
Pathogen Specific, IRF3-Dependent Signaling and Innate Resistance to Human Kidney Infection
The mucosal immune system identifies and fights invading pathogens, while allowing non-pathogenic organisms to persist. Mechanisms of pathogen/non-pathogen discrimination are poorly understood, as is the contribution of human genetic variation in disease susceptibility. We describe here a new, IRF3-dependent signaling pathway that is critical for distinguishing pathogens from normal flora at the mucosal barrier. Following uropathogenic E. coli infection, Irf3−/− mice showed a pathogen-specific increase in acute mortality, bacterial burden, abscess formation and renal damage compared to wild type mice. TLR4 signaling was initiated after ceramide release from glycosphingolipid receptors, through TRAM, CREB, Fos and Jun phosphorylation and p38 MAPK-dependent mechanisms, resulting in nuclear translocation of IRF3 and activation of IRF3/IFNβ-dependent antibacterial effector mechanisms. This TLR4/IRF3 pathway of pathogen discrimination was activated by ceramide and by P-fimbriated E. coli, which use ceramide-anchored glycosphingolipid receptors. Relevance of this pathway for human disease was supported by polymorphic IRF3 promoter sequences, differing between children with severe, symptomatic kidney infection and children who were asymptomatic bacterial carriers. IRF3 promoter activity was reduced by the disease-associated genotype, consistent with the pathology in Irf3−/− mice. Host susceptibility to common infections like UTI may thus be strongly influenced by single gene modifications affecting the innate immune response.
Published in the journal:
. PLoS Pathog 6(9): e32767. doi:10.1371/journal.ppat.1001109
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1001109
Summary
The mucosal immune system identifies and fights invading pathogens, while allowing non-pathogenic organisms to persist. Mechanisms of pathogen/non-pathogen discrimination are poorly understood, as is the contribution of human genetic variation in disease susceptibility. We describe here a new, IRF3-dependent signaling pathway that is critical for distinguishing pathogens from normal flora at the mucosal barrier. Following uropathogenic E. coli infection, Irf3−/− mice showed a pathogen-specific increase in acute mortality, bacterial burden, abscess formation and renal damage compared to wild type mice. TLR4 signaling was initiated after ceramide release from glycosphingolipid receptors, through TRAM, CREB, Fos and Jun phosphorylation and p38 MAPK-dependent mechanisms, resulting in nuclear translocation of IRF3 and activation of IRF3/IFNβ-dependent antibacterial effector mechanisms. This TLR4/IRF3 pathway of pathogen discrimination was activated by ceramide and by P-fimbriated E. coli, which use ceramide-anchored glycosphingolipid receptors. Relevance of this pathway for human disease was supported by polymorphic IRF3 promoter sequences, differing between children with severe, symptomatic kidney infection and children who were asymptomatic bacterial carriers. IRF3 promoter activity was reduced by the disease-associated genotype, consistent with the pathology in Irf3−/− mice. Host susceptibility to common infections like UTI may thus be strongly influenced by single gene modifications affecting the innate immune response.
Introduction
Despite significant advances in the understanding of genetic variation, common infections are often regarded as too complex for genetic analysis. While single gene defects have a major impact on host susceptibility to classic infections like malaria [1], the extent to which susceptibility to diarrhea, respiratory tract and urinary tract infection (UTI) is genetically controlled remains unclear. Critical to the understanding of host resistance and genetic control is the mucosal route of these infections and the molecular interactions through which mucosal tissues are perturbed. UTI serve as a particularly useful model to identify genetic variants contributing to host susceptibility, as innate immunity controls the antimicrobial defense and molecular mechanisms of host parasite interaction are understood in great detail [2], [3]. The disease response to uropathogenic Escherichia coli is initiated through fimbriae-mediated adherence, and the expression of P fimbriae distinguishes the pathogenic strains from non-virulent bacteria, which colonize the same mucosal sites.
TLRs control the survival of complex organisms by balancing protective against destructive forces of innate immunity. During infection, each TLR recognizes a relatively small number of ligands, including conserved microbial patterns (PAMPs) [4]. The horseshoe-shaped, leucine-rich, extracellular TLR domain and its co-receptors are involved in recognition of proteins, as well as lipids, carbohydrates and nucleic acids [5], [6], [7]. At mucosal sites, where the bulk of microbial challenge occurs, PAMP recognition is non-functional, however, and does not explain how mucosal TLRs distinguish pathogenic microbes from members of the normal flora [8]. Pathogen-specific TLR responses to mucosal pathogens require receptors that exclusively engage virulence ligands and signaling pathways that activate a pathogen-specific defense [8]. For example, uropathogenic E. coli adhere to mucosa via glycosphingolipid receptors for P fimbriae, thereby activating a TLR4-dependent but LPS/CD14-independent innate immune response in epithelial cells [9].
Signaling through cell surface sphingolipids involves ceramide, the membrane anchor and a ubiquitous component of cell membranes [10], [11]. The generation of ceramide within rafts alters their biophysical properties and results in the formation of large ceramide-enriched membrane platforms, clustering receptor molecules and facilitating signal transduction following receptor stimulation [12]. Endogenous SMases, activated by many infectious agents, cleave ceramide from the extracellular choline-rich domain of sphingomyelin [13], [14], [15], [16], [17] and activate the “ceramide-signaling pathway”, which is conserved from yeast to humans [18]. In addition, pathogens that utilize the extracellular domain of glycosphingolipids as receptors may release ceramide after bacterial binding, as first described for P-fimbriated, uropathogenic E. coli [9], [17], [19]. Ceramide activates a TLR4-dependent innate immune response [17], similar to infection-mediated activation, and we have proposed that ceramide acts as a signaling intermediate between the pathogen-specific receptors and TLR4 [9], [17], [19]. The molecular mechanisms in this important signal transduction need to be identified, however.
UTIs affect >150 million adults each year and about 5% of children <12 years of age. Severe kidney infections like acute pyelonephritis (APN) are accompanied by life-threatening urosepsis in about 30% of adults. Children may develop renal scars, which are associated with long-term morbidity including hypertension, complications of pregnancy, and renal failure if scarring is extensive. Despite the urgent need, no tools exist at present to identify children at risk of developing recurrent acute pyelonephritis and renal scarring. Host resistance to UTI is controlled by the innate immune system, through Toll-like receptor (TLR) activation [8], [9], [20]. Previous studies have shown that TLR4-deficient mice develop asymptomatic carriage rather than severe disease [8], [20], [21], suggesting that disturbances in TLR4 signaling may alter the innate immune dependent host defense [22].
This study examined TLR4 activation by ceramide and P fimbriated E. coli and characterized this signaling pathway. We propose that ceramide interacts directly with TLR4, activates TRAM phosphorylation followed by nuclear translocation of IRF3. Furthermore, we show that IRF3 dependent innate immunity is essential for the host defense, as Irf3 knockout mice develop severe kidney infection. Finally, we show that IRF3 promoter polymorphisms are more common in APN prone patients than in those who become asymptomatic bacterial carriers. We propose that this pathway offers a model of how TLR4 may distinguish pathogens from commensals at the mucosal level, through modification of pathogen recognition receptors, adaptors and transcription factors.
Results
Increased susceptibility to acute pyelonephritis in Irf3−/− mice
In a genetic screen of innate immune effector genes downstream of TLR4, we identified IRF3 as a major determinant of host susceptibility. Irf3−/− and Irf3+/+ mice were infected via the urinary tract mucosal route with the uropathogenic E. coli strain CFT073 [23]. The Irf3−/− mice developed more severe disease than wild type (wt) Irf3+/+ mice. Acute mortality was higher (50% after 24 hours) and bacterial clearance was significantly impaired, with higher bacterial counts in urine, kidneys and bladders (Figure 1A, p<0.001). Abscess formation was also more extensive in Irf3−/− than in wt mice (day 7 post-infection, Figure 1B–C, p<0.001). In Irf3−/− mice, abscesses were diffuse, destroying large tissue areas while in wt mice abscesses were morphologically distinct from surrounding healthy tissue (Figure 1B).

Renal abscess formation is caused, in part, by an imbalance between neutrophil recruitment and exit from the tissues [24], [25]. The kinetics of early neutrophil recruitment did not differ between wild type and Irf3−/− mice (Figure 1A), but later, neutrophil recruitment subsided in wt mice but remained elevated in Irf3−/− mice. In tissue sections from Irf3−/− mice, neutrophils were detected throughout the abscesses and P-fimbriated bacteria were interspersed among the neutrophils, as shown by PapG adhesin-specific antibody detection (Figure 1D, for negative control, see Figure S1). Wt mice, in contrast, had discrete, neutrophil aggregates with fewer bacteria. The results suggest that Irf3 is essential for a functioning innate immune defense against UTI, to maintain tissue integrity and to clear mucosal E. coli infection.
To examine if the IRF3-dependent immune response discriminates uropathogenic E. coli from non-pathogenic bacteria, we inoculated wt and Irf3−/− mice with the prototypical asymptomatic bacteriuria strain E. coli 83972, which lacks functional UTI-associated virulence factors, including P fimbriae [26], [27], [28], [29]. Both wt and Irf3−/− mice cleared infection rapidly, with no difference in bacterial counts (Figure 1E) and no significant neutrophil recruitment (data not shown). As P fimbriae are essential virulence factors, present in up to 100% of E. coli strains causing urosepsis [30], [31], we subsequently examined if P fimbriae activate the IRF3 pathway. The asymptomatic carrier strain E. coli 83972 was transformed with a chromosomal copy of the pap gene cluster. We compared disease severity and bacterial counts between wt and Irf3−/− mice infected with E. coli 83972pap. The Irf3−/− mice developed acute, symptomatic disease with sepsis and had dramatically increased bacterial numbers in bladders, kidneys and spleens (Figure 1E, p<0.05), compared to wt mice, which were resistant to infection with E. coli 83972pap.
The results show that the IRF3-dependent response distinguishes pathogenic E. coli from non-pathogenic strains and suggest that the expression of a single virulence factor like P fimbriae enables the host to recognize a potential pathogen and to activate this response.
Ceramide/TLR4 interactions and TRAM phosphorylation
P fimbriae bind to glycosphingolipid receptors and trigger ceramide release [9]. To investigate the mechanism of pathogen-specific TLR4/IRF3 signaling activation, we examined if ceramide and TLR4 interact after ceramide release from membrane glycosphingolipids. We treated A498 kidney epithelial cells with sphingomyelinase (SMase) for one hour, to release ceramide (r-ceramide) from the extracellular phosphocholine domain of sphingomyelin [32] (Figure 2A–D). We labeled TLR4 and native ceramide with specific primary antibodies followed by Alexa fluor-488 (donor) and Alexa fluor-568 (acceptor)-labeled secondary antibodies, respectively. In unstimulated cells (no SMase treatment), where ceramide remains bound to sphingomyelin, we detected no FRET signal. After SMase treatment, we recorded a significant FRET signal (Figure 2A–D, 50% FRET-positive cells compared to 8% for unstimulated cells, p<0.05), with most of the FRET-positive regions localized in the plasma membrane. LPS and soluble CD14 (sCD14) stimulation, in contrast, did not stimulate a FRET signal above background (p>0.05, compared to unstimulated cells). sCD14 was used, as the uroepithelial cells lack membrane-bound CD14 and respond poorly to LPS [33]. These results suggest that ceramide interacts with TLR4 after release from membrane glycosphingolipids.

To examine the ceramide-induced TLR4 signaling pathway, we used RNA interference to suppress specific genes (Figure 2E, siRNA used for transfection are listed in Table S1 in Supporting Information S1; for knockdown efficiency compared to control cells transfected with irrelevant siRNA, see Figure S2). First, suppression of TLR4 expression abrogated the innate immune response to r-ceramide (p<0.001), confirming that this pathway is TLR4 dependent. Secondly, TRAM siRNA inhibited the responses to r-ceramide (p<0.05 compared to the siRNA control). MyD88-specific siRNA did not alter the ceramide response (p>0.05 compared to the siRNA control) but did reduce the response to LPS+sCD14 (p<0.05), as did TLR4 - and TRAM-specific siRNAs.
To further investigate ceramide-induced TLR4 signaling, TRAM phosphorylation (TRAM-P) was quantified by confocal microscopy, using polyclonal phospho-specific anti-TRAM antibodies (Figure 2F–G and Figure S3). We detected an increase in TRAM-P staining in cells exposed to r-ceramide or exogenous, water-soluble C6 ceramide; staining had a granular appearance and was most intense in the perinuclear area. By Western blot analysis (Figure 2H), a band corresponding to TRAM-P was increased in cells exposed to C6 and r-ceramide compared to unstimulated cells but total TRAM levels were not altered. LPS+sCD14 triggered weaker TRAM phosphorylation, as shown by confocal microscopy (p<0.001 compared to r-ceramide) and by Western blot. The results indicate that ceramide triggers TRAM phosphorylation more efficiently than LPS+sCD14. As TRAM phosphorylation was virtually absent in unstimulated cells, this pathway may need to be activated by exogenous or endogenous stimuli.
Kinase phosphorylation downstream of ceramide/TLR4
To define signaling downstream of ceramide/TLR4 and TRAM, we examined kinase phosphorylation, using phosphoarrays specific for 46 protein kinases and substrates (Figure 3A). Ceramide release stimulated the phosphorylation of twelve protein kinases: p27T198, eNOS, CREB, Fyn (all 2.3-fold), Hck, PLCγ1, Jun (all 2.1-fold), Pyk2 (2-fold), ERK1/2 and Src (1.9-fold), RSK1/2/3 (1.8-fold), p27T157 (1.7-fold), and p53 (1.6-fold). Antibacterial effectors included eNOS, which regulates nitric oxide and related antibacterial effector functions [34] and Hck, a Src-family tyrosine kinase associated with secretory lysosomes in neutrophils and phagosome-lysosome fusion [35]. A number of the significantly phosphorylated proteins activate IRF3 - and AP1-dependent transcription. PLCγ1 catalyzes the formation of inositol 1,4,5-trisphosphate and diacylglycerol from phosphatidylinositol 4,5-bisphosphate, leading to PKC activation and CREB (cAMP response element binding) phosphorylation [36], [37]. CREB is then phosphorylated and binds to CBP (CREB-binding protein), which preferentially associates with phosphorylated IRF3 [38], [39], leading to IRF3. Fyn is a Src family tyrosine kinase implicated in the activation of PKA, a protein kinase involved in CREB phosphorylation [40]. Jun in combination with Fos bind to and are a part of the AP-1 transcription factor complex [41], which induces the transcription of proinflammatory cytokines. Pyk2 activation is highly correlated with the stimulation of c-Jun N-terminal kinase (JNK). Identified phosphorylation targets also included ERKs (ERK1/2, extracellular signal-regulated kinases) which activate downstream protein kinases and transcription factors, including IRF3 and AP-1 [42].

CREB phosphorylation in r-ceramide-activated cells was confirmed by confocal microscopy (Figure 3D, E, p<0.001 compared to control), but was not detected in LPS-stimulated cells. We obtained similar results using antibodies specific for phosphorylated Fos (Figure 3D, E). JNK phosphorylation, in contrast, was similar after r-ceramide and LPS+sCD14 stimulation (Figure 3D, E), suggesting that JNK signaling was not ceramide-specific (p<0.001 compared to the control). The results suggest that ceramide-induced TLR4 signaling causes rapid phosphorylation/transcription of proteins involved in IRF3 and AP-1 transcription, including CREB, Fyn, PLCγ, MAP kinases, ERK1/2 and Fos/Jun (Figure S4). LPS+sCD14, in contrast, caused a weaker phosphorylation response, comprising p27T198 (2-fold), eNOS (1.8-fold), PLCγ1 (1.7-fold), Pyk2 (1.7-fold), and Jun (1.6-fold), but not the remaining targets that were phosphorylated in response to r-ceramide (Figure 3A).
Transcriptional activation in response to ceramide
Innate immune activation in response to ceramide was further examined by TLR SuperArrays and compared to LPS+sCD14 (Figure 3B, Figure S5). After one hour, five genes in A549 cells had responded to r-ceramide: Fos (6.5-fold) and Jun (2.1-fold), IL-8 (4.4-fold), IL-6 (2.9-fold) and IL-1α (2-fold). The response showed a similar, restricted repertoire in A498 carcinoma cells after three hours (Figure 3C). r-Ceramide upregulated TRAM, Fos and Jun transcription (10.2-, 2.2 - and 2.4-fold, respectively). Ceramide activated IL-6 transcription (6.2-fold), MAP3K1 and MAP2K3 (5.9 - and 2.5-fold), as well as IL-8 and CSF2 transcription levels (about 3-fold). In contrast, LPS+sCD14 did not significantly stimulate Fos (1.5-fold), Jun (1.2-fold) or IL-1α after one hour. After three hours, only IL-8 transcription was higher in response to LPS+sCD14 than to r-ceramide. The transcriptional profile confirmed the difference between ceramide and LPS+sCD14 activated cells, consistent with a different transcription factor usage.
IRF3 translocation to the nucleus in response to ceramide/TLR4
IRF3 is an interferon regulatory transcription factor and following TLR4 activation, phosphorylated IRF3 homodimers translocate from the cytosol to the nucleus [43], [44], [45], [46]. By confocal microscopy (Figure 4A, C) we observed that r-ceramide triggered IRF3 translocation to the nucleus (p<0.001 compared to unstimulated cells, 90 min). We confirmed the results in a human bladder epithelial cell line (J82, Figure S6A) in which ceramide release caused rapid IRF3 translocation. In cells exposed to LPS+sCD14, the nuclear IRF3 translocation was weak (p>0.05 compared to control) and fewer dimers were formed after exposure to LPS+sCD14. In the bladder epithelial cells, the IRF3 response to LPS+sCD14 or LPS alone was low. LPS+sCD14-induced NF-κB p65 translocation, but the NF-κB response to r-ceramide or C6 ceramide was weak (Figure 4 B, D). For a broader field of view see Figure S6B.

Signaling through p38 MAPK has previously been shown to stimulate proinflammatory responses, including IL-8, IL-6 and TNF [47], [48], [49]. The activation of MAP3K1 and MAP2K3 by r-ceramide exposure of A549 cells suggested that this pathway might be involved upstream of IRF3. Pretreatment of the cells with a p38 inhibitor (SB202190) reduced the IL-8 response to r-ceramide (Figure 4E) and prevented nuclear translocation of IRF3 (Figure 4F, G). NF-κB p65 translocation was not affected by the p38 inhibition (Figure 4F, G). The results suggest that ceramide/TLR4 activates IRF3 - rather than NF-κB-dependent transcription, and that the IRF3 response involves p38 MAPK-dependent mechanisms. CREB-phosphorylation was also markedly reduced after p38 inhibition, as shown by confocal microscopy (>99% in A498 cells, >50% in A549 cells, Figure 4H) and Western blots (Figure S7).
Previous work has suggested that the phosphorylation of TRAM is mediated by PKC-epsilon, which is activated downstream of TLR4 [50]. Given that PKC-epsilon is also essential for IRF3 activation, this pathway was examined, using the pan-PKC inhibitor Bisindolylmaleimide II. The inhibitor reduced the response to PMA, which was used as a PKC dependent, positive control. In contrast, the response to ceramide was not impaired (Figure S8).
To further examine the relationship of the ceramide/TLR4 pathway to the classical IRF3 activation pathway, cells were transfected with TBK1 siRNA and responses were compared to irrelevant siRNA transfected cells. In parallel, the cells were transfected with TLR4 and TRAM siRNAs (Knock down efficiency for TLR4 and TRAM was >90% and 64% for TBK1, Figure S9). IRF3-P responses to r-ceramide were reduced by the TLR4 and TRAM siRNAs but were less affected by suppression of TBK1 expression. The response to LPS+sCD14 showed a similar pattern (Figure S9).
These results suggest that the pathway of IRF3 activation identified here has several new features, including p38 dependence and PKC independence. The involvement of TBK1 needs further study.
Bacterial fimbriae and IRF3 translocation in infected human renal tubular epithelial cells
To examine if the IRF3 response is triggered in a pathogen-specific manner involving P fimbriae, we stimulated primary cultures of human CD14+ renal tubular epithelial cells (HRTEC) with isogenic P-fimbriated (E. coli S1918pap) or type 1-fimbriated (E. coli S1918fim) E. coli strains and examined IRF3 by confocal microscopy. Non-fimbriated E. coli S1918 was used as a control. E. coli S1918pap induced higher nuclear IRF3 translocation and IRF3 phosphorylation than E. coli S1918, consistent with results in ceramide-stimulated cells (Figure 5A and Figure S10, p<0.01 for a broader view). There was less IRF3 translocation in response to E. coli S1918fim or to the non-fimbriated control E. coli S1918. All three strains stimulated an NF-κB response, but NF-κB translocation was higher in cells infected with P-fimbriated E. coli compared to type 1-fimbriated E. coli (Figure 5A, p<0.05). Uninfected cells showed no evidence of nuclear IRF3 - or NF-κB translocation. The same phenomenon was observed in A498 kidney epithelial cells (Figure S11). In addition, preliminary Western blot analysis of IRF3P in infected cells suggested that S1918pap and S1918fim stimulated a higher response than S1918 (Figure S11).
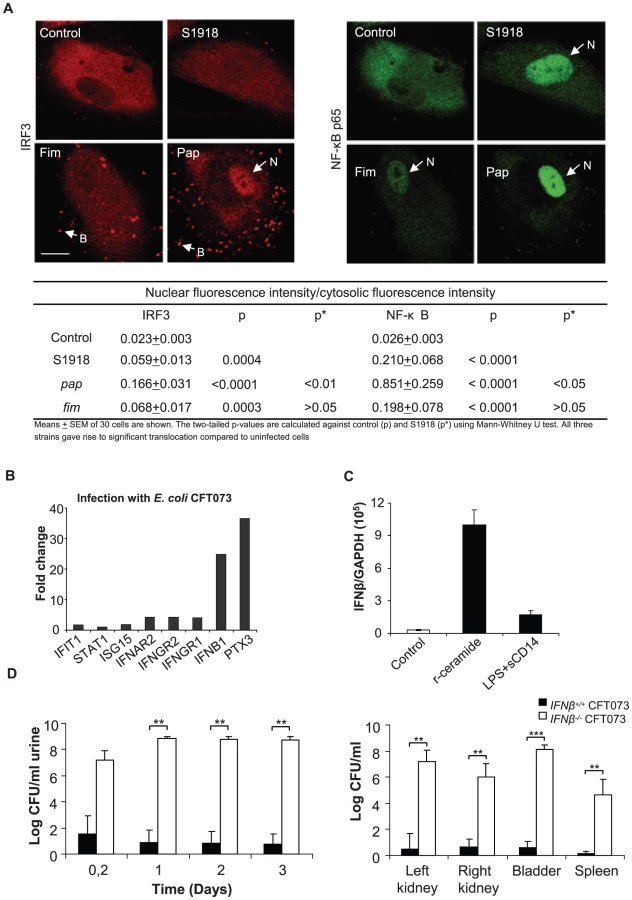
Increased susceptibility to UTI in Ifnβ−/− mice
IRF3 target gene expression was examined by microarray analysis. We infected A498 kidney epithelial cells in vitro with virulent CFT073 or non-pathogenic E. coli (4 hours, 108 CFU/ml) and complementary RNA was hybridized to Illumina whole genome microarrays. There was a dramatic IFNβresponse to infection (24-fold above uninfected control cells, ≥log 2 cut off) (Figure 5B). By Ingenuity Pathway analysis, we detected significant activation of several members of the interferon-signaling pathway such as IFIT1, STAT1 ISG15, IP-10 and IFNAR2. A weaker ISG15 response was observed (1.8-fold above background). By RT-PCR, a strong IFNβ response to r-ceramide was confirmed in human kidney cells (A498, Figure 5 B and C).
To examine if the effects of IRF3 on host susceptibility are IFNβ-dependent, we infected Ifnβ−/− mice with E. coli CFT073 and examined parameters of disease and bacterial persistence (Figure 5E). Bacterial clearance was drastically impaired in Ifnβ−/−mice compared to wt controls (Figure 5E, p<0.001 between Ifnβ−/−and wt mice in urine, kidneys and bladders). The Ifnβ−/− mice also developed abscesses. Positive spleen cultures confirmed systemic spread of infection in these mice, which also developed symptomatic disease and were sacrificed on day three (Figure 5E). The results suggest that IFNβ is activated by infection and that IFNβ might be an essential effector molecule in IRF3-dependent bacterial clearance.
IRF3 promoter polymorphisms in patients with urinary tract infection
To examine if UTI susceptibility is associated with differences in IRF3 promoter efficiency, IRF3 promoter sequence variation was studied in two highly UTI-prone patient populations. Sample 1 comprised children in southern Sweden, with a consistent UTI pattern over several years: either severe recurrent kidney infections (APN; n = 21) or asymptomatic carriage of E. coli with no prior symptomatic infection (primary asymptomatic bacteriuria, ABU, n = 16). These children were identified after prospective, long-term follow-up of a larger patient group. Sample 2 comprised adults in western Sweden, with a history of childhood UTI (n = 82). They were enrolled in a prospective study of febrile UTI (APN) in the 1970s and were recently re-evaluated, after about 30 years, to investigate UTI morbidity and long-term effects on health and kidney function. Both samples included additional patients who developed ABU secondary to an APN episode (secondary ABU, n = 16 in sample 1 and n = 61 in sample 2). Controls were children without UTI or related morbidity (n = 27) and adult blood donors (n = 62) from the same areas.
DNA sequencing of IRF3 promoters from UTI patients revealed variation at the −925 and −776 positions. SNPs −925 and −776 were linked in the study population (r2 = 1.0) but the IRF3 genotype varied with UTI severity (Figure 6A–B). Genotype counts for −925 and −776 were in Hardy Weinberg Equilibrium across both case and control samples apart from the APN group (χ2 = 47, p<0.001), indicating effects of genetic drift in the APN group. We observed significant differences for the two studied markers between cases and controls in allelic or genotypic models. In sample 1, most of the APN patients were homozygous for the two positions (A/A–C/C, 79% vs. 25% in primary ABU, Figure 6A p = 0.0017). The results in APN patients were confirmed in sample 2, with 75% homozygous and 13% heterozygous SNPs compared to 53% and 37% in adult controls. The differences were confirmed when the two samples were combined, as shown in Figure 6B. Furthermore, the minor allele frequency was decreased in APN compared to primary ABU (p = 0.0103) and controls (p = 0.0239) (Figure 6B). The minor allele frequency for paediatric UTI patients, adult UTI patients and the relevant controls are demonstrated in Supplemental Table S4 in Supporting Information S1.

The IRF3 genotype of the secondary ABU patients resembled the APN groups in both samples, consistent with their prior APN episodes.
The acute pyelonephritis-associated promoter SNPs reduced transcription efficiency
To examine if the IRF3 promoter variation influences transcription efficiency, we cloned promoters from one patient with APN and one with ABU into a luciferase reporter vector. We then changed the APN haplotype at positions −925, −776 to the predominating ABU haplotype (A-C to G-T), or the ABU haplotype to the APN haplotype (G-T to A-C) by site-directed mutagenesis. We then transfected A498 human kidney epithelial cells with the different promoter constructs and determined luciferase activity (Figure 6C). The promoter was functional in these cells, resulting in luciferase activity above the vector control.
Transcriptional activity from the APN promoter (A-C) was about 50% lower compared to the ABU (A-T) promoter (Figure 6C). This difference could be attributed to the polymorphic sites, as the promoter activity increased when the APN A-C haplotype was mutated to G-T and decreased when the ABU G-T haplotype was mutated to A-C (p<0.001). This difference was confirmed by cloning the IRF3 promoters from three additional APN (A-C) and three ABU (G-T) patients (p = 0.001). The results show that the IRF3 promoter efficiency is reduced by the SNPs occurring in about 80% of APN patients, consistent with the human SNPs reducing IRF3 expression and increasing the risk for APN.
Discussion
IRF3 was originally described as a transcription factor controlling interferon responses to viral infection [51]. More recently, the involvement of IRFs in antibacterial defense and immunoregulation by TLRs has received more attention, since NF-κB, IRF3 and AP-1 form transcriptional complexes that regulate innate immune responses in monocytes [52]. The relevance of IRF3 to human pathology has not been investigated, however. We show that IRF3 is activated in a pathogen-specific manner by P-fimbriated, uropathogenic E. coli, through a new signaling pathway involving TLR4, TRAM, CREB and p38. In the absence of IRF3, acute morbidity and extensive tissue damage are dramatically augmented, consistent with the need for this pathway to maintain a functional antimicrobial defense. Host susceptibility to common infections like UTI may thus be strongly influenced by single gene modifications affecting the innate immune response.
Mucosal pathogens exploit the extracellular domains of sphingolipids as receptors for AB toxins such as Shiga and cholera toxin, as well as attachment ligands for Pseudomonas aeruginosa, HIV gp120 and uropathogenic E. coli [53], [54], [55]. This study provides evidence that P-fimbriated E. coli, SMase and exogenous, free ceramide all activate the IRF3-dependent innate immune response. Soluble, exogenous ceramide and SMase were used in parallel in these experiments to ensure that the synthetic, short-chained form of ceramide and the endogenous, long-chained form adequately represented the membrane-anchored species in intact cells. SMase is contaminated by low amounts of LPS, but these trace amounts were insufficient to activate an innate immune response in the CD14-negative mucosal cells used in this study [9], [14]. FRET analysis showed that ceramide is approximated to TLR4 in the cell membrane, suggesting that a direct interaction with TLR4 and/or the early adaptors trigger this pathway. Such ceramide-induced TLR4/IRF3 signaling might offer a general mechanism for host sensing of pathogens that perturb membrane sphingolipids in mucosal cells.
To provide further evidence that ceramide signaling via TLR4 to IRF3 discriminates virulent from commensal bacteria, we infected Irf3−/− mice with a commensal-like E. coli strain from a patient with asymptomatic bacteriuria. This strain did not trigger a significant response and was cleared efficiently, suggesting that the IRF3 pathway was not alerted. In constrast, a P-fimbriated transformant triggered rapid, septic infection in the Irf3−/− but not in wt mice linking this virulence factor that recognizes glycosphingolipid surface receptors, to the IRF3-dependent host response. In contrast, IRF3 was not activated by a type 1 fimbriated isogenic strain, suggesting a preference for glycosphingolipid rather than glycoprotein receptors. This does not negate the previous finding that FimH acts as an immune inducer, protecting against viral infection associated with TLR4 and type 1 interferon signaling, but suggests that the mechanisms differ [56]. The results confirm the pathogen specificity of the IRF3 response and the role of P fimbriae as a virulence ligand triggering this response. As a consequence of this selective IRF3 activation, the uropathogenic or P-fimbriated, commensal E. coli strains influenced epithelial gene transcription in a pathogen-specific manner.
IRF3 phosphorylation in response to ceramide was controlled by TLR4 and TRAM, as shown using specific siRNA knock down. Activation was not TBK1 or PKC dependent, however, suggesting alternative activation compared to previously described mechanisms of IRF3 activation [50]. A schematic of the identified kinases and targets is given in supplemental Figure S4. Although this signaling pathway has not been entirely deciphered, a strong involvement of TRAM and CREB was detected as well as involvement of p38 MAPK-dependent events. In this model, IRF3 activation was not controlled by PKC dependent mechanisms, however. The involvement of TBK1 is not clear, but preliminary experiments did not provide evidence that TBK1 controlled IRF3 phosphorylation in this pathway. In addition, P-fimbriated E. coli strains and ceramide significantly activated NF-κB, thus providing a broad basis for the innate immune response to the intact, complex pathogen. Importantly, the IRF3 response differed after LPS+sCD14 stimulation, further suggesting that pathogen recognition and pattern recognition agonists trigger partially different signaling pathways.
The phenotype of Irf3−/− mice predicted that reduced IRF3 expression could also increase human susceptibility to severe kidney infection. In support of this hypothesis, there were marked promoter sequence differences between children with ABU or APN in a long-term prospective study and we confirmed an association of polymorphisms to disease severity in adult patients who were followed for about 30 years after their first febrile UTI episode. In the past, we have shown that genetic variation affecting innate immunity modifies human UTI susceptibility [2]. Chemokine receptor expression and neutrophil function are modified by CXCR1 expression, and promoter variants reducing TLR4 expression are coupled to asymptomatic bacteriuria [22], [57], [58], [59], [60]. The present study adds IRF3 to this short list of polymorphic innate immune response genes that distinguish asymptomatic carriers from APN-prone patients.
The human IRF3 promoter has a number of transcription factor binding sites, including a HOX box, three SP1 sites, NF1, USF, SRF and IRF1-like site and functional elements are within a 113-nucletide long fragment, containing one Sp-1 site, the IRF1-like site, NF1 and HOX box. SNP −925 is located within this region, indicating a possible role in promoter efficiency. IRF3 promoter SNPs were first described in patients with systemic lupus erythematosus (SLE) [61]. It was speculated that the A-C haplotype increased IRF3 transcription and that the G-T haplotype might protect against SLE by reducing type I IFN production. The effect of the Irf3 deletion on disease susceptibility in mice suggested, however, that risk might be associated with reduced, rather than increased, IRF3 function. This idea was supported by luciferase reporter assays designed to test the IRF3 promoter sequences typical of APN - or ABU-prone individuals.
UTIs are among the most common bacterial infections in man, and remain a major cause of morbidity and mortality [62], [63]. A subset of disease-prone individuals is at risk for recurrent severe pyelonephritis and renal dysfunction. Therefore, there is a need to identify and treat these patients, preferably in infancy, when many of them experience their first febrile UTI episode. Although predictive diagnostic tools have been suggested [58], [59], [60], the present study identifies IRF3 for the first time as an innate immune response gene involved in UTI. Thus, IRF3 may be a new molecular target in the diagnosis of UTI susceptibility, potentially creating more precise approaches for detection and prevention of severe, recurrent kidney infection and associated debilitating morbidity.
Methods
Ethics statement
For research involving humans, informed written consent was obtained from all participants or their parents/guardians. The study was approved by the Ethics Committee of the medical faculty, Lund University, Sweden (LU106-02, LU236-99).
All the animal experiments were performed with the permission of the Animal Experimental Ethics Committee at the Lund District Court, Sweden (numbers M166-04 and M87-07). Experimental UTI was performed in a level P2 biohazard laboratory within the MIG animal facility and was governed by the following directive, law, ordinance and provisions: Council Directive EG 86/609/EEC, the Swedish Animal Welfare Act (Djurskyddslag: 1988 : 534) and the Swedish Animal Welfare Ordinance (Djurskyddsförordning: 1988 : 539). Provisions regarding the use of animals for scientific purposes: DFS 2004 : 15, DFS 2005::4, SJVFS 2001 : 91, SJVFS 1991 : 11.
Reagents
SMase (Staphylococcus aureus), bovine serum albumin, SDS, LPS (Salmonella typhimurium), C6 ceramide, SB202190 and Bisindolylmaleimide II were from Sigma Aldrich, St Louis, MO, USA. Soluble CD14 (sCD14) was from Biometec, Greifswald and IL-8 was quantified by Immulite 100, Siemens, Germany. Lipofectamine 2000 transfection reagent was from Invitrogen. siRNA downregulation (Supplemental Table S1 in Supporting Information S1) was validated by qRT-PCR, using primers: TLR4 (Hs00152939, Applied Biosystems), MyD88 (QT00203490, Qiagen), TRAM (QT00033341, Qiagen), TBK1 (QT00078393). TBK1 siRNA (sc-39058), a pool of 3 target-specific 19–25 nt siRNAs was from Santa Cruz Biotechnology (USA). Human Phospho-Kinase Array Kit ARY003 was from R&D Systems, Abingdon, Oxford, UK. Transcriptome analysis of r-ceramide or LPS+sCD14 activated cells (1 or 3 hours) was by Superarray (PAHS018, SaBioscience) for 84 TLR signaling pathway genes. IRF3 promoter SNPs were identified by Pyrosequencing using the PSQ 96 SNP Reagent Kit (Biotage, Uppsala, Sweden, Supplemental Table S1 in Supporting Information S1). Rabbit anti-human TLR4 primary antibodies were from eBioscience, CA, USA, mouse anti-ceramide primary antibodies, clone MID 15B4 from ALEXIS Corporation, Lausen, Switzerland. Rabbit anti-human primary antibodies against CREB-P (Ser 133), Fos-P (Thr 232), JNK-P (Thr 183/Tyr 185) and IRF3 and mouse anti-human-NF-κB p65 antibodies from Santa Cruz Biotechnology (USA), rabbit anti-IRF3-P (Ser 396) antibodies were from Cell Signaling Technology. Rabbit anti-human-TRAM-P (raised against the N-terminal end of the protein, aa 7–21 containing phosphoSer at aa 16) and TRAM antibodies were from FabGennix Inc., Frisco, TX, USA, NIMP-R14 rat anti-mouse neutrophil specific antibodies from Abcam, Cambridge, USA, polyclonal rabbit antiserum raised against a peptide within the PapG adhesin (CRPSAQSLEIKHGDL) was used to detect P-fimbriated E. coli. Alexa 488 anti-rat IgG, Alexa 568 anti-rabbit IgG, Alexa 568 anti-mouse IgM and Alexa 488 anti-mouse-IgG secondary antibodies were from Invitrogen, Eugene, Oregon, USA. Swine anti-rabbit immunoglobulins-HRP secondary antibodies were from DAKO A/S, Glostrup, Denmark and Santa Cruz Biotechnology (USA). FRET and fluorescence microscopy was by LSM510 META confocal microscope (Carl Zeiss, Oberkochen, Germany).
Cell cultures
The human lung adenocarcinoma A549 (ATCC CCL-185) and kidney carcinoma A498 (ATCC HTB-44) epithelial cell lines were grown in RPMI 1640 supplemented with 1 mM sodium pyruvate, 1 mM non-essential amino acids, 50 µM/ml gentamicin, and 5% FBS. Human renal tubular epithelial cells (HRTEC) were isolated as described [64]. Cells were maintained at 37°C+5% CO2 in a humidified atmosphere, split weekly and exposed to P-fimbriated, type I fimbriated or non-fimbriated E. coli, 0.1–1 U/ml of SMase, freshly prepared C6 ceramide or LPS+sCD14. IL-8 secretion was quantified by Immulite 100 (Siemens, Bad Nauheim, Germany).
FRET
LSM 510 Meta confocal laser-scanning microscopy was used for FRET acceptor photobleaching and imaging of epithelial cells. Cell stimulation/infection was in 8-well chamber slides (LabTek, Nunc, RPMI+5% foetal calf serum). The cells were first stimulated with r-ceramide (1U of SMase/ml) or LPS+sCD14 (10+1µg/ml), fixed with 3.7% formaldehyde and stained with a mouse antibody specific for native ceramide (sphingosine-trans-D-erythro-2-amino-4-octadecene-1.3-diol) and with a rabbit polyclonal antibody, specific for the extracellular domain of TLR4 (amino acids 6–169). Secondary antibodies to TLR4 and free ceramide were conjugated with Alexa-488 (donor) and Alexa-568 (acceptor), respectively. FRET efficiency was estimated in percent of fluorescence increase calculated by: FRET efficiency = ((IDA-IDB)/IDA)×100% where IDA is the donor intensity after bleaching and IDB the donor intensity before bleaching.
siRNA transfection
A549 human epithelial cells in 24-well plates (TPP) were siRNA transfected, using Lipofectamine 2000 (Supplemental Table S1 in Supporting Information S1). Knockdown efficiency was validated by qRT-PCR. After a 72 h incubation, transfected cells were stimulated with SMase (1 U/well) or LPS+sCD14 (10+1µg/ml). Supernatants were collected after 24 h.
RT-PCR array
Total extracted mRNAs were converted to cDNA using RT2 First Strand Kit (SABioscience Corporation, Fredrick, MA, USA). The transcriptomic profile of cells exposed to r-ceramide or LPS+sCD14 was examined using a RT-PCR-based superarray, containing 84 genes involved in TLR signaling (SABiosciences, PAHS018). Gene expression levels were calculated by the ΔCt method and normalized to five housekeeping genes. RT-PCR was used to determine the efficiency of siRNA knockdown. The TaqMan system was used to quantify TLR4 and GAPDH cDNA and the QuantiTect SYBR Green systems was used to quantify other genes of interest. cDNA was quantified by RT-PCR using a Rotor gene 2000 instrument (Corbett Life Science, Sydney, Australia) and normalized against GAPDH.
Phospho-kinase array
Protein phosphorylation was quantified using the Human Phospho-Kinase Array Kit (Proteome Prolifer Array, R&D Systems, Abingdon, Oxford, UK). Protein extracts were prepared from 100% confluent A549 cells cultured in 6-well plates and treated with 1U/well SMase or LPS+sCD14 (10+1µg/ml). Untreated cells were used as control. The signals were detected with the ECL Plus Western Blotting Detection System (GE Healthcare).
SDS-PAGE and immunoblotting
In order to detect phosphorylated TRAM, A549 cells grown in 6-well plates were stimulated with 1U/mL SMase, 0.1 µg/mL LPS+sCD14 (10+1µg/ml), 15 µg/mL C6 (Sigma) or RPMI medium alone for 45 and 90 min. Cells were lysed in ice-cold buffer (10 mM HEPES-KOH, 5 mM EDTA, 0.5% Nonidet P-40 and 10 mM KCl, pH 7.9) containing a protease inhibitor mix (Complete, Roche Diagnostics GmbH, Mannheim, Germany) and 1mM Na3VO4. After 10 min incubation, lysates were centrifuged for 5 min at 12000 g at 4°C and protein concentrations in the collected supernatants were quantified using the DC protein assay kit (Bio-Rad, Hercules, USA). Proteins in the lysates were separated by SDS-PAGE (4–12% NuPAGE Bis-Tris gels, Invitrogen) on ice with NuPAGE MES SDS running buffer (Invitrogen). Proteins were transferred to a polyvinylidene difluoride (PVDF) membrane using NuPAGE transfer buffer (Invitrogen) and the membrane was probed with phospospecific primary antibodies followed by HRP-labeled, swine anti-rabbit IgG. Bound antibodies were visualized with the ECL Plus Western Blotting Detection System.
Native PAGE and immunoblotting
IRF3 dimerization was detected by native PAGE and immunoblotting. A549 cells cultured in 6-well plates were stimulated with medium alone, 1 U/mL SMase and 0.1 µg/mL or LPS+sCD14 (10+1µg/ml), for 90 min. Whole cell lysates in a buffer containing 50 mM Tris HCl, pH 7.5, 400 mM NaCl, 1mM EDTA, 1% Nonidet P-40 were separated by electrophoresis on a 7.5% native Tris-glycine gel [65]. Membranes were incubated with primary antibodies against human IRF3 (Fl-425, Santa-Cruz) diluted 1∶1000 and anti-rabbit IgG-HRP (1∶1000). IRF3 monomers and dimers were detected with the ECL Plus Western Blotting Detection System.
DNA microarray analysis
In brief, A498 cells (n = 350000) were seeded in 6-well plates and infected with CFT073 (108 CFU/ml), total RNA was extracted (Trizol, Invitrogen, USA) and cleaned by a Qiagen RNeasy. RNA was reverse-transcribed to biotin-labeled cRNA using a TargetAmp Nano-g Biotin-aRNA Labeling kit (Epicentre Biotechnologies, Madison, USA). Labeled cRNAs were hybridized onto an Illumina HumanHT-12 Expression Beadchip for 16 hours at 58°C. The arrays were then washed and stained (Illumina Wash Protocol) and scanned using a BeadArray Scanner 500GX. The background-subtracted data were pre-processed to correct negative and non-significant intensities. Pre-processed data was normalized using the cross-correlation [66] and genes with a log fold change of 2 were identified as differentially expressed. Data was preprocessed using RMA implemented in the free software packages R and Bioconductor (http://www.r-project.org). For more details, see Yadav et al.
Differentially expressed genes were categorized using the Functional Annotation Clustering Tool in the Database for Annotation, Visualization and Integrated Discovery (DAVID) [67] and the EASE score (a modified enrichment score derived from Fisher exact P-value) was used to judge the enrichment. To further study signaling pathways altered by CFT073, the differentially expressed genes were submitted for Ingenuity Pathway Analysis (Ingenuity Systems, Redwood City, CA).
Bacteria and growth conditions
Escherichia coli S1918 [68] (KanR) lacks genes encoding known adhesins and was used as a recipient strain for recombinant plasmid pPIL 110-75 (AmpR) carrying the papAD1100 gene cluster (pap+) [69] or PKL-4 carrying the entire fim gene cluster from E. coli PC31 (type I+) [68]. The papGX deletion mutant of E. coli strain 83972 (ABU 83972ΔpapGX) was generated using the lambda red homologous recombination technique [70]. Clones with the reconstituted pap determinant were screened using PCR and verified by DNA sequencing. Primers used for reconstitution of a functional pap gene cluster in E. coli 83972 are shown in Supplemental Table S5 in Supporting Information S1. The growth rates of the reconstituted mutant strain and the 83972 wild type strain were shown to be identical. In addition, the ability of the reconstituted mutant to agglutinate sheep blood erythrocytes was also confirmed by agglutination assays.
Experimental urinary tract infection (UTI)
Mice were bred at the MIG animal facilities, Lund, Sweden. Female C57BL/6 wild type or Irf3−/− (from T. Taniguichi) and Ifnβ−/− (from F. Ivars, Lund University) mice were used at 9–15 weeks. After anesthesia (Isofluorane), mice were infected by intravesical inoculation with E. coli CFT073 (109 CFU in 0.1 mL) through a soft polyethylene catheter (outer diameter 0.61 mm; Clay Adams, Parsippany, NJ, USA). Animals were sacrificed while under anesthesia, kidneys and bladders were removed and prepared for hematoxylin-eosin staining or immunohistochemistry. Viable counts in homogenized tissues (Stomacher 80, Seward Medical, UAC House, London, UK) were determined after overnight growth on tryptic soy agar plates at 37°C. Urine samples collected prior to and daily after infection were cultured and recruited neutrophils were quantified in uncentrifuged urine by use of a hemocytometer.
Histology and immunostaining
Kidney sections were examined by immunohistochemistry [71]. Tissue sections were dried and permeabilized in 0.2% Triton X-100, 5% goat normal serum (DAKO) in PBS, incubated with NIMP-R14 rat anti-mouse neutrophil specific antibodies (1∶200) and a polyclonal rabbit antiserum to the Pap G adhesin (1∶200) to detect P-fimbriated E. coli and to Alexa 488 anti-rat IgG and Alexa 568 anti-rabbit IgG secondary antibodies and nuclei were counterstained with DAPI (0.05 µM). After mounting, coverslipped slides were examined by fluorescence microscopy (AX60, Olympus Optical, Hamburg, Germany) at the Department of Pathology, Lund University, Sweden.
Confocal fluorescence immunocytochemistry was performed on cells grown to 70–80% confluence on 8-well chamber slides. After stimulation, cells were fixed and permeabilized with 0.25% Triton X-100, 5% FBS in PBS and incubated with primary antibodies diluted 1∶50 in 5% FBS in PBS overnight at 4°C. Alexa fluor-labeled secondary antibodies were applied for 1 hour at RT in the dark. In order to control specific staining of neutrophils and bacteria, slides were stained with only secondary antibodies (Figure S1). Slides were covered with mounting medium (M1289, Sigma) and cover glasses and the cells were examined with a LSM510 META confocal microscope.
UTI-prone patients and pyrosequencing
The IRF3 promoter from patients with UTI or healthy controls was sequenced (PSQ96, Biotage, Uppsala, Sweden) and examined for −925 and −776 polymorphisms [61]. Genomic DNA was extracted from heparinized peripheral blood using the QIAamp DNA Blood midi kit. More detailed descriptions of inclusion critera and diagnosis are provided in [58], [59], [72]. The IRF3 promoter SNPs (−925; −776) were genotyped using Pyrosequencer PSQ 96 after PCR amplification of chromosomal DNA and a second biotinylated PCR for each SNP (for primers see Supplemental Table S2 in Supporting Information S1).
Transient transfection and dual luciferase reporter assay
The promoter sequences from extracted chromosomal DNA derived from APN and ABU patients were PCR-amplified using the Infusion primers 5′ IRF3 NheI and 3′IRF3 NcoI (Supplemental Table S3 in Supporting Information S1) and Phusion hot start polymerase according to the manufacturer (Finnzymes Oy, Finland). Amplicons were introduced by recombination, using the Infusion cloning technique (Clontech), into a NheI - and NcoI-cleaved and gel-purified luciferase reporter vector, pGL3 basic (Promega). The recombinant DNA was transformed into E. coli and recombinant clones were screened for the presence of cloned promoter insert. Plasmids of the correct size were further analyzed by DNA sequencing using the Big Dye terminator v3.1 cycle sequencing chemistry and ABI capillary sequence.
A quick change Multi Site-directed Mutagenesis kit (Stratagene) was used according to the manufacturer's instructions to create the various IRF3 promoter constructs (Supplemental Table S3 in Supporting Information S1). 5498 human kidney epithelial cells were cultured in 24-well plates at a density of 2.5×105 cells per well. The cells were transiently transfected with wild type or mutant IRF3 promoter driven firefly luciferase constructs (pGL3) together with a constitutively expressed internal control construct with Renilla luciferase-thymidine kinase promoter (pRL-TK, Promega) using Fugene HD (Roche) Transfection reagent at 4∶2 ratio. Luciferases were measured using the Dual Luciferase Reporter System Assay (Promega) with a Glomax Integrated Luminometer (Promega). Firefly luciferase data were normalized against transfection efficiency of Renilla luciferase and expressed as a ratio.
Statistics
Student's t test or Wilcoxon's rank-sum test were used for paired comparisons, Mann-Whitney test was applied for unpaired comparisons. P values below 0.05 were considered to indicate statistical significance. Deviations from Hardy-Weinberg equilibrium (HWE) for genotypes at individual loci in patients and controls, as well as differences in genotype and allele distributions between groups, were assessed using the χ2 test. Fisher's exact test was used where appropriate.
List of ID numbers for genes and proteins of mouse and humans
Gene ID number for human TLR4 is 7099, human MyD88 is 4615, human TRIF is 148022, human TRAM is 353376, human TBK1 is 29110, human CREB is 1385, human IRF3 is 3661, mouse Irf3 is 54131, human IFNB is 3456 and mouse Ifnb is 15977.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. ChotivanichK
UdomsangpetchR
PattanapanyasatK
ChierakulW
SimpsonJ
2002 Hemoglobin E: a balanced polymorphism protective against high parasitemias and thus severe P falciparum malaria. Blood 100 1172 1176
2. RagnarsdottirB
FischerH
GodalyG
Gronberg-HernandezJ
GustafssonM
2008 TLR - and CXCR1-dependent innate immunity: insights into the genetics of urinary tract infections. Eur J Clin Invest 38 Suppl 2 12 20
3. SvanborgC
BergstenG
FischerH
GodalyG
GustafssonM
2006 Uropathogenic Escherichia coli as a model of host-parasite interaction. Curr Opin Microbiol 9 33 39
4. MogensenTH
2009 Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev 22 240 273, Table of Contents
5. KimHM
ParkBS
KimJI
KimSE
LeeJ
2007 Crystal structure of the TLR4-MD-2 complex with bound endotoxin antagonist Eritoran. Cell 130 906 917
6. BellJK
BotosI
HallPR
AskinsJ
ShiloachJ
2005 The molecular structure of the Toll-like receptor 3 ligand-binding domain. Proc Natl Acad Sci U S A 102 10976 10980
7. KobeB
KajavaAV
2001 The leucine-rich repeat as a protein recognition motif. Curr Opin Struct Biol 11 725 732
8. FischerH
YamamotoM
AkiraS
BeutlerB
SvanborgC
2006 Mechanism of pathogen-specific TLR4 activation in the mucosa: fimbriae, recognition receptors and adaptor protein selection. Eur J Immunol 36 267 277
9. HedlundM
SvenssonM
NilssonA
DuanRD
SvanborgC
1996 Role of the ceramide-signaling pathway in cytokine responses to P-fimbriated Escherichia coli. J Exp Med 183 1037 1044
10. HannunY
BellR
1989 Functions of sphingolipids and sphingolipid break-down products in cellular regulation. Science 247 500 507
11. KolesnickR
1991 Sphingomyelin and derivatives as cellular signals. Prog Lipid Res 30 1 38
12. ZhangY
LiX
BeckerKA
GulbinsE
2009 Ceramide-enriched membrane domains–structure and function. Biochim Biophys Acta 1788 178 183
13. HannunYA
1996 Functions of ceramide in coordinating cellular responses to stress. Science 274 1855 1859
14. HedlundM
NilssonÅ
DuanRD
SvanborgC
1998 Sphingomyelin, glycosphingolipids and ceramide signalling in cells exposed to P fimbriated Escherichia coli. Mol Microbiol 29 1297 1306
15. GrassmeH
JendrossekV
RiehleA
von KurthyG
BergerJ
2003 Host defense against Pseudomonas aeruginosa requires ceramide-rich membrane rafts. Nat Med 9 322 330
16. van BlitterswijkWJ
van der LuitAH
VeldmanRJ
VerheijM
BorstJ
2003 Ceramide: second messenger or modulator of membrane structure and dynamics? Biochem J 369 199 211
17. FischerH
EllstromP
EkstromK
GustafssonL
GustafssonM
2007 Ceramide as a TLR4 agonist; a putative signalling intermediate between sphingolipid receptors for microbial ligands and TLR4. Cell Microbiol
18. HannunYA
1994 The sphingomyelin cycle and the second messenger function of ceramide. J Biol Chem 269 3125 3128
19. HedlundM
DuanRD
NilssonA
SvenssonM
KarpmanD
2001 Fimbriae, transmembrane signaling, and cell activation. J Infect Dis 183 Suppl 1 S47 50
20. FrendeusB
WachtlerC
HedlundM
FischerH
SamuelssonP
2001 Escherichia coli P fimbriae utilize the Toll-like receptor 4 pathway for cell activation. Mol Microbiol 40 37 51
21. HagbergL
HullR
HullS
McGheeJR
MichalekSM
1984 Difference in susceptibility to gram-negative urinary tract infection between C3H/HeJ and C3H/HeN mice. Infect Immun 46 839 844
22. RagnarsdottirB
JonssonK
UrbanoA
Gronberg-HernandezJ
LutayN
2010 Toll-like receptor 4 promoter polymorphisms: common TLR4 variants may protect against severe urinary tract infection. PLoS One 5 e10734
23. WelchRA
BurlandV
PlunkettG3rd
RedfordP
RoeschP
2002 Extensive mosaic structure revealed by the complete genome sequence of uropathogenic Escherichia coli. Proc Natl Acad Sci U S A 99 17020 17024
24. GodalyG
BergstenG
HangL
FischerH
FrendeusB
2001 Neutrophil recruitment, chemokine receptors, and resistance to mucosal infection. J Leukoc Biol 69 899 906
25. GodalyG
HangL
FrendeusB
SvanborgC
2000 Transepithelial neutrophil migration is CXCR1 dependent in vitro and is defective in IL-8 receptor knockout mice. J Immunol 165 5287 5294
26. KlemmP
RoosV
UlettGC
SvanborgC
SchembriMA
2006 Molecular characterization of the Escherichia coli asymptomatic bacteriuria strain 83972: the taming of a pathogen. Infect Immun 74 781 785
27. RoosV
SchembriMA
UlettGC
KlemmP
2006 Asymptomatic bacteriuria Escherichia coli strain 83972 carries mutations in the foc locus and is unable to express F1C fimbriae. Microbiology 152 1799 1806
28. KlemmP
HancockV
SchembriMA
2007 Mellowing out: adaptation to commensalism by Escherichia coli asymptomatic bacteriuria strain 83972. Infect Immun 75 3688 3695
29. ZdziarskiJ
SvanborgC
WulltB
HackerJ
DobrindtU
2008 Molecular basis of commensalism in the urinary tract: low virulence or virulence attenuation? Infect Immun 76 695 703
30. JohnsonJR
RobertsPL
StammWE
1987 P fimbriae and other virulence factors in Escherichia coli urosepsis: association with patients' characteristics. J Infect Dis 156 225 229
31. OttoG
SandbergT
MarklundBI
UllerydP
SvanborgC
1993 Virulence factors and pap genotype in Escherichia coli isolates from women with acute pyelonephritis, with or without bacteremia. Clin Infect Dis 17 448 456
32. BallouLR
LaulederkindSJ
RosloniecEF
RaghowR
1996 Ceramide signalling and the immune response. Biochim Biophys Acta 1301 273 287
33. SamuelssonP
HangL
WulltB
IrjalaH
SvanborgC
2004 Toll-like receptor 4 expression and cytokine responses in the human urinary tract mucosa. Infect Immun 72 3179 3186
34. RabelinkTJ
LuscherTF
2006 Endothelial nitric oxide synthase: host defense enzyme of the endothelium? Arterioscler Thromb Vasc Biol 26 267 271
35. Astarie-DequekerC
CarrenoS
CougouleC
Maridonneau-PariniI
2002 The protein tyrosine kinase Hck is located on lysosomal vesicles that are physically and functionally distinct from CD63-positive lysosomes in human macrophages. J Cell Sci 115 81 89
36. XueL
FirestoneGL
BjeldanesLF
2005 DIM stimulates IFNgamma gene expression in human breast cancer cells via the specific activation of JNK and p38 pathways. Oncogene 24 2343 2353
37. MaoLM
TangQ
WangJQ
2007 Protein kinase C-regulated cAMP response element-binding protein phosphorylation in cultured rat striatal neurons. Brain Res Bull 72 302 308
38. ChriviaJC
KwokRP
LambN
HagiwaraM
MontminyMR
1993 Phosphorylated CREB binds specifically to the nuclear protein CBP. Nature 365 855 859
39. LinR
GeninP
MamaneY
HiscottJ
2000 Selective DNA binding and association with the CREB binding protein coactivator contribute to differential activation of alpha/beta interferon genes by interferon regulatory factors 3 and 7. Mol Cell Biol 20 6342 6353
40. MayrB
MontminyM
2001 Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat Rev Mol Cell Biol 2 599 609
41. SanyalS
SandstromDJ
HoefferCA
RamaswamiM
2002 AP-1 functions upstream of CREB to control synaptic plasticity in Drosophila. Nature 416 870 874
42. ServantMJ
GrandvauxN
HiscottJ
2002 Multiple signaling pathways leading to the activation of interferon regulatory factor 3. Biochem Pharmacol 64 985 992
43. AuWC
MoorePA
LowtherW
JuangYT
PithaPM
1995 Identification of a member of the interferon regulatory factor family that binds to the interferon-stimulated response element and activates expression of interferon-induced genes. Proc Natl Acad Sci U S A 92 11657 11661
44. FitzgeraldKA
RoweDC
BarnesBJ
CaffreyDR
VisintinA
2003 LPS-TLR4 signaling to IRF-3/7 and NF-kappaB involves the toll adapters TRAM and TRIF. J Exp Med 198 1043 1055
45. HiscottJ
PithaP
GeninP
NguyenH
HeylbroeckC
1999 Triggering the interferon response: the role of IRF-3 transcription factor. J Interferon Cytokine Res 19 1 13
46. KawaiT
TakeuchiO
FujitaT
InoueJ
MuhlradtPF
2001 Lipopolysaccharide stimulates the MyD88-independent pathway and results in activation of IFN-regulatory factor 3 and the expression of a subset of lipopolysaccharide-inducible genes. J Immunol 167 5887 5894
47. LeeJC
LaydonJT
McDonnellPC
GallagherTF
KumarS
1994 A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature 372 739 746
48. HanJ
RichterB
LiZ
KravchenkoV
UlevitchRJ
1995 Molecular cloning of human p38 MAP kinase. Biochim Biophys Acta 1265 224 227
49. SymonsA
BeinkeS
LeySC
2006 MAP kinase kinase kinases and innate immunity. Trends Immunol 27 40 48
50. McGettrickAF
BrintEK
Palsson-McDermottEM
RoweDC
GolenbockDT
2006 Trif-related adapter molecule is phosphorylated by PKC{epsilon} during Toll-like receptor 4 signaling. Proc Natl Acad Sci U S A 103 9196 9201
51. TaniguchiT
OgasawaraK
TakaokaA
TanakaN
2001 IRF family of transcription factors as regulators of host defense. Annu Rev Immunol 19 623 655
52. HondaK
TaniguchiT
2006 IRFs: master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat Rev Immunol 6 644 658
53. KiarashA
BoydB
LingwoodCA
1994 Glycosphingolipid receptor function is modified by fatty acid content. Verotoxin 1 and verotoxin 2c preferentially recognize different globotriaosyl ceramide fatty acid homologues. J Biol Chem 269 11138 11146
54. LingwoodCA
1993 Verotoxins and their glycolipid receptors. Adv Lipid Res 25 189 211
55. SvensonSB
HultbergH
KalleniusG
KorhonenTK
MollbyR
1983 P-fimbriae of pyelonephritogenic Escherichia coli: identification and chemical characterization of receptors. Infection 11 61 67
56. Ashkar AAMK
CoombesBK
GylesCL
MackenzieR
2008 FimH adhesin of type 1 fimbriae is a potent inducer of innate antimicrobial responses which requires TLR4 and type 1 interferon signalling. PLoS Pathog 4 e1000233
57. FrendeusB
GodalyG
HangL
KarpmanD
LundstedtAC
2000 Interleukin 8 receptor deficiency confers susceptibility to acute experimental pyelonephritis and may have a human counterpart. J Exp Med 192 881 890
58. LundstedtAC
LeijonhufvudI
RagnarsdottirB
KarpmanD
AnderssonB
2007 Inherited susceptibility to acute pyelonephritis: a family study of urinary tract infection. J Infect Dis 195 1227 1234
59. LundstedtAC
McCarthyS
GustafssonMC
GodalyG
JodalU
2007 A genetic basis of susceptibility to acute pyelonephritis. PLoS ONE 2 e825
60. RagnarsdottirB
SamuelssonM
GustafssonMC
LeijonhufvudI
KarpmanD
2007 Reduced toll-like receptor 4 expression in children with asymptomatic bacteriuria. J Infect Dis 196 475 484
61. AkahoshiM
NakashimaH
SadanagaA
MiyakeK
ObaraK
2008 Promoter polymorphisms in the IRF3 gene confer protection against systemic lupus erythematosus. Lupus 17 568 574
62. KuninC
1987 Urinary tract infections. Detection, prevention and management, 5th ed Baltimore Williams & Wilkins
63. FoxmanB
ManningSD
TallmanP
BauerR
ZhangL
2002 Uropathogenic Escherichia coli are more likely than commensal E. coli to be shared between heterosexual sex partners. Am J Epidemiol 156 1133 1140
64. KarpmanD
HåkanssonA
PerezM-TR
IsakssonC
CarlemalmE
1998 Apoptosis of renal cortical cells in the hemolytic uremic syndrome: in vivo and in vitro studies. Infect Immun 66 636 644
65. IwamuraT
YoneyamaM
YamaguchiK
SuharaW
MoriW
2001 Induction of IRF-3/-7 kinase and NF-kappaB in response to double-stranded RNA and virus infection: common and unique pathways. Genes Cells 6 375 388
66. ChuaSW
VijayakumarP
NissomPM
YamCY
WongVV
2006 A novel normalization method for effective removal of systematic variation in microarray data. Nucleic Acids Res 34 e38
67. DennisGJr
ShermanBT
HosackDA
YangJ
GaoW
2003 DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol 4 P3
68. KlemmP
JørgensenB
van DieI
de ReeH
BergmansH
1985 The fim genes responsible for the synthesis of type 1 fimbriae in Escherichia coli, cloning and genetic organization. Mol Gen Genet 199 410 414
69. van DieI
BergmansH
1984 Nucleotide sequence of the gene encoding the F72 fimbrial subunit of a uropathogenic Escherichia coli. Gene 32 83 90
70. DatsenkoKA
WannerBL
2000 One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97 6640 6645
71. BucE
VartanianMD
DarchaC
DechelotteP
PezetD
2005 Guanylyl cyclase C as a reliable immunohistochemical marker and its ligand Escherichia coli heat-stable enterotoxin as a potential protein-delivering vehicle for colorectal cancer cells. Eur J Cancer 41 1618 1627
72. MartinellJ
JodalU
Lidin-JansonG
1990 Pregnancies in women with and without renal scarring after urinary tract infections in childhood. Br Med J 300 840 844
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2010 Číslo 9
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Měli bychom postcovidový syndrom léčit antidepresivy?
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- Structure of the Extracellular Portion of CD46 Provides Insights into Its Interactions with Complement Proteins and Pathogens
- The Length of Vesicular Stomatitis Virus Particles Dictates a Need for Actin Assembly during Clathrin-Dependent Endocytosis
- Inhibition of TIR Domain Signaling by TcpC: MyD88-Dependent and Independent Effects on Virulence
- The Coevolution of Virulence: Tolerance in Perspective