes were defined as Foxp3-regulated if they were differentially expressed in both Naive TH cells vs splenic TReg and also in TH::control vs TH::Foxp3. A gene was defined as dysregulated in a particular condition if (i) it was initially defined as Foxp3-regulated as above, (ii) it was found to be differentially expressed between TH::Foxp3 and the condition of interest and (iii) its change in gene expression was in the direction of TH::control. Conversely, a gene was defined as maintaining its Foxp3-like regulation in a particular condition if (i) it was initially defined as Foxp3-regulated as above and (ii) it was not dysregulated.
Clustering/PCA
Euclidean distances between samples were calculated from rlog transformed gene expression values for Foxp3-regulated genes (using the dist R function). Hierarchical clustering was performed using these distances with the heatmap.2 function (from the gplots R package). Principal component analysis was performed using rlog transformed gene expression values for Foxp3-regulated genes in each sample.
Gene ontology enrichment and gene function analysis
The ‘Comprehensive list of Immune-Related Genes’ downloaded from https://immport.niaid.nih.gov [32, accessed 9th April 2015] and mouse orthologs found using Ensemble BioMart (http://www.ensembl.org/biomart/). Genes that appeared in this list of orthologs were classified as ‘immunology-related’.
GO term enrichment analyses were performed using the tool available at http://amigo.geneontology.org/rte. Terms were considered enriched if they had a P value < 0.01. The manually-curated list of immunology keywords was downloaded from the ImmPort project and used to search the lists of enriched GO terms—terms containing a keyword were classified as ‘Immunology-related’.
Antibody staining for flow cytometry and microscopic study
For cell surface antibody staining, cells were washed in PBS supplemented with 2% FBS and incubated with fluorophore conjugated antibodies (APC-αCD25, dilution V:V: 1 : 600; PE-αCD25, 1 : 600; PE-αrCD8a, 1 : 200; PB-αCD4, 1 : 400; Fc-Block (αCD16/32), 1 : 500; APC-αCD62L, 1 : 2500; APC-αLAG3, 1 : 200; FITC-αGITR, 1 : 300) at 4°C for 10 min. For intracellular staining, cells were then fixed and permeabilized using Foxp3 intracellular staining kit (eBioscience) before being stained with corresponding antibodies (APC-αCTLA4, 1 : 300; APC-αFoxp3 1 : 100; FITC-αHA, 1 : 250; FITC-αFLAG, 1 : 200; APC-αIL-2, 1 : 50; PE-αIgG2a, 1 : 100; FITC-αIgG2a, 1 : 250; APC-αIgG2a 1 : 100). After three washes in permeabilizing buffer stained cells were resuspended in PBS and analysed on an LSRII flow cytometer (BD Biosciences Immunocytometry Systems). Data were analyzed with FlowJo software. For localization analysis, Foxp3 and mutant transduced 293T cell were cultured on cover slides and fixed and permeabilized with Foxp3 Intracellular Staining Kit (eBioscience). After blocking with 5% BSA in PBS, cells were stained with AlexaFluor488-αFoxp3 antibody (Dilution 1 : 500 in permeabilization buffer for 2 h. After three washes using cell permeabilization buffer, cells were mounted on slides and imaged using a Zeiss LSM 710 confocal microscope.
siRNA knockdown in primary T cells
Accell self-delivery siRNA (GE, Dharmacon siRNA) was used to knock down Class I HDACs. Foxp3 transduced TH cells were treated with 1.5 μM control siRNA and a cocktail containing 1.5 μM HDAC1, 1.5 μM HDAC2 & 1.5 μM HDAC3 siRNA in siRNA delivery medium with 10 ng/ml IL2 and 2% FBS (GE, Dharmacon siRNA) for 96 h. Then the cells were re-stimulated with cell activation cocktail plus protein transport inhibitor for 12 h, permeabilized and stained for IL2.
Comparative genomics
All sequences were retrieved from the NCBI, UCSC and Ensembl databases before October 2010. Protein sequences were aligned using Muscle as implemented in the Geneious program package [61]. The coiled coil, zinc finger and forkhead domains of Foxp3 were predicted using the SMART and COILS2 programs at http://smart.embl-heidelberg.de/smart/. The nuclear localization signal within the FKH was determined based on the presence or absence of a RKKR motif [33]. The REL test implemented on the Datamonkey website [62] was used with codon-aligned Foxp3 sequences from mouse, cat, cow, macaque, dog, horse, human, pig, rat, rhesus macaque and sheep. Prior to running the REL test, a model selection tool was executed to select the best nucleotide substitution model. All codons showing evidence of purifying selection with the REL test (based on non-synonymous over synonymous nucleotide replacements (dN>dS)) also replicated in an independent SLAC and FEL test. All three tests agreed when using a Bayes factor cutoff value of >40. The plots were generated using EMBOSS plotcon with a window size of 20.
CFSE proliferation assay
CD4+CD25- TH cells were harvested by centrifugation for 10 min at 400xg and resuspended at 2x107 cells/ml in prewarmed PBS in a 15mL falcon tube. The cells were then mixed 1 : 1 with pre-warmed 10 μM CFSE solution in PBS and incubated for 15 min at 37°C. Stained cells were washed twice with ice-cold PBS-FBS before being resuspended in pre-warmed cRPMI. Cells were then plated at 2x105 cells per well in αCD3ε coated (0.6μg/ml) and soluble αCD28 (1μg/ml) U-bottomed 96-well plates together with 2x105 Th cells transduced with wild-type Foxp3 or its mutants. Cells were cultured at 37°C, 5%CO2 and the proliferation of the labelled cells was assessed by determination CFSE dilution by FACS after 72 h.
In vivo functional assessment of Foxp3 and mutants ectopic expression in TH cells
A colitis induction model [63] was adopted for the assessment of the in vivo suppressive competence of T cells transduced with Foxp3 and its mutants. Three days prior to adoptive transfer, CD4+CD25- were isolated from 8–10 week old wild-type Balb/c female mice. These cells were transduced with either wild type Foxp3, a mutant version of Foxp3 or the control. After transduction, cells were rested for one day in cRPMI supplemented with mIL-2 (5 ng/ml). On the day of adoptive transfer the transduced cells were FACS sorted to >98% purity and CD4+CD25-CD45RBhi (>98% pure) T cells were isolated from spleens of 8–10 week old female Balb/c mice by MACS followed by FACS sorting. Weight-matched female C.B.-17 SCID mice between seven and eight weeks of age each received an intraperitoneal (i.p.) injection of 2x105 CD4+CD25-CD45RBhi cells along with 2x105 transduced cells. The weight of the mice during the study was recorded by trained animal technicians at the MRC-ARES facility (Cambridge, UK). Mice reaching 20% weight loss (or defined welfare end-points) were humanely sacrificed. Five to seven weeks post-transfer the remaining mice were humanely sacrificed.
Quantitative real-time PCR
Transduced cells were MACS sorted to >90% purity. Cells were either rested for 48 hours without activation or re-activated with plate-coated αCD3ε (5 μg/ml) and soluble αCD28 (1μg/ml) for 6 h. Total RNA was isolated using the RNeasy micro kit (Qiagen), and was reversely transcribed to cDNA with random hexamer priming using the SuperScript III kit (Invitrogen). The cDNA was used for quantitative PCR with SYBR green reagent (Thermo Scientific) according to manufacturer's instruction. Real-time qPCR was performed using the ABI/ViiA7 detection system (PE Applied Biosystems). Melting curves of the amplified products were obtained to determine the specificity of the amplification reaction. Cycling conditions were 2 min at 50°C, 5 min at 95°C followed by 40 cycles of 15s at 95°C, 60s at 60°C. Data was analyzed using the ΔΔCt method. For relative expression level detection, the data were normalized to CD3ε expression. For ChIP-qPCR, data were normalized to the input DNA. Primers were as detailed in S5 Table.
Co-immunoprecipitation and western blotting
HEK293T cells were co-transfected with equal amounts of HA-tagged Foxp3 or its mutant plasmids and FLAG-tagged interaction protein plasmid using phosphate transfection kit (Invitrogen). 24 h post-transfection, 293T cells were lysed with hypertonic lysis buffer (20 mM Tris-HCl pH 7.4, 0.4 M NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, Roche Complete Protease Inhibitor Cocktail) and shaken for 15 min at 4°C. Cell lysate was pelleted by centrifugation for 15 min at 16000 g. The supernatant was collected and diluted with an equal volume of hypotonic dilution buffer (20 mM Tris-HCl pH 7.4, 1 mM EDTA, 1 mM EGTA, 0.1% Triton X-100, 20% glycerol, Roche Complete Protease Inhibitor Cocktail). 10% cell lysate was retained at -20°C for gene expression verification. The remaining lysate was incubated with αFLAG M2 Affinity Gel (Invitrogen) or αHA affinity gel (Invitrogen) for 2 h with rotation at 4°C. Unbound protein was removed by washing three times with high salt wash buffer (20 mM Tris-HCl pH7.4, 0.5 M NaCl, 1 mM EDTA, 1 mM EGTA, 0.1% Triton X-100, 10% glycerol, protease inhibitors). Precipitated protein was eluted from the affinity gel with 150 μg/ml 3XFLAG or 100 μg/ml HA peptide in PBS buffer. Protein samples were separated by SDS-PAGE using NuPAGE 4–12% bis-tris acrylamide gels. After electrophoresis, proteins were transferred from the gel to PVDF membrane (Millipore). After incubating with corresponding primary antibodies and HRP-conjugated secondary antibodies, the development of signal was performed using chemiluminescence ECL Standard Western Blotting Detection Reagents (GE). For co-IP experiments in primary T cells, CD4+CD25 - TH cells were transduced with virus carrying the appropriate tag-fused protein followed by immunoprecipitation as described above.
GST pull-down assay
BL21 chemically competent E. coli (NEB) were transformed with pGEX-6P-1 containing HDAC cDNA. The expression of GST-fused protein was induced by adding 0.1 mM IPTG followed by incubation for 6 h at 20°C. Cells were lysed using Bugbuster protein extraction reagent (Novagen) at RT for 20 min with gentle shaking. The lysate was centrifuged for 15 min at 16000xg at 4°C and the supernatant was mixed with glutathione sepharose 4B beads for overnight incubation at 4°C. The beads were washed five times with PBS and then eluted using 20 mM reduced glutathione. The protein sample was then desalted with a protein desalting column (Thermo Scientific). pCDNA3.1(+) plasmid containing Foxp3 cDNA was used as template for in vitro transcription and translation (TNT coupled reticulocyte lysate system, Promega). Five μl of in vitro synthesized protein was incubated with 10 μg of the affinity purified GST-tagged HDAC protein in BC100 buffer (20mM Tris pH7.9, 100mM NaCl, 10%glycerol, 0.2mM EDTA, 0.1% Triton X-100, complete protease inhibitor cocktail) on a rotator overnight at 4°C. The proteins were pulled down using Glutathione sepharose 4B beads, followed by five washes with BC100 buffer. The bound proteins were eluted with reduced SDS loading buffer and the eluted materials were resolved on SDS-PAGE. The presence of Foxp3 and GST-proteins were detected with αFoxp3 (Rat), αGST(Goat) and corresponding secondary antibodies.
Chromatin-immunoprecipitation (ChIP)
Virus-transduced TH cells were enriched using PE-αCD8a (1 : 200) together with αPE magnetic beads (Miltenyi Biotec) using the AutoMACS separator. Transduced cells were MACS sorted to >90% purity. The expression of HA tag and Foxp3 was confirmed by intracellular staining and flow cytometry. Cells were either rested 48 hours without activation or re-activated with plate-coated αCD3ε (5 μg/ml) and soluble αCD28 (1 μg/ml) for 6 hours. Chromatin - immunoprecipitation was performed using the EZ-ChIP kit (Millipore). Briefly, cells were fixed and protein/DNA was crosslinked in 1% formaldehyde (Sigma). After quenching with 0.125 M glycine, fixed cells were lysed in SDS lysis buffer and the lysate was sonicated to 200 to 600 bp fragments using the Bioruptor UCD-400 (Diagenode) with 20 cycles of 30 s On, 30 s off. After pre-clearing with protein G agarose beads (Invitrogen), sonicated samples were diluted in ChIP dilution buffer and incubated with either αIgG or corresponding ChIP-grade antibodies overnight. Protein G agarose beads were added to precipitate antibody-protein-DNA complexes. After five washes with the buffers provided by the manufacturer, the protein/DNA complex was eluted and the crosslinking was reversed by incubating at 65°C overnight. After digestion with RNase A and proteinase K to remove RNA and protein contents, the precipitated DNA was purified using QIAquick PCR purification kit (Qiagen). The purified DNA was either amplified by PCR reaction and analyzed in 2% agarose gel or quantified directly by qPCR. HA-GFP transduced cells served as control for non-specific binding in anti-HA ChIP and to determine the basal level in anti-HDAC and anti-Acetyl H3 ChIP. In anti-acetyl H3 ChIP of the resting and activated T cells, normal rabbit IgG was used as control for non-specific background signal.
Accession codes
RNA-seq data: ArrayExpress E-MTAB-2988
Supporting Information
Zdroje
1. Wing K, Sakaguchi S (2010) Regulatory T cells exert checks and balances on self tolerance and autoimmunity. Nat Immunol 11 : 7–13. doi: 10.1038/ni.1818 20016504
2. Kim JM, Rasmussen JP, Rudensky AY (2007) Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat Immunol 8 : 191–197. 17136045
3. Lahl K, Loddenkemper C, Drouin C, Freyer J, Arnason J, et al. (2007) Selective depletion of Foxp3(+) regulatory T cells induces a scurfy-like disease. J. Exp. Med. 204 : 57–63. 17200412
4. Chen T, Darrasse-Jeze G, Bergot AS, Courau T, Churlaud G, et al. (2013) Self-specific memory regulatory T cells protect embryos at implantation in mice. J Immunol 191 : 2273–2281. doi: 10.4049/jimmunol.1202413 23913969
5. Izcue A, Coombes JL, Powrie F (2006) Regulatory T cells suppress systemic and mucosal immune activation to control intestinal inflammation. Immunol Rev 212 : 256–271. 16903919
6. Campbell DJ, Ziegler SF (2007) FOXP3 modifies the phenotypic and functional properties of regulatory T cells. Nat Rev Immunol 7 : 305–310. 17380159
7. Fontenot JD, Rasmussen JP, Williams LM, Dooley JL, Farr AG, et al. (2005) Regulatory T cell lineage specification by the forkhead transcription factor foxp3. Immunity 22 : 329–341. 15780990
8. Dhamne C, Chung Y, Alousi AM, Cooper LJ, Tran DQ (2013) Peripheral and thymic foxp3(+) regulatory T cells in search of origin, distinction, and function. Front Immunol 4 : 253. doi: 10.3389/fimmu.2013.00253 23986762
9. Ohkura N, Hamaguchi M, Morikawa H, Sugimura K, Tanaka A, et al. (2012) T Cell Receptor Stimulation-Induced Epigenetic Changes and Foxp3 Expression Are Independent and Complementary Events Required for Treg Cell Development. Immunity 37 : 785–799. doi: 10.1016/j.immuni.2012.09.010 23123060
10. Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, et al. (2001) Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet 27 : 68–73. 11138001
11. Lahl K, Mayer CT, Bopp T, Huehn J, Loddenkemper C, et al. (2009) Nonfunctional Regulatory T Cells and Defective Control of Th2 Cytokine Production in Natural Scurfy Mutant Mice. J. Immunl. 183 : 5662–5672.
12. Li B, Samanta A, Song X, Iacono KT, Brennan P, et al. (2007) FOXP3 is a homo-oligomer and a component of a supramolecular regulatory complex disabled in the human XLAAD/IPEX autoimmune disease. Int Immunol 19 : 825–835. 17586580
13. Rudra D, deRoos P, Chaudhry A, Niec RE, Arvey A, et al. (2012) Transcription factor Foxp3 and its protein partners form a complex regulatory network. Nat Immunol 13 : 1010–1019. doi: 10.1038/ni.2402 22922362
14. Bandukwala HS, Wu Y, Feuerer M, Chen Y, Barboza B, et al. (2011) Structure of a domain-swapped FOXP3 dimer on DNA and its function in regulatory T cells. Immunity 34 : 479–491. doi: 10.1016/j.immuni.2011.02.017 21458306
15. Marson A, Kretschmer K, Frampton GM, Jacobsen ES, Polansky JK, et al. (2007) Foxp3 occupancy and regulation of key target genes during T-cell stimulation. Nature 445 : 931–935. 17237765
16. Song X, Li B, Xiao Y, Chen C, Wang Q, et al. (2012) Structural and biological features of FOXP3 dimerization relevant to regulatory T cell function. Cell Rep 1 : 665–675. doi: 10.1016/j.celrep.2012.04.012 22813742
17. Zheng Y, Josefowicz SZ, Kas A, Chu TT, Gavin MA, et al. (2007) Genome-wide analysis of Foxp3 target genes in developing and mature regulatory T cells. Nature 445 : 936–940. 17237761
18. Wu Y, Borde M, Heissmeyer V, Feuerer M, Lapan AD, et al. (2006) FOXP3 controls regulatory T cell function through cooperation with NFAT. Cell 126 : 375–387. 16873067
19. Ono M, Yaguchi H, Ohkura N, Kitabayashi I, Nagamura Y, et al. (2007) Foxp3 controls regulatory T-cell function by interacting with AML1/Runx1. Nature 446 : 685–689. 17377532
20. Pan F, Yu H, Dang EV, Barbi J, Pan X, et al. (2009) Eos mediates Foxp3-dependent gene silencing in CD4+ regulatory T cells. Science 325 : 1142–1146. doi: 10.1126/science.1176077 19696312
21. Loizou L, Andersen KG, Betz AG (2011) Foxp3 interacts with c-Rel to mediate NF-kappaB repression. PLoS One 6: e18670. doi: 10.1371/journal.pone.0018670 21490927
22. Zhang F, Meng G, Strober W (2008) Interactions among the transcription factors Runx1, RORgammat and Foxp3 regulate the differentiation of interleukin 17-producing T cells. Nat Immunol 9 : 1297–1306. doi: 10.1038/ni.1663 18849990
23. Li B, Samanta A, Song X, Iacono KT, Bembas K, et al. (2007) FOXP3 interactions with histone acetyltransferase and class II histone deacetylases are required for repression. Proc Natl Acad Sci U S A 104 : 4571–4576. 17360565
24. Chen C, Rowell EA, Thomas RM, Hancock WW, Wells AD (2006) Transcriptional regulation by Foxp3 is associated with direct promoter occupancy and modulation of histone acetylation. J Biol Chem 281 : 36828–36834. 17028180
25. Bettelli E, Dastrange M, Oukka M (2005) Foxp3 interacts with nuclear factor of activated T cells and NF-kappa B to repress cytokine gene expression and effector functions of T helper cells. Proc Natl Acad Sci U S A 102 : 5138–5143. 15790681
26. Andersen KG, Butcher T, Betz AG (2008) Specific immunosuppression with inducible Foxp3-transduced polyclonal T cells. PLoS Biol 6: e276. doi: 10.1371/journal.pbio.0060276 18998771
27. Hebenstreit D, Fang M, Gu M, Charoensawan V, van Oudenaarden A, et al. (2011) RNA sequencing reveals two major classes of gene expression levels in metazoan cells. Mol Syst Biol 7 : 497. doi: 10.1038/msb.2011.28 21654674
28. Andersen KG, Nissen JK, Betz AG (2012) Comparative Genomics Reveals Key Gain-of-Function Events in Foxp3 during Regulatory T Cell Evolution. Front Immunol 3 : 113. doi: 10.3389/fimmu.2012.00113 22590469
29. Stubbington MJT, Mahata B, Svensson V, Deonarine A, Nissen JK, et al. (2015) An atlas of mouse CD4+ T cell transcriptomes. Biology Direct. 10 : 14. doi: 10.1186/s13062-015-0045-x 25886751
30. Samstein RM, Arvey A, Josefowicz SZ, Peng X, Reynolds A, et al. (2012) Foxp3 Exploits a Pre-Existent Enhancer Landscape for Regulatory T Cell Lineage Specification. Cell 151 : 153–166. doi: 10.1016/j.cell.2012.06.053 23021222
31. Wang Z, Gerstein M, Snyder M (2009) RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 10 : 57–63. doi: 10.1038/nrg2484 19015660
32. Bhattacharya S, Andorf S, Gomes L, Dunn P, Schaefer H, et al. (2014) ImmPort: disseminating data to the public for the future of immunology. Immunol. Res. 58 : 234–239. doi: 10.1007/s12026-014-8516-1 24791905
33. Hancock WW, Ozkaynak E (2009) Three Distinct Domains Contribute to Nuclear Transport of Murine Foxp3. Plos One 4: e7890. doi: 10.1371/journal.pone.0007890 19924293
34. Lin W, Haribhai D, Relland LM, Truong N, Carlson MR, et al. (2007) Regulatory T cell development in the absence of functional Foxp3. Nat. Immunol. 8 : 359–368. 17273171
35. Camperio C, Caristi S, Fanelli G, Soligo M, Del Porto P, et al. (2012) Forkhead Transcription Factor FOXP3 Upregulates CD25 Expression through Cooperation with RelA/NF-kappa B. Plos One 7: e48303. doi: 10.1371/journal.pone.0048303 23144749
36. Metzler B, Burkhart C, Wraith DC (1999) Phenotypic analysis of CTLA-4 and CD28 expression during transient peptide-induced T cell activation in vivo. Int Immunol 11 : 667–675. 10330272
37. de la Rosa M, Rutz S, Dorninger H, Scheffold A (2004) Interleukin-2 is essential for CD4+CD25+ regulatory T cell function. Eur J Immunol 34 : 2480–2488. 15307180
38. Boucheron N, Tschismarov R, Goeschl L, Moser MA, Lagger S, et al. (2014) CD4(+) T cell lineage integrity is controlled by the histone deacetylases HDAC1 and HDAC2. Nat Immunol 15 : 439–448. doi: 10.1038/ni.2864 24681565
39. Baine I, Basu S, Ames R, Sellers RS, Macian F (2013) Helios induces epigenetic silencing of IL2 gene expression in regulatory T cells. J Immunol 190 : 1008–1016. doi: 10.4049/jimmunol.1200792 23275607
40. Lee SM, Gao B, Fang DY (2008) FoxP3 maintains Treg unresponsiveness by selectively inhibiting the promoter DNA-binding activity of AP-1. Blood 111 : 3599–3606. doi: 10.1182/blood-2007-09-115014 18223166
41. Bandyopadhyay S, Dure M, Paroder M, Soto-Nieves N, Puga I, et al. (2007) Interleukin 2 gene transcription is regulated by Ikaros-induced changes in histone acetylation in anergic T cells. Blood 109 : 2878–2886. 17148585
42. Bandyopadhyay S, Montagna C, Macian F (2012) Silencing of the Il2 gene transcription is regulated by epigenetic changes in anergic T cells. Eur J Immunol 42 : 2471–2483. doi: 10.1002/eji.201142307 22684523
43. Kay BK, Williamson MP, Sudol P (2000) The importance of being proline: the interaction of proline-rich motifs in signaling proteins with their cognate domains. Faseb Journal 14 : 231–241. 10657980
44. van der Lee R, Buljan M, Lang B, Weatheritt RJ, Daughdrill GW, et al. (2014) Classification of Intrinsically Disordered Regions and Proteins. Chem Rev 114 : 6589–6631. doi: 10.1021/cr400525m 24773235
45. Sojka DK, Hughson A, Fowell DJ (2009) CTLA-4 is required by CD4(+)CD25(+) Treg to control CD4(+) T-cell lymphopenia-induced proliferation. Eur J Immunol 39 : 1544–1551. doi: 10.1002/eji.200838603 19462377
46. Bachetta R, Gambineri E, Passerini L, Barzaghi F, Mannurita SC, et al. (2012) Ipex Syndrome: Clinical and Immunological Findings. Update from the Italian Study Group of Ipex. J Clin Immunol 32 : 79–79.
47. Katoh H, Zheng P, Liu Y (2013) FOXP3: Genetic and epigenetic implications for autoimmunity. J Autoimmun 41 : 72–78. doi: 10.1016/j.jaut.2012.12.004 23313429
48. van Loosdregt J, Vercoulen Y, Guichelaar T, Gent YYJ, Beekman JM, et al. (2010) Regulation of Treg functionality by acetylation-mediated Foxp3 protein stabilization. Blood 115 : 965–974. doi: 10.1182/blood-2009-02-207118 19996091
49. Chen ZJ, Barbi J, Bu SR, Yang HY, Li ZY, et al. (2013) The Ubiquitin Ligase Stub1 Negatively Modulates Regulatory T Cell Suppressive Activity by Promoting Degradation of the Transcription Factor Foxp3. Immunity 39 : 272–285. doi: 10.1016/j.immuni.2013.08.006 23973223
50. Morawski PA, Mehra P, Chen CX, Bhatti T, Wells AD (2013) Foxp3 Protein Stability Is Regulated by Cyclin-dependent Kinase 2. J Biol Chem 288 : 24494–24502. doi: 10.1074/jbc.M113.467704 23853094
51. Tao R, de Zoeten EF, Ozkaynak E, Chen CX, Wang LQ, et al. (2007) Deacetylase inhibition promotes the generation and function of regulatory T cells. Nat Med 13 : 1299–1307. 17922010
52. Akimova T, Beier UH, Liu YJ, Wang LQ, Hancock WW (2012) Histone/protein deacetylases and T-cell immune responses. Blood 119 : 2443–2451. doi: 10.1182/blood-2011-10-292003 22246031
53. Su L, Creusot RJ, Gallo EM, Chan SM, Utz PJ, et al. (2004) Murine CD4(+) CD25(+) regulatory T cells fail to undergo chromatin remodeling across the proximal promoter region of the IL-2 gene. J Immunol 173 : 4994–5001. 15470042
54. Levine AG, Arvey A, Jin W, Rudensky AY (2014) Continuous requirement for the TCR in regulatory T cell function. Nat Immunol 15 : 1070–1078. doi: 10.1038/ni.3004 25263123
55. Kendal AR, Chen Y, Regateiro FS, Ma J, Adams E, et al (2011) Sustained suppression by Foxp3+ regulatory T cells is vital for infectious transplantation tolerance. J Exp Med. 208 : 2043–2053. doi: 10.1084/jem.20110767 21875958
56. Bloor S, Ryzhakov G, Wagner S, Jonathan P, Butler G, et al. (2008) Signal processing by its coil zipper domain activates IKK gamma. Proc Natl Acad Sci U S A 105 : 1279–1284. doi: 10.1073/pnas.0706552105 18216269
57. Wu TD, Nacu S (2010) Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics 26 : 873–881. doi: 10.1093/bioinformatics/btq057 20147302
58. Anders S, Pyl PT, Huber W (2014) HTSeq—A Python framework to work with high-throughput sequencing data. bioRxiv.
59. Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-Seq data with DESeq2. bioRxiv.
60. Hebenstreit D, et al. (2011) EpiChIP: gene-by-gene quantification of epigenetic modification levels. Nucleic Acids Res 39: e27. doi: 10.1093/nar/gkq1226 21131282
61. Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, et al. (2012) Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28 : 1647–1649. doi: 10.1093/bioinformatics/bts199 22543367
62. Pond SLK, Frost SDW (2005) Datamonkey: rapid detection of selective pressure on individual sites of codon alignments. Bioinformatics 21 : 2531–2533. 15713735
63. Liu H, Hu B, Xu D, Liew FY (2003) CD4+CD25+ regulatory T cells cure murine colitis: the role of IL-10, TGF-beta, and CTLA4. J Immunol 171 : 5012–5017. 14607897
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2015 Číslo 6
Nejčtenější v tomto čísle
- Non-reciprocal Interspecies Hybridization Barriers in the Capsella Genus Are Established in the Endosperm
- Translational Upregulation of an Individual p21 Transcript Variant by GCN2 Regulates Cell Proliferation and Survival under Nutrient Stress
- Exome Sequencing of Phenotypic Extremes Identifies and as Interacting Modifiers of Chronic Infection in Cystic Fibrosis
- The Human Blood Metabolome-Transcriptome Interface