
Prehospital immune responses and development of multiple organ dysfunction syndrome following traumatic injury: A prospective cohort study
In this prospective cohort study, Jon Hazeldine and colleagues investigate the immune and inflammatory response of trauma patients immediately after and in the hours and days following injury.
Published in the journal:
. PLoS Med 14(7): e32767. doi:10.1371/journal.pmed.1002338
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pmed.1002338
Summary
In this prospective cohort study, Jon Hazeldine and colleagues investigate the immune and inflammatory response of trauma patients immediately after and in the hours and days following injury.
Introduction
Although the major and immediate cause of death following severe trauma is haemorrhage, many trauma victims later die following complications such as multiorgan dysfunction or sepsis, with the individual’s immune response to injury significantly influencing the chances of developing these life-threatening conditions [1]. Two opposing clinical syndromes characterise the immune and inflammatory response to traumatic injury: systemic inflammatory response syndrome (SIRS), characterised by elevated levels of circulating proinflammatory cytokines and immune cell activation, and compensatory anti-inflammatory response syndrome (CARS), characterised by raised anti-inflammatory cytokines and immuneparesis [2]. The immune response that develops during the SIRS and CARS responses is complex and involves the innate and adaptive arms of the immune system, with significant alterations apparent in the composition, phenotype, and/or function of the circulating immune cell pools. For example, following major injury, marked alterations have been described in the antimicrobial functions of neutrophils [3–6], the surface phenotype of monocytes [7,8], and the absolute number of circulating lymphocytes [9].
The current paradigm for how major injury influences the immune system is based almost entirely upon the analysis of blood samples obtained from patients post-hospital admission and several hours postinjury. Indeed, with the exception of a small number of studies in which research samples were acquired at the scene of injury [10–12], the literature is saturated with studies in which emergency departments or intensive care units (ICUs) have served as the site of initial sample collection, an approach that has resulted in significant interstudy variation in time to first blood sampling [4,6,7,13–17]. Thus, whilst we have a detailed understanding of the alterations that occur in the immune system during the acute immune response to injury, our knowledge of trauma-induced changes in immunity during the ultra-early postinjury phase (particularly within the first hour) is limited. Indeed, of the above-mentioned prehospital based studies, only 1 investigated immune function, reporting a significant impairment in lipopolysaccharide (LPS)-induced cytokine production by whole blood leukocytes within minutes of injury, suggesting that trauma patients are immune suppressed even prior to hospital admission [10]. Unfortunately, the group performed no additional assays to investigate the function of specific immune cells, nor did they examine whether traumatic injury resulted in any immediate alterations to the composition or surface phenotype of the circulating immune cell pool. Such a study would provide much-needed and novel data relating to the immune status of trauma patients prior to their arrival at hospital and would provide the evidence base for early intervention to improve patient outcomes or stratification for treatment.
Results presented in a series of recent prospective observational cohort studies suggest that patients who experience poor clinical outcomes following traumatic injury elicit a more robust and prolonged immune/inflammatory response than those who report better outcomes [18–22]. These data, coupled with studies that have shown that elevated proinflammatory cytokines [22–26], impaired leukocyte function [13,27], and altered monocyte phenotype [28,29] are associated with and/or predictive of mortality, multiple organ dysfunction/failure, and sepsis, suggest a potential role for immune monitoring in identifying patients at risk of poor outcome. Common to all these studies has been the acquisition and analysis of post-hospital admission blood samples, meaning the data collected relates to the immune and inflammatory response during the acute postinjury phase. Recently, in a cohort of 40 patients, from whom blood samples were acquired within 2 hours of injury, Manson et al. [9] reported an increased percentage of CD56DIM natural killer (NK) cells and a reduced frequency of γδ–low T lymphocytes in those subjects who subsequently developed multiple organ dysfunction syndrome (MODS). Thus, it would appear that immunological events activated prior to hospital admission may impact upon patient recovery [9].
Here, via the analysis of blood samples acquired from 89 adult trauma patients within 1 hour of injury (mean time to sampling; 42±1 minutes postinjury), we have analysed the composition, phenotype, and/or function of the innate and adaptive arms of the immune system to provide a comprehensive insight into the immediate cellular immune response to trauma. Using these data, an exploratory analysis was performed to test for potential relationships between the ultra-early immune/inflammatory response to injury and the development of MODS. In addition to the samples collected within 1 hour of injury, we also analysed the immune status of patients 4–12 and 48–72 hours postinjury, intervals chosen to mimic the time periods in which previous trauma studies had acquired their first postinjury research blood samples [6,16,18–22].
Methods
Study design and setting
A prospective observational study was undertaken at a regional trauma network in the West Midlands, United Kingdom. The study aimed to characterise the relationship between biomarkers, brain injury severity, and outcome. Patients were enrolled into the study during prehospital emergency evacuation and were followed up at the major trauma centre to which they were conveyed, the Queen Elizabeth Hospital Birmingham. Research ethics committee approval was granted before the study was started (Brain Biomarkers After Trauma Cohort Study; reference 13/WA/0399). No patients in the study received prehospital blood products.
Patient selection
On a 24/7 basis between 15 May 2014 and 16 December 2016, prehospital emergency care teams acquired blood samples from adult trauma patients (≥18 years) with a suspected injury severity score (ISS) ≥ 8 within 1 hour of injury (defined as the time of phone call to emergency services). A screening log of all trauma patients was prospectively recorded in order to reduce the risk of selection bias. All patients had complete follow-up data for their hospital stay.
Capacity and consent
Because of the nature of their injuries, patients were unlikely to be able to provide informed consent to enrol in the study. Recruitment into the study was therefore undertaken under the guidance of the Mental Health Capacity Act for research in emergency situations, in accordance with the Declaration of Helsinki. If the patient lacked capacity, a written agreement for study participation was sought from a legal consultee, with written consent obtained from the patient after they regained capacity. In cases in which the patient did not regain capacity to consent, data were retained in accordance with the legal consultee’s assent.
Prehospital enrolment and blood sampling
As this study involved the acquisition of blood samples during the prehospital evacuation of trauma patients, regional training for prehospital personnel was undertaken before the study was started. They were instructed to sample any patient with significant injuries and a suspected ISS greater than 8 that warranted immediate transfer to a major trauma centre and were provided with information on how to acquire, store, and hand over blood samples for research. Peripheral venous blood was either obtained during the intravenous cannulation of patients or by venepuncture. Blood tubes were stored in the ambulance at room temperature until arrival at hospital, when they were placed into a study-specific refrigerator. All samples were collected and the analysis was begun within 1 hour of deposition by the same laboratory researcher on a 24/7 basis in order to minimise heterogeneity in blood preparation and storage. Further samples were taken at 4–12 and 48–72 hours postinjury by research nursing staff, who delivered samples directly to the laboratory. At all time points, blood samples were collected into 3 separate BD Vacutainers (Becton Dickinson, Oxford, UK) containing lithium heparin, z-serum clotting activator, or 1/10 volume of 3.2% trisodium citrate. Patients were excluded from the study if they were deemed unlikely to survive transportation to hospital. Patients who had prehospital blood samples taken >1 hour postinjury, a confirmed ISS < 8, or a previous diagnosis of neurodegenerative disease were also subsequently excluded from the study. Data obtained from isolated traumatic brain injury (TBI) patients and subjects who received steroid treatment were not included in the final analysis.
Healthy controls
Blood was sampled from 116 adults who served as healthy controls (HCs). The mean age and gender of the HC cohort were not significantly different from those of the patient cohort (S1 Table). HCs were volunteers who were not taking any regular medication for a diagnosed illness and did not have an acute episode of infection. Healthy subjects were excluded if they were taking any medication that would modify immune responses, such as steroids.
Data collection
Clinical and demographic data were obtained from electronic medical records as well as a contemporaneous history provided by the next of kin. Data regarding mortality, length of ICU and hospital stay, ISS, new ISS (NISS), and abbreviated injury scale scores were obtained from the Trauma Audit Research Network (a UK-based centralised network that records injury details).
Haematological analysis
Whole blood cell counts were performed on citrated whole blood using a Sysmex XN-1000 haematology analyser (Sysmex UK, Milton Keynes, UK), which defines immature granulocytes (IGs) as promyelocytes, myelocytes, and metamyelocytes. Instrument performance was ensured by daily measurement of quality control material (XN Check) and participation in an external quality assurance scheme (UKNEQAS, Watford, UK).
Neutrophil oxidative burst
The ability of neutrophils to generate reactive oxygen species (ROS) in response to stimulation with 1.62 μM phorbol 12-myristate 13-acetate (PMA) was assessed using the commercially available PhagoBURST kit (BD Biosciences, Oxford, UK) in 100 μl aliquots of heparinised whole blood. Ten thousand neutrophils, gated according to forward scatter (FS)/sideward scatter (SS) properties, were analysed on an Accuri C6 flow cytometer. Data were evaluated using CFlow software (BD Biosciences) and are presented as the percentage of neutrophils that produced ROS as well as their mean fluorescence intensity (MFI).
Neutrophil phenotyping
Fifty μl aliquots of whole blood were stained for 30 minutes on ice with the following mouse antihuman monoclonal antibodies or their concentration-matched isotype controls: 8 μg/ml fluorescein isothiocyanate (FITC)-labelled CD62L (clone DREG56; eBioscience), 4 μg/ml CXCR1-FITC (clone eBIO8F1-1-4; eBioscience), 20 μg/ml R-phycoerythrin (PE)-labelled CD88 (clone S5/1; BioLegend, London, UK), 2 μg/ml CXCR2-PE (clone eBio5E8-C7-F10; eBioscience), 20 μg/ml CD63-PE (clone CLB-180; Life Technologies, Cheshire, UK), 4 μg/ml APC-labelled CD11b (clone ICRF44, BioLegend), or 2 μg/ml CD16-APC (clone 3G8, BD Biosciences). Post-incubation, red blood cells were lysed (BD PharmLyse, BD Biosciences) and samples analysed using an Accuri C6 flow cytometer, where receptor expression on 10,000 neutrophils was recorded as median fluorescence intensity (MedFI).
To determine neutrophil responsiveness to formylated peptides, heparinised blood was stimulated for 5 minutes (37°C, 5% CO2) with 1 μM formyl-methionine-leucine-phenylalanine (fMLF), after which CD62L and CD11b immunostaining was performed.
Detection of citrullinated histone H3 (CitH3) in platelet-free plasma (PFP)
PFP was prepared by double centrifugation of citrated whole blood. Blood was centrifuged at 2,000 x g for 20 minutes at 4°C, and the top two-thirds of platelet-poor plasma (PPP) was removed. PPP was then centrifuged at 13,000 x g for 2 minutes at 4°C, after which PFP was collected and stored at −80°C. CitH3 in PFP was measured by western blotting as described previously [3].
Monocyte and lymphocyte phenotyping
Fifty or one hundred μl aliquots of heparinised whole blood were stained on ice for 30 minutes with combinations of the following mouse antihuman monoclonal antibodies or their concentration-matched isotype controls: 1 μg/ml CD14-FITC (clone TUK4; Dako, Cambridgeshire, UK), 0.5 μg/ml CD16-FITC (clone eBioCB16; eBioscience), 2.5 μg/ml CCR7-FITC (clone 150503; R&D Systems, Abingdon, Oxford, UK), 1 μl CD8-PE (clone UCHT-4; ImmunoTools, Friesoythe, Germany), 0.5 μg/ml CD14-PE (clone 61D3; eBioscience), 2 μg/ml CD19-PE (clone HIB19; eBioscience), 5 μl CD56-PE (clone AF12-7H3; Miltenyi Biotec, Surrey, UK), 0.07 μg/ml HLA-DR-PE (clone LN3; eBioscience), 0.1 μg/ml CD45-APC (clone HI100; BioLegend), 10 μg/ml CD86-APC (clone IT2.2; BioLegend), 10 μg/ml toll like receptor (TLR)4-APC (clone HTA125; eBioscience), 0.625 μg/ml TLR2-APC (clone 11G7; BD Biosciences), 4 μg/ml Pacific Blue (PcB)-labelled CD3 (clone UCHT1; BD Biosciences), 1.5 μg/ml CD4-PcB (clone OKT4; eBioscience), 4 μg/ml CD14-PcB (clone M5E2; BioLegend), or 2 μg/ml PE-Cy7-labelled CD3 (clone UCHT1; eBioscience). Postincubation, red blood cells were lysed (BD PharmLyse, BD Biosciences) and samples fixed for 20 minutes at room temperature (RT) with 50 μl of fixation medium (Life Technologies). After a single wash in phosphate-buffered saline, samples were analysed on a CyAnADP bench top cytometer (Dako) and data evaluated using Summit v4.3 software (Dako). Monocytes were defined as CD14+, B cells as CD19+, NK cells as CD3−56+, and NKT cells as CD3+56−. T cells were defined as CD3+ cells and divided into 4 subsets based on the differential surface expression of the protein tyrosine phosphatase isoform CD45RA and the chemokine receptor CCR7. These subsets were denoted as naive (CD45RA+ CCR7+), central memory (CD45RA− CCR7+), effector memory (CD45RA− CCR7−), and terminally differentiated effector memory (TEMRA; CD45RA+ CCR7−) cells. Receptor expression on a minimum of 1,000 monocytes was recorded as both the percentage of antigen-positive cells and MedFI. B cell, T cell, NK cell, and NKT cell frequencies were determined in a total of 5,000 lymphocytes. These frequency values were used alongside whole blood cell counts from the Sysmex XN-1000 haematology analyser to calculate the absolute numbers of immune cells.
LPS stimulation of whole blood and cytokine/chemokine quantification
Four hundred μl aliquots of heparinised whole blood were stimulated with 1 or 10 ng/ml LPS from Escherichia coli (serotype 0111:B4; Sigma-Aldrich, Dorset, UK) or vehicle control for 4 hours (37°C, 5% CO2). Postincubation, samples were centrifuged at 461 x g for 8 minutes at 4°C, after which supernatants were collected and stored at −80°C until analysed. Following the manufacturer’s instructions, concentrations of tumour necrosis factor-alpha (TNF-α), interleukin (IL)-6, IL-8, IL-10, and monocyte chemoattractant protein-1 (MCP-1) were quantified using a commercially available magnetic bead 5-plex assay (BioRad, Hertfordshire, UK). Data were analysed using BioPlex software (BioRad), and cytokine/chemokine concentrations were normalised to monocyte counts.
Cytokine and cortisol measurements
Blood collected into BD vacutainers containing z-serum clotting activator was left at RT for 30 minutes prior to centrifugation at 1,620 x g for 10 minutes at 4°C, after which serum was removed and stored at −80°C until analysed. Following the manufacturer’s instructions, concentrations of IL-1 receptor antagonist (IL-1Ra), IL-6, IL-8, IL-10, TNF-α, granulocyte-colony stimulating factor (G-CSF), and MCP-1 were measured using a commercially available magnetic bead multiplex immunoassay (BioRad), whilst cortisol concentrations were measured by an enzyme-linked immunosorbent assay (IBL international, Hamburg, Germany).
Outcomes
The primary outcome of interest was the development of MODS, which was defined as a Sequential Organ Failure Assessment score of 6 or more, on 2 or more consecutive days, at least 48 hours postadmission [9]. Secondary outcomes were mortality and ICU-free days and hospital-free days (as calculated by 30 minus the number of days the patient stayed in hospital).
Statistical analysis
The current study is an exploratory investigation using a small convenience sample of trauma patients in order to generate hypotheses. There was no hypothesised effect upon which to power the study. Data were checked for normality using the Shapiro-Wilk test. A one-way ANOVA with Bonferroni post hoc test or a Kruskal-Wallis test with Dunn’s post hoc test was used to assess differences between patients and HCs. Relationships between continuous variables were assessed using a Pearson’s correlation. Comparisons of MODS versus no MODS patients were made on 34 variables; differences in continuous variables were assessed by Mann-Whitney U tests or independent samples t tests, whilst Chi-squared tests were performed to compare categorical variables. The resulting p-values from these 34 tests were compared to their Benjamini-Hochberg critical values to control for a false discovery rate of 5% [30]. Binary logistic regression analyses were used to explore the relationships between immune parameters and the development of MODS. In these models, the reference level of MODS was coded as “No MODS” (versus “MODS”). Model performance was measured through the proportion of variation explained by the model via R2 statistics and Brier scores, the level of calibration using the le Cessie-van Houwelingen goodness-of-fit test, and the level of discrimination using the concordance (or C) statistic [31]. Bias-corrected estimates of the C statistic were produced to account for model overfitting [32]. This internal validation consisted of 9,999 bootstrap resamples. Odds ratios (ORs) were calculated for the immune parameters in each model. The analysis was performed using the statistical software packages SPSS (IBM, New York, United States), R version 3.3.2 (http://www.r-project.org) together with the ggplot2, effects, and rms packages, and GraphPad Prism software (GraphPad Software, California, US) on data that were available for each given time point. The threshold for significance was considered to be p ≤ 0.05, with nominal p-values reported with no adjustment for multiple testing unless otherwise stated. In all figures, the horizontal line displayed in the data points collected from HCs depicts the median value.
Results
Patient enrolment and demographics
Fig 1 shows a flow diagram of patient enrolment and sampling. A total of 892 adult trauma patients were screened for inclusion into the study. Of these, 89 patients (mean age 41 years, range 18–90 years, 75 males) with a mean ISS of 24 (range 9–66) were enrolled prospectively (Table 1), with blood samples acquired from all patients ≤1 hour postinjury (mean time to sample 42 minutes, range 17–60 minutes).


Traumatic injury results in an immediate and persistent leukocytosis
Analysis of whole blood cell counts revealed a significant leukocytosis within minutes of traumatic injury that remained at the 4–12 - and 48–72-hour time points (Table 2). Underlying the immediate leukocytosis were significant elevations in monocyte, neutrophil, IG, and lymphocyte counts (Table 2), with the lymphocytosis driven by a significant increase in the absolute number of B cells, NK cells, NKT cells, and both CD4+ and CD8+ T cells (Table 3). Further phenotypical analysis of lymphocyte subsets revealed an immediate post-trauma elevation in the numbers of CD56DIM cytotoxic NK cells as well as CD4+ and CD8+ effector memory T cells and CD4+ and CD8+ central memory subsets (Table 3). For CD8+ T cells, a significant increase in highly differentiated TEMRA cells was also seen (Table 3).


In the acute postinjury phase, a significant bifurcation was seen in the innate and adaptive immune cell responses. The monocytosis, neutrophilia, and elevated IG counts persisted at the 4–12 - and 48–72-hour postinjury time points, whereas the lymphocyte counts were significantly lower than the values for HCs. This trauma-induced lymphopenia was comprehensive with reduced numbers of CD56DIM and CD56BRIGHT NK cells, TEMRA CD4+ and CD8+ T cells, and both naive and effector memory CD4+ T cells (Table 3).
Neutrophil ROS production
In response to stimulation with the protein kinase C activator PMA, ROS production by neutrophils isolated from trauma patients within minutes of injury was comparable to that recorded for HCs (Fig 2A and 2B). However, at the 4–12-hour and 48–72-hour time points, a significant reduction in both the percentage of ROS producing neutrophils (Fig 2A) and the oxidative capacity of each cell was observed (Fig 2B).

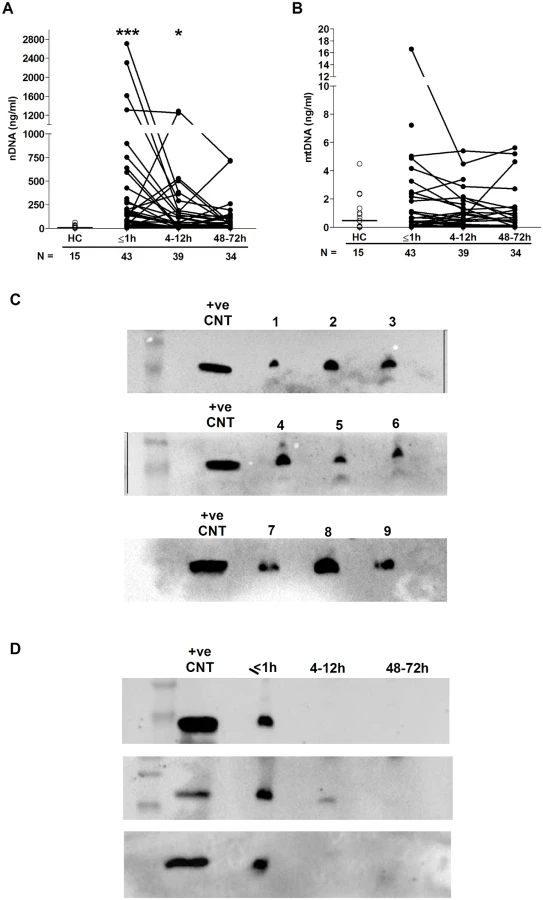
Sterile traumatic injury triggers the immediate release of nuclear DNA with evidence of neutrophil extracellular trap (NET) generation
Relative to HCs, we detected significantly elevated concentrations of nuclear but not mitochondrial DNA in plasma samples collected ≤1 hour and 4–12 hours postinjury (Fig 3A and 3B). We hypothesised that 1 possible source of this nuclear DNA was neutrophils via their production of extracellular traps, a defence mechanism in which neutrophils extrude their DNA into the extracellular environment. To test this, we screened plasma samples for CitH3, a protein that decorates the nuclear DNA backbone of NETs [33]. Western blotting revealed the presence of CitH3 in samples acquired within 1 hour of injury, but not in those obtained at the 4–12 - or 48–72-hour postinjury time points (Fig 3C and 3D).
Neutrophil surface phenotype and responsiveness to damage-associated molecular pattern (DAMP) stimulation following injury
To study the activation status of neutrophils ex vivo, we measured, in the absence of external stimulation, the surface density of the adhesion molecules CD62L and CD11b as well as the chemokine receptors CXCR1 and CXCR2. Compared to values for HCs, CD11b and CXCR2 surface expression was significantly increased and decreased, respectively, on neutrophils isolated from blood samples acquired within 1 hour of injury, with no differences detected for CD62L or CXCR1 expression (Table 4). At the 4–12 - and 48–72-hour time points, neutrophils exhibited significantly reduced CD62L and CXCR2 expression, which was accompanied by increased CD11b expression (Table 4). CXCR1 surface expression was significantly reduced relative to HC values only at the 4–12-hour postinjury time point (Table 4). An assessment of CD88 expression, the receptor for the complement factor C5a, revealed a significant trauma-induced reduction in surface density 48–72 hours postinjury (Table 4).

By combining CD16 expression data with our analysis of CD62L, we could determine the presence of CD16BRIGHT CD62LDIM neutrophils, a highly mature neutrophil subset with immunosuppressive properties [34]. In samples collected within 1 hour of trauma, as well as 4–12 and 48–72 hours postinjury, we found a significantly increased frequency and absolute number of CD16BRIGHT CD62LDIM neutrophils relative to the values recorded for HCs (Fig 4A–4C).

To examine the responsiveness of neutrophils to agents released as a result of tissue injury—so-called damage-associated molecular patterns (DAMPs)—we measured CD11b and CD62L surface density following 5 minutes of stimulation with the formylated peptide fMLF. Relative to baseline readings, the fold increase in CD11b expression that occurred post-fMLF stimulation was significantly lower for neutrophils isolated from trauma patients within minutes of injury as well as 4–12 and 48–72 hours postinjury when compared to HCs (Table 4). At all 3 postinjury time points, neutrophils from trauma patients shed significantly less CD62L following fMLF stimulation when compared to neutrophils from HCs (Table 4).
Traumatic injury is associated with immediate alterations in LPS-induced cytokine production by whole blood leukocytes that persist into the acute postinjury phase
Analysis of blood samples acquired within 1 hour of injury revealed an immediate impairment in LPS-induced cytokine production by whole blood leukocytes. Relative to HCs, significantly lower concentrations of IL-6, TNF-α, and MCP-1 were present in supernatants collected from blood samples obtained within minutes of injury following a 4-hour stimulation with 1 or 10 ng/ml LPS (Table 5). In addition, we detected a significant reduction in IL-10 secretion by leukocytes when challenged with 1 ng/ml LPS (Table 5). However, traumatic injury had no immediate impact upon LPS-induced IL-8 production (Table 5). With the exception of IL-10 secretion in response to 10 ng/ml LPS, significantly lower concentrations of IL-6, IL-8, IL-10, TNF-α, and MCP-1 were detected in supernatants of blood samples acquired from patients 4–12 hours postinjury following stimulation with 1 or 10 ng/ml LPS (Table 5). At the 48–72-hour postinjury time point, significant trauma-induced impairments in LPS-stimulated cytokine production were recorded only for IL-6, IL-10, and TNF-α (Table 5).

Changes to the surface phenotype of monocytes are evident within minutes of injury
Categorising monocytes as CD14++16− or CD14++16+, we found—in blood samples collected within 1 hour of injury—a significant increase, relative to HCs, in the absolute number of both subsets (Table 6). This trauma-induced elevation persisted 4–12 and 48–72 hours postinjury, with a significantly increased and decreased frequency of CD14++16+ and CD14++16− monocytes, respectively, observed at the latter time point (Table 6).

Surface expression of the antigen-presenting molecule human leukocyte antigen DR (HLA-DR) is commonly used as a marker of the immune competence of circulating monocytes. When measured as either the percentage of positive cells or surface density, we detected no difference in HLA-DR expression between monocytes isolated from trauma patients within minutes of injury and HCs (Table 6). However, at the 4–12 - and 48–72-hour time points, a significant trauma-induced reduction in both parameters of HLA-DR expression was observed (Table 6).
Furthermore, compared to the values recorded for HCs, trauma patients presented in the immediate aftermath of injury with a significantly higher absolute number of CD14+HLA-DRlow/− monocytes (−41; 95% confidence interval [CI] −61.00 to −25.00; p < 0.001, Fig 5C), a subset that has been shown to have immune suppressive properties [35]. Elevated counts remained at the 4–12-hour (−281; 95% CI −329 to −246; p < 0.001) and 48–72-hour (−308; 95% CI −380 to −244; p < 0.001) time points, when significantly increased frequencies of CD14+HLA-DRlow/− monocytes were also observed (Fig 5A–5C).

Analysis of TLR expression revealed an immediate and significant trauma-induced increase in the surface density of TLR2 and TLR4, which in the case of TLR4 was accompanied by an increased percentage of receptor positive monocytes (Table 6). In subsequent 4–12-hour samples, both TLR2 and TLR4 expression returned to a level comparable to HCs (Table 6). Whilst this remained the case for TLR2 at the 48–72-hour time point, a significant increase in both the percentage of TLR4+ monocytes and TLR4 surface density was found (Table 6).
Compared to the values of HCs, we found surface density of the costimulatory molecule CD86 was significantly higher on monocytes isolated from trauma patients within 1 hour of injury (Table 6). At the 4–12 - and 48–72-hour postinjury time points, CD86 surface density returned to a level comparable to that of HCs, although the frequency of CD86+ monocytes was significantly lower (Table 6).
Traumatic injury results in an immediate and sustained systemic inflammatory response
Quantification of cytokine and chemokine concentrations in patient serum samples revealed an immediate postinjury elevation in pro - and anti-inflammatory mediators. Relative to HCs, concentrations of IL-6, IL-8, G-CSF, IL1-Ra, TNF-α, and IL-10 were all significantly greater in blood samples acquired in the immediate aftermath of trauma, with these elevations persisting at the 4–12-hour time point (Fig 6A–6F). By 48–72 hours postinjury, whilst serum IL-10 and TNF-α concentrations had returned to levels seen in HCs, IL-6, IL-8, G-CSF, and IL1-Ra levels remained significantly increased (Fig 6A–6F). At no postinjury time point did we observe any significant difference in serum MCP-1 concentrations between patients and HCs (S1 Fig). An assessment of serum cortisol levels revealed a significant trauma-induced increase in circulating concentrations at all 3 postinjury time points when compared to HCs (Fig 7).


Specific immune responses in the prehospital setting are associated with the development of MODS
As previous analysis of post-hospital admission blood samples had revealed that trauma-induced alterations in lymphocyte number and monocyte function/phenotype were associated with the development of MODS [9,36,37], we performed an exploratory data analysis to examine whether, in samples acquired within minutes of injury, any potential associations existed between these facets of the immune response or other measures of immune activation and the development of MODS. From our cohort of 89 patients, 12 were excluded from analysis due to either mortality (n = 3) or hospital discharge (n = 9) within 72 hours of injury, as a length of stay (LOS) ≥ 4 days was required for patients to meet the definition of MODS adopted in this study (S2 Fig). Of the 77 remaining patients, 40 (52%) developed MODS (Table 1). A higher incidence of head, face/neck, and spinal injuries was present amongst patients in the MODS group, who also exhibited significantly higher ISS and NISS as well as lower admission Glasgow coma scale (GCS) scores and base excess (an indicator of haemorrhagic shock) when compared to those patients who did not develop MODS (Table 1). In terms of outcomes, both ICU and hospital-free days were significantly lower for MODS patients, in whom a higher incidence of mortality was also recorded (Table 1).
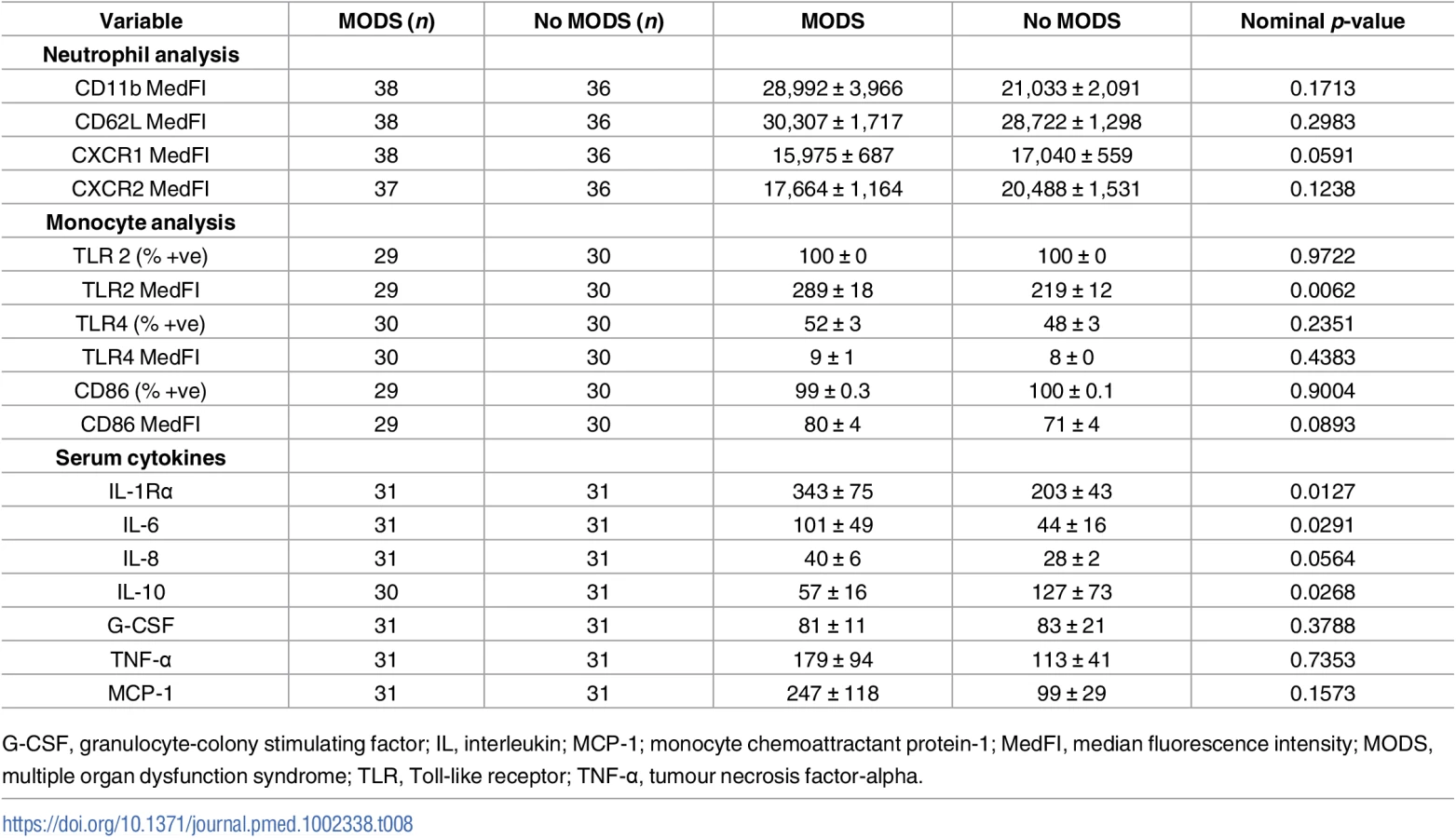
For all immune and inflammatory parameters assessed (Tables 7 and 8), only NKT and CD3+8+ cell numbers were found to be significantly different between the 2 patient groups after controlling for a false discovery rate of 5%, with elevated values present in samples acquired within 1 hour of injury from those patients who later developed MODS (mean difference of 383, 95% CI 153–614, nominal p = 0.0006 for NKT number and mean difference of 509, 95% CI 193–811, nominal p = 0.0015 for CD3+8+; Table 7).

Patients who developed MODS had a significantly higher ISS and a lower base excess at admission (Table 1). Thus, binary logistic regression analysis was performed to account for any potential effects of injury severity and haemorrhagic shock on the relationship we had identified between NKT and CD3+8+ number and the development of MODS. Our analysis found that only NKT cell counts exhibited an independent association with MODS after adjusting for ISS and base excess (Table 9). To examine this output in further detail, we explored the effect that increasing the value of NKT cell number by quartiles had on developing MODS. As shown in Table 10, we found that increasing the value of NKT cell number from its first quartile (84.5 x 106/L) to the second quartile (185.5 x 106/L) corresponded to an OR of developing MODS of 1.63 (95% CI 1.081–2.463), whilst increasing the value of NKT number from its second quartile (185.5 x 106/L) to the third quartile (416.8 x 106/L) corresponded to an OR of developing MODS of 3.06 (95% CI 1.195–7.880).


Discussion
Here, via the analysis of 89 blood samples obtained from patients within 1 hour of injury, the so-called “Golden Hour”, we have provided for the first time, to our knowledge, a detailed description of the ultra-early changes that occur in the composition, function, and/or phenotype of the innate and adaptive arms of the immune system post-trauma. We have shown that accompanying the well-documented “cytokine storm” of raised pro - and anti-inflammatory cytokines was a complex immune response that encompassed features of both immune activation and suppression. For example, in the background of leukocytosis, ex vivo phenotyping revealed increased and decreased surface expression of CD11b and CXCR2, respectively, on neutrophils and increased surface TLR2, TLR4, and CD86 expression on monocytes, whilst the presence of nuclear DNA and CitH3 in plasma samples suggested the rapid induction of neutrophil function in the form of NET generation. These signs of immediate immune activation occurred alongside alterations consistent with immuneparesis, such as elevated numbers of suppressive CD16BRIGHT CD62LDIM neutrophils and CD14+HLA-DRlow/− monocytes, a lack of responsiveness of neutrophils to fMLF stimulation, and impaired leukocyte cytokine production following LPS stimulation. Thus, together with the results of previous genomic and functional studies [10,18], our data challenge the traditional SIRS:CARS concept in which a sequential immune and inflammatory response to injury is suggested and in which the CARS response is proposed to follow the SIRS response in order to restore homeostasis [38]. Rather, the increasing consensus is that the SIRS and CARS responses are largely concomitant features of the postinjury immune response, and our study reveals that these are evident within minutes of trauma.
Analysis of neutrophil function across our 3 postinjury time points revealed a significant reduction in PMA-induced ROS production and a reduced responsiveness to bacterial peptide stimulation, with the latter defect demonstrated by reduced shedding of CD62L following fMLF treatment. These aberrations were accompanied by significantly elevated numbers and frequencies of both highly mature CD16BRIGHT CD62LDIM neutrophils and IGs, with the emergence of IGs into the circulation borne most likely from a stress-induced early mobilisation of immature precursors from the bone marrow. The increase in mature neutrophil numbers is most likely a result of glucocorticoid-induced demargination [39], as up to 50% of neutrophils are marginated in the endovascular lining in the noninjured state and can be very rapidly mobilised into the circulation to produce the neutrophilia of trauma. Compared to their CD16BRIGHT CD62LBRIGHT counterparts, CD16BRIGHT CD62LDIM neutrophils exhibit reduced ROS production, a trait shared by IGs, who also show reduced responsiveness to fMLF challenge [40–42]. Thus, the heterogeneous neutrophil pool consisting of highly mature subsets and IGs that developed postinjury could be 1 factor underlying the altered neutrophil function we describe here. In addition to reduced ROS generation and responsiveness to fMLF, immature neutrophils exhibit impaired phagocytosis, chemotaxis, and bacterial killing [40] aberrations, which in the case of phagocytosis and chemotaxis, mirror the ex vivo behaviours described for neutrophils isolated from trauma patients [3–5]. Thus, the release of IGs into the circulation post-trauma may impact upon many facets of neutrophil antimicrobial defence, and given that IG numbers peaked in samples acquired in the immediate aftermath of injury, defects in neutrophil function may be apparent within minutes of trauma.
At all 3 postinjury time points, trauma patients presented with significantly elevated numbers and/or frequencies of suppressive CD14+HLA-DRlow/− monocytes and CD16BRIGHT CD62LDIM neutrophils. A rich source of IL-10 and arginase [43,44], CD14+ HLA-DR-/low monocytes have been shown in vitro to inhibit T cell activation, suppress NK cell cytotoxicity, and promote the expansion of T regulatory cells [45–47], whilst via their generation of ROS, CD16BRIGHT CD62LDIM neutrophils have been reported to suppress T cell proliferation [34]. Thus, as proposed for other conditions in which systemic immune suppression has been described [43], the emergence into circulation of immune modulatory subsets could offer 1 potential mechanistic explanation for the development of immuneparesis after injury. On this theme of post-trauma immuneparesis, data are beginning to emerge that suggest that in addition to triggering cell activation, prior exposure to DAMPs can induce a state of functional tolerance in immune cells. In monocytes, it has been shown that preconditioning with heat shock protein-70, a nuclear-derived DAMP whose plasma concentrations are significantly elevated within minutes of injury [10], culminates in impaired cytokine production upon secondary LPS challenge [48], whilst for neutrophils, prior exposure to fMLF, which activates the same signalling pathways as mitochondrial-derived N-formyl peptides, results in aberrant responses in subsequent functional assays [5]. Thus, in line with previous studies [5,10], we speculate that via immune cell activation, an immediate trauma-induced elevation in circulating DAMPS may induce a state of functional tolerance that culminates in impaired functional responses in vitro.
At the 4–12 - and 48–72-hour postinjury time points, we detected a significant reduction in PMA-induced neutrophil ROS production, an impairment that has previously been described for neutrophils isolated from patients following thermal [49] and isolated TBI [50]. In the case of thermal injury, the reduction in ROS production was attributed to a deficiency in p47-phox and p67-phox [49], 2 cytosolic components of NADPH oxidase, a multi-subunit enzyme that initiates the respiratory burst. In the only human-based study to our knowledge that has examined NADPH oxidase expression in immune cells post-trauma, Liao and colleagues reported elevated expression of the membrane-residing subunit gp91-phox in leukocyte homogenates from TBI patients [4]. However, the group did not investigate the expression of other membrane or cytosolic subunits of NADPH oxidase, nor did they study neutrophils in isolation [4]. Thus, it is currently unclear as to whether, as reported for burns patients [49], reduced expression of NADPH oxidase subunits could underlie the impairment in PMA-induced ROS generation we found postinjury. A critical step in the activation of NADPH oxidase is the phosphorylation of its subunits. A pharmacological mimetic of diacylglycerol, PMA works in conjunction with intracellular calcium to activate the classical isoforms of the serine/threonine protein kinase, protein kinase C (PKC), whose substrates include the NADPH oxidase subunits gp91-phox, p47-phox, and p67-phox [51–53]. Interestingly, blunted intracellular calcium mobilisation following inflammatory agonist stimulation has been reported for neutrophils isolated from major trauma patients [54]. Thus, an additional/alternative explanation for the impaired ROS production in response to PMA postinjury could be reduced PKC activation due to a lack of intracellular calcium.
Recently, in the setting of critical illness, Hirose et al. [55] failed to detect CitH3, a protein that decorates the DNA backbone of NETs [33], in blood samples obtained from trauma patients at the time of their admission to an ICU. The authors proposed that its absence reflected the fact that samples had been acquired too soon following the onset of critical illness for NET formation to have occurred [55]. However, our preliminary data showing the presence of CitH3 in plasma samples collected from patients within minutes of trauma suggest their samples were obtained too late (rather than too early), since NET production appears to be a feature unique to the ultra-early immune response to trauma. Our observation of immediate NET formation raises the question as to how a feature of neutrophil antimicrobial defence involved in pathogen entrapment and neutralisation can be triggered so quickly following sterile injury. Recently, it was reported that neutrophils treated in vitro with the nuclear-derived DAMP high-mobility group box 1 (HMGB1) generate NETs within 1 hour of stimulation [56], and elevated plasma levels of this DAMP have been recorded within 30 minutes of major injury [57]. Thus, an immediate release of HMGB1 from damaged tissue may be 1 mechanism underlying the rapid formation of NETs post-trauma.
Our study highlights how differences in sample timing can influence our understanding of the postinjury immune response. For instance, elevated absolute numbers of B, NK, NKT, CD3+, CD3+4+, and CD3+8+ cells were recorded in blood samples acquired from patients within minutes of injury, thereby revealing an immediate post-trauma leukocytosis. However, at the 4–12-hour time point, patients presented in a state of lymphopenia, with significantly reduced numbers of circulating CD56BRIGHT NK cells as well as CD3+ and CD3+4+ T cells. Similarly, we found features of the ultra-early immune response, such as the presence of plasma-residing CitH3 and increased surface densities of CD16 on neutrophils and TLR2, TLR4, and CD86 on monocytes, were absent from samples acquired from patients 4–12 hours postinjury. Conversely, only in blood samples obtained in the hours following injury were defects in neutrophil ROS generation upon PMA stimulation and reduced HLA-DR expression by monocytes observed, thereby demonstrating that not all aspects of trauma-induced changes in immunity occur within minutes of injury.
Manson et al. recently reported that lymphocyte activation within 2 hours of injury was associated with later development of MODS [9]. Based on this observation, we performed exploratory analysis of our dataset to investigate whether any features of the ultra-early immune response to injury were related to patient outcome. After controlling for a false discovery rate of 5%, we identified a potential association between elevated absolute numbers of NKT and CD3+8+ cells within minutes of injury and the subsequent development of MODS. However, after adjusting for ISS and base excess, only NKT cell counts exhibited an independent association with MODS. It must be stressed that because of our small patient cohort and the fact that our study was not designed from the outset to test this relationship directly, this association may be limited to our dataset, and thus, we cannot generalise our data to all trauma patients from whom blood samples could be acquired within 1 hour of injury. Thus, rather than a conclusive finding, our observation has generated a hypothesis that future adequately powered prospective cohort studies should investigate in order to establish whether the immune response elicited within minutes of trauma is indeed associated with poor clinical outcome.
The relatively high level of MODS for this study cohort may at first seem unusual since inclusion criteria for the study was ISS > 8. However, the mean ISS for the study cohort was 24, representing a relatively high injury burden. The statistically significant difference between the ISS for those who developed organ failure (mean ISS 31) versus those who did not (mean ISS 18) is in keeping with what might be expected in the trauma population. It is also important to note that none of our patients received blood products in the prehospital setting, thereby ruling out the possibility that the immediate dysfunction we observed was a consequence of the immune modulatory effects of blood transfusion [58,59].
The major limitation of our study is its relatively small sample size and the fact that it was conducted at a single major trauma centre, meaning the results generated require validation in larger independent cohorts. That said, given the logistical difficulties associated with obtaining prehospital samples, our study has provided novel data that should serve to stimulate future prehospital-based research to answer questions we did not address here. For example, although we performed phenotypic analysis, we did not assess T or B cell function, nor did we examine the phagocytic capacity of innate immune cells. Moreover, we did not examine any aspect of immune cell signalling, which may have provided a mechanistic insight into the immune changes we observed. A previous study reported that reduced immune function 48–96 hours postinjury was accompanied by impaired intracellular signalling [17], and our finding of reduced PMA-induced ROS production by neutrophils postinjury points to a potential impairment in signalling pathways distal to the plasma membrane. In addition, our study recruited a very heterogeneous patient group that included subjects with and without head injuries, though we excluded those with isolated head injuries in this study. Interestingly, a higher incidence of head injury was observed amongst patients who developed MODS, and thus, future studies enrolling a larger number of patients should consider patients with extracranial injuries in the presence or absence of head injury as separate groups. Finally, our study design was at risk of selection bias, since only patients that underwent venepuncture during prehospital evacuation could be enrolled. This is a procedure that takes a few minutes and may have only been performed at certain times or during prehospital treatment that would allow such an addition to standard care. This may have inadvertently increased the risk of selection bias towards less severely injured patients, although maintenance of a screening log helped reduce this risk.
In summary, our study has highlighted the dynamic nature of the immune response to trauma and shown at the functional and phenotypic level that immune alterations consistent with activation and suppression are evident within 1 hour of injury, thus supporting the idea of an immediate and concomitant induction of the SIRS and CARS responses post-trauma.
Supporting Information
Zdroje
1. Giannoudis PV. Current concepts of the inflammatory response after major trauma: an update. Injury. 2003; 34 : 397–404. 12767787
2. Hazeldine J, Hampson P, Lord JM. The diagnostic and prognostic value of systems biology research in major traumatic and thermal injury: a review. Burns and Trauma. 2016. doi: 10.1186/s41038-016-0059-3 27672669
3. Hampson P, Dinsdale RJ, Wearn CM, Bamford AL, Bishop JR, Hazeldine J, et al. Neutrophil Dysfunction, Immature Granulocytes, and Cell-free DNA are Early Biomarkers of Sepsis in Burn-injured Patients: A Prospective Observational Cohort Study. Ann.Surg. 2016. doi: 10.1097/SLA.0000000000001807 27232244
4. Liao Y, Liu P, Guo F, Zhang ZY, Zhang Z. Oxidative burst of circulating neutrophils following traumatic brain injury in human. PLoS ONE. 2013. 8: e68963. doi: 10.1371/journal.pone.0068963 23894384
5. Li H, Itagaki K, Sandler N, Gallo D, Galenkamp A, Kaczmarek E, et al. Mitochondrial damage-associated molecular patterns from fractures suppress pulmonary immune responses via formyl peptide receptors 1 and 2. J.Trauma Acute.Care Surg. 2015;78 : 272–279. doi: 10.1097/TA.0000000000000509 25757111
6. Marks W, Dropiewska G, Bryl E, Dudek R, Wieruszewski J, Stasiak M, et al. Immunomonitoring in patients with early moderate and severe head trauma. Cent. Eur. J. Public Health. 2013;38 : 494–499.
7. Perez-Barcena J, Regueiro V, Crespi C, Pierola J, Oliver A, Llompart-Pou JA, et al. Expression of toll-like receptors 2 and 4 is upregulated during hospital admission in traumatic patients: lack of correlation with blunted innate immune responses. Ann.Surg. 2010;251 : 521–527. doi: 10.1097/SLA.0b013e3181cc8f84 20134316
8. Laudanski K, Miller-Graziano C, Xiao W, Mindrinos MN, Richards DR, De A, et al. Cell-specific expression and pathway analyses reveal alterations in trauma-related human T cell and monocyte pathways. Proc.Natl.Acad.Sci.U.S.A 2006;103 : 15564–15569. doi: 10.1073/pnas.0607028103 17032758
9. Manson J, Cole E, De'Ath HD, Vulliamy P, Meier U, Pennington D, et al. Early changes within the lymphocyte population are associated with the development of multiple organ dysfunction syndrome in trauma patients. Crit Care 2016;20 : 176. doi: 10.1186/s13054-016-1341-2 27268230
10. Timmermans K, Kox M, Vaneker M, van den Berg M, John A, van LA, et al. Plasma levels of danger-associated molecular patterns are associated with immune suppression in trauma patients. Intensive Care Med. 2016;42 : 551–561. doi: 10.1007/s00134-015-4205-3 26912315
11. Gebhard F, Pfetsch H, Steinbach G, Strecker W, Kinzl L, Bruckner UB. Is interleukin 6 an early marker of injury severity following major trauma in humans? Arch.Surg. 2000;135 : 291–295. 10722030
12. Arand M, Melzner H, Kinzl L, Bruckner UB, Gebhard F. Early inflammatory mediator response following isolated traumatic brain injury and other major trauma in humans. Langenbecks Arch.Surg. 2001;386 : 241–248. 11466564
13. Hietbrink F, Koenderman L, Althuizen M, Pillay J, Kamp V, Leenen LP. Kinetics of the innate immune response after trauma: implications for the development of late onset sepsis. Shock 2013;40 : 21–27. doi: 10.1097/SHK.0b013e318295a40a 23603769
14. Visser T, Hietbrink F, Groeneveld KM, Koenderman L, Leenen LP. Isolated blunt chest injury leads to transient activation of circulating neutrophils. Eur.J.Trauma Emerg.Surg. 2011;37 : 177–184. doi: 10.1007/s00068-010-0041-x 21837259
15. West SD, Goldberg D, Ziegler A, Krencicki M, Du Clos TW, Mold C. Transforming growth factor-beta, macrophage colony-stimulating factor and C-reactive protein levels correlate with CD14(high)CD16+ monocyte induction and activation in trauma patients. PLoS ONE. 2012;7: e52406. doi: 10.1371/journal.pone.0052406 23285029
16. Wolach B, Sazbon L, Gavrieli R, Broda A, Schlesinger M. Early immunological defects in comatose patients after acute brain injury. J.Neurosurg. 2001;94 : 706–711. doi: 10.3171/jns.2001.94.5.0706 11354400
17. Kasten KR, Goetzman HS, Reid MR, Rasper AM, Adediran SG, Robinson CT, et al. Divergent adaptive and innate immunological responses are observed in humans following blunt trauma. BMC.Immunol. 2010;11 : 4. doi: 10.1186/1471-2172-11-4 20100328
18. Xiao W, Mindrinos MN, Seok J, Cuschieri J, Cuenca AG, Gao H, et al. A genomic storm in critically injured humans. J.Exp.Med. 2011;208 : 2581–2590. doi: 10.1084/jem.20111354 22110166
19. Vanzant EL, Hilton RE, Lopez CM, Zhang J, Ungaro RF, Gentile LF, et al. Advanced age is associated with worsened outcomes and a unique genomic response in severely injured patients with hemorrhagic shock. Crit Care 2015. 19 : 77. doi: 10.1186/s13054-015-0788-x 25880307
20. Orr SK, Butler KL, Hayden D, Tompkins RG, Serhan CN, Irimia D. Gene Expression of Proresolving Lipid Mediator Pathways Is Associated With Clinical Outcomes in Trauma Patients. Crit Care Med. 2015;43 : 2642–2650. doi: 10.1097/CCM.0000000000001312 26488221
21. Namas RA, Vodovotz Y, Almahmoud K, Abdul-Malak O, Zaaqoq A, Namas R, et al. Temporal Patterns of Circulating Inflammation Biomarker Networks Differentiate Susceptibility to Nosocomial Infection Following Blunt Trauma in Humans. Ann.Surg. 2016; 263 : 191–198. doi: 10.1097/SLA.0000000000001001 25371118
22. Jeschke MG, Gauglitz GG, Finnerty CC, Kraft R, Mlcak RP, Herndon DN. Survivors versus nonsurvivors postburn: differences in inflammatory and hypermetabolic trajectories. Ann.Surg. 2014;259 : 814–823. doi: 10.1097/SLA.0b013e31828dfbf1 23579577
23. Kraft R, Herndon DN, Finnerty CC, Cox RA, Song J, Jeschke MG. Predictive Value of IL-8 for Sepsis and Severe Infections After Burn Injury: A Clinical Study. Shock 2015;43 : 222–227. doi: 10.1097/SHK.0000000000000294 25514427
24. Cuschieri J, Bulger E, Schaeffer V, Sakr S, Nathens AB, Hennessy L, et al. Early elevation in random plasma IL-6 after severe injury is associated with development of organ failure. Shock 2010;34 : 346–351. doi: 10.1097/SHK.0b013e3181d8e687 20844410
25. Ferreira LC, Regner A, Miotto KD, Moura S, Ikuta N, Vargas AE, et al. Increased levels of interleukin-6, -8 and -10 are associated with fatal outcome following severe traumatic brain injury. Brain Inj. 2014;28 : 1311–1316. doi: 10.3109/02699052.2014.916818 24830571
26. Maier B, Lefering R, Lehnert M, Laurer HL, Steudel WI, Neugebauer EA, et al. Early versus late onset of multiple organ failure is associated with differing patterns of plasma cytokine biomarker expression and outcome after severe trauma. Shock 2007;28 : 668–674. 18092384
27. Muszynski JA, Nofziger R, Greathouse K, Nateri J, Hanson-Huber L, Steele L, et al. Innate immune function predicts the development of nosocomial infection in critically injured children. Shock 2014;42 : 313–321. doi: 10.1097/SHK.0000000000000217 24978895
28. Gouel-Cheron A, Allaouchiche B, Guignant C, Davin F, Floccard B, Monneret G. Early interleukin-6 and slope of monocyte human leukocyte antigen-DR: a powerful association to predict the development of sepsis after major trauma. PLoS ONE 2012;7: e33095. doi: 10.1371/journal.pone.0033095 22431998
29. Cheron A, Floccard B, Allaouchiche B, Guignant C, Poitevin F, Malcus C, et al. Lack of recovery in monocyte human leukocyte antigen-DR expression is independently associated with the development of sepsis after major trauma. Crit Care 2010;14: R208. doi: 10.1186/cc9331 21092108
30. Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society Series B (Methodological) 1995. 57 : 289–300.
31. Steyerberg EW, Vickers AJ, Cook NR, Gerds T, Gonen M, Obuchowski N, et al. Assessing the performance of prediction models: a framework for traditional and novel measures. Epidemiology 2010;21 : 128–138. doi: 10.1097/EDE.0b013e3181c30fb2 20010215
32. Steyerberg EW, Harrell FE Jr, Borsboom GJ, Eijkemans MJ, Vergouwe Y, Habbema JD. Internal validation of predictive models: efficiency of some procedures for logistic regression analysis. J.Clin.Epidemiol. 2001;54 : 774–781. 11470385
33. Liu CL, Tangsombatvisit S, Rosenberg JM, Mandelbaum G, Gillespie EC, Gozani OP, et al. Specific post-translational histone modifications of neutrophil extracellular traps as immunogens and potential targets of lupus autoantibodies. Arthritis Res.Ther. 2012;14: R25. doi: 10.1186/ar3707 22300536
34. Pillay J, Kamp VM, van HE, Visser T, Tak T, Lammers JW, et al. A subset of neutrophils in human systemic inflammation inhibits T cell responses through Mac-1. J.Clin.Invest 2012; 122 : 327–336. doi: 10.1172/JCI57990 22156198
35. Laborde RR, Lin Y, Gustafson MP, Bulur PA, Dietz AB. Cancer Vaccines in the World of Immune Suppressive Monocytes (CD14(+)HLA-DR(lo/neg) Cells): The Gateway to Improved Responses. Front Immunol. 2014;5 : 147. doi: 10.3389/fimmu.2014.00147 24772111
36. Kirchhoff C, Biberthaler P, Mutschler WE, Faist E, Jochum M, Zedler S. Early down-regulation of the pro-inflammatory potential of monocytes is correlated to organ dysfunction in patients after severe multiple injury: a cohort study. Crit Care 2009; 13: R88. doi: 10.1186/cc7914 19519886
37. Rainer TH, Ng MH, Lam NY, Chan TY, Cocks RA. Role of monocyte L-selectin in the development of post-traumatic organ failure. Resuscitation 2001;51 : 139–149. 11718969
38. Gentile LF, Cuenca AG, Efron PA, Ang D, Bihorac A, McKinley BA, et al. Persistent inflammation and immunosuppression: a common syndrome and new horizon for surgical intensive care. J.Trauma Acute.Care Surg. 2012;72 : 1491–1501. doi: 10.1097/TA.0b013e318256e000 22695412
39. Nakagawa M, Terashima T, D'yachkova Y, Bondy GP, Hogg JC, van Eeden SF. Glucocorticoid-induced granulocytosis: contribution of marrow release and demargination of intravascular granulocytes. Circulation 1998;98 : 2307–2313. 9826319
40. Drifte G, Dunn-Siegrist I, Tissieres P, Pugin J. Innate immune functions of immature neutrophils in patients with sepsis and severe systemic inflammatory response syndrome. Crit Care Med. 2013;41 : 820–832. doi: 10.1097/CCM.0b013e318274647d 23348516
41. Pillay J, Ramakers BP, Kamp VM, Loi AL, Lam SW, Hietbrink F, et al. Functional heterogeneity and differential priming of circulating neutrophils in human experimental endotoxemia. J.Leukoc.Biol. 2010;88 : 211–220. doi: 10.1189/jlb.1209793 20400675
42. Sauce D, Dong Y, Campillo-Gimenez L, Casulli S, Bayard C, Autran B, et al. Reduced Oxidative Burst by Primed Neutrophils in the Elderly Individuals Is Associated With Increased Levels of the CD16bright/CD62Ldim Immunosuppressive Subset. J.Gerontol.A Biol.Sci.Med.Sci. 2017;72 : 163–172. doi: 10.1093/gerona/glw062 27069096
43. Lin Y, Gustafson MP, Bulur PA, Gastineau DA, Witzig TE, Dietz AB. Immunosuppressive CD14+HLA-DR(low)/ - monocytes in B-cell non-Hodgkin lymphoma. Blood 2011;117 : 872–881. doi: 10.1182/blood-2010-05-283820 21063024
44. Vuk-Pavlovic S, Bulur PA, Lin Y, Qin R, Szumlanski CL, Zhao X, et al. Immunosuppressive CD14+HLA-DRlow/ - monocytes in prostate cancer. Prostate 2010;70 : 443–455. doi: 10.1002/pros.21078 19902470
45. Hoechst B, Ormandy LA, Ballmaier M, Lehner F, Kruger C, Manns MP, et al. A new population of myeloid-derived suppressor cells in hepatocellular carcinoma patients induces CD4(+)CD25(+)Foxp3(+) T cells. Gastroenterology 2008;135 : 234–243. doi: 10.1053/j.gastro.2008.03.020 18485901
46. Hoechst B, Voigtlaender T, Ormandy L, Gamrekelashvili J, Zhao F, Wedemeyer H, et al. Myeloid derived suppressor cells inhibit natural killer cells in patients with hepatocellular carcinoma via the NKp30 receptor. Hepatology 2009;50 : 799–807. doi: 10.1002/hep.23054 19551844
47. Xiu B, Lin Y, Grote DM, Ziesmer SC, Gustafson MP, Maas ML, et al. IL-10 induces the development of immunosuppressive CD14(+)HLA-DR(low/-) monocytes in B-cell non-Hodgkin lymphoma. Blood Cancer J. 2015; 5: e328. doi: 10.1038/bcj.2015.56 26230952
48. Aneja R, Odoms K, Dunsmore K, Shanley TP, Wong HR. Extracellular heat shock protein-70 induces endotoxin tolerance in THP-1 cells. J.Immunol. 2006;177 : 7184–7192. 17082636
49. Rosenthal J, Thurman GW, Cusack N, Peterson VM, Malech HL, Ambruso DR. Neutrophils from patients after burn injury express a deficiency of the oxidase components p47-phox and p67-phox. Blood 1996;88 : 4321–4329. 8943869
50. Junger WG, Rhind SG, Rizoli SB, Cuschieri J, Baker AJ, Shek PN, et al. Prehospital hypertonic saline resuscitation attenuates the activation and promotes apoptosis of neutrophils in patients with severe traumatic brain injury. Shock 2013;40 : 366–374. doi: 10.1097/SHK.0000000000000038 24088993
51. Benna JE, Dang PM, Gaudry M, Fay M, Morel F, Hakim J, et al. Phosphorylation of the respiratory burst oxidase subunit p67(phox) during human neutrophil activation. Regulation by protein kinase C-dependent and independent pathways. J.Biol.Chem. 1997; 272 : 17204–17208. 9202043
52. Nauseef WM, Volpp BD, McCormick S, Leidal KG, Clark RA. Assembly of the neutrophil respiratory burst oxidase. Protein kinase C promotes cytoskeletal and membrane association of cytosolic oxidase components. J.Biol.Chem. 1991;266 : 5911–5917. 1848559
53. Raad H, Paclet MH, Boussetta T, Kroviarski Y, Morel F, Quinn MT, et al. Regulation of the phagocyte NADPH oxidase activity: phosphorylation of gp91phox/NOX2 by protein kinase C enhances its diaphorase activity and binding to Rac2, p67phox, and p47phox. FASEB J. 2009;23 : 1011–1022. doi: 10.1096/fj.08-114553 19028840
54. Tarlowe MH, Kannan KB, Itagaki K, Adams JM, Livingston DH, Hauser CJ. Inflammatory chemoreceptor cross-talk suppresses leukotriene B4 receptor 1-mediated neutrophil calcium mobilization and chemotaxis after trauma. J.Immunol. 2003;171 : 2066–2073. 12902512
55. Hirose T, Hamaguchi S, Matsumoto N, Irisawa T, Seki M, Tasaki O, et al. Presence of neutrophil extracellular traps and citrullinated histone H3 in the bloodstream of critically ill patients. PLoS ONE. 2014;9: e111755. doi: 10.1371/journal.pone.0111755 25392950
56. Huang H, Tohme S, Al-Khafaji AB, Tai S, Loughran P, Chen L, et al. Damage-associated molecular pattern-activated neutrophil extracellular trap exacerbates sterile inflammatory liver injury. Hepatology 2015;62 : 600–614. doi: 10.1002/hep.27841 25855125
57. Cohen MJ, Brohi K, Calfee CS, Rahn P, Chesebro BB, Christiaans SC, et al. Early release of high mobility group box nuclear protein 1 after severe trauma in humans: role of injury severity and tissue hypoperfusion. Crit Care 2009;13: R174. doi: 10.1186/cc8152 19887013
58. Torrance HD, Vivian ME, Brohi K, Prowle JR, Pearse RM, Owen HC, et al. Changes in gene expression following trauma are related to the age of transfused packed red blood cells. J.Trauma Acute.Care Surg. 2015;78 : 535–542. doi: 10.1097/TA.0000000000000534 25710424
59. Torrance HD, Brohi K, Pearse RM, Mein CA, Wozniak E, Prowle JR, et al. Association between gene expression biomarkers of immunosuppression and blood transfusion in severely injured polytrauma patients. Ann.Surg. 2015; 261 : 751–759. doi: 10.1097/SLA.0000000000000653 24670848
Štítky
Interní lékařstvíČlánek vyšel v časopise
PLOS Medicine
2017 Číslo 7
- Alternativní léčebné možnosti u hypercholesterolemie při intoleranci statinů
- Vliv kombinace nutraceutik na remodelaci levé komory srdeční u osob s metabolickým syndromem
- Nutraceutika a jejich ovlivnění mírného kardiometabolického rizika
- Princip účinku medu v léčbě chronických i infikovaných ran
- Superoxidovaný roztok a jeho využití v léčbě ran
Nejčtenější v tomto čísle
- Signatures of inflammation and impending multiple organ dysfunction in the hyperacute phase of trauma: A prospective cohort study
- Multidrug-resistant gonorrhea: A research and development roadmap to discover new medicines
- Patient-reported outcomes and survival in multiple sclerosis: A 10-year retrospective cohort study using the Multiple Sclerosis Impact Scale–29
- Ammonium tetrathiomolybdate following ischemia/reperfusion injury: Chemistry, pharmacology, and impact of a new class of sulfide donor in preclinical injury models