
Is Genetically Diverse in Animals and Appears to Have Crossed the Host Barrier to Humans on (At Least) Two Occasions
Chlamydia pneumoniae is a common human and animal pathogen associated with a wide range of diseases. Since the first isolation of C. pneumoniae TWAR in 1965, all human isolates have been essentially clonal, providing little evolutionary insight. To address this gap, we investigated the genetic diversity of 30 isolates from diverse geographical locations, from both human and animal origin (amphibian, reptilian, equine and marsupial). Based on the level of variation that we observed at 23 discreet gene loci, it was clearly evident that the animal isolates were more diverse than the isolates of human origin. Furthermore, we show that C. pneumoniae isolates could be grouped into five major genotypes, A-E, with A, B, D and E genotypes linked by geographical location, whereas genotype C was found across multiple continents. Our evidence strongly supports two separate animal-to-human cross species transfer events in the evolutionary history of this pathogen. The C. pneumoniae human genotype identified in the USA, Canada, Taiwan, Iran, Japan, Korea and Australia (non-Indigenous) most likely originated from a single amphibian or reptilian lineage, which appears to have been previously geographically widespread. We identified a separate human lineage present in two Australian Indigenous isolates (independent geographical locations). This lineage is distinct and is present in Australian amphibians as well as a range of Australian marsupials.
Published in the journal:
. PLoS Pathog 6(5): e32767. doi:10.1371/journal.ppat.1000903
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1000903
Summary
Chlamydia pneumoniae is a common human and animal pathogen associated with a wide range of diseases. Since the first isolation of C. pneumoniae TWAR in 1965, all human isolates have been essentially clonal, providing little evolutionary insight. To address this gap, we investigated the genetic diversity of 30 isolates from diverse geographical locations, from both human and animal origin (amphibian, reptilian, equine and marsupial). Based on the level of variation that we observed at 23 discreet gene loci, it was clearly evident that the animal isolates were more diverse than the isolates of human origin. Furthermore, we show that C. pneumoniae isolates could be grouped into five major genotypes, A-E, with A, B, D and E genotypes linked by geographical location, whereas genotype C was found across multiple continents. Our evidence strongly supports two separate animal-to-human cross species transfer events in the evolutionary history of this pathogen. The C. pneumoniae human genotype identified in the USA, Canada, Taiwan, Iran, Japan, Korea and Australia (non-Indigenous) most likely originated from a single amphibian or reptilian lineage, which appears to have been previously geographically widespread. We identified a separate human lineage present in two Australian Indigenous isolates (independent geographical locations). This lineage is distinct and is present in Australian amphibians as well as a range of Australian marsupials.
Introduction
Members of the family Chlamydiaceae are obligate intracellular pathogens of a wide range of animals, birds and humans. Of the nine currently recognised species (Chlamydia pneumoniae, Chlamydia trachomatis, Chlamydia psittaci, Chlamydia suis, Chlamydia pecorum, Chlamydia abortus, Chlamydia felis, Chlamydia muridarum and Chlamydia caviae), C. pneumoniae has an extremely diverse host range (like C. psittaci), being reported in humans [1], horses [2], reptiles [3], amphibians [3]–[5] and several Australian marsupials, including koalas [6] and bandicoots [7].
C. pneumoniae exposure is widespread in humans, with sero-prevalence studies reporting 50% infection levels by age 20 and reaching 80% in the elderly [8]. In humans, C. pneumoniae infections can range from asymptomatic to severe respiratory disease, including pneumonia. Less common presentations include bronchitis, pharyngitis, laryngitis and sinusitis, making up 5% of cases [9]. In addition to respiratory infections in humans, C. pneumoniae has also been associated with atherosclerosis and stroke [8], [10], myocarditis [11], multiple sclerosis [12] and Alzheimer's disease [13].
Despite the widespread prevalence of C. pneumoniae in humans, all isolates studied to date are extremely similar at the DNA level. Four C. pneumoniae human isolates have had their full genome sequenced: AR39 [14], CWL029 [15], J138 [16] and TW183 [17]. Genomic comparisons revealed a highly conserved (>99.9%) gene order and organisation, with few deletions and less than 300 single nucleotide polymorphisms (SNPs) distinguishing the isolates. This near clonality of C. pneumoniae human isolates that are temporally and geographically separate, has been taken to indicate that human infections are a relatively recent event [18] and that the efficient respiratory spread of the agent explains how 60–80% of adults worldwide have been infected at least once in their lifetime [19].
In addition to infections in humans, C. pneumoniae infections have been reported from a range of animals from an equally diverse range of body sites, including respiratory (frog, snake, bandicoot, koala, horse), liver and spleen (frog, iguana, chameleon), heart (turtle, snake, frog), conjunctival (koala) and urogenital tract (koala) [3]–[5], [7], [20].
While there have been some previous genetic studies on the C. pneumoniae animal isolates, these have generally been restricted to partial sequencing of three genes, 16S ribosomal (r) RNA, ompA (encoding the major outer membrane protein) and omcB (encoding a large cysteine-rich protein) [4]–[6], [21]. Recently, Rattei et al. [18] examined the relationship of 38 C. pneumoniae isolates, from measurement of genetic diversity within a representative set of 232 synonymous (s)SNPs (no amino acid change; reduced evolutionary pressure). Although only two animal isolates (koala and frog) were examined, two major points have emerged from this study (i) there were 15 genotypes and four major clusters among the isolates, and (ii) the animal lineages were basal to human lineages, suggesting recent transmission to human through successive bottlenecks 150,000 years ago (based on an Escherichia coli molecular clock). Myers et al. [22] recently sequenced the full genome of the C. pneumoniae koala respiratory isolate, LPCoLN. C. pneumoniae koala LPCoLN was largely homologous to the previously sequenced C. pneumoniae human isolates, although it has several key differences. There are 6,213 SNPs between the koala and human isolates, and importantly, there are several examples of genes that are full-length in the koala LPCoLN isolate, but which have become truncated and fragmented in the human isolates [22]. These data strongly suggest that the koala strain is ancestral to the sequenced human isolates, and points to an animal-to-human cross host transmission event in the (recent) past.
The full genome sequence of C. pneumoniae koala LPCoLN has also enabled us to select additional target genes as the basis for an extended genetic and phylogenetic comparison between a broad range of C. pneumoniae animal (19) and human (11) isolates. The 30 C. pneumoniae isolates that we compared in the current study can be grouped into three categories: (i) seven archival samples (frogs BMTF - type 1 and BMTF-type 2, snakes Pufadd and Burpyth, turtle GST, chameleon cham and iguana Iguana) for which material was no longer available, and three isolates (human LKK1, bandicoot WBB and frog CPXT1) with sequences available in GenBank, restricting comparisons to the already published gene sequences, usually partial 16S rRNA, ompA and omcB, (ii) five C. pneumoniae isolates which have had their entire full genome sequenced (koala LPCoLN and humans AR39, J138, CWL029 and TW183), (iii) 15 isolates which were either available as viable cultures (frog GBF, and humans WA97001 and IOL207) or recoverable tissue material (frogs DE177 and 2040.3, bandicoots B10, B26 and B37, koala EBB, potoroo Pot37, horse N16, and humans SH-511, 1979, TOR1 and A03) which were subjected to gene-specific PCR and sequencing.
Results/Discussion
Nucleotide diversity reveals distinct geographical lineages
We had access to a total of 19 C. pneumoniae animal isolates (seven marsupial, six amphibian, five reptilian and one equine) and 11 C. pneumoniae human isolates (Table S1). We targeted 23 genes from these 30 isolates for analysis. However, due to technical difficulties with some of the DNA preparations, we could not obtain reliable sequence for all genes from all the isolates. C. pneumoniae genotypes were assigned by sequence and phylogenetic analyses of nucleotide sequences. Bootstrapped phylogenetic trees are shown in Figure S1. Nucleotide and amino acid (aa) alignments for each gene are presented in Figures S2, S3, S4, S5, S6, S7, S8, S9, S10, S11, S12, S13, S14, S15, S16, S17, S18, S19, S20, S21, S22 and S23. We propose, on the basis of 21 sSNPs from various genomic regions, that the C. pneumoniae isolates that were sequenced successfully could be assigned to five common genotypes, A-E (Table S2). The same genotypes were assigned to isolates whose Genbank sequences were included in the analysis (Table S2). The five genotypes show a distinct geographic distribution among the isolates examined (Table S2). Genotype A was common among Australian animals. Genotype B may be indicative of African isolates. Genotype C has a worldwide distribution and was the most common genotype identified. Genotype D was unique to Australian Indigenous isolates. Genotype E was the most variable C. pneumoniae genotype and comprised the sole isolate from the United Kingdom. Below is a gene-by-gene comparison to highlight the potential evolutionary relationships among C. pneumoniae isolates.
Genes showing significant size variation/truncation/deletion between isolates
The CPK_ORF00679 (LPCoLN locus designation: CPK) gene encodes a Lamin 2-like protein, which is a conserved hypothetical protein identified in C. pneumoniae and C. felis. This gene can be used as a target gene for molecular differentiation of C. pneumoniae. Sequence analysis of eight C. pneumoniae human (AR39, CWL029, TW183, J138, TOR1, WA97001, SH-511 and 1979) and four C. pneumoniae animal isolates (koala LPCoLN, bandicoot B26, frog DE177 and horse N16) revealed a distinct size variation at the 5′ end (Figure S2). Three animal isolates (bandicoot B26, koala LPCoLN and frog DE177) had the full-length (833 bp) gene with only five sSNPs among them. Primer walking was used to determine the nucleotide sequence of horse N16 and the Indigenous human isolates when initial attempts with the degenerate primers failed to amplify a product, indicating more distantly related sequences were present. Comparison of the eight C. pneumoniae human isolates revealed the absence of 251 bp at the 5′ end of six non-Indigenous human isolates, and only two non-synonymous (n)SNPs (an amino acid change) across the gene. Interestingly, both Australian Indigenous human isolates (SH-511 and 1979) have the 251 bp extended 5′ region of the gene (translating an 83 aa segment), which is identical to the extended 5′ sequence of bandicoot B26, koala LPCoLN and frog DE177 isolates (Figure 1). A second variable region of the gene included a 15 bp indel (insertion/deletion), translating the sequence IADRF position 244-248 aa. This five aa indel is present in isolates koala LPCoLN, bandicoot B26, frog DE177, horse N16, and humans SH-511 and 1979 (Figure 1).

The second length polymorphic gene, Membrane Attack Complex/Perforin (MACPF), remains uncharacterised in Chlamydia species, however, its role in other pathogens suggests that it may be virulence-related. Analysis of the partial-length sequence (1,939 bp) revealed only one sSNP differentiating frog DE177 from koala LPCoLN and bandicoots B26 and B37 (Figure S3). PCR confirmed the long version of the gene to be present in horse N16 (data not shown), with 99% identity over the available 1,303 bp to frog DE177, koala LPCoLN and bandicoots B26 and B37 (15 SNPs; data not shown). PCR and sequence analysis of nine C. pneumoniae human isolates (AR39, AR39-2 ‘gene sequenced in our laboratory’, CWL029, J138, TW183, TOR1, WA97001, SH511 and 1979) confirmed a length polymorphism (compared to the animal isolates), as the result of a common 840 bp internal deletion. Within this region there was a seven nucleotide indel TGATCCT between positions 1,053–1,055 bp and 1,059–1,062, found in 10 isolates (bandicoots B26 and B37, koala LPCoLN, frog DE177, and humans AR39-2, IOL207, TOR1, WA97001, SH511 and 1979). The indel sequence was absent from the four published C. pneumoniae human genomes (AR39, CWL029, J138 and TW183) (Figure S3). A third polymorphism for this gene was evident in Australian Indigenous human isolates SH511 and 1979, which lacked an additional 270 bp fragment between positions 1,333–1,602 bp. The characterisation and identification of polymorphisms within the MACPF should serve as a useful marker in future genetic investigation. For example, the MACPF gene sequence can differentiate C. pneumoniae animal isolates from Indigenous human and non-Indigenous human sources.
A significant difference between C. pneumoniae and other chlamydial species is the absence of tryptophan biosynthesis genes. Despite this absence, C. pneumoniae encodes a functional aromatic amino acid (tryptophan) hydroxylase (AroAA-Hs) [23]. The C. pneumoniae aromatic amino acid hyrdoxylase has a unique, extended 5′ region among human isolates, which is absent from koala LPCoLN [23]. To investigate whether the 5′ region may be host specific for C. pneumoniae human isolates, we examined the sequence of a bandicoot isolate (B26) and four additional C. pneumoniae human isolates (TOR1, WA97001, SH551 and 1979). Sequence analysis confirmed the 244 bp extended 5′ region in all five isolates (identical sequence) (Figure S4). Therefore, the extended 5′ region does not appear to be human host-specific, but may play an important role in regulating its function.
Polymorphic outermembrane protein (pmp) genes
The pmps are an important group of Chlamydia surface proteins that have been considered as potential vaccine candidates. We selected three variable pmps (unknown function) for analysis; (i) pmpE/F2, (ii) pmpE/F3, and (iii) pmpG6. Partial-length sequence comparisons (2,169 bp) of pmpE/F2 revealed four unique features (Figure S5), including (i) a total of 64 scattered SNPs (23 nSNPs; 41 sSNPs) differentiating koala LPCoLN and bandicoot B26 from all other isolates, (ii) 10 SNPs (8nSNPs; 2 sSNPs) for frog DE177, six of which were shared with LPCoLN, and four of these were also shared with human SH511 and human 1979 isolates, (iii) four shared SNPs (3 nSNPs; 1 sSNP) between Australian Indigenous human isolates SH511 and 1979, and (iv) one nSNP differentiating human J138 from human isolates AR39, CWL029, J138, TW183, TOR1, and WA97001.
A partial alignment (922 bp) of pmpE/F3, revealed four features (Figure S6), including (i) a total of 20 SNPs (8 nSNPs; 12 sSNPs) unique to koala LPCoLN, (ii) two shared SNPs (1 nSNP; 1 sSNP) among koala LPCoLN and frog DE177, (iii) four nSNPs unique to Australian Indigenous human isolates SH511 and 1979, and (iv) no polymorphisms among non-Indigenous human isolates (AR39, TW183, CWL029, J138, TOR1 and WA9001).
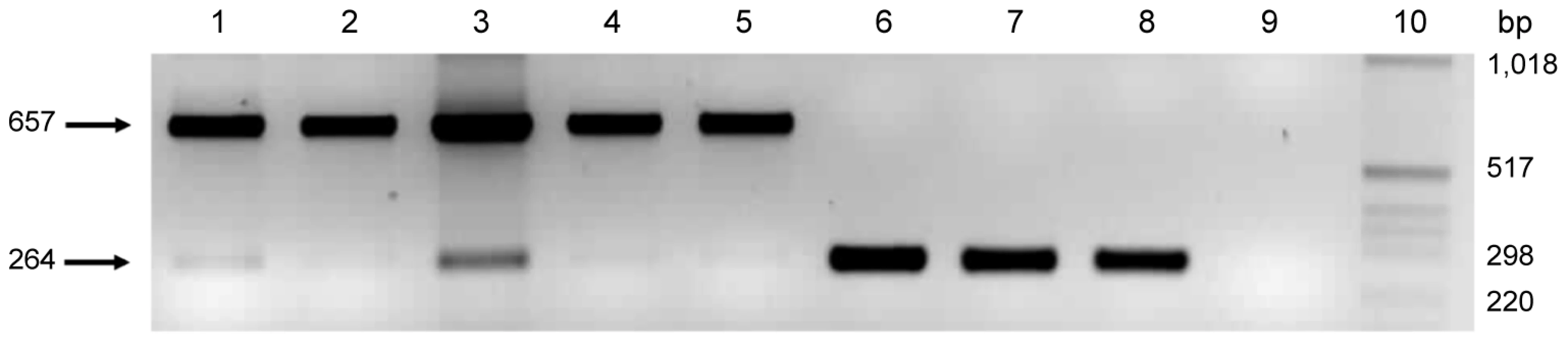
The role of pmpG6 is unknown, but owing to the variable number of tandem repeats there may be a functional role in vivo. Koala LPCoLN and two C. pneumoniae human isolates, TW183 and CWL029, have three tandem repeats of (i) 396 bp, (ii) 393 bp, and (iii) 387 bp, whereas human isolates J138 and AR39 only have two tandem repeats (missing ii – 393 bp). Primers were designed to flank the variable repeat region where a 657 bp product was indicative of 3 tandem repeats, and a 264 bp product was indicative of only 2 tandem repeats (Figure 2). We examined eight C. pneumoniae isolates by PCR, including koala LPCoLN and human AR39 positive controls. Five isolates were confirmed to have three tandem repeats (koala LPCoLN, bandicoot B26, frog DE177, and humans SH511 and 1979) and three isolates were identified with only two tandem repeats (humans AR39, WA9701 and TOR1) (Figure 2). This gene can be used as a marker for intra-species variation, differentiating isolates by the size of their PCR product.
Previously well-studied genes
The 16S rRNA and ompA genes have been extensively used to distinguish and phylogenetically group chlamydial species [24]–[27]. Although not as informative as our newly identified target genes, our analysis showed that 16S rRNA, ompA and omcB were evolving in much the same way as our target genes. A portion of the 16S rRNA gene sequence has been previously determined for several C. pneumoniae animal isolates including koala LPCoLN, horse N16, frogs DE177, CPXT1, GBF, BMTF-type 1 and BMTF-type 2, snakes Pufadd and Burpyth, turtle GST, chameleon cham, iguana Iguana, and human isolates AR39, CWL029, TW183, J138, LKK1 and IOL207 [3]–[5], [14]–[17], [22], [28]–[31]. We expanded this comparison to include the additional animal isolates frog 2040.3, bandicoots B10, B26 and B37, potoroo Pot37, koala EBB, and human isolates TOR1, WA97001, SH511 and 1979 (Figure S7). As expected from previous studies, the 16S rRNA sequences were highly conserved between all animal and human isolates. However, the minor differences (SNPs) might indicate some interesting trends in C. pneumoniae evolution. Analysis of a 215 bp segment revealed seven SNPs. Of these, two were unique to horse N16 (position 88; G/A and 100; A/C). Frog DE177 had a 2 bp insertion (position 84; T and 85; T), while frog BMTF-type 1 and snake Puffadd shared one unique bp insertion (position 28; A). The two remaining SNPs (position 21; A/G and 51; A/G) differentiated bandicoot (B10, B26, B37 and WBB), koala (LPCoLN and EBB), frog (GBF, 2040.3, DE177 and CPXT1), horse N16 and Australian Indigenous human isolates (SH511 and 1979) from frog (BMTF-type 1 and BMTF-type 2), iguana (Iguana), snake (Puffadd) and non-Indigenous human isolates.
The C. pneumoniae ompA gene is much less variable, unlike other species in which several variable domain regions flank diversity. We examined the ompA variable domain 4 partial-length (258 bp) sequence of 10 human and 16 animal C. pneumoniae isolates from diverse geographical and anatomical locations. From the 258 bp sequence, we identified a total of 27 SNPs (12 nSNPs; 15 sSNPs). Horse N16 had the highest number of SNPs (relative to all other isolates) with 19 unique SNPs (7 nSNPs; 12 sSNPs). All six marsupial isolates (koalas LPCoLN and EBB, bandicoots B26, B37 and WBB, and potoroo Pot37) were identical in sequence, whereas all 10 human isolates (AR39, CWL029, TW183, J138, TOR1, WA97001, IOL207, LKK1, SH511 and 1979) were identical to one another (Figure S8). Region 221–251 bp was the most polymorphic, with a total of 16 SNPs, five of which differentiate marsupial (B26, B37, WBB, EBB, LPCoLN and Pot37) and frog (CPXT1, DE177 and GBF) isolates from frog (BMTF-type 1), snake (Burpyth and puffadd), lizard (chameleon and iguana), turtle (GST), horse (N16) and human isolates (AR39, CWL029, J138, TW183, IOL207, LKK1, TOR1, WA97001, 1979 and SH511) at nucleotide positions 225 (A/G), 227 (G/C), 231 (A/G), 236 (C/T) and 244 (G/A/C).
The omcB gene encodes a large cysteine-rich protein, which offers stabilisation through disulphide cross-linkage [32]. Five SNPs were identified from the partial-length (386 bp) comparison of 10 isolates (Figure S9). Three SNPs (2 nSNP; 1 sSNP) were associated with koala LPCoLN, frog DE177 and frog GBF isolates. One sSNP was shared between koala LPCoLN and frog GBF, while another sSNP was unique to horse N16.
Two highly conserved genes from within the plasticity zone include accC and pfk, encoding acetyl-CoA carboxylase, biotin carboxylase and diphosphate-fructose-6-phosphate 1-phosphotransferase, respectively. A partial length (302 bp) comparison of accC revealed only four SNPs, two (position 31; C/A and 171; C/T) differentiated seven non-Indigenous human isolates from two Australian Indigenous human isolates and nine animal isolates. A third SNP (position 185; G/A) was detected in horse N16 and two human isolates (AR39 and J138). The fourth SNP (position 226; T/C) was found in horse N16 and frog 2040.3 isolates (Figure S10). A partial length (263 bp) comparison of pfk also revealed four SNPs, two (1 nSNP; 1 sSNP) were unique to horse N16, while two additional SNPs (C/T and G/A) were conserved in koala LPCoLN, bandicoot B37 and frog GBF isolates (Figure S11).
Hypothetical genes or unique genes showing characteristics specific to C. pneumoniae
The CP_1042 (AR39 locus designation: CP) gene encodes a hypothetical protein, which is unique to the C. pneumoniae human genome. The four fully-sequenced C. pneumoniae human isolates (AR39, CWL029, J138 and TW183) have the full-length (558 bp) gene, whereas koala LPCoLN is missing a 361 bp segment at the 5′ end of the gene; the remaining nucleotides do not translate a protein due to truncation mutations. In order to determine the extent to which genetic diversity occurs in other animal isolates, we examined a frog (DE177), bandicoot (B26), koala (EBB) and horse (N16) isolate, as well as four additional human isolates (TOR1, WA97001, SH511 and 1979) across the 558 bp gene (Figure S12). Like koala LPCoLN, PCR (primers designed for the full-length gene) was unable to detect a product for bandicoot B26 and koala EBB isolates. Interestingly, frog DE177 and horse N16 isolates have the full-length gene, indicating that CP_1042 is not unique to C. pneumoniae human isolates. Horse N16 is highly variable with a total of 56 unique SNPs, one of which is shared with frog DE177 and Australian Indigenous human isolates, SH511 and 1979 (Figure S12). All eight human isolates have the full-length gene, with only two SNPs differentiating the non-Indigenous and Australian Indigenous isolates. The fragmentation in the marsupial isolates appears to be host-specific and may not be essential for growth in the marsupial host.
The CP_0880 gene encodes a hypothetical protein, which is variable among C. pneumoniae animal isolates, making it difficult to obtain reliable sequence for frog DE177 and horse N16 isolates despite many attempts at different primer set combinations. It was possible to align a partial-length sequence (845 bp) for one bandicoot (B26), two koala (LPCoLN and EBB) and eight human isolates (AR39, CWL029, TW183, J138, TOR1, WA97001, SH511 and 1979) (Figure S13). Of the eight human isolates, four SNPs (2 nSNPs; 2 sSNPS) were detected in SH511 and 1979 Australian Indigenous isolates. Bandicoot B26 and koala LPCoLN isolates were identical in sequence and had diverged from all other isolates, the result of 110 unique SNPs. Of these SNPs, 74 led to an amino acid change, while two SNPs (1nSNP; 1sSNP) were shared with Australian Indigenous human SH511 and 1979 isolates.
The CP_0505 gene encodes a hypothetical protein. Sequence analysis (Figure S14) of the partial-length (723 bp) gene revealed four features (i) 13 SNPs (8 nSNPs; 5 sSNPs) unique to koala LPCoLN and bandicoots B26 and B37, (ii) 42 SNPs (24 nSNPs; 18 sSNPs) unique to horse N16, (iii) two SNPs (1 nSNP; 1 sSNP) unique to Australian Indigenous SH511 and 1979 isolates, while a third sSNP was shared with bandicoots B26 and B37, koala LPCoLN and horse N16 isolates, and (iv) a single bp deletion distinguished frog DE177 from the non-Indigenous isolates (identical sequence).
The SctC gene encodes a type III secretion system protein, which has a C. pneumoniae-specific (approximately 700 bp) 5′ region. Type III secretion system proteins are well-conserved among Chlamydia species (and other bacteria), therefore, it was not surprising to find little variation among the C. pneumoniae isolates. Overall, there were five SNPs. Two nSNPs (position 232; A/C and 273; A/C) distinguished bandicoot B26, koala LPCoLN, frog DE177 and Indigenous human isolates SH511 and 1979 from the non-Indigenous human isolates AR39, CWL029, J138, TW183, TOR1 and WA97001 (identical sequences). Frog DE177 had one sSNP (position 342; T/C). Koala LPCoLN and bandicoot B26 had the same nSNP (position 429; G/T). Australian Indigenous human isolates SH511 1979 shared one nSNP (position 469; A/G) (Figure S15). This 482 bp gene segment might be a useful marker for C. pneumoniae detection and differentiation of animal isolates from Indigenous and non-Indigenous human isolates, on the basis of five key SNPs.
The CPK_ORF00201 gene (termed HAF) encodes a HAF family auto-transporter beta domain protein. Nucleotide comparisons of the fully-sequenced C. pneumoniae isolates (koala LPCoLN and human AR39, CWL029, TW183 and J138) revealed a truncated (two separate genes) homolog among human isolates; absence of a 120 bp internal segment. A partial-length (1,412 bp) comparison with seven previously un-sequenced C. pneumoniae isolates (bandicoot B26, frog DE177, horse N16, human WA97001, TOR1, SH511 and 1979) revealed an intact, full-length gene in all seven isolates (Figure S16). One possible explanation for this difference may be due to sequencing error in the C. pneumoniae human genomes; the missing segment is revealed in a BLAST search. Limited sequence was available for horse N16, despite many attempts with different primer sets, suggesting that there may be more sequence variation in the region that could not be amplified. There were, however, nine unique SNPs (4 nSNPs; 5 sSNPs) detected in the horse N16 isolate. It is worth mentioning that horse N16, bandicoot B26, koala LPCoLN, frog DE177 and both Australian Indigenous human isolates (SH511 and 1979) had two common SNPs at nucleotide positions 761/T and 1,020/T, while six non-Indigenous human isolates had C/C at these positions. These polymorphisms could be used to distinguish animal and Indigenous human isolates from non-Indigenous human isolates.
Strain-specific genes of C. pneumoniae
Three genes present in the C. pneumoniae human genome, but absent from the koala LPCoLN genome include guaA (GMP synthase), guaB (IMP dehydrogenase) and add (AMP adenosine deaminase). This three-gene cluster, located in the plasticity zone, was also confirmed by PCR in the frog (DE177) and horse (N16) isolates. A comparison of guaB (Figure S17) revealed just three SNPs differentiating frog DE177 from the non-Indigenous human C. pneumoniae isolates (AR39, CWL029, TW183, J138, TOR1 and WA97001), while no polymorphisms were evident in the guaA gene (Figure S18). Reliable add sequence was not available for frog DE177 (Figure S19). The horse N16 isolate had four unique SNPs for guaB (Figure S17); however, reliable sequence was not available for guaA and the presence of add was not definitive in the horse N16 isolate. The guaBA-add cluster was identified (identical sequence) in human isolates TOR1, WA97001 and IOL207 (limited sequence) (Figures S17, S18 and S19). Both Australian Indigenous human isolates (SH511 and 1979) have the guaB gene with six SNPs (4 nSNPs; 2 sSNPs) to the non-Indigenous human isolates; two SNPs were shared with frog DE177 and horse N16 isolates (Figure S17). Since PCR failed to amplify any product for guaA and add genes in the Australian Indigenous human isolates (using two sets of primers for each gene from either 5′ or 3′ regions), these two genes are either absent from the genome, or are highly divergent and undetectable with our primer sets. Three additional animal isolates (koala EBB, bandicoots B26 and B37) also lack the guaBA-add cluster which suggests that it is not a core component required for C. pneumoniae survival.
The CPK_ORF00678 gene encodes a hypothetical protein, which is located within the plasticity zone of the koala LPCoLN genome. This gene was not identified in any of the four fully-sequenced C. pneumoniae human isolates (AR39, CWL029, J138 and TW183) or subsequent human isolates (WA97001, TOR1, SH511 and 1979), but was detected in three additional animal isolates (bandicoot B26, frog DE177 and horse N16) (Figure S20). A partial-length sequence comparison (698 bp) revealed 10 SNPs (4 nSNPs; 6 sSNPs) differentiating frog DE177 from koala LPCoLN and bandicoot B26 isolates (identical sequence). Four of these SNPs were also shared with horse N16 (Figure S20). Of the animal isolates, horse N16 showed greater sequence divergence with 11 unique SNPs and four indels (positions 73–84, 151–305, 387–395 and 419–425). The absence of CPK_ORF00678 from the C. pneumoniae human genome suggests that this gene may have been selected for a specific function in the animal host, and thus, provides a new animal C. pneumoniae-specific target gene.
Koala LPCoLN encodes a plasmid (7,655 bp) with eight ORFs (open reading frames) [22]. This plasmid is absent from C. pneumoniae human isolates [14]–[17]. Thomas et al. [33] detected a plasmid in the horse N16 isolate, and so it was of interest to see whether additional C. pneumoniae isolates have a plasmid. Primers (Table S3) were designed to investigate three plasmid ORFs: (i) site-specific recombinase II; SSR2, (ii) replicative DNA helicase dnaB; helicase, and (iii) a conserved hypothetical protein; PGP3D. Seventeen isolates were examined by PCR for the presence of a plasmid, including three bandicoots (B10, B26 and B37), three frogs (GBF, DE177 and 2040.3), two koalas (LPCoLN and EBB), one potoroo (Pot37), one horse (N16) and seven human isolates (AR39, IOL207, WA97001, A03, TOR1, SH511 and 1979). All three plasmid ORFs were confirmed by PCR in each of the 10 animal isolates, although no human isolate was found to have SSR2 (Figure S21), helicase (Figure S22) or PGP3D (Figure S23) ORFs. Of the three ORFs, SSR2 showed evidence of a length difference when compared to the horse N16 isolate. This isolate was missing a 165 bp segment between positions 66–230 bp, while bandicoots B10 and B37, frogs DE177, 2040.3, GBF and koala LPCoLN sequences were identical and contained the whole 369 bp segment under examination (Figure S21).
Phylogenetic relationships among C. pneumoniae isolates
Despite the importance and widespread prevalence of C. pneumoniae, there has been little phylogenetic analysis to assist evolutionary and epidemiological investigations. Therefore, in order to examine in-depth the evolutionary relationships within C. pneumoniae, we examined several previously studied isolates in addition to 10 novel isolates using 22 target genes selected from various regions of the genome. We constructed rooted phylogenetic trees from all available C. pneumoniae isolates by Neighbour-Joining and UPGMA (Unweighted Pair Group Method with Arithmetic mean) methods. As both trees were of similar structure, only the Neighbour-Joining tree is presented (Figure S1). The phylogenetic patterns that we observed were congruent in 19 out of the 22 genes that we analysed. This gives us confidence that our interpretations are sound and suggests that horizontal gene transfer is not a major contributor to genetic changes in C. pneumoniae. These phylogenetic analyses led us to propose several key new findings. The Australian Indigenous human isolates (genotype D) formed a unique group in 10 out of the 15 trees analysed. These could be clearly distinguished from the non-Indigenous human isolates (genotype C) which formed a tight cluster, regardless of their geographical origin. The evolutionary position of horse N16 (genotype E) is somewhat less confident. Horse N16 was distinct from all other isolates in at least 9 out of the 13 trees examined. However, the bootstrap values were low at 54–78%. It will be necessary to obtain additional equine isolates to confirm the correct lineage of this strain. The Australian marsupial isolates formed a tight grouping (genotype A) with all genes analysed, even when they were obtained from different geographical regions within Australia. The Australian marsupial isolates shared common nucleotide substitutions with the amphibian and horse isolates originating from Australia, Africa and the United Kingdom, as did the Australian Indigenous human isolates. Analysis of the Australian frog isolates resulted in the identification of two distinct genotypes. Genotype C was identified in two captive frog isolates from New South Wales (BMTF-type 1 and BMTF - type 2). A third free-range frog (GBF) isolate, also from New South Wales, was identical to the sequence derived from Australian marsupial isolates, from independent regions. The third isolate was therefore clustered into genotype A. The degree of variation in the limited sequence analysed was unusual given the geographical distance from other world regions. Moreover, two African frog isolates from different regions were clustered together, in genotype B. However, a third African isolate from a chameleon was grouped with the international cluster in genotype C, comprising American snakes, an American turtle, a Central American iguana, Australian frogs, as well as the non-Indigenous human isolates. Members of genotype C are both common and present across the globe, possibly indicating that it is a relatively recent expansion, as proposed by Rattei et al. [18].
Conclusions
Sequence data suggests that two separate animal to human transmission events have occurred
Our aim was to determine whether our target genes could distinguish C. pneumoniae isolates from human and animal sources and to investigate their genetic diversity. Previous analyses have shown that C. pneumoniae human isolates are essentially clonal and do not provide much evolutionary insight. To our knowledge, all previous analyses of C. pneumoniae human have focussed on non-Indigenous isolates: AR39 (USA), CWL029 (USA), TW183 (Taiwan), J138 (Japan), LKK1 (Korea), A03 (USA), TOR1 (Canada) and IOL207 (Iran). Our analysis also included a non-Indigenous human isolate from Australia (WA97001), and more importantly, we were able to analyse two Australian Indigenous human isolates (SH511 and 1979) which originated from geographically separate communities.
Our collective sequence data (using 23 target genes from 30 isolates) and phylogenetic analyses strongly support both the whole genome findings of Myers et al. [22] as well as the synonymous SNP conclusions of Rattei et al. [18] in that the extant C. pneumoniae human isolates have derived from C. pneumoniae animal isolates. Our data extends both of these studies, however, and strongly suggests two (or more) lineages of C. pneumoniae. One lineage involves amphibian isolates (DE177, CPXT1, BMTF-type 1 and BMTF-type 2), which subsequently ‘evolved’ to C. pneumoniae infections in reptiles (Iguana, Pufadd, Burpyth, GST, cham) and via as yet undiscovered intermediates, to the dominant C. pneumoniae human clone present in the world today (AR39, TW183, CWL029, J138, TOR1, A03, IOL207, LKK1, and WA97001). The second presumably also started with amphibian infections (as above), but then diverged into other amphibian and reptilian infections (frogs GBF and 2040.3). At this point (there are probably intermediate hosts and C. pneumoniae strains that are either extinct or as yet, undiscovered) two sub-lines are evident. One lineage has resulted in the widespread C. pneumoniae infections seen in Australian marsupials today (koala LPCoLN, koala EBB, bandicoot B10, bandicoot B26, bandicoot B37, bandicoot WBB and potoroo Pot37) while the second lineage has crossed the animal-human barrier to infect Australian Aboriginals (SH511 and 1979). The key genomic data, which supports these lineages and their divergence is summarised in Supporting Information Text S1.
Although the evolutionary time-frame of C. pneumoniae is unclear, we have described patterns of genetic differentiation whereby C. pneumoniae profiles from the Australian Indigenous human populations appear to be negatively correlated (for the most part) with the genetic differentiation in non-Indigenous human populations. The possibility that C. pneumoniae was an established pathogen in Australia before the settlement of Europeans in 1788, is one possible theory considering the characteristic diseases of a hunter-gatherer population have been described as those with low morbidity, long illness and long infection stages, such as chlamydial diseases [34]. From ancient times, hunter-gatherer Aboriginal communities have lived in close proximity to animals, and may have been predisposed to C. pneumoniae infected animals. However, if C. pneumoniae had been present in the Australian Indigenous population prior to European settlement, the differences observed in our target genes would suggest that the Australian Indigenous isolates have evolved independently from the non-Indigenous isolates. This trend was also observed in an Australian Indigenous and non-Indigenous cohort of Haemophillus influenzae isolates from Australia [35] where the genetic diversity and the time span from European settlement was not likely to support the amount of differentiation observed. A likely explanation for the observed diversity in our human isolates would be that C. pneumoniae may have been more recently introduced to urban non-Aboriginals from infected animals, or alternatively via native or non-native inhabitants in other regions around the world, and the infections were not derived from the Indigenous Australians; the United Kingdom horse (N16) and African frog (DE177) isolates also had indels in common to the Australian marsupials (koalas LPCoLN and EBB, bandicoots B10, B26, B37 and WBB, and potoroo Pot37) and Australian Indigenous human isolates, despite being geographically isolated from the Australian population. There is convincing evidence that zoonotic transmission of C. pneumoniae can/has occur/ed. The finding that a genotype common to non-human and human hosts was dispersed in human carotid plaques [36] is not by itself conclusive evidence of zoonotic transmission, but rather highlights the need for consideration of its zoonotic potential, particularly for animal handlers and laboratory personnel.
In the present study, we observed five genotypes (A-E) based on genetic and phylogenetic data. These data revealed dominant genotypes in various regions of the world. Genotype A was prevalent among Australian animals, genotype B was identified in Africa, genotype C had the broadest distribution and was worldwide, genotype D was predominant among Indigenous Australians, while genotype E was unique to the United Kingdom. Although a very limited number of isolates were used in this study, the results clearly showed genetic diversity associated with host type and geographic locations. Based on the large number of polymorphisms between horse N16 and all other C. pneumoniae isolates, we believe that we have identified sufficient genetic differences that would accommodate the classification of horse N16 into a subspecies of C. pneumoniae, designated C. pneumoniae subsp. equi, following the proposal of Pettersson et al. [29]. These findings of high genetic variation may represent mosaic genotypes in the population and may need to be considered as a subspecies variant within C. pneumoniae.
Materials and Methods
Chlamydia pneumoniae isolates
30 isolates from 11 human, 7 marsupial, 5 reptilian, 6 amphibian and 1 equine host were analysed (Table S1).
The C. pneumoniae Pot37 isolate was initially obtained from a pharyngeal swab of a Gilbert's potoroo (Potorous gilbertii) from Perth, Western Australia in 2006. The C. pneumoniae EBB isolate was isolated from a pharyngeal swab of a koala (Phascolarctos cinereus) from Queensland, Australia in 2005.
All analyses involving the Australian Indigenous human samples were approved by the Queensland University of Technology and the Menzies School of Health Research, Human Research Ethics Committee. All human samples were obtained from a previous study, in which informed written consent for the collection of samples and subsequent analysis was correctly obtained and ethics approval obtained. All animal samples were obtained from previous studies and experiments were performed according to national and institutional guidelines with ethics, biosafety and animal care committee approval (QUT IBC No. 1413/2A, QUT IBC No. 0900000267, QUT IBC No. 928A).
Strategy for selection of genes used for comparisons
Recently, we examined five complete C. pneumoniae genomes, including koala LPCoLN and the four available C. pneumoniae human (AR39, TW183, CWL029 and J138) genomes (Mitchell et al., unpublished data). This approach allowed us to identify regions of high SNP accumulation and to select candidate genes for comparison. In this study, we selected 13 of these genes for comparative analysis using a broader selection of isolates, while 1 additional length variable gene, 1 polymorphic gene, 5 highly conserved genes and three plasmid genes were also selected for analysis. Our target genes could be grouped into six categories:
-
three genes (CPK_ORF00679, AroAA-Hs and MACPF) showing length polymorphisms, of at least 100 bp between C. pneumoniae koala and human isolates. Aromatic amino acid hydroxylases (AroAA-Hs) hydroxylate phenylalanine, tyrosine, and tryptophan into tyrosine, dihydroxyphenylalanine, and 5-hydroxytryptophan, respectively [23]. CPK_ORF00679 and MAC/perforin (MACPF) genes have an unknown role, although the MACPF may be a potential virulence gene based on the function in other intracellular pathogens.
-
three polymorphic outer membrane protein genes; two (pmpE/F2 and pmpE/F3) have regions with more than 20 SNPs per 1 kbp between C. pneumoniae koala and human isolates, and one (pmpG6) varies in the number of tandem repeats.
-
three previously well-studied ‘conserved’ genes: 16S rRNA belongs to a small unit of ribosomes, ompA is a major outermembrane protein and omcB is a large cysteine-rich outer membrane protein; and two highly conserved genes (accC and pfk) located within the plasticity zone – accC (acetyl-CoA carboxylase, biotin carboxylase) is involved in fatty acid/phospholipid metabolism and pfk (diphosphate-fructose-6-phosphate 1-phosphotransferase) is involved in glycolysis/gluconeogenesis.
-
three hypothetical genes (CP_1042, CP_0880 and CP_0505) with an unknown function.
-
two C. pneumoniae-specific genes (SctC and HAF). SctC (highly conserved) forms part of the type III secretion system and has a unique 700 bp at the 5′ region of the gene. HAF is predicted to be a HAF family-autotransporter (beta domain) protein.
-
seven strain-specific genes: guaA (GMP synthase), guaB (IMP dehydrogenase) and add (AMP adenosine deaminase) are located in the plasticity zone and were absent from koala LPCoLN; CPK_ORF00678 is a hypothetical gene that is absent from all four full-sequenced C. pneumoniae human isolates, and; the plasmid is an extra-chromosomal element identified in the koala LPCoLN genome. Three plasmid genes were examined: (i) site-specific recombinase II; SSR2, (ii) replicative DNA helicase dnaB; helicase, and (iii) a conserved hypothetical protein; PGP3D.
The strategy involved PCR-based amplification of six human isolates and nine animal isolates, followed by sequencing of the PCR products. Additional sequences for 16S rRNA, ompA and omcB were retrieved for previously sequenced human and animal C. pneumoniae isolates (refer to accession numbers). The analysis included; (i) gene by gene sequence alignments to identify SNPs and indels, and (ii) generation of bootstrapped phylogenetic trees.
PCR and sequencing
Oligonucleotide primers (Sigma-Aldrich, Castle Hill, Australia) were designed based on homology of C. pneumoniae LPCoLN and AR39 sequences (GenBank accession numbers CP001713 and AE002161) using Primer3 v. 0.4.0 (denoted by an asterisk) (http://biotools.umassmed.edu/bioapps/primer3_www.cgi) [37] and two primers already published (Table S3). The primer pairs, expected PCR product sizes and annealing temperatures are summarised, and several of the target genes have internal primers to sequence a particular region because of sequence variation or to enable sequencing of whole PCR products (Table S3). PCR reactions were performed in a final volume of 50 µl, including 5.0 µl 10X PCR reaction buffer (Roche, Castle Hill, Australia), 1.0 µl PCR nucleotide mix (Roche, Castle Hill, Australia), 2.0 µl of each (10 µM) primer (Sigma-Aldrich, Castle Hill, Australia), 0.2 µl of 5 U/µl Taq DNA polymerase (Roche, Castle Hill, Australia), 2.0 µl of template and PCR grade water to a final volume of 50 µl. Amplification conditions consisted of an initial denaturation at 94°C, followed by 30 cycles of 1 min at 94°C, 1 min at the specified annealing temperature (refer to Table S3), 1 min at 72°C, and a final extension for 10 min at 72°C. PCR products were separated by electrophoresis and visualised by ethidium bromide (10 µg/ml) staining of a 2% agarose gel in Tris Borate EDTA (TBE) buffer.
PCR products were purified with a PureLink PCR purification kit (Invitrogen, Australia). To confirm sequence confirmation, DNA sequencing was performed in both directions using a BigDye terminator Cycle Sequencing Ready Reaction Kit and an automated DNA sequencer AB 3730xl (Australian Genome Research Facility, University of Queensland, Australia). A third sequence was obtained if required.
Sequences and phylogeny
Nucleotide sequences were translated to amino acids using blastx (www.ncbi.nlm.nih.gov/blast). Nucleotide and derived amino acid sequences were then trimmed to a uniform length. Sequence alignments were generated with Geneious version 4.7 using the pairwise alignment default settings (Nucleotide Cost Matrix = 65% similarity 5.0/−4.0, Protein Cost Matrix = Blosum62, Gap Open = 12.0, Gap Extension = 3.0, Alignment Type = global alignment with free end gaps) [38; http://www.geneious.com]. The amino acid colour scheme is standard where each amino acid has an assigned colour. Phylogenetic trees were generated with Geneious version 4.7 using the pairwise alignment default settings (described above), and a bootstrap consensus phylogenetic tree was constructed by Neighbor-Joining or UPGMA analysis and Jukes-Cantor correction using 1,000 bootstrap replicates [38]. Bootstrap values greater than or equal to 50% are shown at the nodes.
Nucleotide and amino acid sequence accession numbers
The nucleotide and amino acid sequence of the target genes have been deposited in the GenBank database, and the accession numbers are as follows: CPK_ORF00679 GQ918195 to GQ918201; MACPF GQ918233 to GQ918240; AroAA-Hs GQ918166 to GQ918170; pmpE/F2 GQ507462 to GQ507467; pmpE/F3 GQ918162 to GQ918165; 16S rRNA GQ507433 to GQ507442; ompA GQ918216 to GQ918222; accC GQ918224 to GQ918232, GU013548; pfk GQ918243 to GQ918254; CP_1042 GQ918189 to GQ918194; CP_0880 GQ507456 to 507461; CP_0505 GQ507449 to GQ507455; SctC GQ507443 to GQ507448; HAF GQ918202 to GQ918208; guaA GQ918156 to GQ918158; guaB GQ918209 to GQ918215; add GQ918154 to GQ918155; CPK_ORF00678 GQ918159 to GQ918161; SSR2 GQ918171 to GQ918176; helicase GQ918183 to GQ918188; PGP3D GQ918177 to GQ918182.
Supporting Information
Zdroje
1. GraystonJT
1965 Immunisation against trachoma. Pan Am Health Organ Sci Publ 147 549
2. StoreyCC
LusherM
YatesP
RichmondSJ
1993 Evidence for Chlamydia pneumoniae of non-human origin. J Gen Microbiol 139 2621 2626
3. BodettiTJ
JacobsonE
WanC
HafnerL
PospischilA
2002 Molecular evidence to support the expansion of the host range of Chlamydophila pneumoniae to include reptiles as well as humans, horses, koalas and amphibians. Syst App Microbiol 25 146 152
4. BergerL
VolpK
MathewsS
SpeareR
TimmsP
1999 Chlamydia pneumoniae in a free-ranging giant barred frog (Mixophyes iteratus) from Australia. J Clin Microbiol 37 2378 2380
5. HotzelH
GrossmannE
MutschmannF
SachseK
2001 Genetic characterization of a Chlamydophila pneumoniae isolate from an African frog and comparison to currently accepted biovars. Syst Appl Microbiol 24 63 66
6. WardropS
FowlderA
O'CallaghanP
GiffardP
TimmsP
1999 Characterization of the koala biovar of Chlamydia pneumoniae at four gene loci–ompAVD4, ompB, 16S rRNA, groESL spacer region. Syst Appl Microbiol 22 22 27
7. KutlinA
RoblinPM
KumarS
KohlhoffS
BodettiT
2007 Molecular characterization of Chlamydophila pneumoniae isolates from Western barred bandicoots. J Med Microbiol 56 407 417
8. GraystonJT
2000 Background and current knowledge of Chlamydia pneumoniae and atherosclerosis. J Infect Dis 181 Suppl 3 S402 S410
9. KuoCC
JacksonLA
CampbellLA
GraysonJT
1995 Chlamydia pneumoniae TWAR. Clin Microbiol Rev 8 451 461
10. SaikkuP
LeinonenM
MattilaK
EkmanMR
NieminenMS
1988 Serological evidence of an association of a novel Chlamydia, TWAR, with chronic coronary heart disease and acute myocardial infarction. Lancet 2 983 986
11. WesslenL
PahlsonC
FrimanG
FohlmanJ
LindquistO
1992 Myocarditis caused by Chlamydia pneumoniae (TWAR) and sudden unexpected death in a Swedish elite orienteer. Lancet 340 427 428
12. SriramS
MitchellW
StrattonC
1998 Multiple sclerosis associated with Chlamydia pneumoniae infection of the CNS. Neurology 50 571 572
13. BalinBJ
GerardHC
ArkingEJ
AppeltDM
BraniganPJ
1998 Identification and localization of Chlamydia pneumoniae in the Alzheimer's brain. Med Microbiol Immunol 187 23 42
14. ReadTD
BrunhamRC
ShenC
GillSR
HeidelbergJF
2000 Genome sequences of Chlamydia trachomatis MoPn and Chlamydia pneumoniae AR39. Nucleic Acids Res 6 1397 1406
15. KalmanS
MitchellW
MaratheR
LammelC
FanJ
1999 Comparative genomes of Chlamydia pneumoniae and C. trachomatis. Nature Genetics 21 385 389
16. ShiraiM
KirakawaH
KimotoM
TabuchiM
KishiF
2000 Comparison of whole genome sequences of Chlamydia pneumoniae J138 from Japan and CWL029 from USA. Nucleic Acids Res 12 2311 2314
17. GengMM
SchuhmacherA
MuehldorferI
BenschKW
SchaeferKP
2003 The genome sequence of Chlamydia pneumoniae TW183 and comparison with other Chlamydia strains based on whole genome sequence analysis. Genbank Accession No AE009440
18. RatteiT
OttS
GutackerM
RuppJ
MaassM
2007 Genetic diversity of the obligate intracellular bacterium Chlamydophila pneumoniae by genome-wide analysis of single nucleotide polymorphisms: evidence for highly clonal population structure. BMC Genomics 8 355
19. PeelingRW
BrunhamRC
1996 Chlamydiae as pathogens: new species and new issues. Emerg Infect Dis 2 307 319
20. WillsJM
WatsonG
LusherM
MairTS
WoodD
1990 Characterisation of Chlamydia psittaci isolated from a horse. Vet Microbiol 24 11 19
21. GlassickT
GiffardP
TimmsP
1996 Outer membrane protein 2 gene sequence indicates that Chlamydia pecorum and Chlamydia pneumoniae cause infection in koalas. Syst Appl Microbiol 19 457 464
22. MyersGSA
MathewsSA
EppingerM
MitchellC
O'BrienKK
2009 Evidence that human Chlamydia pneumoniae was zoonotically acquired. J Bacteriol 191 7225 7233
23. AbromaitisS
HeftyPS
StephensRS
2009 Chlamydia pneumoniae encodes a functional aromatic amino acid hydroxylase. FEMS Immunol Med Microbiol 55 196 205
24. ZhangYX
FoxJG
HoY
ZhangL
StillsHFJr
1993 Comparison of the major outer-membrane protein (MOMP) gene of mouse pneuomonitis (MoPn) and hamster SFPD strains of Chlamydia trachomatis with other Chlamydia strains. Mol Biol Evol 10 1327 1342
25. StothardDR
BoguslawskiG
JonesRB
1998 Phylogenetic analysis of the Chlamydia trachomatis major outer membrane protein and examination of potential pathogenic determinants. Infect Immun 66 3618 3625
26. BrunelleBW
SensabaughGF
2006 The ompA gene in Chlamydia trachomatis differs in phylogeny and rate of evolution from other regions of the genome. Infect Immun 74 578 585
27. EverettKD
BushRM
AndersenAA
1999 Emended description of the order Chlamydiales, proposal of Parachlamydiaceae fam. nov. and Simkaniaceae fam. nov., each containing one monotypic genus, revised taxonomy of the family Chlamydiaceae, including a new genus and five new species, and standards for the identification of organisms. Int J Syst Bacteriol 49 415 440
28. ReedKD
RuthGR
MeyerJA
ShuklaSK
2000 Chlamydia pneumoniae infection in a breeding colony of African clawed frogs (Xenopus tropicalis). Emerg Infect Dis 6 196 199
29. PetterssonB
AnderssonA
LeitnerT
OlsvikO
UhlenM
1997 Evolutionary relationships among members of the genus Chlamydia based on 16S ribosomal DNA analysis. J Bacteriol 179 4195 4205
30. WilsonPA
PhippsJ
SamuelD
SaundersNA
1996 Development of a simplified polymerase chain reaction-enzyme immunoassay for the detection of Chlamydia pneumoniae. J Appl Bacteriol 80 431 438
31. KwonSJ
LeeKJ
ChoenDS
KimKS
LeeSJ
2004 The sequence of 16S rRNA of Korean isolate Chlamydia pneumoniae LKK-1. Unpublished 2004, Submitted (February-2004) to the EMBL/GenBank/DDBJ databases
32. HatchTP
AllanI
PearceJH
1984 Structural and polypeptide differences between envelopes of infective and reproductive life cycle forms of Chlamydia spp. J Bacteriol 157 13 20
33. ThomasNS
LusherM
StoreyCC
ClarkeIN
1997 Plasmid diversity in Chlamydia. Microbiol 143 1847 1854
34. GrayA
1988 Society and culture: demographic and social history.
JuppJ
The Australian people: An encyclopedia of the nation, its people and their origins Oakleigh Cambridge University press 88
35. MoorPE
CollignonPC
GilbertGL
1999 Pulsed-field gel electrophoresis used to investigate genetic diversity of Haemophilus influenzae type b isolates in Australia shows differences between Aboriginal and non-Aboriginal isolates. J Clin Microbiol 37 1524 1531
36. CochraneM
WalkerP
Gibbs
TimmsP
2005 Multiple genotypes of Chlamydia pneumoniae identified in human carotid plaque. Microbiol 151 2285 2290
37. RozenS
SkaletskyHJ
2000 Primer3 on the WWW for general users and for biologist programmers.
KrawetzS
MisenerS
Bioinformatics Methods and Protocols: Methods in Molecular Biology Totowa, NJ Humana Press 365 386
38. DrummondAJ
AshtonB
CheungM
HeledJ
KearseM
2007 Geneious version 4.7. Available: http://www.geneious.com/. Accessed 13 August 2009
39. DwyerRS
TreharneJD
JonesBR
HerringJ
1972 Chlamydial infection. Results of micro-immunofluorescence tests for the detection of type-specific antibody in certain chlamydial infections. Br J Vener Dis 48 452 459
40. ColesKA
TimmsP
SmithDW
2001 Koala biovar of Chlamydia pneumoniae infects human and koala monocytes and induces increased uptake of lipids in vitro. Infect Immun 69 7894 7897
41. Dreses-WerringloerU
BhuiyanM
ZhaoY
GerardHC
Whittum-HudsonJ
2008 Initial characterization of Chlamydophila (Chlamydia) pneumoniae cultured from the late-onset Alzheimer brain. Int J Med Microbiol 299 187 201
42. RamirezJA
1996 Isolation of Chlamydia pneumoniae from the coronary artery of a patient with coronary atherosclerosis. Ann Intern Med 125 979 982
43. LeeSJ
NamEC
WonJ
ParkWS
KimWJ
2003 Characterization of the first Korean isolate of a Chlamydia pneumoniae strain. J Infect Dis Japan 56 62 64
44. HuttonS
DoddH
1993 C. pneumoniae successfully isolated. Current topics 42 43
45. AscheVL
HuttonSI
DouglasFP
1993 Serological evidence of the three chlamydial species in an Aboriginal community in the Northern Territory. Med J Aust 158 606 604
46. WarrenK
SwanR
BodettiT
FriendT
HillS
2005 Ocular Chlamydiales infections of Western barred bandicoots (Perameles bougainville) in Western Australia. J Zoo Wildl Med 36 100 102
47. BlumerC
ZimmermannDR
WeilenmannR
VaughanL
PospischilA
2007 Chlamydiae in free-ranging and captive frogs in Switzerland. Vet Pathol 44 144 150
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2010 Číslo 5
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Měli bychom postcovidový syndrom léčit antidepresivy?
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- Quorum Sensing Inhibition Selects for Virulence and Cooperation in
- The Role of Intestinal Microbiota in the Development and Severity of Chemotherapy-Induced Mucositis
- Crystal Structure of HIV-1 gp41 Including Both Fusion Peptide and Membrane Proximal External Regions
- Susceptibility to Anthrax Lethal Toxin-Induced Rat Death Is Controlled by a Single Chromosome 10 Locus That Includes