
Persistent Growth of a Human Plasma-Derived Hepatitis C Virus Genotype 1b Isolate in Cell Culture
HCV (hepatitis C virus) research, including therapeutics and vaccine development, has been hampered by the lack of suitable tissue culture models. Development of cell culture systems for the growth of the most drug-resistant HCV genotype (1b) as well as natural isolates has remained a challenge. Transfection of cultured cells with adenovirus-associated RNAI (VA RNAI), a known interferon (IFN) antagonist and inhibitor of dsRNA-mediated antiviral pathways, enhanced the growth of plasma-derived HCV genotype 1b. Furthermore, persistent viral growth was achieved after passaging through IFN-α/β-deficient VeroE6 cells for 2 years. Persistently infected cells were maintained in culture for an additional 4 years, and the virus rescued from these cells induced strong cytopathic effect (CPE). Using a CPE-based assay, we measured inhibition of viral production by anti-HCV specific inhibitors, including 2′-C-Methyl-D-Adenosine, demonstrating its utility for the evaluation of HCV antivirals. This virus constitutes a novel tool for the study of one of the most relevant strains of HCV, genotype 1b, which will now be available for HCV life cycle research and useful for the development of new therapeutics.
Published in the journal:
. PLoS Pathog 6(5): e32767. doi:10.1371/journal.ppat.1000910
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1000910
Summary
HCV (hepatitis C virus) research, including therapeutics and vaccine development, has been hampered by the lack of suitable tissue culture models. Development of cell culture systems for the growth of the most drug-resistant HCV genotype (1b) as well as natural isolates has remained a challenge. Transfection of cultured cells with adenovirus-associated RNAI (VA RNAI), a known interferon (IFN) antagonist and inhibitor of dsRNA-mediated antiviral pathways, enhanced the growth of plasma-derived HCV genotype 1b. Furthermore, persistent viral growth was achieved after passaging through IFN-α/β-deficient VeroE6 cells for 2 years. Persistently infected cells were maintained in culture for an additional 4 years, and the virus rescued from these cells induced strong cytopathic effect (CPE). Using a CPE-based assay, we measured inhibition of viral production by anti-HCV specific inhibitors, including 2′-C-Methyl-D-Adenosine, demonstrating its utility for the evaluation of HCV antivirals. This virus constitutes a novel tool for the study of one of the most relevant strains of HCV, genotype 1b, which will now be available for HCV life cycle research and useful for the development of new therapeutics.
Introduction
Hepatitis C virus (HCV), a member of the Flaviviridae family, is an enveloped, positive-sense RNA virus that infects approximately 170 million people worldwide. Chronic HCV infection can lead to serious liver disease, including cirrhosis and hepatocellular carcinoma. Current therapy with pegylated interferon (IFN) and ribavirin is expensive, associated with serious side effects and only effective in about 50% of treated patients. Of the six major genotypes of HCV, the relatively IFN-resistant genotypes 1a and 1b predominate in the United States, Japan and Western Europe [1].
Recent developments have advanced the HCV research field whereby a single virus isolate (cloned from a patient with a rare case of fulminant hepatitis C), JFH-1, or derivatives of that isolate have been shown to robustly replicate in the human hepatoma cell line, Huh7 [2], [3]. Full-length replicons constructed by adding the structural coding regions from another genotype 2a virus, J6 [2], were shown to not only replicate in culture, but to efficiently produce infectious viral particles [2]–[6]. Replication of the J6/JFH-1 virus in Huh7 cells was more robust in a derivative cell line, termed Huh7.5, which was selected from replicon-containing Huh7 cells after curative treatment with IFN [6], [7]. An infectious system based on the use of a Vero cell line and the pHCV-WHU-1 consensus clone (genotype 1b) was reported to produce high levels of HCV genome (>108 copies/ml) with the aid of T7 polymerase provided by recombinant vaccinia virus vTF7-3 [8].
While the current cell culture systems utilize viruses that were initially replicon-derived from the JFH-1 isolate [2]–[4], [9]–[15], from HCV genotype 1b consensus clones [8], [16] or from the HCV genotype 1a prototype virus (H77-S) [10], there remains the need for a system that would be permissive for a wide variety of HCV strains found in nature. Human hepatocytes (including fetal hepatocytes) have been reported to support virus replication after RNA transfection or infection with patient sera [17], [18]. However, the use of primary cells has several technical limitations because they proliferate poorly in vitro and divide only a few times. Primary cultures could be maintained for longer periods of time only if the cells were immortalized by introducing oncogenes, a procedure that typically results in changes of the hepatocyte characteristics and function [17].
One approach to overcoming the obstacle of limited HCV growth in culture is to identify the mechanism of restriction. Activation of alpha/beta interferon (IFN-α/β) production is a key step in the innate response to viral infection and to the presence of double-stranded RNA (dsRNA) synthesized during replication of many viruses [19]. Several cellular dsRNA-binding proteins have been implicated in the IFN-response to infection. For instance, we have previously identified the adenosine deaminase that acts on dsRNA (ADAR1) as an IFN-α/β-induced protein that is a potent inhibitor of HCV replicon growth in cell culture [20]. ADAR1 converts adenosines in viral RNA to inosine [21], rendering the RNA inactive [20]. Both ADAR1 and the IFN-induced dsRNA-activated protein kinase (PKR) are inhibited by the small adenovirus-associated RNA (VA RNAI) [20], [22], [23]. When VA RNAI is transfected into replicon containing Huh7 cells, it increases replication by 40-fold [20], suggesting that these IFN-induced proteins impose critical limitations to HCV replication.
In this study, we achieved growth of an HCV genotype 1b isolate by inoculating IFN-deficient cells with human plasma from an infected patient. Viral replication was stimulated further with the addition of VA RNAI, and led to the creation of a cell line persistently infected with HCV. More interestingly, the virus isolated from these cultures has the potential to induce cytopathic effects in the persistently infected VeroE6 cells and cause massive cell death in Huh7.5 cells.
Results
Construction of a persistently infected cell line, LB-piVe
Based on our previous finding that VA RNAI enhanced HCV replication in the replicon system [20], we hypothesized that virus growth in cell culture may also be inhibited by IFN-induced pathways. Our approach was to employ VeroE6 cells, which contain a homozygous-allelic deletion of the IFN-α/β genes [24], [25], yet retain the ability to express IFN-induced genes such us ADAR1 and PKR, which can be activated during virus infection. Cells were transfected with a plasmid encoding VA RNAI (pVA; [26], [27]), and then inoculated once with HCV genotype 1b infectious human plasma, LB [28], [29] (Figure 1) or with genotype 1a infectious chimpanzee serum [30] (see Text S1 and Table S1). Normal human serum was used as a negative control. Infected cells were passaged (division ratio of 1∶6) every seven days for 20 weeks with weekly pVA re-transfection (Table 1). HCV RNA was detected sporadically in the virus-infected cells after week 20. Nevertheless, passages were continued weekly in the absence of VA RNAI. Surprisingly, after 2 years of passage in culture in the absence of VA RNAI, HCV RNA was detected consistently, indicating that the virus was able to establish a persistent infection. No virus was detected after 20 weeks in the control experiment that was infected with normal human serum. The possibility that the positive PCR results were due to RNA carry-over is extremely low since the cells had been diluted ∼1.94×1096 after 2 years in culture. LB-plasma persistently infected VeroE6 cells (LB-piVe cells) were screened with anti-human - and anti-monkey-specific primers to ensure that the cultures were not contaminated with human cells (data not shown).


Sequence analysis showed that the persistent virus (LB-piVe virus) shares 99.7% amino acid homology with the parental genotype 1b virus and contains only 10 amino acid changes in the nonstructural region. Sequences have been deposited into GenBank. A representative nucleotide sequence of the LB-piVe virus, aligned with the parental virus sequence and a prototype genotype 1b virus is shown in Figure S1. The complete sequence alignment and reverse genetics studies are being conducted and will be presented for publication in the future.
LB-piVe cells express HCV viral antigens
To visualize HCV antigen expression, we stained LB-piVe fixed cells with polyclonal anti-HCV serum [30] (Figure 1A and B) or anti-NS5A monoclonal antibodies (Figure 1C and D). To increase the sensitivity of the immunofluorescence assay, we enriched the cell culture by selecting virus-containing, antigen-expressing cells (Figure 1B and D) using a cell panning procedure (see Materials and Methods). LB-piVe cells expressed HCV antigens in both perinuclear and cytoplasmic regions of the cells as expected (Figure 1A–D, right panels). The results suggest that the addition of VA RNAI may broaden cellular tropism by allowing persistent growth and replication of HCV from plasma in non-hepatic VeroE6 cells.
Western blot analysis of LB-piVe (after two rounds of cell panning) and J6/JFH-1-infected cell extracts demonstrates that HCV proteins were expressed at detectable levels (Figure 1E and F). Because the proportion of immunofluorescent cells was low, we then compared the levels of viral RNA in filter-clarified supernatants from LB-piVe panned cells versus J6/JFH-1-infected Huh7.5 cells (Figure 1G). J6/JFH-1 yielded 9.2×107 RNA copies/ml, while LB-piVe yielded 1×104 RNA copies/ml; (see Figure 1G). Persistent infection could only be maintained at a low viral titer, as attempts to obtain the higher viral yields by cell panning (Figure 1B and D) resulted in viral instability due to cell cytolysis (data not shown).
Quantitation of viral titers by a CPE (cytopathic effect)-based assay
Interestingly, we observed evidence of CPE in LB-piVe cells after 2 years in culture (Figure 2A, right). To demonstrate that the virus from the persistently-infected cells was infectious, filter-clarified culture supernatants from LB-piVe cells were used to inoculate naïve Huh7.5 cells. The infected Huh7.5 cells demonstrated enhanced CPE compared to the parental LB-piVe cells, and resulted in gross cell death after 5 days (Figure 2B, right). Viral antigens were detected at 3 days post-transfer of supernatants by immunostaining the infected Huh7.5 cells (Figure 2C, right) and also by immunoblotting Huh7.5 cell extracts (Figure 2D) with anti-NS5A antibody. The level of CPE observed in Huh7.5 cells (Figure 2E, micrographs) was directly related to the amount of viral RNA in the inoculum (Figure 2E, histogram). Taken together, our results show that viral infectivity can be transferred from the persistently infected cell line, LB-piVe, to naïve hepatic cells and that the level of CPE correlates with the level of input viral RNA.

Based on these unique characteristics of LB-piVe, we developed a CPE-based end-point dilution assay for quantification of viral titers. Naïve Huh7.5 cells were plated in 96-well plates and then infected with serial dilutions of virus-containing filter-clarified supernatants (see Materials and Methods). Five days post-infection (dpi), cells were observed by light microscopy and those wells showing CPE were assigned a positive result. The 50% tissue culture infectious dose (TCID50) was calculated using the method of Reed and Muench [31].
Virus neutralization
To further confirm that the CPE was linked to virus infection, we employed the end-point dilution assay (based on visualization of cell death) to study virus neutralization. Huh7.5 cells were first incubated with antibodies to the putative viral receptor CD81 [32]–[34] and then infected with serial dilutions of filter-clarified supernatants of LB-piVe (Figure 3A) or J6/JFH-1 (Figure 3B). Viral titers were determined as described in Materials and Methods. This study showed that anti-CD81 antibodies reduced genotype 1b LB-piVe viral titers by ∼1×log10 (Figure 3A), similar to that observed for the genotype 2a virus J6/JFH-1 (Figure 3B). Pre-incubation of LB-piVe virus with HCV-specific immunoglobulin intravenous (HCIGIV) [35] (Figure 3C) or anti-E2 monoclonal antibodies [36] (Figure 3D) also inhibited virus growth similarly. However, pre-incubation of LB-piVe virus or J6/JFH-1 with normal IGIV or an isotype-matched negative control antibody did not affect viral titers. It may be noted that the anti-E2 monoclonal antibodies were generated to genotype 1a recombinant E2 proteins, including the hypervariable region. Consequently, their ability to neutralize a genotype 1b virus could be limited to some extent as reflected by the 60% decrease in viral titers observed. These neutralization experiments demonstrate that Huh7.5 cell death resulted from the transfer of virus from the LB-piVe cells, and that infection and viral spread in Huh7.5 cells was blocked by the addition of HCV-specific antibodies.
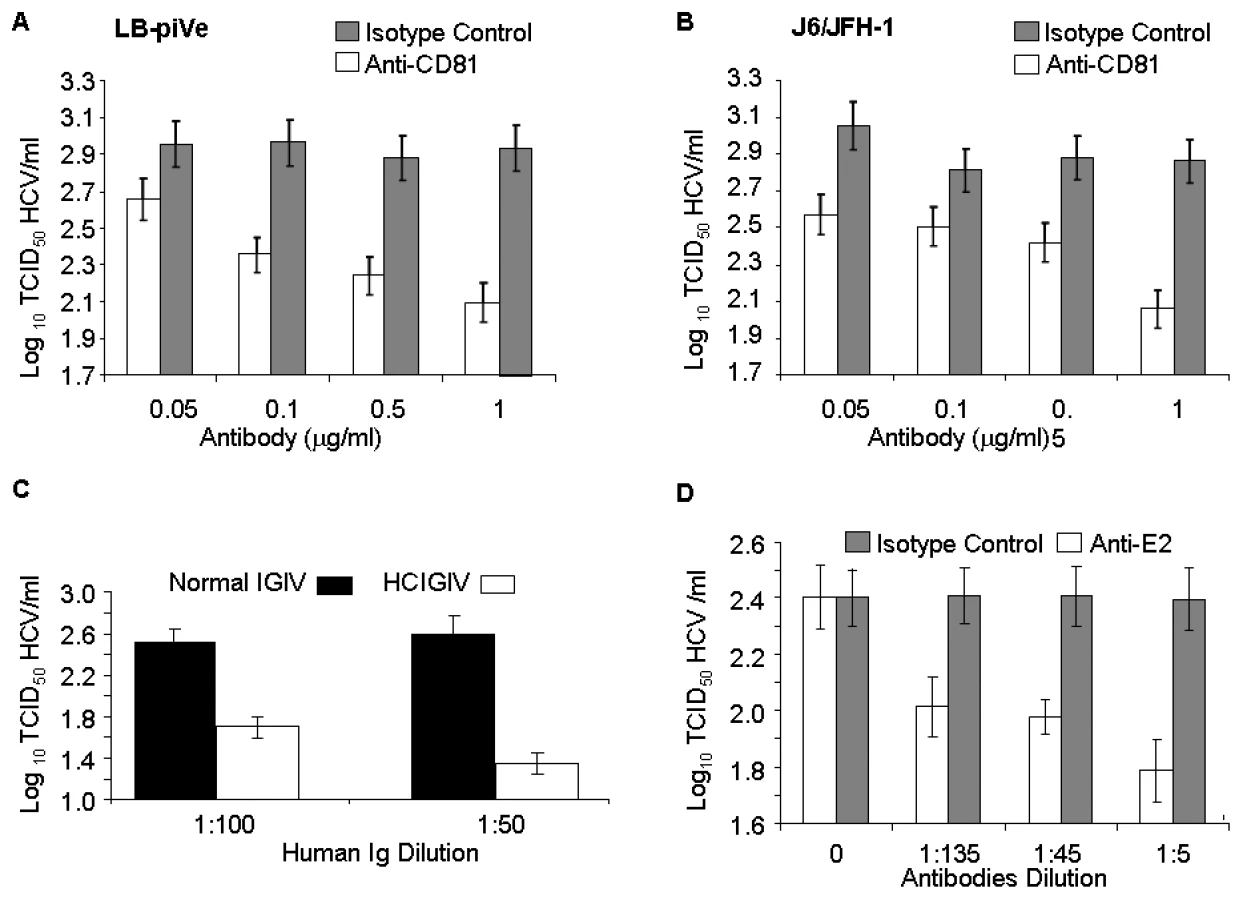
We then explored the utility of the CPE-based assay to screen therapeutics by treating virus-infected cells with HCV inhibitors. We used a well characterized inhibitor of the HCV polymerase, 2′-C-Methyl-D-Adenosine (2′-C-Me-A) [37]. J6/JFH-1 and LB-piVe infected cells were incubated with complete growth medium containing a range of 2′-C-Me-A, from 0.05 to 1 µM. Titers were determined as described in Materials and Methods. The results showed that LB-piVe growth was reduced after treatment with 2′-C-Me-A (Figure 4A), with a 50% effective concentration (EC50) value in the nanomolar range, comparable to that observed for J6/JFH-1 (Figure 4B). Additionally, we tested an HCV-specific small inhibitory RNA to knock-down viral titer (siRNA 313; [38], for details see Materials and Methods). In this assay, siRNA 313 inhibited CPE caused by LB-piVe virus by ∼80% (Figure 4C), while J6/JFH-1 was inhibited by >90% (Figure 4D).

When LB-piVe cells were treated with IFN, the virus continued to replicate. We measured LB-piVe viral titers in cells that were treated with 0, 10, 100 or 1000 IU/mL of IFN for 24, 48 and 72 hr (Figure 4E). LB-piVe titer decreased slightly only at 72 hr with 100 or 1000 IU/mL, but the values were not significantly lower than for 48hr. In contrast, J6/JFH-1 titer decreased by 5-fold when treated with 10 IU/mL for 48 hr and there was no detectable virus with 1000 IU/mL (Figure 4F). To ensure that the LB-piVe cells had not become insensitive to IFN treatment, we measured LB-piVe titers in Huh7.5 cells that were also treated with IFN (Figure 4G). There was no significant effect on LB-piVe titers by treating the Huh7.5 cells with IFN for 5 days at 10 IU/mL and no change after treatment with 1000 IU/mL. A decrease of 0.2 log10 was observed when comparing 10 IU/mL vs 1000 IU/mL over 5 days (Figure 4G). This small difference may be attributed to the long incubation period. These results indicate that the LB-piVe virus and not the persistently infected cells are relatively IFN resistant as would be expected for a natural genotype 1b virus isolate. To ensure that the virus had not acquired IFN resistance through passage in culture, we compared the effects of IFN treatment on the parental virus (LB) with the persistent virus in VeroE6 cells (Figure 4H). There was little effect after treating the LB virus for 24, 48 or 72 hr, demonstrating that this parental genotype 1b strain was relatively IFN resistant, as expected.
These inhibition studies demonstrated that the LB-piVe virus was sensitive to HCV-specific inhibitors and that the CPE-based assay provides an easy and quantitative method for measuring the efficacy of antiviral compounds. Furthermore, the LB-piVe virus behaved like the wild-type parental virus and maintained its relative IFN resistance.
Limitations on HCV growth are alleviated by VA RNAI
VeroE6 cells express the putative receptors for HCV [39]. To elucidate the general growth properties of HCV in these cells, we tested their ability to support replication of the IFN-sensitive genotype 2a virus (Figure 5). Cells were mock infected (Figure 5A and B, left) or J6/JFH-1-infected (Fig. 5A and B, right), and immunostained at 4 dpi with anti-NS5A antibody. J6/JFH-1 was infectious and replicated in many Huh7.5 cells (Figure 5B, right) and fewer VeroE6 cells (Figure 5A, right). An increase in the number, size and intensity of the foci in J6/JFH-1-infected Huh7.5 cells was observed in the presence of wild-type VA RNAI (WT) (Figure 5C, right panel) but not mutant VA RNAI (dl1) (Figure 5C, center panel), demonstrating that J6/JFH-1 growth was improved by VA RNAI. These data suggest that while the paracrine and autocrine IFN pathways may be defective in VeroE6 cells, additional cellular factors antagonized by VA RNAI are limiting for HCV growth. We also determined the viral titer of the J6/JFH-1-infected Huh7.5 cells, which were transfected with mutant VA RNAI (dl1) or wild-type VA RNAI (WT) (Figure 5D). The results showed that wild-type VA RNAI led to an increase in J6/JFH-1 viral titers by >1.5 log units (Figure 5D, WT), and further confirms that VA RNAI enhances both growth and replication of this genotype 2a virus, probably through inhibition of dsRNA-activated pathways.
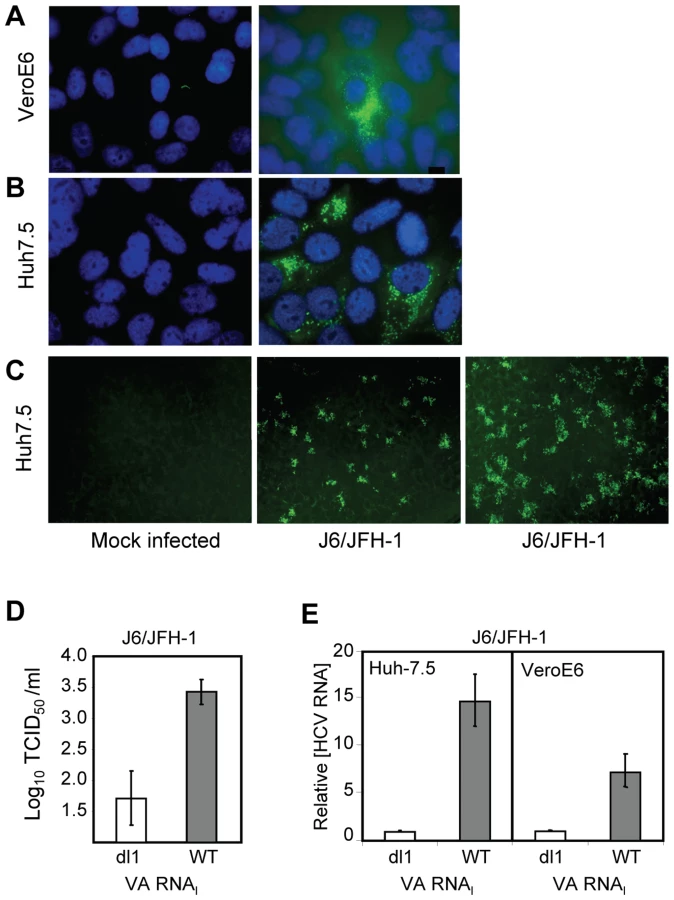
The amount of J6/JFH-1 RNA was quantitated (relative to GAPDH) by real-time RT-PCR. Replication of J6/JFH-1 increased by 15-fold (±3-fold SEM) in Huh7.5 cells with VA RNAI, whereas VeroE6 cells containing VA RNAI yielded 7-fold (±2-fold SEM) more viral RNA than cells with mutant (dl1) VA RNAI (Figure 5E). While the exact mechanism is unknown, a 15-fold increase in viral RNA suggests that the fate of viral RNA in the cells may be affected by the presence of VA RNAI, consistent with our previous findings that ADAR1 was inhibited in replicon cells containing VA RNAI [20]. J6/JFH-1 RNA titers were enhanced by VA RNAI twofold in Huh7.5 cells (Figure 5E, left) over VeroE6 cells (Figure 5E, right), illustrating preferential growth of J6/JFH-1 in Huh7.5 cells and demonstrating that HCV genotype 2a growth, in addition to genotypes 1a and 1b, is enhanced by VA RNAI.
VA RNAI may increase HCV replication through RNA stability
VA RNAI allowed the establishment of a persistently infected cell line and increased growth of LB-piVe (Figure 6A) and J6/JFH-1 (Figure 5A–E). To evaluate the effects of VA RNAI on the parental virus during the first few days of infection, we examined its effect on HCV RNA stability by comparing the relative increase in viral RNA in VeroE6 cells to that in Huh7.5 cells that were inoculated with the same HCV-positive human plasma (LB, [28], [29]) that was used to establish the persistently infected cell line LB-piVe. Naïve VeroE6 and Huh7.5 cells were transfected with pVA before inoculation with LB plasma (transient infection) (Figure 6B–D). RNA was extracted from cell lysates on the days indicated (Figure 6B, C) and HCV RNA was measured by quantitative RT-PCR. The relative amount of HCV RNA in pVA-transfected cells versus pVA-untransfected cells (Figure 6B) increased 60-fold after 8 days of transient infection in VeroE6 cells. However, in transiently infected Huh7.5 cells, the relative amount of HCV RNA did not increase with the addition of VA RNAI over 8 days (Figure 6B), consistent with our inability to obtain a persistently infected Huh7.5 cell line. It may be noted here that the CT values of GAPDH employed as a normalization control in these experiments, were consistent among cells in the presence or absence of pVA. Thus, the dramatic increase in viral RNA in VeroE6 cells may be due to factors other than the variation in transcript levels of GAPDH reported in liver cells [40]–[43]. Furthermore, when the results were expressed in terms of absolute HCV RNA copy number (using an HCV RNA standard curve and measuring copies per ml; Figure 6C), the number of RNA copies remained stable in Huh7.5 cells, suggesting that the level of replication may be equal to the degradation of HCV RNA, with or without the addition of VA RNAI. In contrast, a precipitous decline in HCV RNA copy number was observed in transiently infected VeroE6 cells in the absence of VA RNAI, while the levels remained relatively stable in cells that contained VA RNAI (Figure 6C), thus indicating that VA RNAI has an effect on viral RNA over 8 days in VeroE6 cells. We speculate that this effect may be due to; (i) altering the RNA synthesis rate, (ii) altering the degradation rate of HCV RNA molecules other than the input RNA, or (iii) inhibition of an RNA degradation pathway.

Incubation of transiently infected cells with an RNA polymerase inhibitor helped to assess the level of viral RNA in the absence of viral replication. We treated the parental virus with 2′-C-Me-A, which resulted in similar HCV RNA levels when cells were transfected with either WT - or mutant-VA RNAI (Figure 6D). Initially there was a decrease in viral RNA (time 0 = input RNA). VA RNAI was not able to stimulate replication in the presence of an inhibitor of HCV polymerase. Additionally, the level of viral RNA did not increase in the presence of wild-type VA RNAI, suggesting that viral RNA stability was also not affected by the presence of VA RNAI in the absence of replication. Figure 6 shows that in the presence of VA RNAI, viral RNA titer goes down and then levels off; while in the absence of wild-type VA RNAI it continues to decrease (Figure 6C). When the experiment is done in the presence of 2′,C-Me-A, the RNA titer decreases and levels off, independent of VA RNAI (Figure 6D). This is consistent with the mechanism of 2′,C-Me-A, which inhibits new RNA synthesis, however, in this experiment the viral RNA is not degraded 100-fold (c.f., Figure 6C and 6D). We interpret these data as follows: 1) in the absence of viral replication (in 2′,C-Me-A-treated cells), there is less degradation of the viral RNA; 2) in the absence of viral replication (in 2′,C-Me-A-treated cells) there is also the absence of dsRNA (positive strand plus negative strand); and 3) therefore, dsRNA-activated proteins, including ADAR1, would not be activated, leaving VA RNAI with no effect on stability. This is consistent with our ongoing studies that show that only wild-type VA RNAI (that which can bind PKR or ADAR1) is capable of stimulating the replicon (Taylor, unpublished results). Taken together, these results suggest that VA RNAI in the early stages of infection may affect the stability of the viral RNA, either by altering the degradation rate of new HCV RNA molecules or by inhibition of an RNA degradation pathway that may be modulated by viral replication. However, we also cannot exclude the possibility that VA RNAI alters the HCV RNA synthesis rate in the absence of a polymerase inhibitor.
Discussion
In this study, we have demonstrated that the addition of VA RNAI, a known IFN antagonist and inhibitor of dsRNA-mediated antiviral pathways, permitted the persistent growth of a plasma-derived HCV in a cell line that lacks IFN genes. Most of the current knowledge of HCV biology and pathogenesis has been derived from the use of the unique JFH-1 cell culture system, which now allows the study of the complete virus life cycle, including entry, assembly and release. The limitation of this model, however, is that robust viral growth is restricted only to hepatic-derived cell lines such as Huh7.5 and Huh7 cells [44] and only by a genotype 2a replicon-derived virus. The establishment of an alternative model to characterize other HCV genotypes from infected individuals is still needed and is critical for the development of efficient viral therapies to control the disease.
By passaging genotype 1b virus-infected VeroE6 cells for 20 weeks in the presence of VA RNAI and more than 2 years without VA RNAI, we generated a persistently infected cell line that expresses HCV antigens at levels high enough to be detected by immunofluorescence and Western blot (Figure 1A–F, Table 1). We found that the LB-piVe virus is highly cytotoxic, and is capable of inducing massive Huh7.5 cell death (Figure 2B, C and E); indicating that the virus produced in the persistently infected cells is infectious to hepatocytes. CPE could be blocked by antibodies to CD81 (Figure 3A), by anti-HCV-specific immunoglobulins (Figure 3C) and by anti-E2 monoclonal antibodies (Figure 3D), confirming the link between cell death and viral infection. While neutralization was not as potent using the anti-E2 monoclonal antibodies, we believe that this may be due to the antibodies being raised against recombinant genotype 1a proteins. The genotype differences in the E2 proteins (including hypervariable domains) may be reflected in loss of epitope recognition, thus explaining the 0.6 log10 decrease in viral titer.
The LB-piVe virus-mediated CPE has the advantage that it can be assessed visually, and quantified easily and rapidly. This represents a significant improvement over the current genotype 2a HCVcc systems that utilize FFU assays, RT-PCR or reporter assays for quantitation, which are both laborious and time-consuming [45]. In addition, we have demonstrated the utility of this system in virus neutralization studies (Figure 3A, C and D) and in testing virus inhibition by well characterized HCV-specific antivirals (Figure 4A, C, E and G).
CPE was observed in VeroE6 cells and more-exaggerated CPE was found when filterable supernatants were used to infect Huh7.5 cells. While it was possible to enhance viral titer by panning the LB-piVe cells, and effectively increasing the number of virus-infected cells, the new culture could not survive after several passages. We suspect that the virus cannot be maintained in a culture that demonstrates massive CPE, such as that seen in Huh7.5 cells. This may be the reason that we were unable to obtain persistently infected Huh7.5 cell line, while VeroE6 cells can support persistent HCV infection due to a low-level display of CPE.
HCV - associated cell death has also been reported in Huh7.5.1 cells after infection with JFH-1 when HCV RNA levels reached a maximum [46]. Gene expression profiling of HCV-infected Huh7.5 cells showed both the presence of activated caspase-3 and induction of cell death-related genes, suggesting an association of virus infection with cytopathic effects. Although not yet resolved, it has been postulated that HCV could mediate direct apoptosis by deregulating the cell cycle, which may contribute to liver injury in infected individuals [46]. While still requiring further studies and more comparisons between human pathology and cell culture, we suggest that the LB-piVe system may very well mimic a natural HCV infection in humans and could represent a useful tool to study the intricate process of viral pathogenesis.
VeroE6 cells were also permissive for replication of genotype 2a J6/JFH-1 virus [2] (Figure 5A–C). VA RNAI boosted replication and spread in these cells, as shown by the increase in the HCV RNA yield (Figure 5D, E). This may be attributable to an increase in viral RNA stability and possibly reflects the type of interplay between host and virus. The presence of VA RNAI allowed for broadened cell tropism by HCV to include non-hepatic cells (Tables 1 and S1), perhaps due to its ability to circumvent the IFN-induced antiviral response. The full-extent of the mechanisms employed by VA RNAI towards overcoming the negative effects of IFN is currently unknown. VA RNAI is important to adenovirus infection and confers virus stability in the presence of IFN and IFN-induced proteins. It has been suggested that VA RNAI has an effect on HCV RNA stability by inhibition of the IFN-induced protein, ADAR1 [20]. When we compared the relative amount of HCV RNA in VA RNAI-transfected cells versus -untransfected cells (Figure 6B, C) that were infected with HCV-positive human plasma LB, we observed a 60-fold increase in VeroE6 cells. Interestingly, a precipitous decline in HCV RNA was observed in these cells in the absence of VA RNAI (Figure 6C). Thus, VA RNAI has an effect in the VeroE6 cells, at least during the first 8 days of infection. We have yet to evaluate possible defects in the RIG-I pathway observed previously in Huh7.5 cells and likely to play a role in early infection [47]. The fact that we did not observe any increase in the relative amount of HCV RNA in Huh7.5 cells after VA RNAI transfection followed by infection with the parental genotype 1b serum-derived virus (Figure 6B and C), was unexpected. We suspect that the relatively stable amount of viral RNA reflects extremely low viral replication of the LB virus in Huh7.5 cells. These findings are supported by the evidence that we could not establish a persistently infected cell-line with Huh7.5 cells, suggesting that VeroE6 cells were more permissive for persistent infection, perhaps due to the lack of IFN genes.
VA RNAI was not able to rescue the virus during 2′-C-Me-A treatment and does not stimulate replication nor does it protect the virus from an antiviral that targets the HCV polymerase. We used this RNA polymerase inhibitor to evaluate RNA stability in the absence of viral RNA replication. Since VA RNAI only increased the HCV RNA in the presence of viral replication, we believe that it may perhaps inhibit cellular factors that are activated during viral replication (e.g., dsRNA-binding proteins) and cause instability of the virus. It's possible that VA RNAI interacted with, and therefore blocked ADAR1 and PKR pathways. This would be consistent with our previous findings showing that the HCV replicon was stimulated by knock-down or inhibition of ADAR1 or PKR [20]. Additionally, the inhibition of RNA replication (including loss of negative strand RNA) should inhibit the formation of dsRNA intermediates, thus avoiding the activation of dsRNA-activated proteins that can lead to viral instability [48]–[54]. Again, we cannot exclude the possibility that VA RNAI enhanced viral replication in the absence of the polymerase inhibitor. Taken together, these data suggest that VA RNAI may possibly contribute to establishing a persistent infection in VeroE6 cells; however, the presence of VA RNAI alone is not enough to overcome the cellular antiviral response in Huh7.5 cells. IFN-deficient VeroE6 cells probably provide a more ideal environment for a virus that is, usually, IFN responsive. We suspect that this may be due to the decreased expression of IFN-induced proteins which may actively inhibit HCV replication [20]. Both Huh7.5 cells and VeroE6 cells express PKR and ADAR1, but only the VeroE6 cells lack the IFN genes that induce these proteins. We found that even the persistent virus was stimulated by the presence of VA RNAI, suggesting that some of the dsRNA-activated proteins were still expressed and were inhibitory to the virus.
In patients, in general, genotype 2 and 3 viruses are more sensitive to current antiviral therapy than the genotype 1 viruses [55]. Genotype 1b is thought to be the most IFN resistant and the most prevalent in North America, Europe and Japan. However, the HCV replicons (genotype 1b) and J6/JFH1 virus are sensitive to IFN in cell culture. It is not clear why viruses respond to IFN differently in vivo versus in vitro. Since HCV grows well in VeroE6 cells, especially when assisted by VA RNAI, we suggest that endogenous IFNs may limit HCV replication in cell culture.
We suspect that the LB-piVe virus, like the parental LB from which it was derived, was relatively resistant to IFN (Figure 4E, H), a property that has not yet been reported in infectious cell culture (Figure 4G). While it warrants further investigation, it may be possible that we were able to obtain this virus because VA RNAI was present in the early stages of infection and inhibited the antiviral response generated by viral RNA replication. Our results on the enhancement of virus replication by VA RNAI are clearly consistent with evasion of the antiviral response, and correlate with the observation that susceptibility of human primary hepatocytes to HCV infection could be improved by impairing expression of other IFN signaling factors such us interferon regulatory factor-7 (IRF-7) [17]. We suggest that in the early stages of cell culture infection, before viral proteins are in sufficient quantity, the innate immune pathways are active and control infection (RIG-I, PKR, ADAR1, RNaseL, etc.). However, once the virus is given a chance to accumulate, it can overcome these mechanisms of host control, either through the E2, NS5A or NS3 proteins [56]–[58].
Our findings have raised some interesting questions. Future studies with IFN-sensitive viruses, complemented with known IFN-resistant HCV proteins (such as NS5A and E2) using sequences from the LB-piVe virus, are planned. Additionally, the LB-piVe virus will be ideal for evaluating the genes responsible for conferring IFN resistance. We plan to construct an infectious clone and a replicon based on this virus with the aim of evaluating individual genes. At the same time, alignments with the IFN-resistant parental strain of LB with IFN-sensitive genotype 1b replicons may enable the identification of important amino acids that determine IFN resistance. Transient transfection experiments complementing the IFN-sensitive replicons will be among the experiments that will provide insights into the identification of the features that may confer IFN resistance by this genotype 1b virus.
In summary, here we demonstrate that wild-type HCV genotype 1b viruses from human plasma can replicate in African green monkey kidney cells, VeroE6, and that replication of viral genotypes 1a and 2a can be stimulated by the presence of VA RNAI. This is a new approach to culturing HCV and the first report of a cell culture system that represents a convenient assay for studying genotype 1b. This is an improvement in terms of utility for research, as the virus can be titrated without employing error-prone, quantitative RT-PCR methods nor arduous immunocytochemistry-based focus forming assays. The availability of the LB-piVe virus raises an exciting possibility; potentially opening a new era of HCV research through the use of a new model system. Moreover, a persistently infected cell line that exhibits CPE provides a novel assay that may be conducive to high throughput development and screening of new antivirals.
Materials and Methods
Plasmids
pVA containing the adenovirus 2 virus-associated RNA I (VA RNAI) sequence, and VA RNAI mutant dl1 (pVAdl1) plasmids, were provided by M. B. Mathews [26], [27].
Cell culture and transfections
VeroE6 cells (ATCC) were maintained in complete Dulbecco's modified Eagle's medium (DMEM; Invitrogen, Carlsbad, CA) containing 10% heat-inactivated Fetal Bovine Serum (FBS; Hyclone) at 37°C with 5% CO2. Huh7.5 cells were provided by C.M. Rice (Rockefeller University, NY) and maintained in complete DMEM containing 10% FBS and non-essential amino acids (Invitrogen). FBS was screened by RT-PCR to ensure the absence of bovine viral diarrhea virus (BVDV). Multiple lots of VeroE6 cells were infected to check for reproducibility. Cells were cultured before transfection in T25 flasks or 6-well plates at a density to provide an overnight confluence of 35%, and transfected with 15–30 µg plasmid vector pVA [26], [27] using DMRIE-C per the manufacturer's specifications (Invitrogen, Carlsbad, CA).
Hepatitis viruses and titer determination
HCV genotype 1b
Cells transfected with pVA were inoculated with 200 µl of plasma containing 107 RNA copies/ml from a genotype 1b-infected patient (LB, [28], [29]) at week 0. Seven dpi, cells were divided 1∶6 and transferred to T25 flasks. The next day, cells were transfected with pVA. The passage and transfection process was repeated, without re-infection, weekly for 20 weeks (weekly results shown in Table 1). After 20 weeks, cells were divided 1∶6 weekly without transfection or re-infection for two years. These LB-persistently infected VeroE6 cells were then known as LB-piVe cells. A cytopathic effect (CPE)-based end-point dilution assay was developed for quantification of LB-piVe virus titer. LB-piVe cells were grown for 8 days. Flasks (T75) containing culture medium were frozen (at −80°C) and thawed 3 times. This mixture was cleared by centrifugation and subsequent filtering (0.45 µm), and filter-clarified culture supernatants were obtained. To measure LB-piVe titers, naïve Huh7.5 cells were plated at a density of 5×103 per well in 96-well plates to obtain 60% confluence after 24 hr, and then infected with serial dilutions of filter-clarified supernatants (8 replicates per dilution). Cells were observed by light microscopy at 5 dpi. Wells showing CPE were assigned a positive result. Alternatively, cells were fixed and stained with Crystal Violet (0.1%). The 50% tissue culture infectious dose (TCID50) was calculated using the method of Reed and Muench [31].
HCV genotype 2a
Genotype 2a virus J6/JFH-1 [2] was provided by C.M. Rice (Rockefeller University, NY) and was titrated by an end-point dilution assay in 96-well plates. Briefly, virus inocula were serially diluted and used to infect 8 replicate wells of naïve Huh7.5 cells growing in microtiter plates. Four dpi, the cells were washed, fixed with cold methanol, probed with a mouse anti-NS5A antibody (Abcam, Cambridge, MA) and a fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse IgG (H+L) (KPL Inc., Gaithersburg, MD), and finally quantitated using an indirect immunofluorescence assay (see below) [59]. Wells were scored positive if at least 1 positive cell was detected. The TCID50 was calculated using the method of Reed and Muench [31]. This procedure was followed for the experiments shown in Figures 3B and 5D. Alternatively, stained foci were counted in triplicate wells (Figures 4D and F), and titers were calculated as the mean number of foci per ml (FFU/ml).
Preparation of HCV virus stocks
Virus stocks for J6/JFH-1 were prepared by inoculating 1×108 Huh7.5 cells with 1 ml culture supernatant (103 FFU/ml) in serum-free medium [2]. Inoculated cells were grown at 37°C for 12 days. Filter-clarified culture supernatants were obtained as described above. LB-piVe stocks were prepared by growing the persistently infected cells for 8 days at 37°C in T75 flasks and supernatants were collected as for J6/JFH-1.
Evaluation of infected cultures
Indirect immunofluorescence assays
Mock - and HCV-infected cells grown in eight-well Permanox chamber slides (Nunc Inc., Denmark) at 37°C were washed with PBS, fixed with cold acetone for 30 min, and air dried. After incubation with 2% fetal bovine serum in PBS (to block nonspecific binding), cells were probed with mouse anti-NS5A antibodies (Abcam, Cambridge, MA), anti-Core monoclonal antibodies (Affinity Bioreagents, Golden, CO) or Ch1536 serum [30], followed by washing and staining with FITC-conjugated goat anti-mouse IgG (H+L). Slides were mounted with VECTASHIELD mounting medium (Vector laboratories, Burlingame, CA) containing 4′, 6-diamindino-2-phenylindole (DAPI). Fluorescent micrographs were taken with a Zeiss Axiovert microscope at a magnification of 200× (Figures 2A and B; 3G; 4C) or 1000× (Figures 1A–G; 2C; 4A and B) with an oil-immersion objective.
Western blot analysis
To detect intracellular NS5A, 3×106 infected cells were lysed in 0.2 ml lysis buffer [50 mM Tris, pH 8; 150 mM NaCl; 1% Nonidet P-40; 0.5% Deoxycolate; 0.1% (w/v) sodium dodecyl sulfate (SDS)] containing protease inhibitors (Complete protease inhibitor cocktail; Roche Applied Science, Indianapolis, IN). Cell extracts were clarified by centrifugation, denatured by boiling in Tris-Glycine-SDS sample buffer [60] and resolved on Novex 4–20% Tris-glycine polyacrylamide gels (Invitrogen, Carlsbad, CA) using Tris-Glycine-SDS running buffer [60]. Subsequently, proteins were transferred to Hybond ECL membranes (Amersham, Piscataway, NJ). Membranes were incubated with PBS containing 5% (w/v) nonfat milk and 0.05% Tween-20 (polyoxyethylene sorbitan monolaurate) to reduce nonspecific binding, and then probed with anti-HCV NS5 antibody (Austral Biologicals, San Ramon, CA, at 1∶1000), or anti-glyceraldehyde-3-phosphate dehydrogenase antibody (GAPDH, Trevigen, Gaithersburg, MD, at 1∶3,000). After washing with PBS-tween, membranes were probed with horse radish peroxidase-conjugated secondary antibodies (Kirkegaard & Perry Laboratories, Inc, Gaithersburg, MD), and antigens were detected with SuperSignal West Femto Maximum Sensitivity Substrate (Pierce, Rockford, IL).
Cell panning
Polystyrene petri dishes were coated overnight with a 2.5 µg/ml solution of Anti-Human Fc antibodies (KPL Inc., Gaithersburg, MD) in 0,05M Tris-HCl pH = 9.5. Dishes were then washed 3 times with PBS, and incubated 1 hr at room temperature (RT) with a 1∶1000 dilution of an experimental 5% immunoglobulin intravenous (IGIV) preparation made from of anti-HCV positive plasma (HCIGIV) or a 5% IGIV preparation made from anti-HCV negative plasma donations [35]. LB-piVe cells were grown for 5 days in a T150 flask, and then detached by incubation with 0.5 mM EDTA in PBS at 37°C for 30 min. After centrifugation, cells were washed, resuspended in 10 ml of PBS containing 5% FBS and distributed into the panning plates. Following an incubation of 2 hr at RT which allowed antigen-expressing cells to attach, the plates were washed three times gently with PBS/5% FBS and recovered and grown in complete 10% FBS DMEM supplemented with non-essential amino acids (Invitrogen, Carlsbad, CA). This protocol was performed 3 consecutive times.
LB-piVe sequence analysis
LB-piVe filter-clarified culture supernatants (300 µl; prepared as described above) were used to extract viral RNA with Trizol LS (as per the manufacturer's directions; Invitrogen, Carlsbad, CA) followed by alcohol precipitation. cDNA was synthesized using primer 9325R (5′-TAGGCACCACATGAACCAG-3′) and AffinittyScript Multiple Temperature Reverse Transcriptase (Stratagene, La Jolla, CA) for one hour at 55°C followed by 15 minutes at 70°C. A first round PCR product was generated with primers (300 nM each) 6038S (5′-CAGCAATACTGCGTCGGCACGT-3′) and D1-1R (5′-TTCTTGGATTTCCGCAGGATCTCC-3′) using TaKaRa LA Taq HS (Clontech Laboratories, Inc., Madison, WI) with the following parameters: 2 minutes at 95°C, 40 cycles with 30 seconds at 95°C, 1 minute at 53°C, 3 minutes at 72°C, and a final extension of 10 minutes at 72°C. For the nested-PCR, 5 µl (1/10) of the first PCR sample was added to a new tube containing 45 µl of TaKaRa LA Taq HS PCR reaction mixture and primers (300 nM each) 6144S (5′-CACTATGTGCCTGAGAGCGACGCC-3′) and D1-2R (5′-TCTCTGACTCCACGCGGGTGATGT-3′). The reaction was carried out using the following parameters: 2 minutes at 95°C, 40 cycles with 30 seconds at 95°C, 1 minute at 56°C, 3 minutes at 72°C, and a final extension of 10 minutes at 72°C. The PCR product was gel purified by using the NucleoSpinR Extract II kit ( Macherey-Nagel, Inc. Easton, PA) and sub-cloned into pCRII-TOPO (Invitrogen, Carlsbad, CA). After transformation of E. coli competent bacteria, 10 clones were selected. Both strands of the cloned-PCR product were subjected to direct sequencing by using M13 Forward and Reverse primers. Sequencing reactions were performed with the ABI Prism BigDye Terminator version 3.1 Cycle-Sequencing Kit (Applied Biosystems, Foster City, CA) according to the manufacturer's protocol and analyzed by using the ABI Prism 3100 system (Applied Biosystems, Foster City, CA). Sequences have been deposited into the National Center for Biotechnology Institutes GenBank.
Real-time PCR assays
Determination of HCV RNA titers in filter-clarified culture supernatants
Viral RNA was extracted from 300 µl of LB-piVe filter-clarified culture supernatants (prepared as described above) with Trizol LS (as per the manufacturer's directions; Invitrogen, Carlsbad, CA), followed by alcohol precipitation. RNA was quantitated using a NanoDrop 1000 Spectrophotometer (Thermo Fisher Scientific Inc., Waltham, MA). cDNA was synthesized using primer 200R (5′-CAAGAAAGGACCCGGTCGTC-3′) and AffinittyScript Multiple Temperature Reverse Transcriptase (Stratagene, La Jolla, CA) for one hour at 42°C followed by 15 minutes at 70°C. cDNA samples were tested in triplicate in a 25 µl reaction. Reactions contained 5 µl cDNA, Premix Ex Taq reaction mixture (Clontech Laboratories, Inc., Madison, WI), 300 nM each of primers 124S (5′-CCCTCCCGGGAGAGCCATAG-3′) and 200R, and 200 nM of probe (6FAM - 5′-TCTGCGGAACCGGTGAGTACACC-3′-TAMRA, Applied Biosystems, Foster City, CA). Real-time PCR analysis was performed in an ABI 7300 Sequence Detection System as follows: 1 minute at 95°C, 5 cycles with 20 seconds at 95°C and 1 minute at 60°C, 40 cycles with 20 seconds at 95°C, 30 seconds at 60°C and 31 seconds at 72°C.
RNA standards, run in triplicate, were prepared as described previously [61].
Determination of HCV RNA titers in cell extracts
Total cellular RNA was extracted from approximately 106 infected Huh7.5 or VeroE6 cells, using the RNeasy minikit, following the manufacturer's recommendations (Qiagen, Valencia, CA), and quantitated using a NanoDrop 1000 Spectrophotometer (Thermo Fisher Scientific Inc., Waltham, MA). RNA was analyzed by RT-PCR using primers in the HCV 5′ end extending to the Core area [62]. Determination of HCV RNA titers in cells infected with genotype 1b plasma (LB) was performed by real-time RT-PCR analysis as described previously [63]. HCV RNA titers in J6/JFH-1 infected cells were quantitated following identical procedures as for LB, but using a fluorescent probe (FAM-labeled) coding for nucleotides 335–358 designed from published sequences [63]. Relative quantitation of HCV RNA was performed with the Comparative CT Method, using glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as endogenous control, following the manufacturer's protocols and recommendations (Applied Biosystems, Foster City, CA).
Inhibition of infection by anti-CD81 antibodies
Huh7.5 cells were plated at a density of 5×103 per well in 96-well plates to obtain 60% confluence after 24 hr. Cells were incubated with anti-CD81 (BD Pharmingen, San Diego, CA) or isotype-matched control anti-flag M2 (Sigma, St. Louis, MO) antibodies for 1 hr at 37°C.and subsequently infected with serial dilutions of J6/JFH-1 or LB-piVe filter-clarified supernatants. After 6 hr at 37°C, cells were washed and supplemented with fresh media. Three dpi, J6/JFH-1-infected cells were immunostained with anti-Core antibodies [59]. Wells were scored positive if at least 1 positive cell was detected. LB-piVe-infected cells were observed by light microscopy at 5 dpi. Wells showing CPE were assigned a positive result and titers were calculated as described above.
Neutralization of LB-piVe by anti-E2 antibodies and HCV-specific immunoglobulins (HCIGIV)
8×102 TCID50 LB-piVe were treated for 1hr at 37°C with 5-fold dilutions of a cocktail of anti-E2 monoclonal antibodies [36] or 5µg/ml of isotype-matched control anti-flag M2 (Sigma, St. Louis, MO) antibody. The anti-E2 monoclonal antibodies were produced by hybridomas obtained after immunization of BALB/c mice with E1 and E2 glycoproteins expressed in insect cells [36]. Huh7.5 cells growing in 96-well plates were inoculated with serial dilutions of the neutralization reaction products and incubated for 5 days. LB-piVe titers were determined by a CPE-based end-point dilution assay. To test LB-piVe virus neutralization by immunoglobulins prepared from human plasma, virus was incubated with HCIGIV [35] or HCV-negative IGIV [35], before titrating on naïve Huh7.5 cells.
Inhibition of viral replication with 2′-C-Methyl-D-Adenosine
2′-C-Methyl-D-Adenosine (2′-C-Me-A) was obtained from Carbosynth Ltd. (Berkshire, UK) and resuspended at 100 mM [37] in dimethylsulfoxide (DMSO). 5×103 Huh7.5 cells were infected with filter-clarified supernatants containing 100 FFU of J6/JFH-1 for 12 hours, washed, and incubated with complete growth medium containing a range of 0.05 to 1 µM 2′-C-Me-A. Three dpi, J6/JFH-1 infected cells were immunostained with anti-Core antibodies [59]. Fluorescent foci were counted in triplicate wells, and titers were calculated as the mean number of foci per ml (FFU/ml). To measure inhibition of LB-piVe growth, 5×105 LB-piVe cells were grown in T25 flasks. After 3 days, 2′-C-Me-A was added to the growth media and cells were incubated for 3 additional days. Filter-clarified culture supernatants from treated LB-piVe cells were titrated in a CPE-based end-point dilution assay as described above. HCV growth in the absence of 2′-C-Me-A was set at 100%. The percentage reduction in the inhibitor treated cells relative to the untreated control was plotted against 2′-C-Me-A concentrations, employing GraphPad Prism 3.0 software. 50% effective concentration (EC50) value values were interpolated from the resulting curves. To measure the level of viral RNA in the presence of 2′-C-Me-A, Vero E6 cells growing in 6-well plates were transfected with either WT - or mutant-VA RNAI. Four hours post-transfection, cells were treated with media containing 1 µM 2′-C-Me-A overnight and then infected with the parental LB virus. Total cellular RNA was extracted at 0, 2, 4, 6 and 8 days post-infection. Determination of HCV RNA titers was performed by real-time RT-PCR analysis as described previously [62].
Inhibition of viral replication by HCV-specific siRNA
A chemically synthesized irrelevant oligo, termed siIRR [64] [5′-AAGGACUUCCAGAAGAACAUCTT-3′] and an HCV-specific oligo, termed si313 [38] [5′-CCCGGGAGGUCUCGUAGACTT-3′ ], were obtained from Dharmacon, Lafayette, CO. Huh7.5 cells (2×104) were transfected with 100 nM of siIRR or si313 using DharmaFECT Transfection reagent 1 (Dharmacon, Lafayette, CO) following the manufacturer's protocol and recommendations. One day after siRNA transfection, cells were infected with 200 FFU of J6/JFH-1 or 200 TCID50 LB-piVe. Three dpi, J6/JFH-1 infected cells were washed, fixed and stained using an indirect immunofluorescence assay with anti-Core antibodies [59]. Fluorescent foci were counted in triplicate wells, and titers were calculated as the mean number of foci per ml (FFU/ml). LB-piVe infected cells were detected by observing CPE by light microscopy. Culture supernatants of LB-piVe infected cells were titrated by a CPE-based end-point dilution assay on naïve Huh7.5 cells as described above. Viral titers were calculated using the method of Reed and Muench [31].
Inhibition of viral replication by IFN
LB-piVe cells were grown in T25 flasks at a density to provide an overnight confluence of 40%, and transfected with pVA. One day post-transfection, cells were treated with 0, 10, 100 and 1000 IU/ml of Universal Type 1 IFN (PBL Interferon Source, Piscataway, NJ) for 24, 48 and 72 hr. Filter-clarified culture supernatants were prepared and titrated by a CPE-based end-point dilution assay on naïve Huh7.5 cells as described above. Viral titers were calculated using the method of Reed and Muench [31]. Alternatively, LB-piVe titers were determined in naïve Huh7.5 in the presence of 0, 10, and 1000 IU/ml of IFN.
Effect of VA RNAI on J6/JFH-1
To show the effect of VA RNAI on J6/JFH-1, Huh7.5 cells were transfected with plasmid vectors pVA or pVAdl [26], [27], and infected the following day with 105 TCID50. J6/JFH-1 (filter-clarified culture supernatants from Huh7.5 cells that were transfected with wild-type or mutant VA RNAI plasmids). The infected Huh7.5 cells were titrated by end-point dilution in 96-well plates as described above. Wells were scored positive if at least 1 positive cell was detected. The TCID50 was calculated using the method of Reed and Muench [31].
Accession numbers
Sequences can be accessed from GenBank through the NCBI website: H77 (AF009606); JFH-1 (AB047639); adenovirus 2 (AC000007); LB-piVe (FJ976045, FJ976046 and FJ976047).
Supporting Information
Zdroje
1. SimmondsP
BukhJ
CombetC
DeleageG
EnomotoN
2005 Consensus proposals for a unified system of nomenclature of hepatitis C virus genotypes. Hepatology 42 962 973
2. LindenbachBD
EvansMJ
SyderAJ
WolkB
TellinghuisenTL
2005 Complete replication of hepatitis C virus in cell culture. Science 309 623 626
3. WakitaT
PietschmannT
KatoT
DateT
MiyamotoM
2005 Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med 11 91 96
4. HellerT
SaitoS
AuerbachJ
WilliamsT
MoreenTR
2005 An in vitro model of hepatitis C virion production. Proc Natl Acad Sci USA 102 2579 2583
5. LindenbachBD
PlossA
VanwolleghemT
SyderAJ
McKeatingJA
2006 Cell culture-grown hepatitis C virus is infectious in vivo and can be recultured in vitro. Proc Natl Acad Sci USA 103 3805 3809
6. ZhongJ
GastaminzaP
ChengG
KapadiaS
KatoT
2005 Robust hepatitis C virus infection in vitro. Proc Natl Acad Sci U S A 102 9294 9299
7. SumpterRJr
LooY
FoyE
LiK
YoneyamaM
2005 Regulating intracellular antiviral defense and permissiveness to hepatitis C virus RNA replication through a cellular RNA helicase, RIG-I. J Virol 79 2689 2699
8. GuoJ
YanR
XuG
LiW
ZhengC
2009 Construction of the Vero cell culture system that can produce infectious HCV particles. Mol Biol Rep 36 111 120
9. PietschmannT
KaulA
KoutsoudakisG
ShavinskayaA
KallisS
2006 Construction and characterization of infectious intragenotypic and intergenotypic hepatitis C virus chimeras. Proc Natl Acad Sci U S A 103 7408 7413
10. YiM
VillanuevaRA
ThomasDL
WakitaT
LemonSM
2006 Production of infectious genotype 1a hepatitis C virus (Hutchinson strain) in cultured human hepatoma cells. Proc Natl Acad Sci USA 103 2310 2315
11. GottweinJM
ScheelTK
HoeghAM
LademannJB
Eugen-OlsenJ
2007 Robust hepatitis C genotype 3a cell culture releasing adapted intergenotypic 3a/2a (S52/JFH1) viruses. Gastroenterology 133 1614 1626
12. JensenTB
GottweinJM
ScheelTK
HoeghAM
Eugen-OlsenJ
2008 Highly efficient JFH1-based cell-culture system for hepatitis C virus genotype 5a: failure of homologous neutralizing-antibody treatment to control infection. J Infect Dis 198 1756 1765
13. ScheelTK
GottweinJM
JensenTB
PrentoeJC
HoeghAM
2008 Development of JFH1-based cell culture systems for hepatitis C virus genotype 4a and evidence for cross-genotype neutralization. Proc Natl Acad Sci U S A 105 997 1002
14. GottweinJM
ScheelTK
JensenTB
LademannJB
PrentoeJC
2009 Development and characterization of hepatitis C virus genotype 1–7 cell culture systems: role of CD81 and scavenger receptor class B type I and effect of antiviral drugs. Hepatology 49 364 377
15. MateuG
DonisRO
WakitaT
BukhJ
GrakouiA
2008 Intragenotypic JFH1 based recombinant hepatitis C virus produces high levels of infectious particles but causes increased cell death. Virology 376 397 407
16. PietschmannT
ZayasM
MeulemanP
LongG
AppelN
2009 Production of infectious genotype 1b virus particles in cell culture and impairment by replication enhancing mutations. PLoS Pathog 5 e1000475
17. AlyHH
WatashiK
HijikataM
KanekoH
TakadaY
2007 Serum-derived hepatitis C virus infectivity in interferon regulatory factor-7-suppressed human primary hepatocytes. J Hepatol 46 26 36
18. FarquharMJ
McKeatingJA
2008 Primary hepatocytes as targets for hepatitis C virus replication. J Viral Hepat 15 849 854
19. JacobsBL
LanglandJO
1996 When two strands are better than one: the mediators and modulators of the cellular responses to double-stranded RNA. Virology 219 339 349
20. TaylorDR
PuigM
DarnellME
MihalikK
FeinstoneSM
2005 New antiviral pathway that mediates hepatitis C virus replicon interferon sensitivity through ADAR1. J Virol 79 6291 6298
21. MorseDP
BassBL
1997 Detection of inosine in messenger RNA by inosine-specific cleavage. Biochemistry 36 8429 8434
22. O'MalleyRP
MarianoTM
SiekierkaJ
MathewsMB
1986 A mechanism for the control of protein synthesis by adenovirus VA RNAI. Cell 44 391 400
23. LeiM
LiuY
SamuelCE
1998 Adenovirus VAI RNA antagonizes the RNA-editing activity of the ADAR adenosine deaminase. Virology 245 188 196
24. DiazMO
ZieminS
Le BeauMM
PithaP
SmithSD
1988 Homozygous deletion of the alpha - and beta 1-interferon genes in human leukemia and derived cell lines. Proc Natl Acad Sci U S A 85 5259 5263
25. MoscaJD
PithaPM
1986 Transcriptional and posttranscriptional regulation of exogenous human beta interferon gene in simian cells defective in interferon synthesis. Mol Cell Biol 6 2279 2283
26. ClarkePA
Pe'eryT
MaY
MathewsMB
1994 Structural features of adenovirus 2 virus-associated RNA required for binding to the protein kinase DAI. Nucleic Acids Res 22 4364 4374
27. GunneryS
MathewsMB
1995 Functional mRNA can be generated by RNA polymerase III. Mol Cell Biol 15 3597 3607
28. SaldanhaJ
HeathA
LelieN
PisaniG
NublingM
2000 Calibration of HCV working reagents for NAT assays against the HCV international standard. The Collaborative Study Group. Vox Sang 78 217 224
29. YuMW
FarshidM
MasonBL
TanD
KochWH
1998 Formulation of a hepatitis C virus RNA panel for standardization of nucleic acid testing of blood, plasma, and their derived products. Hepatology 28 566A
30. KolykhalovAA
AgapovEV
BlightKJ
MihalikK
FeinstoneSM
1997 Transmission of hepatitis C by intrahepatic inoculation with transcribed RNA. Science 277 570 574
31. ReedLJ
MuenchH
1938 A simple method of estimating fifty percent end points. Am J Hyg 27 493 497
32. CormierEG
TsamisF
KajumoF
DursoRJ
GardnerJP
2004 CD81 is an entry coreceptor for hepatitis C virus. Proc Natl Acad Sci U S A 101 7270 7274
33. FlintM
Loomis-PriceLD
ShottonC
DubuissonJ
MonkP
1999 Characterization of hepatitis C virus E2 glycoprotein interaction with a putative cellular receptor, CD81. J Virol 73 6235 6244
34. PileriP
UematsuY
CampagnoliS
GalliG
FalugiF
1998 Binding of hepatitis C virus to CD81. Science 282 938 941
35. YuMY
BartoschB
ZhangP
GuoZP
RenziPM
2004 Neutralizing antibodies to hepatitis C virus (HCV) in immune globulins derived from anti-HCV-positive plasma. Proc Natl Acad Sci U S A 101 7705 7710
36. DubuissonJ
HsuHH
CheungRC
GreenbergHB
RussellDG
1994 Formation and intracellular localization of hepatitis C virus envelope glycoprotein complexes expressed by recombinant vaccinia and Sindbis viruses. J Virol 68 6147 6160
37. Le PogamS
JiangWR
LevequeV
RajyaguruS
MaH
2006 In vitro selected Con1 subgenomic replicons resistant to 2′-C-methyl-cytidine or to R1479 show lack of cross resistance. Virology 351 349 359
38. ChevalierC
SaulnierA
BenureauY
FlechetD
DelgrangeD
2007 Inhibition of hepatitis C virus infection in cell culture by small interfering RNAs. Mol Ther 15 1452 1462
39. GermiR
CranceJM
GarinD
GuimetJ
Lortat-JacobH
2002 Cellular glycosaminoglycans and low density lipoprotein receptor are involved in hepatitis C virus adsorption. J Med Virol 68 206 215
40. DiamondDL
SyderAJ
JacobsJM
SorensenCM
WaltersKA
2010 Temporal proteome and lipidome profiles reveal hepatitis C virus-associated reprogramming of hepatocellular metabolism and bioenergetics. PLoS Pathog 6 e1000719
41. PetrikJ
ParkerH
AlexanderGJ
1999 Human hepatic glyceraldehyde-3-phosphate dehydrogenase binds to the poly(U) tract of the 3′ non-coding region of hepatitis C virus genomic RNA. J Gen Virol 80 (Pt 12) 3109 3113
42. RomanowskiT
SikorskaK
BielawskiKP
2008 GUS and PMM1 as suitable reference genes for gene expression analysis in the liver tissue of patients with chronic hepatitis. Med Sci Monit 14 BR147 BR152
43. WaxmanS
WurmbachE
2007 De-regulation of common housekeeping genes in hepatocellular carcinoma. BMC Genomics 8 243
44. von HahnT
RiceCM
2008 Hepatitis C virus entry. J Biol Chem 283 3689 3693
45. IroM
WitteveldtJ
AngusAG
WoerzI
KaulA
2009 A reporter cell line for rapid and sensitive evaluation of hepatitis C virus infectivity and replication. Antiviral Res
46. WaltersKA
SyderAJ
LedererSL
DiamondDL
PaeperB
2009 Genomic analysis reveals a potential role for cell cycle perturbation in HCV-mediated apoptosis of cultured hepatocytes. PLoS Pathog 5 e1000269
47. SaitoT
OwenDM
JiangF
MarcotrigianoJ
GaleMJr
2008 Innate immunity induced by composition-dependent RIG-I recognition of hepatitis C virus RNA. Nature 454 523 527
48. SaundersLR
BarberGN
2003 The dsRNA binding protein family: critical roles, diverse cellular functions. FASEB J 17 961 983
49. TaylorDR
LeeSB
RomanoPR
MarshakDR
HinnebuschAG
1996 Autophosphorylation sites participate in the activation of the double-stranded-RNA-activated protein kinase PKR. Mol Cell Biol 16 6295 6302
50. ClemensMJ
1997 PKR–a protein kinase regulated by double-stranded RNA. Int J Biochem Cell Biol 29 945 949
51. ScaddenAD
SmithCW
1997 A ribonuclease specific for inosine-containing RNA: a potential role in antiviral defence? EMBO J 16 2140 2149
52. ScaddenAD
SmithCW
2001 Specific cleavage of hyper-edited dsRNAs. EMBO J 20 4243 4252
53. EspertL
DegolsG
GongoraC
BlondelD
WilliamsBR
2003 ISG20, a new interferon-induced RNase specific for single-stranded RNA, defines an alternative antiviral pathway against RNA genomic viruses. J Biol Chem 278 16151 16158
54. TaylorDR
SilbersteinE
2009 Innate immunity and hepatitis C virus: eluding the host cell defense. Front Biosci 14 4950 4961
55. PawlotskyJM
2003 Mechanisms of antiviral treatment efficacy and failure in chronic hepatitis C. Antiviral Res 59 1 11
56. GaleMJJr
KorthMJ
TangNM
TanSL
HopkinsDA
1997 Evidence that hepatitis C virus resistance to interferon is mediated through repression of the PKR protein kinase by the nonstructural 5A protein. Virology 230 217 227
57. TaylorDR
ShiST
RomanoPR
BarberGN
LaiMM
1999 Inhibition of the interferon-inducible protein kinase PKR by HCV E2 protein. Science 285 107 110
58. VyasJ
EliaA
ClemensMJ
2003 Inhibition of the protein kinase PKR by the internal ribosome entry site of hepatitis C virus genomic RNA. RNA 9 858 870
59. ZhangP
WuCG
MihalikK
Virata-TheimerML
YuMY
2007 Hepatitis C virus epitope-specific neutralizing antibodies in Igs prepared from human plasma. Proc Natl Acad Sci U S A 104 8449 8454
60. LaemmliUK
1970 Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227 680 685
61. MajorME
MihalikK
FernandezJ
SeidmanJ
KleinerD
1999 Long-term follow-up of chimpanzees inoculated with the first infectious clone for hepatitis C virus. J Virol 73 3317 3325
62. PuigM
MihalikK
YuMY
FeinstoneSM
MajorME
2002 Sensitivity and reproducibility of HCV quantitation in chimpanzee sera using TaqMan real-time PCR assay. J Virol Methods 105 253 263
63. OkamotoH
OkadaS
SugiyamaY
KuraiK
IizukaH
1991 Nucleotide sequence of the genomic RNA of hepatitis C virus isolated from a human carrier: comparison with reported isolates for conserved and divergent regions. J Gen Virol 72 (Pt 11) 2697 2704
64. RandallG
GrakouiA
RiceCM
2003 Clearance of replicating hepatitis C virus replicon RNAs in cell culture by small interfering RNAs. Proc Natl Acad Sci U S A 100 235 240
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2010 Číslo 5
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Měli bychom postcovidový syndrom léčit antidepresivy?
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- Quorum Sensing Inhibition Selects for Virulence and Cooperation in
- The Role of Intestinal Microbiota in the Development and Severity of Chemotherapy-Induced Mucositis
- Crystal Structure of HIV-1 gp41 Including Both Fusion Peptide and Membrane Proximal External Regions
- Susceptibility to Anthrax Lethal Toxin-Induced Rat Death Is Controlled by a Single Chromosome 10 Locus That Includes