
Cyclin-Dependent Kinase-Like Function Is Shared by the Beta- and Gamma- Subset of the Conserved Herpesvirus Protein Kinases
The UL97 protein of human cytomegalovirus (HCMV, or HHV-5 (human herpesvirus 5)), is a kinase that phosphorylates the cellular retinoblastoma (Rb) tumor suppressor and lamin A/C proteins that are also substrates of cellular cyclin-dependent kinases (Cdks). A functional complementation assay has further shown that UL97 has authentic Cdk-like activity. The other seven human herpesviruses each encode a kinase with sequence and positional homology to UL97. These UL97-homologous proteins have been termed the conserved herpesvirus protein kinases (CHPKs) to distinguish them from other human herpesvirus-encoded kinases. To determine if the Cdk-like activities of UL97 were shared by all of the CHPKs, we individually expressed epitope-tagged alleles of each protein in human Saos-2 cells to test for Rb phosphorylation, human U-2 OS cells to monitor nuclear lamina disruption and lamin A phosphorylation, or S. cerevisiae cdc28-13 mutant cells to directly assay for Cdk function. We found that the ability to phosphorylate Rb and lamin A, and to disrupt the nuclear lamina, was shared by all CHPKs from the beta - and gamma-herpesvirus families, but not by their alpha-herpesvirus homologs. Similarly, all but one of the beta and gamma CHPKs displayed bona fide Cdk activity in S. cerevisiae, while the alpha proteins did not. Thus, we have identified novel virally-encoded Cdk-like kinases, a nomenclature we abbreviate as v-Cdks. Interestingly, we found that other, non-Cdk-related activities reported for UL97 (dispersion of promyelocytic leukemia protein nuclear bodies (PML-NBs) and disruption of cytoplasmic or nuclear aggresomes) showed weak conservation among the CHPKs that, in general, did not segregate to specific viral families. Therefore, the genomic and evolutionary conservation of these kinases has not been fully maintained at the functional level. Our data indicate that these related kinases, some of which are targets of approved or developmental antiviral drugs, are likely to serve both overlapping and non-overlapping functions during viral infections.
Published in the journal:
. PLoS Pathog 6(9): e32767. doi:10.1371/journal.ppat.1001092
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1001092
Summary
The UL97 protein of human cytomegalovirus (HCMV, or HHV-5 (human herpesvirus 5)), is a kinase that phosphorylates the cellular retinoblastoma (Rb) tumor suppressor and lamin A/C proteins that are also substrates of cellular cyclin-dependent kinases (Cdks). A functional complementation assay has further shown that UL97 has authentic Cdk-like activity. The other seven human herpesviruses each encode a kinase with sequence and positional homology to UL97. These UL97-homologous proteins have been termed the conserved herpesvirus protein kinases (CHPKs) to distinguish them from other human herpesvirus-encoded kinases. To determine if the Cdk-like activities of UL97 were shared by all of the CHPKs, we individually expressed epitope-tagged alleles of each protein in human Saos-2 cells to test for Rb phosphorylation, human U-2 OS cells to monitor nuclear lamina disruption and lamin A phosphorylation, or S. cerevisiae cdc28-13 mutant cells to directly assay for Cdk function. We found that the ability to phosphorylate Rb and lamin A, and to disrupt the nuclear lamina, was shared by all CHPKs from the beta - and gamma-herpesvirus families, but not by their alpha-herpesvirus homologs. Similarly, all but one of the beta and gamma CHPKs displayed bona fide Cdk activity in S. cerevisiae, while the alpha proteins did not. Thus, we have identified novel virally-encoded Cdk-like kinases, a nomenclature we abbreviate as v-Cdks. Interestingly, we found that other, non-Cdk-related activities reported for UL97 (dispersion of promyelocytic leukemia protein nuclear bodies (PML-NBs) and disruption of cytoplasmic or nuclear aggresomes) showed weak conservation among the CHPKs that, in general, did not segregate to specific viral families. Therefore, the genomic and evolutionary conservation of these kinases has not been fully maintained at the functional level. Our data indicate that these related kinases, some of which are targets of approved or developmental antiviral drugs, are likely to serve both overlapping and non-overlapping functions during viral infections.
Introduction
Kinases catalyze the transfer of phosphate groups onto targeted substrates. These phosphorylation events are fundamental to proper cellular function and viability and help control enzyme activity, signaling cascades, and protein trafficking, as well as a multitude of other pathways and processes [1]. The enzymatic activity of many kinases can be inhibited both potently and specifically with small molecules, making them suitable drug targets for clinical applications. In fact, kinases are now targets for lung and breast cancer chemotherapies [2], and importantly, for herpesviral infections as well [3], [4].
The human herpesviruses (HHVs) are large, enveloped viruses with double-stranded DNA genomes. Based on sequence homology and cellular tropisms, they are divided into three families, alpha, beta, and gamma. The alpha-herpesviruses are Herpes Simplex Virus type 1 (HSV-1, HHV-1), type 2 (HSV-2, HHV-2), and Varicella Zoster Virus (VZV, HHV-3). These viruses complete productive (lytic) replication cycles in epithelial cells, and establish life-long latency in sensory neurons. They cause recurrent and sometimes painful skin lesions and, in rare cases, meningitis [5], [6]. The beta-herpesviruses are Human Cytomegalovirus (HCMV, HHV-5), and the roseolaviruses Human Herpesviruses 6A and 6B (HHV-6), and Human Herpesvirus 7 (HHV-7). These viruses are classified as lymphotropic because lymphocytes likely serve as a latent reservoir, but they replicate productively in many cell types. Beta-herpesviruses cause severe disease in patients with immature or compromised immune function, and may exacerbate certain chronic ailments in otherwise healthy patients [7], [8]. The gamma-herpesviruses are Epstein-Barr Virus (EBV, HHV-4) and Kaposi's Sarcoma Associated Herpesvirus (KSHV, HHV-8). These viruses are also characterized by a lymphocyte tropism, but can also infect epithelial and endothelial cells. They are causally associated with human cancers [9], [10].
As obligate intracellular parasites, viruses must manipulate cellular processes to facilitate their own replication. Because protein phosphorylation events are so crucial to cellular activity, it is not surprising that they are key targets of regulation during viral infections. While many (perhaps all) families of viruses manipulate the activity of cellular kinases, only two families, the poxviruses [11], [12] and the herpesviruses [13], [14], [15] encode viral proteins with confirmed kinase activity. However, rotavirus, a member of the Reoviridae family of double stranded RNA viruses, encodes a protein called NSP5 that may be a functional kinase [16].
At least eighteen human herpesvirus proteins are reported to possess protein kinase activity. Sixteen of these are grouped into three distinct families, (Us3, UL13 and thymidine kinase) based on amino acid sequence homology (Table 1). The other two human herpesvirus proteins reported to have kinase activity are HHV-2 ICP10 [17] and HHV-5 pp65 [18]. These proteins do not appear to be members of any known viral kinase family. Among the conserved kinase families, only alpha-herpesviruses encode the Us3 family of kinases [14] that, among other functions, prevent apoptosis [19], [20] and disrupt the nuclear lamina [21], [22], [23]. Both the alpha - and gamma-herpesviruses encode thymidine kinase family members that, as their name implies, phosphorylate nucleosides, including thymidine [24]. Importantly, viral thymidine kinases can also phosphorylate unnatural nucleoside analogs (such as ganciclovir and its derivatives) that act as chain terminators for viral DNA replication, and constitute an important subset of anti-herpesviral drugs [24]. The third family of herpesviral kinases was originally termed the UL13 family [25], [26]. However, because these represent the only homologous kinases (at the level of genome position and amino acid sequence) found in every human herpesvirus, they were renamed the CHPKs for conserved herpesvirus-encoded protein kinases [13], [27]. The individual members of the human CHPKs are named UL13 (HHV-1 and -2), ORF47 (HHV-3), BGLF4 (HHV-4), UL97 (HHV-5), U69 (HHV-6 and -7), and ORF36 (HHV-8) (Table 2).

The CHPKs are not absolutely required for viral replication in cell culture, but deletion mutants are severely attenuated for viral growth [28], [29], [30], [31], [32]. They are expressed with early-late kinetics and are incorporated into virions [33], [34], [35], [36], [37]. Demonstrated or postulated functions for the CHPKs during viral replication include tegument disassembly [38], [39], modulation of gene expression [28], [31], [40], stimulation of viral DNA replication [31], [41], [42], [43], and facilitating capsid nuclear egress in part through disruption of the nuclear lamina [31], [44], [45]. Recently, the CHPK encoded by the UL97 gene of HHV-5 (HCMV) was shown to directly phosphorylate the cellular retinoblastoma (Rb) tumor suppressor protein both in vivo and in vitro, on residues that are normally targeted by the cellular cyclin-dependent kinase (Cdk) proteins that control cell cycle progression [46], [47]. UL97 was also found to phosphorylate lamin A/C proteins in vitro on Cdk phosphorylation sites [45], and to rescue the G1-to-S cell cycle defect of Saccharomyces cerevisiae cells lacking Cdk function. Significantly, this yeast complementation assay demonstrated that UL97 can functionally substitute for cellular Cdks [46], indicating that the kinase has Cdk activity, and marking UL97 as the first identified v-Cdk, an abbreviation for the term virally-encoded Cdk-like kinase.
Here we show that the CHPKs encoded by the beta - and gamma-herpesviruses are all capable of inducing Rb phosphorylation in vivo on residues that inactivate the cell cycle inhibitory and tumor suppressor function of this protein. They can also induce lamin A phosphorylation and disrupt the nuclear lamina. Importantly, all the beta - and gamma-herpesvirus CHPKs, with the exception of the HHV-8 (KSHV) ORF36 protein, displayed authentic Cdk function in the yeast complementation assay. The alphaherpesvirus CHPKs were unable to phosphorylate Rb or lamin A, efficiently disrupt the nuclear lamina, or act as Cdks in S. cerevisiae. When we assayed all eight kinases for additional non-Cdk functions against cellular proteins that were previously reported for UL97, we found that despite the evolutionary conservation of these proteins, functional conservation was poor. Altogether, our study identifies a subset of the CHPKs as viral Cdk-like kinases (v-Cdks), but also indicates that the positional homology and amino acid similarity of these protein kinases does not always translate into common substrates or similar biological activities for these proteins.
Results
Analysis of the steady state levels and sub-cellular localization of ectopically-expressed, epitope-tagged conserved herpesvirus protein kinases (CHPKs)
The CHPKs are grouped as a kinase family based on their conserved genome location and limited sequence homology. While some common functions for select members of the CHPK family have been identified, a thorough functional comparison of this group of kinases has not been reported. Upon the revelation that the HHV-5 (HCMV) CHPK, the UL97 protein, was a viral Cdk ortholog [46], we initiated experiments to determine if the other CHPKs were also v-Cdks. We obtained an expression plasmid for the HHV-2 CHPK which, upon transfection into mammalian cells, produces an active kinase that is tagged with the hemagglutinin (HA) epitope [48]. We then generated individual plasmids that express HA epitope-tagged derivatives of the other seven CHPKs (Table 2). Previous reports indicate that epitope-tagging does not eliminate the kinase activity of the seven CHPKs for which such an analysis has been conducted [48], [49], [50], [51], [52], [53], [54], [55]. The activity of an epitope-tagged CHPK of HHV-7 has not been previously examined. Verification of the correct CHPK sequence by direct analysis (data not shown) and our finding that each tagged kinase scored positive in at least one activity assay (Table 3) provide confidence that our expression plasmids produced active kinases. Although we cannot rule out changes in substrate specificity or specific activity due to the epitope tag, we do note that tagged CHPKs can complement the growth defects of HHV-4 and HHV-5 mutant viruses lacking the untagged (wild type) CHPK gene [52], [56]. Importantly, the epitope tag allows us to monitor the expression and localization of these eight different kinases with the same antibody. Individual transfection of these eight plasmids into human U-2 OS cells allowed for the production of the viral proteins which, when detected on Western blots with an HA antibody, migrated near the predicted molecular weight (Table 2) of the full-length protein [34], [35], [57], [58], [59], [60], [61]. Adjusting the amount of transfected plasmid DNA allowed us to identify experimental conditions under which the steady state protein levels achieved for six of these eight different proteins was consistent and comparable. However, the CHPKs encoded by HHV-2 and HHV-5 often accumulated to lower levels. Therefore, Western blot expression controls are presented for each individual experiment.

The sub-cellular localization of the transfected CHPKs in U-2 OS cells was examined by fluorescence microscopy (Fig. 1A) and quantitated (Fig. 1B) by visually determining the percentage of cells with primarily nuclear localization of the kinase, primarily cytoplasmic localization, or localization to both cellular compartments. All of the kinases were found in both the nucleus and cytoplasm in at least 25% of the transfected cells. The alpha-herpesvirus CHPKs (HHV-1, -2, and -3) generally showed both strong cytoplasmic and faint nuclear staining. Certain members of the beta - and gamma-herpesvirus CHPKs showed predominantly nuclear staining (HHV-4, -5, and -6), while others (HHV-7 and -8) were more likely to be found in both cellular compartments. These observed sub-cellular localizations are in agreement with previously published data [34], [35], [59], [60], [62], [63].

Expression of the beta - and gamma - CHPKs leads to Rb phosphorylation
The retinoblastoma (Rb) and lamin A/C proteins are phosphorylated by the cyclin-dependent kinases (Cdks) that control cell cycle progression. Rb is a tumor suppressor responsible for regulating the G1/S cell cycle checkpoint [64]. Phosphorylation of Rb on specific Cdk-consensus sites results in the dissociation of protein complexes between Rb, histone deacetylases (HDACs), and E2F transcription factors and allows for the expression of E2F-responsive genes that drive progression through G1 and entry into the S-phase of the cell cycle [64]. Certain E2F-responsive genes also play critical roles in nucleotide biosynthesis and DNA replication. Many DNA viruses partially rely on cellular machinery for the replication of their genomes, and therefore target Rb for inactivation during infection [65], [66]. For example, during HHV-5 (HCMV) infection, the UL97 CHPK is necessary and sufficient for Rb inactivation through phosphorylation on Cdk consensus sites [46]. Rb inactivation appears to be a critical function of UL97 for efficient viral replication [67].
In vivo phosphorylation of Rb by an ectopically-expressed kinase can be easily and reliably determined only in human Saos-2 cells. These Rb-null osteosarcoma cells fail to phosphorylate ectopically-expressed Rb unless a cyclin protein (cellular or viral) or UL97 is included in the transfection [46], [68], [69]. Rb phosphorylation in Saos-2 cells can be observed on Western blots by an electrophoretic mobility shift of the protein to higher molecular weights, as well as with phospho-specific antibodies that detect Rb proteins only when phosphorylated on certain Cdk consensus sites. We found that the CHPKs encoded by all of the beta - and gamma-herpesviruses (HHV-4, -5, -6, -7, and -8) were able to phosphorylate Rb when co-transfected into Saos-2 cells (Fig. 2A). Phosphorylation was detected by both the shift in molecular weight and with three independent phospho-specific antibodies that detect Rb only when phosphorylated on specific Cdk consensus sites that regulate association with the E2F proteins [70], [71]. Interestingly, the beta-herpesvirus CHPKs induced the phosphorylation of Rb-inactivating residues to a substantially higher degree than the gamma-herpesvirus proteins (Fig. 2A). The alpha-herpesvirus proteins (HHV-1, -2, and -3) were unable to phosphorylate Rb (Fig. 2A), as judged by both electrophoretic mobility and the lack of reactivity with the phospho-specific antibodies. The ability of the beta - and gamma-herpesvirus CHPKs to phosphorylate Rb required that each protein was an active kinase. Substitution of the catalytic lysine to create kinase-inactive mutants inhibited the ability of the CHPKs to phosphorylate Rb, shown by both molecular weight shift as well as with the phospho-Rb-specific antibodies (Fig. 2B).

There was no correlation between steady state expression level and the ability to phosphorylate Rb (Fig. 2A). CHPKs that accumulated to high levels either did (e.g. HHV-4 and HHV-7) or did not (e.g. HHV-3) phosphorylate Rb. Likewise, CHPKs that accumulated only to low levels either did (e.g. HHV-5) or did not (e.g. HHV-2) phosphorylate Rb. To further explore any potential relationship between expression level and Rb phosphorylation, we decreased the amount of HHV-4 and HHV-7 CHPK expression plasmid transfected in an attempt to equalize, as much as possible, the steady-state expression levels of these Rb-phosphorylating kinases to the non-Rb-phosphorylating HHV-2 kinase. We found that lower steady state levels of the HHV-4 and HHV-7 CHPK still led to Rb phosphorylation (Fig. 2C), as seen by the shift in apparent molecular weight. While substantial kinase overexpression may result in the phosphorylation of non-physiologic substrates (false-positives), and low expression levels may prevent detection of the phosphorylation of true substrates (false-negatives), the data we present here are consistent with the conclusion that the beta - and gamma-herpesvirus CHPKs, but not the alpha-herpesvirus CHPKs, result in Rb phosphorylation when ectopically expressed.
The beta - and gamma - CHPKs induce lamin A phosphorylation and lamina disruption
Like Rb, lamin A/C is also a Cdk substrate phosphorylated by the CHPKs of HHV-4 and HHV-5 on Cdk consensus sites [45], [72]. Lamins are intermediate filament proteins that line the inner nuclear membrane as part of the nuclear lamina [73]. During mitosis, the nuclear lamina is disassembled after phosphorylation of lamin A/C by Cdk1 [73]. During viral infections, the nuclear lamina likely represents a physical barrier to herpesviral capsids that must leave the nucleus through envelopment at the inner nuclear membrane. Thus, it has been proposed that in order to gain access to the inner nuclear membrane, herpesviruses must disrupt the nuclear lamina [74]. In cells infected with HHV-1, HHV-4, and HHV-5, lamina disruption is achieved through phosphorylation of lamin A/C on Cdk consensus sites [23], [45], [72].
We found that beta - and gamma-herpesvirus CHPKs (HHV-4, -5, -6, -7, and -8), but not those of the alpha-herpesviruses (HHV-1, -2, and -3), were able to efficiently disrupt the nuclear lamina in transfected U-2 OS cells (Fig. 3A). The nuclear lamina was visualized by fluorescence microscopy in cells co-transfected with expression plasmids for the CHPKs and human lamin A fused to GFP. Western blots of the ectopically-expressed proteins are shown in Figure S1. Non-confocal fluorescent images of the transfected cells show a dim GFP signal throughout the body of the nucleus that is brighter where lamin A/C concentrates at the nuclear periphery (see the panel with empty vector co-transfected cells, Fig. 3A, bottom right). Co-expression (visualized by indirect immunofluorescence with an HA antibody) of the beta - and gamma-herpesvirus CHPKs, and to a lesser extent the HHV-2 CHPK, caused a redistribution of GFP from the ring-like signal around the nuclear perimeter into large punctate spots found throughout the nucleus (Fig. 3A) in the majority of cells imaged. For these CHPKs, the percentage of kinase-positive cells showing disrupted nuclear lamina was statistically different than cells transfected with GFP-lamin A and an empty vector control (Fig. 3B). The HHV-1 and HHV-3 alphaherpesvirus CHPKs did not visibly disrupt the nuclear lamina at a frequency distinguishable from empty vector transfected cells.

We also examined the ability of ectopically-expressed CHPKs to direct the phosphorylation of the endogenous lamin A protein at serine-22. Phosphorylation of this and other lamin A residues by cellular Cdk1 initiates the process of lamina breakdown during mitosis [75]. Matching our results from the GFP-lamin experiments (Fig. 3A and 3B), the alpha-herpesvirus CHPKs were unable to induce serine-22 phosphorylation (Fig. 3C and 3F), whereas the beta - (Figure 3D and 3F) and gamma-herpesvirus CHPKs (Fig. 3E and 3F) did induce serine-22 phosphorylation. Kinase-inactive mutants of the beta - and gamma-herpesvirus CHPKs were unable to induce serine-22 phosphorylation. Western blots of the ectopically-expressed CHPKs are shown in Figure S2, and quantitation of the data is shown in Figure 3F.
The HHV-2 CHPK showed low level lamina disruption that was statistically different from an empty vector control (Fig. 3B). A similar level of serine-22 phosphorylation was observed, although the difference from the empty vector control did not reach statistical significance. Thus, although the HHV-2 CHPK may retain some ability to phosphorylate lamin A, (perhaps more efficiently in the tail region) and to disrupt the nuclear lamina [76], we conclude that these activities are, in general, absent from the alpha-herpesvirus CHPKs, but present in the beta - and gamma-herpesvirus CHPKs. Although experiments of this type are often cited as evidence that CHPK-mediated lamin A/C phosphorylation directly leads to lamina disruption, we note that they cannot rule out other mechanisms for lamina disruption upon CHPK expression or herpesvirus infection (such as lamin B or lamin receptor phosphorylation) in addition to or instead of lamin A/C phosphorylation.
Beta - and gamma-herpesvirus CHPKs function as cyclin dependent kinases (Cdks)
The first human Cdk was cloned by genetic complementation of Cdk mutant yeast cells [77]. Unlike higher eukaryotes, S. cerevisiae encodes only a single Cdk, the CDC28 gene. Yeast cells harboring a temperature-sensitive mutant allele of this gene (cdc28-13) exhibit growth arrest in G1 phase upon a shift to the restrictive temperature [78]. Expression of a functional human Cdk in cdc28-13 cells rescues this arrest phenotype [79]. Expression of the HHV-5 (HCMV) CHPK, the UL97 protein, in cdc28-13 yeast cells also rescued the cell cycle arrest phenotype at the restrictive temperature, indicating that it has authentic Cdk activity, and identifying UL97 as the first known viral functional ortholog of a Cdk [46].
We used the same assay to determine if the other CHPKs were also genuine Cdks. Asynchronously growing cdc28-13 yeast cells harboring plasmids expressing the CHPKs under the control of the GAL1 promoter were shifted to the restrictive temperature to inactivate the sole yeast Cdk, and grown in the presence of galactose to induce the expression of the CHPK. Western blots of the ectopically-expressed CHPKs are shown in Figure S3. Five hours later, cell cycle progression through G1 was verified by determining the number of budded cells. In the absence of an ectopic Cdk, cdc28-13 cells grown at the restrictive temperature arrested in the G1 phase of the cell cycle as unbudded cells [46], [78], (Fig. 4). However, as was previously observed [46], ectopic expression of human Cdk1, and to a lesser extent the HHV-5 CHPK permitted cell cycle progression through G1 and into S phase of cdc28-13 cells grown at the restrictive temperature, as evidenced by an increase in the number of budded cells (Fig. 4). This result is indicative of authentic Cdk function. Expression of the other beta-herpesvirus CHPKs (HHV-6 and -7) and the CHPK from the HHV-4 gamma-herpesvirus also rescued the cell cycle defect of cdc28-13 yeast grown at the restrictive temperature (Fig. 4), indicating that they also have genuine Cdk activity. The HHV-8 gamma-herpesvirus CHPK, the ORF36 protein of KSHV, failed to produce a statistically significant increase in cdc28-13 budded cells at the restrictive temperature (Fig. 4), although it did display substantial Cdk activity in one of the three experiments performed. Interestingly, KSHV is the only human herpesvirus to encode a viral cyclin [80]. The KSHV cyclin pairs with and activates human Cdks, perhaps obviating the need for Cdk function of the viral CHPK. As expected from their inability to phosphorylate Rb or lamin A/C, the alpha-herpesvirus CHPKs did not display Cdk activity in this assay. Note that we cannot rule out the possibility that the alpha-herpesvirus proteins might not be active when expressed in yeast cells. In total, the Rb, lamin, and yeast experiments functionally define the beta - and gamma-herpesvirus CHPKs as viral cyclin-dependent-like kinases, or v-Cdks.

The HHV-4 protein (EBV-BGLF4) is the CHPK that most efficiently disrupts PML-NBs
While the CHPKs are grouped as a family based upon their positional homology and limited amino acid similarity, our data indicate that only the beta - and gamma-herpesvirus CHPKs are v-Cdks. These observations indicate that the evolutionary relationship of these eight kinases does not translate to a complete conservation of Cdk-like function. To determine if other functions of these kinases may have been more widely conserved throughout evolution, we screened each kinase for their ability to disrupt PML-NBs, nuclear aggresomes, and cytoplasmic aggresomes, all Cdk-unrelated activities that have been previously attributed to UL97, the HHV-5 (HCMV) CHPK [47].
Promyelocytic Leukemia Nuclear Bodies (PML-NBs) are sub-nuclear structures that appear as multiple punctate spots within the nucleus and are involved in a wide array of cellular activities [81]. During infections with HHV-1, new PML-NBs are generated at the sites where infecting viral genomes enter the nucleus [82]. HHV-5 genomes also co-localize with PML-NBs shortly after infection [83]. Proteins that localize to PML-NBs institute an intrinsic cellular defense against these herpesviruses by silencing viral immediate early (IE) gene expression [84], [85]. Viral proteins delivered to cells upon virion entry and/or expressed as IE genes inactivate this defense and disrupt PML-NBs [86], [87], [88].
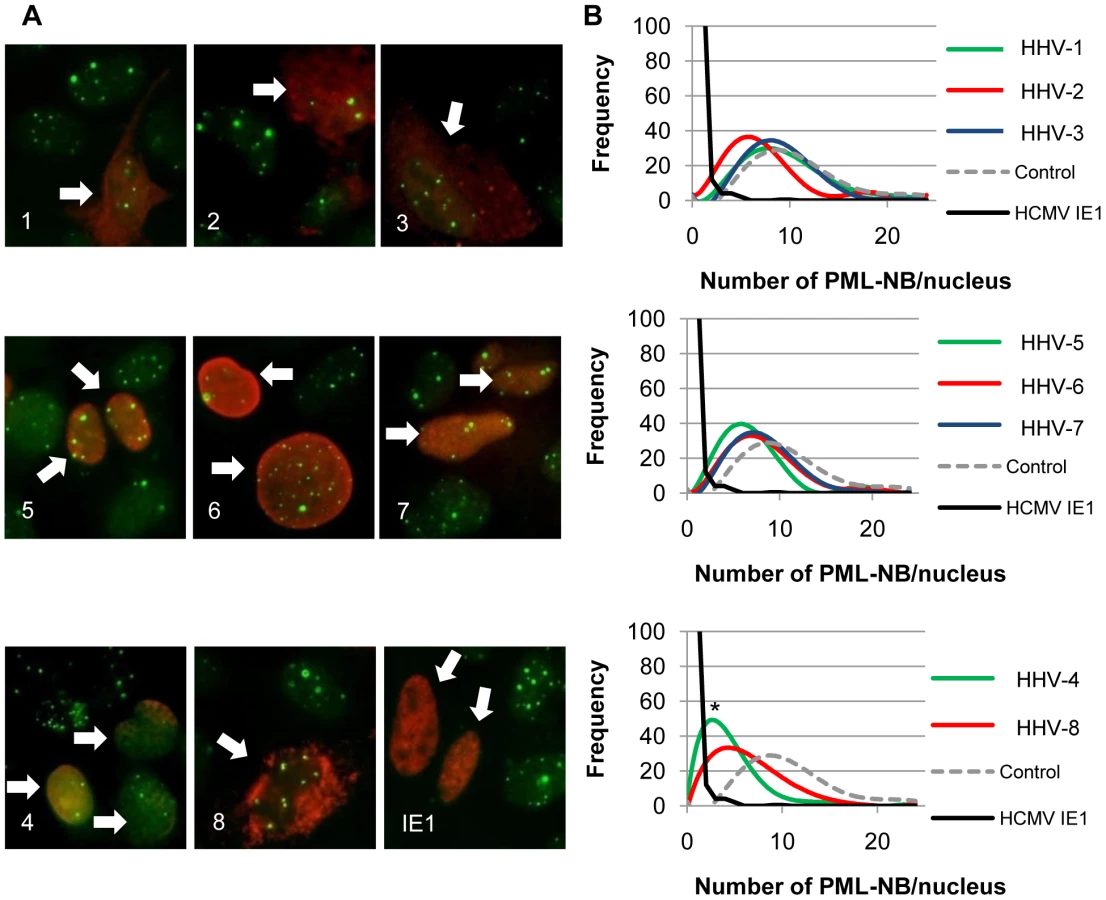
We used U-2 OS cells and indirect immunofluorescent detection of the PML protein (Fig. 5A) to monitor the effects of ectopically-expressed CHPKs on PML-NBs. Western blots of the ectopically-expressed CHPKs are shown in Figure S4. As expected, we found that the number of PML-NBs in U-2 OS cells resembles a Gaussian distribution between 4 and 24 (Fig. 5B), with an average of 11 per cell (Table 3). Our average agrees well with previously reported data [63]. We used HCMV IE1 as a positive control for PML-NB disruption [86], and counted the number of PML-NBs in 100 CHPK-positive nuclei in three separate experiments. With the exception of HHV-1 and HHV-3, expression of all of the other CHPKs resulted in a perceptible shift in the distribution of PML-NBs per cell towards lower numbers (Fig. 5B). Most of the shifts were small, although the HHV-5 and HHV-8 CHPKs showed more substantial effects. However, only the HHV-4 CHPK caused a drop in the average number of PML-NBs per cell that was statistically different than control cells (Table 3).
Disruption of nuclear and cytoplasmic protein aggregates by the CHPKs
High synthesis rates, cellular stresses, and other stimuli can lead to protein aggregation and the formation of aggresomes. Ectopic expression of the HHV-5 (HCMV) CHPK (UL97) in uninfected cells has been shown to disrupt aggregates of viral proteins as well as nuclear and cytoplasmic aggresomes formed by GFP-GCP170*, an artificial stimulator of aggresome formation [47], [53]. Conversely, GFP-GCP170* aggregates have been detected in HHV-1 infected cells [89], implying that the HHV-1 CHPK may not disrupt aggresomes.
To determine if aggresome disruption was a general feature of the CHPKs, we tested the ability of the kinases to disrupt nuclear or cytoplasmic aggresomes formed by the ataxin-1 protein. Ataxin-1 (Atx1) has a poly-glutamine tract that, when expanded beyond 40 amino acids, produces a protein with a non-native conformation that forms aggregates, is cytotoxic, and causes spinocerebellar ataxia type 1 [90]. We co-transfected cells with the CHPKs and FLAG-tagged derivatives of either Atx1(Q82) or Atx1(Q82)-K772T that form aggregates in the nucleus and cytoplasm, respectively [90]. Western blots of the ectopically-expressed proteins are shown in Figure S5. The presence of at least one aggregate or the total absence of aggregates in ataxin-expressing cells (Fig. 6A and 6C) was determined for at least 200 CHPK-positive cells in at least three independent experiments. We found that the HHV-2 and HHV-5 CHPKs resulted in a statistically significant decrease in the number of cells in which ataxin-1 could form both nuclear (Fig. 6A and 6B) and cytoplasmic (Fig. 6C and 6D) aggregates even though these were the CHPKs expressed to the lowest level (Figure S5). CHPKs encoded by HHV-1, -3, -4, and -6 caused a statistically significant decrease in cells with either nuclear or cytoplasmic aggresomes, but not both (Fig. 6). While the HHV-4 CHPK caused a substantial decrease in the percentage of cells containing nuclear aggresomes, the wide variability in the numbers accumulated over three independent experiments resulted in data that did not achieve statistical significance (Fig. 6B). Our data confirm previous results demonstrating that the HHV-5 CHPK can either prevent or disrupt protein aggregation [47], but also indicate that this ability is not well conserved among the eight CHPKs.

In summary, we found that the most conserved activity of the CHPKs is their Cdk-like function. However, this is only found in the beta-and gamma-herpesvirus members, and is absent in the alpha-herpesvirus proteins. The non-Cdk-like activities reported for the HHV-5 CHPK, the HCMV UL97 protein, are poorly conserved among the other family members. Thus the functional conservation between these eight kinases as a whole, much like their amino acid sequence similarity, is limited.
Discussion
Cellular substrates of selected CHPKs that are normally phosphorylated by the Cdks in uninfected cells have been identified. These include Rb [46], [47], lamin A/C [45], [72], translation elongation factor 1 delta [49], the carboxy-terminal domain of RNA Polymerase II [91], the helicase complex component MCM4 [92], the condensin protein involved in chromatin packaging [93], and the Cdk inhibitor p27 [51]. However, only the CHPK encoded by HHV-5, the HCMV UL97 protein, had been shown to be a functional Cdk ortholog [46]. In this study, we examined the remainder of the CHPKs for Cdk-like activity, as well as non-Cdk-like activities previously reported for UL97. We employed the yeast complementation assay because it is the most basic and stringent test for Cdk activity, and the Saos-2 assay because it is the most widely-accepted assay for demonstrating Rb phosphorylation in vivo by a co-transfected protein. For all other experiments, we used U-2 OS cells because they transfect well, do not express a transforming viral oncogene, and represent a generic human cell (the differing cellular tropisms of the eight human herpesviruses prevented us from using a more physiologically relevant cell type). Effects on cellular targets or processes (as opposed to viral ones) were analyzed so that activity against identical (as opposed to just homologous) substrates could be monitored.
Our results indicate that Cdk-like activity is absent in the alpha-herpesvirus CHPKs, as they were unable to induce Rb or lamin A phosphorylation, lamina disassembly, or yeast cdc28-13 cell cycle progression. These results agree well with the observations that Rb is not phosphorylated in cells infected with HHV-1, HHV-2, or HHV-3 [66], but is phosphorylated in cells infected with HHV-4 and HHV-5, [94], [95], representative members of the beta - and gamma-herpesvirus families. Our lamin results are also mostly consistent with previously published data. Although it is clear that lamin A/C becomes phosphorylated in cells infected with representative members of the alpha - [23], beta - [45], and gamma - [72] herpesviruses, different kinase families appear to be responsible for these phosphorylation events. A recent analysis [72] found that, in addition to the HHV-4, HHV-5 [96], and HHV-8 CHPKs, the HHV-1 CHPK was able to disrupt the nuclear lamina in HeLa cells [72], however, we detected no activity of the HHV-1 protein in two independent lamin assays (Fig. 3). It is unclear if expression of the papillomavirus E7 oncoprotein in HeLa cells, a known stimulator of Cdk activity [97] that is not present in the U-2 OS cells used here, could explain this difference between the two studies. Thus, although previous studies [72], [76] and our own data with the HHV-2 CHPK (Fig. 3) indicate that the alpha-herpesvirus CHPKs may retain some ability to phosphorylate lamin A/C and to disrupt the nuclear lamina, we suspect, as others have shown, that the members of the alpha-herpesvirus-specific Us3 family of kinases are largely responsible for this activity in virus-infected cells [21], [22], [23], [98]. Thus, we conclude that the alpha-herpesvirus CHPKs do not mimic cellular Cdk activity. Interestingly, cellular Cdks are active during, and required for efficient alpha-herpesvirus replication [66]. The critical substrates for cellular Cdks in alpha-herpesvirus infected cells remain to be determined.
In contrast, the beta - and gamma-herpesvirus CHPKs were found to possess Cdk-like activity. All of these proteins phosphorylated Rb and disrupted the nuclear lamina, and all but one rescued cell cycle progression in the yeast complementation assay. It is presently undetermined what (if any) advantage encoding and expressing their own Cdk-like protein affords these viruses that utilization of cellular Cdk activity would not. Nevertheless, this analysis, along with our previous study [46] identifies the HHV-4 (EBV-BGLF4), HHV-5 (HCMV-UL97), HHV-6 (U69), HHV-7 (U69), and HHV-8 (KSHV-ORF36) CHPKs as virally-encoded cyclin-dependent-like kinases, or v-Cdks.
Unlike cellular Cdks, the HHV-5 CHPK (HCMV UL97), the first identified v-Cdk, lacks many of the regulatory features that restrict kinase activity during certain stages of the cell cycle or under physiological stresses [46]. For example, UL97 lacks conserved sequences for cyclin binding and appears to have cyclin-independent kinase activity. UL97 has an amino acid substitution at the Cdk site (Thr 160 of Cdk2) of phosphorylation by Cdk activating kinase (CAK), and is active under conditions of CAK inhibition. UL97 lacks amino acid residues important for binding both the Cip/Kip and INK classes of cyclin-dependent kinase inhibitors, and is immune to inhibition by p21WAF1/CIP1 in vivo and in vitro. Finally, UL97 has a phenylalanine substitution for a tyrosine residue (Tyr 15 of Cdk2) found in Cdks which, when phosphorylated in G2 phase or in response to DNA damage, attenuates kinase activity.
A sequence alignment of the other v-Cdks showed that, like UL97, none of these kinases contain the cyclin-binding, cyclin-dependent kinase inhibitor-binding, or CAK phosphorylation motifs found in the cellular Cdks (Fig. 7). Thus, the newly-identified v-Cdks lack many of the amino acids and protein domains responsible for regulating the activity of their cellular functional orthologs. Interestingly, all of the v-Cdks except UL97 and KSHV ORF36 (the HHV-8 CHPK) do contain the tyrosine residue that, in cellular Cdks, allows for the attenuation of kinase activity upon phosphorylation. Whether or not this amino acid substitution affects kinase activity during viral infection remains to be examined.
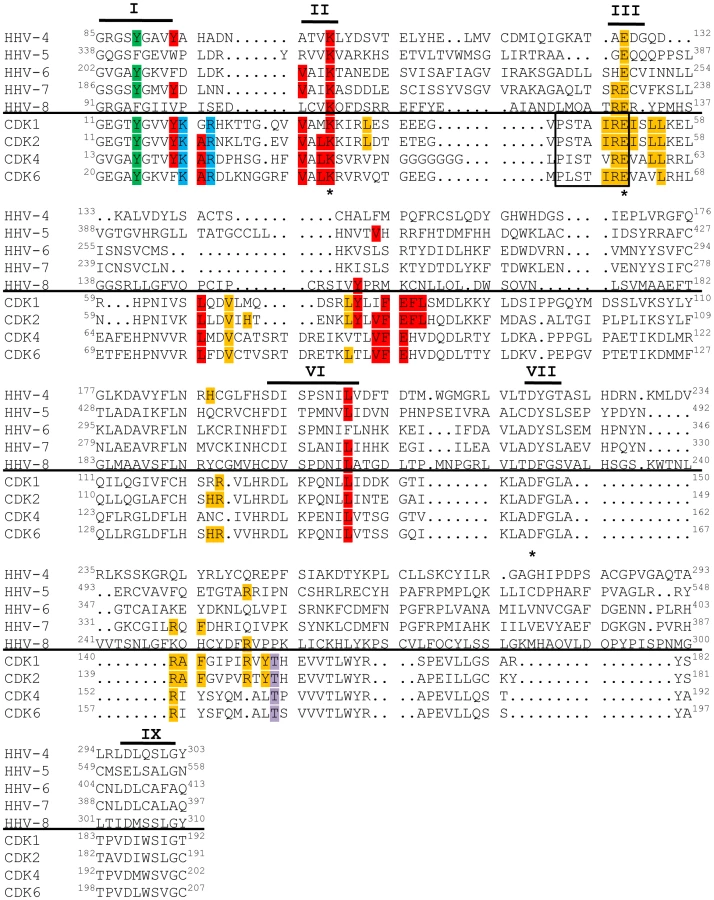
In the past, designation as a Cdk required not only sequence and functional homology to known Cdks, but also experimental evidence that the activity of the kinase was dependent upon association with a cyclin regulatory subunit [99]. However, a recent nomenclature adjustment has grouped all human and murine kinases with sequence and functional similarity to Cdks into the Cdk family, even if cyclin binding has not been observed or is not expected [100]. Thus, while the beta - and gamma-herpesvirus CHPKs do not appear to require cyclin binding for activity, describing these proteins as v-Cdks is in accordance with the accepted classification system because they are related to true Cdks by amino acid sequence (Fig. 7), and because they mimic Cdk activity (Fig. 2, Fig. 3, Fig. 4, Table 3) even though they are likely not regulated in an identical fashion to cellular Cdks.
The lack of conservation of Cdk activity in the alpha-herpesvirus CHPKs was surprising based on the strong evolutionary relationship of this family of proteins, and prompted us to ask if non-Cdk-like functions were more conserved throughout the CHPK family. Interestingly, we found that non-Cdk-like functions were even less well conserved than Cdk activity (Table 3). Only the HHV-4 CHPK mediated a decrease in PML-NB numbers per cell that reached statistical significance, although other kinases, most notably the HHV-5 and HHV-8 CHPKs, clearly appeared to affect these structures. This finding varies considerably from previous reports, perhaps because of different experimental approaches. Our study used a version of the HHV-4 CHPK tagged at the amino-terminus with the 9 amino acid HA epitope tag, while a previous examination of this protein that failed to demonstrate PML-NB disruption [63] used a 69 amino acid SPA tag at the carboxy-terminus. The differences in tag size and location may affect kinase activity, which was not directly examined in the other study [63], but has been demonstrated here (Fig. 2). A study that concluded the HHV-5 CHPK disrupted PML-NBs [47] was conducted in Cos7 cells that express the simian virus 40 (SV40) tumor (T) antigen, and monitored Sp100 localization as a measure of PML-NBs. Our study visualized PML, the sole protein required for PML-NB formation, in U-2 OS cells that do not express a transforming viral oncogene. It is currently unknown which if any of these differences may explain why the subtle effects on PML-NB distribution that we observed upon expression of the HHV-5 CHPK failed to reach statistical significance.
Proteins that efficiently disrupt PML-NBs (HCMV IE1 and EBV Zta) are expressed within infected cells in vitro prior to the time when the CHPKs are expressed [101], [102], so the ability of the HHV-4 and HHV-5 CHPKs to decrease the numbers of PML-NBs when ectopically expressed may have limited relevance during viral infection. Furthermore, because the proteins that localize to PML-NBs and not the PML-NB structures themselves appear to be the mediators of an intrinsic antiviral defense [84], [85], the significance of the ability of these viral proteins to reduce the number of, but not eliminate PML-NBs from cells is unclear, especially since it is unknown whether or not any PML-NB resident proteins are direct substrates of these kinases. Therefore, perhaps unsurprisingly, the ability to disrupt PML-NBs appears to be an activity not well conserved throughout the CHPK family.
Disruption of protein aggregates termed aggresomes is another non-Cdk function attributed to the HHV-5 CHPK. Aggresomes have been hypothesized to represent a cellular anti-viral defense that identifies, initially sequesters, and ultimately disposes of viral proteins to inhibit viral replication [103]. Thus, aggresome disruption could potentially enhance viral infections. However, aggresome formation is also thought to sequester misfolded proteins and perhaps facilitate their degradation by the proteasome or through autophagy [103]. In addition, viruses may commandeer aggresome formation pathways to help form replication or assembly compartments [103]. In these respects, aggresome formation would appear to enhance virus replication.
Different human herpesviruses appear to present examples where aggresome formation has opposing effects on viral replication. Structures morphologically resembling aggresomes form in both the nuclei and cytoplasm of cells infected with HHV-1 and -2 (the herpes simplex viruses) or HHV-5 (HCMV). At early times after HHV-1 infection, aggregates containing viral proteins, cellular chaperones, and proteasomes form in the nucleus [104]. These virus-induced chaperone enriched (VICE) domains are proposed to be sites where misfolded proteins are sequestered away from virus replication compartments, perhaps to facilitate either their proper folding or degradation, as well as to prevent the misfolded proteins from triggering cellular processes deleterious to virus infection such as apoptosis or the unfolded protein response [105]. At late times after HHV-2 infection, similar structures form in the cytoplasm [106]. Disruption of these cytoplasmic aggresome-like structures correlates with decreased HHV-2 yields [106], indicating that the integrity of these structures may positively influence viral infection. In contrast, the disruption of aggresomes may facilitate HHV-5 replication. Cells infected with an HHV-5 mutant lacking its CHPK contain nuclear and cytoplasmic aggresome-like structures that are absent during wild type-infection [53], and produce fewer infectious progeny virions than wild-type infection [32]. These findings have led to the hypothesis that aggresomes may represent a cellular anti-viral defense, and that one role for the HHV-5 CHPK during viral infection is to prevent their formation [47].
Only one CHPK from each family (alpha, HHV-2; beta, HHV-5; and gamma, HHV-4) disrupted cytoplasmic aggresomes. However, disruption of nuclear aggresomes was the one activity that could conceivably be considered as conserved among these kinases, even though the magnitude of this disruption was minimal in some cases. All of the three alpha-herpesvirus CHPKs scored positive for this assay, as did two (HHV-5 and HHV-6) of the three beta-herpesvirus CHPKs. While our statistical analysis argues that both gamma-herpesvirus proteins are devoid of this activity, the HHV-4 CHPK clearly appeared to have an effect on these structures.
Because, for the most part, the non-Cdk-like activities we examined do not segregate to specific viral families (as Cdk activity does), we suggest that cellular proteins that are substrates for individual CHPKs, but not the Cdks, may represent a more divergent set of proteins than might be expected based upon the evolutionary relationship of these kinases. Furthermore, our data suggest that the v-Cdks may have an expanded repertoire of substrates as compared to the alpha-herpesvirus CHPKs. This may not be surprising because the alpha-herpesvirus-specific Us3-family kinases appear to assume at least one v-Cdk role, lamina disruption [21], [22], [23], during viral infection. Whether or not a comprehensive analysis of kinase activity against homologous viral proteins would reveal more conservation of function throughout the CHPK family awaits further examination.
An alternative strategy to assay for functional conservation among the CHPKs would be to determine if other members of the kinase family could complement the growth defect observed in viruses lacking their endogenous CHPK. The few experiments that have been performed support the contention that the v-Cdks have a partially overlapping but also extended substrate range when compared to the alpha-herpesvirus CHPKs. Expression of the HHV-5 CHPK from an HHV-1 genome in which its CHPK was deleted resulted in a complete restoration of progeny virion formation at both high and low multiplicities of infection [107], indicating that this v-Cdk can perform all the necessary functions for viral replication normally carried out by the HHV-1 CHPK. However, when expressed from a recombinant adenovirus, the HHV-1 CHPK was unable to rescue the growth of an HHV-5 CHPK null-mutant virus [108]. Interestingly, that same series of experiments demonstrated that the HHV-4 CHPK could partially complement the growth defect of the HHV-5 CHPK mutant virus, indicating that there is substantially more functional overlap within the v-Cdks than between the v-Cdks and the alpha-herpesvirus CHPKs. A comprehensive genetic analysis of inter-virus CHPK complementation should increase our understanding of the evolutionary path that these viral kinases have traveled, as well as helping to define their critical roles during viral infection.
In all eight human herpesviruses, the CHPK gene is found in a conserved genomic block and is flanked on either side by homologous genes [109], [110], likely indicating that the current CHPK genes are descended from a single primordial precursor. Over time, the functions of this putative CHPK precursor appear to have diversified differently among these eight viruses. Thus while they represent a single historical kinase family, it may be more practical to consider the CHPK family as representing two individual clades, the UL13s and the v-Cdks. An evolutionary tree diagram of the sixteen shared human herpesvirus kinases (Fig. 8) clearly shows their segregation into different families and clades. Whether the putative primordial CHPK was a Cdk whose Cdk-like activity was lost in the alpha-herpesvirus lineage, or whether it was a kinase that evolved to acquire Cdk-like function in the beta - and gamma-herpesviruses is an interesting topic for speculation and debate.

Materials and Methods
Cells and transfections
U-2 OS human osteosarcoma cells were grown in a 5% CO2 atmosphere at 37°C in Dulbecco's modified Eagle's medium (Invitrogen) supplemented with 10% (vol/vol) fetal bovine serum (Gemini), 100 U/ml penicillin, 100 ug/ml streptomycin, and 0.292 mg/ml glutamine (Gibco). Cells (2×106) were seeded on 10 cm plates five hours prior to transfection by the calcium phosphate co-precipitation method. After an overnight incubation, the cells were washed twice with medium and then re-fed with serum-containing media (time zero). Different amounts of CHPK expression plasmid DNA were transfected (HHV-2, -5, -7, 15 µg; HHV-1 and -8, 10 µg; HHV-3, 5 µg; HHV-4, 2µg; HHV-6, 1µg) in order to approximately equalize steady state protein levels. Total DNA levels in transfections were balanced with pGEM7 (Promega). Saos-2 cells were grown as above, seeded at a density of 8×105 cells per 60 mm dish, and transfected 5 hours later with TransIT-2020 (Mirus) according to the manufacturer's instructions. Along with 1µg of Rb expression plasmid, different amounts of CHPK expression plasmid DNA were transfected (HHV-2, -5, -5KD, 1.5 µg; HHV-1, -7, -7KD, -8KD, 1 µg; HHV-8, 0.15 µg; HHV-3, -4KD, -6, -6KD, 0.1 µg; HHV-4, 0.05 µg). Transfection conditions for the experiment in Figure 2C were the same except 0.01 µg was transfected for HHV-4, and 0.2 µg for HHV-7.
CHPK expression plasmids
Kinase alleles (except HHV-2) were amplified by PCR, sequence verified (see Table 2), and subsequently cloned into the pCGN vector that adds an N-terminal hemagglutinin (HA) epitope tag and expresses genes from the HCMV major immediate early promoter. HA-tagged alleles for all of the CHPKs except HHV-5 were also cloned into the yeast expression vector pMSS78 under the control of the yeast GAL1 promoter [111]. The plasmid that expresses V5 epitope-tagged HHV-5 CHPK in yeast has been previously described [46]. Kinase-deficient (KD) mutants (Table 2) were created by standard mutagenesis approaches and confirmed by complete sequencing of the mutant allele. PCR templates were as follows: HHV-1, viral genomic DNA strain KOS, a gift from Curtis Brandt; HHV-3, plasmid pCAGGS ORF47.12 [50], a gift from Charles Grose; HHV-4, plasmid pcDNA3.1-FLAG BGLF4 [112], a gift from Thomas Stamminger; HHV-5, ppUL97-V5 [53], a gift from Mark Prichard; HHV-6 and HHV-7, cosmids pMF147-19 [113] and I6 (unpublished) respectively, gifts from Steven Dewhurst; HHV-8, plasmid pND ORF36 [55], a gift from Paul Luciw. Plasmid pSG5-UL13 3′ HA [48] kindly provided by Lynda Morrison, was used to express a kinase-active C-terminally tagged HHV-2 CHPK from the SV40 promoter.
Sequence alignment and phylogenic tree assembly
The sequence comparison was created with a ClustalW alignment in MEGA 4 [114], and utilized only the kinase domains of the indicated proteins. The parameters were as follows: Pairwise alignment - gap opening penalty 2, gap extension penalty 0.05; multiple alignment - gap opening penalty 5, gap extension penalty 1, utilizing the Blosum matrix. The alignment was heuristically adjusted to align kinase domain III. The phylogenic tree was constructed by the neighbor-joining method, in MEGA 4 [114].
Western blots, indirect immunofluorescence, and antibodies
Equal amounts of protein (determined by Bradford assay) from cell lysates prepared in radioimmunoprecipitation assay (RIPA) buffer with protease inhibitors were analyzed by Western blotting (WB) as previously described [115]. Cells grown on glass coverslips were processed for indirect immunofluorescence (IF) as previously described [116]. Images were produced with a Nikon Eclipse TE2000-S or Zeiss Axiovert 200M microscope. The following antibodies were from commercial sources: HA (IF: Roche 3F10; WB: Covance MMS-101P), PML (Santa Cruz sc-966), Total Rb (Cell Signaling 9309), Rb p-Ser807/811 (Cell Signaling 9308), Rb p-Ser780 (Cell Signaling 9307), Rb p-Thr821 (Biosource 44-582G), Lamin A/C (Novocastra NCL-Lam-A/C) Lamin A p-Ser22 (Cell Signaling 2026), Flag (Sigma F1804), V5 (Invitrogen R960-25) Tubulin (DM 1A Sigma), PGK (Invitrogen 459250). The HCMV IE1 antibody 1B12 has been previously described [117].
Assays
Rb Phosphorylation in Saos-2 cells and Cdk activity in S. cerevisiae cdc28-13 cells were determined as previously described [46]. For the nuclear lamina disruption assays, U-2 OS cells grown on coverslips were co-transfected with expression plasmids for the indicated CHPK and pEGFPhLA-WT [72] that expresses a GFP-lamin A fusion protein (a kind gift of David Gilbert). Coverslips were harvested 24h later, stained for the CHPK, and then visualized by fluorescence microcopy. At least 300 CHPK - and GFP-lamin A-positive cells for each co-transfection were analyzed in three independent experiments. For the lamin A/C serine-22 phosphorylation assay, U-2 OS cells grown on coverslips were transfected with expression plasmids for the indicated CHPK. Cells were then incubated in media containing 0.1% FBS for 36 hours before harvesting the coverslips, staining for the CHPK and lamin A phosphorylated at serine-22, and visualization by fluorescence microscopy. At least 200 (wild type) or 100 (kinase-deficient) CHPK-positive cells were analyzed in three separate experiments. For PML-NB disruption assays, U-2 OS cells grown on coverslips were transfected with an expression plasmid for the indicated CHPK or pCGN-IE1 [117]. Coverslips were harvested 24h later, stained for PML and the CHPK, and then visualized by fluorescence microscopy. The number of PML-NBs in 100 individual CHPK - or IE1-positive cells was counted in each of three independent experiments. For the aggresome disruption assays, U-2 OS cells grown on coverslips were co-transfected with expression plasmids for the indicated CHPK and either p3PK-Flag-Ataxin-1 or p3PK-Flag-Ataxin-1 K722T [90] that express, respectively, nuclear or cytoplasmic versions of the ataxin-1 Q82 protein that spontaneously forms large aggregates fused to GFP (kindly provided by Harry Orr). Coverslips were harvested 24h later, stained for the CHPK, and visualized by fluorescence microscopy. At least 200 (nuclear) or 300 (cytoplasmic) CHPK - and ataxin-positive nuclei were analyzed in three (nuclear) or four (cytoplasmic) separate experiments.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. AlbertsB
2002 Molecular biology of the cell New York Garland Sciencexxxiv, [1548]
2. ArslanMA
KutukO
BasagaH
2006 Protein kinases as drug targets in cancer. Curr Cancer Drug Targets 6 623 634
3. TrofeJ
PoteL
WadeE
BlumbergE
BloomRD
2008 Maribavir: a novel antiviral agent with activity against cytomegalovirus. Ann Pharmacother 42 1447 1457
4. DrewWL
1991 Clinical use of ganciclovir for cytomegalovirus infection and the development of drug resistance. J Acquir Immune Defic Syndr 4 Suppl 1 S42 46
5. RoizmanB
KnipeDM
WhitleyRJ
2007 Herpes Simplex Viruses.
DavidM
KnipePMH
Fields' Virology Philadelphia Lippincott Williams and Wilkins 2502 2601
6. CohenJI
StrausSE
ArvinAM
2007 Varicella-Zoster Virus Replication, Pathogenesis, and Management.
DavidM
KnipePMH
Fields' Virology Philadelphia Lipincott, Williams, and Wilkins 2774 2818
7. MocarskiES
ShenkT
PassRF
2007 Cytomegaloviruses.
DavidM
KnipePMH
Fields' Virology Philadelphia Lipincott, Williams, and Wilkins 2702 2772
8. YamanashiK
MoriY
PellettPE
2007 Human Herpesviruses 6 and 7.
DavidM
KnipePMH
Fields' Virology Philadelphia Lipincott, Williams, and Wilkins 2819 2845
9. GanemD
2007 Kaposi's Sarcoma-associated Herpesvirus.
DavidM
KnipePMH
Fields' Virology Philadelphia Lipincott, Williams, and Wilkins 2847 2888
10. KieffED
RickinsonAB
2007 Epstein-Barr Virus and Its Replication.
DavidM
KnipePMH
Fields' Virology Philadelphia Lippincott, Williams, and Wilkins 2603 2654
11. WiebeMS
TraktmanP
2007 Poxviral B1 kinase overcomes barrier to autointegration factor, a host defense against virus replication. Cell Host Microbe 1 187 197
12. PunjabiA
TraktmanP
2005 Cell biological and functional characterization of the vaccinia virus F10 kinase: implications for the mechanism of virion morphogenesis. J Virol 79 2171 2190
13. GershburgE
PaganoJS
2008 Conserved herpesvirus protein kinases. Biochim Biophys Acta 1784 203 212
14. McGeochDJ
DavisonAJ
1986 Alphaherpesviruses possess a gene homologous to the protein kinase gene family of eukaryotes and retroviruses. Nucleic Acids Res 14 1765 1777
15. GriffinAM
BoursnellME
1990 Analysis of the nucleotide sequence of DNA from the region of the thymidine kinase gene of infectious laryngotracheitis virus; potential evolutionary relationships between the herpesvirus subfamilies. J Gen Virol 71 ( Pt 4) 841 850
16. VendeP
TaraporewalaZF
PattonJT
2002 RNA-binding activity of the rotavirus phosphoprotein NSP5 includes affinity for double-stranded RNA. J Virol 76 5291 5299
17. ChungTD
WymerJP
SmithCC
KulkaM
AurelianL
1989 Protein kinase activity associated with the large subunit of herpes simplex virus type 2 ribonucleotide reductase (ICP10). J Virol 63 3389 3398
18. BrittWJ
AugerD
1986 Human cytomegalovirus virion-associated protein with kinase activity. J Virol 59 185 188
19. BenettiL
MungerJ
RoizmanB
2003 The herpes simplex virus 1 US3 protein kinase blocks caspase-dependent double cleavage and activation of the proapoptotic protein BAD. J Virol 77 6567 6573
20. OggPD
McDonellPJ
RyckmanBJ
KnudsonCM
RollerRJ
2004 The HSV-1 Us3 protein kinase is sufficient to block apoptosis induced by overexpression of a variety of Bcl-2 family members. Virology 319 212 224
21. MorrisJB
HofemeisterH
O'HareP
2007 Herpes simplex virus infection induces phosphorylation and delocalization of emerin, a key inner nuclear membrane protein. J Virol 81 4429 4437
22. LeachN
BjerkeSL
ChristensenDK
BouchardJM
MouF
2007 Emerin is hyperphosphorylated and redistributed in herpes simplex virus type 1-infected cells in a manner dependent on both UL34 and US3. J Virol 81 10792 10803
23. MouF
ForestT
BainesJD
2007 US3 of herpes simplex virus type 1 encodes a promiscuous protein kinase that phosphorylates and alters localization of lamin A/C in infected cells. J Virol 81 6459 6470
24. ShugarD
1999 Viral and host-cell protein kinases: enticing antiviral targets and relevance of nucleoside, and viral thymidine, kinases. Pharmacol Ther 82 315 335
25. SmithRF
SmithTF
1989 Identification of new protein kinase-related genes in three herpesviruses, herpes simplex virus, varicella-zoster virus, and Epstein-Barr virus. J Virol 63 450 455
26. CheeMS
LawrenceGL
BarrellBG
1989 Alpha-, beta - and gammaherpesviruses encode a putative phosphotransferase. J Gen Virol 70 ( Pt 5) 1151 1160
27. KawaguchiY
KatoK
2003 Protein kinases conserved in herpesviruses potentially share a function mimicking the cellular protein kinase cdc2. Rev Med Virol 13 331 340
28. PurvesFC
OgleWO
RoizmanB
1993 Processing of the herpes simplex virus regulatory protein alpha 22 mediated by the UL13 protein kinase determines the accumulation of a subset of alpha and gamma mRNAs and proteins in infected cells. Proc Natl Acad Sci U S A 90 6701 6705
29. HeinemanTC
CohenJI
1995 The varicella-zoster virus (VZV) open reading frame 47 (ORF47) protein kinase is dispensable for viral replication and is not required for phosphorylation of ORF63 protein, the VZV homolog of herpes simplex virus ICP22. J Virol 69 7367 7370
30. MoffatJF
ZerboniL
SommerMH
HeinemanTC
CohenJI
1998 The ORF47 and ORF66 putative protein kinases of varicella-zoster virus determine tropism for human T cells and skin in the SCID-hu mouse. Proc Natl Acad Sci U S A 95 11969 11974
31. GershburgE
RaffaS
TorrisiMR
PaganoJS
2007 Epstein-Barr virus-encoded protein kinase (BGLF4) is involved in production of infectious virus. J Virol 81 5407 5412
32. PrichardMN
GaoN
JairathS
MulambaG
KroskyP
1999 A recombinant human cytomegalovirus with a large deletion in UL97 has a severe replication deficiency. J Virol 73 5663 5670
33. TanakaM
NishiyamaY
SataT
KawaguchiY
2005 The role of protein kinase activity expressed by the UL13 gene of herpes simplex virus 1: the activity is not essential for optimal expression of UL41 and ICP0. Virology 341 301 312
34. IzumiyaY
IzumiyaC
Van GeelenA
WangDH
LamKS
2007 Kaposi's sarcoma-associated herpesvirus-encoded protein kinase and its interaction with K-bZIP. J Virol 81 1072 1082
35. StevensonD
ColmanKL
DavisonAJ
1994 Characterization of the putative protein kinases specified by varicella-zoster virus genes 47 and 66. J Gen Virol 75 ( Pt 2) 317 326
36. WangJT
YangPW
LeeCP
HanCH
TsaiCH
2005 Detection of Epstein-Barr virus BGLF4 protein kinase in virus replication compartments and virus particles. J Gen Virol 86 3215 3225
37. van ZeijlM
FairhurstJ
BaumEZ
SunL
JonesTR
1997 The human cytomegalovirus UL97 protein is phosphorylated and a component of virions. Virology 231 72 80
38. AsaiR
KatoA
KatoK
Kanamori-KoyamaM
SugimotoK
2006 Epstein-Barr virus protein kinase BGLF4 is a virion tegument protein that dissociates from virions in a phosphorylation-dependent process and phosphorylates the viral immediate-early protein BZLF1. J Virol 80 5125 5134
39. MorrisonEE
WangYF
MeredithDM
1998 Phosphorylation of structural components promotes dissociation of the herpes simplex virus type 1 tegument. J Virol 72 7108 7114
40. LongMC
LeongV
SchafferPA
SpencerCA
RiceSA
1999 ICP22 and the UL13 protein kinase are both required for herpes simplex virus-induced modification of the large subunit of RNA polymerase II. J Virol 73 5593 5604
41. KroskyPM
BaekMC
JahngWJ
BarreraI
HarveyRJ
2003 The human cytomegalovirus UL44 protein is a substrate for the UL97 protein kinase. J Virol 77 7720 7727
42. MarschallM
FreitagM
SuchyP
RomakerD
KupferR
2003 The protein kinase pUL97 of human cytomegalovirus interacts with and phosphorylates the DNA polymerase processivity factor pUL44. Virology 311 60 71
43. WolfDG
CourcelleCT
PrichardMN
MocarskiES
2001 Distinct and separate roles for herpesvirus-conserved UL97 kinase in cytomegalovirus DNA synthesis and encapsidation. Proc Natl Acad Sci U S A 98 1895 1900
44. KroskyPM
BaekMC
CoenDM
2003 The human cytomegalovirus UL97 protein kinase, an antiviral drug target, is required at the stage of nuclear egress. J Virol 77 905 914
45. HamirallyS
KamilJP
Ndassa-ColdayYM
LinAJ
JahngWJ
2009 Viral mimicry of Cdc2/cyclin-dependent kinase 1 mediates disruption of nuclear lamina during human cytomegalovirus nuclear egress. PLoS Pathog 5 e1000275
46. HumeAJ
FinkelJS
KamilJP
CoenDM
CulbertsonMR
2008 Phosphorylation of retinoblastoma protein by viral protein with cyclin-dependent kinase function. Science 320 797 799
47. PrichardMN
SztulE
DailySL
PerryAL
FrederickSL
2008 Human cytomegalovirus UL97 kinase activity is required for the hyperphosphorylation of retinoblastoma protein and inhibits the formation of nuclear aggresomes. J Virol 82 5054 5067
48. Cano-MonrealGL
TavisJE
MorrisonLA
2008 Substrate specificity of the herpes simplex virus type 2 UL13 protein kinase. Virology 374 1 10
49. KawaguchiY
KatoK
TanakaM
KanamoriM
NishiyamaY
2003 Conserved protein kinases encoded by herpesviruses and cellular protein kinase cdc2 target the same phosphorylation site in eukaryotic elongation factor 1delta. J Virol 77 2359 2368
50. KenyonTK
LynchJ
HayJ
RuyechanW
GroseC
2001 Varicella-zoster virus ORF47 protein serine kinase: characterization of a cloned, biologically active phosphotransferase and two viral substrates, ORF62 and ORF63. J Virol 75 8854 8858
51. IwahoriS
MurataT
KudohA
SatoY
NakayamaS
2009 Phosphorylation of p27Kip1 by Epstein-Barr virus protein kinase induces its degradation through SCFSkp2 ubiquitin ligase actions during viral lytic replication. J Biol Chem
52. KamilJP
CoenDM
2007 Human cytomegalovirus protein kinase UL97 forms a complex with the tegument phosphoprotein pp65. J Virol 81 10659 10668
53. PrichardMN
BrittWJ
DailySL
HartlineCB
KernER
2005 Human cytomegalovirus UL97 Kinase is required for the normal intranuclear distribution of pp65 and virion morphogenesis. J Virol 79 15494 15502
54. AnsariA
EmeryVC
1999 The U69 gene of human herpesvirus 6 encodes a protein kinase which can confer ganciclovir sensitivity to baculoviruses. J Virol 73 3284 3291
55. HamzaMS
ReyesRA
IzumiyaY
WisdomR
KungHJ
2004 ORF36 protein kinase of Kaposi's sarcoma herpesvirus activates the c-Jun N-terminal kinase signaling pathway. J Biol Chem 279 38325 38330
56. MengQ
HagemeierSR
KunyCV
KalejtaRF
KenneySC
Simian virus 40 T/t antigens and lamin A/C small interfering RNA rescue the phenotype of an Epstein-Barr virus protein kinase (BGLF4) mutant. J Virol 84 4524 4533
57. NgTI
OgleWO
RoizmanB
1998 UL13 protein kinase of herpes simplex virus 1 complexes with glycoprotein E and mediates the phosphorylation of the viral Fc receptor: glycoproteins E and I. Virology 241 37 48
58. DaikokuT
ShibataS
GoshimaF
OshimaS
TsurumiT
1997 Purification and characterization of the protein kinase encoded by the UL13 gene of herpes simplex virus type 2. Virology 235 82 93
59. GershburgE
MarschallM
HongK
PaganoJS
2004 Expression and localization of the Epstein-Barr virus-encoded protein kinase. J Virol 78 12140 12146
60. MichelD
PavicI
ZimmermannA
HauptE
WunderlichK
1996 The UL97 gene product of human cytomegalovirus is an early-late protein with a nuclear localization but is not a nucleoside kinase. J Virol 70 6340 6346
61. De BolleL
MichelD
MertensT
ManichanhC
AgutH
2002 Role of the human herpesvirus 6 u69-encoded kinase in the phosphorylation of ganciclovir. Mol Pharmacol 62 714 721
62. IsegawaY
MiyamotoY
YasudaY
SemiK
TsujimuraK
2008 Characterization of the human herpesvirus 6 U69 gene product and identification of its nuclear localization signal. J Virol 82 710 718
63. SalsmanJ
ZimmermanN
ChenT
DomagalaM
FrappierL
2008 Genome-wide screen of three herpesviruses for protein subcellular localization and alteration of PML nuclear bodies. PLoS Pathog 4 e1000100
64. WeinbergRA
1995 The retinoblastoma protein and cell cycle control. Cell 81 323 330
65. HeltAM
GallowayDA
2003 Mechanisms by which DNA tumor virus oncoproteins target the Rb family of pocket proteins. Carcinogenesis 24 159 169
66. HumeAJ
KalejtaRF
2009 Regulation of the retinoblastoma proteins by the human herpesviruses. Cell Div 4 1
67. KamilJP
HumeAJ
JurakI
MungerK
KalejtaRF
2009 Human papillomavirus 16 E7 inactivator of retinoblastoma family proteins complements human cytomegalovirus lacking UL97 protein kinase. Proc Natl Acad Sci U S A 106 16823 16828
68. HindsPW
MittnachtS
DulicV
ArnoldA
ReedSI
1992 Regulation of retinoblastoma protein functions by ectopic expression of human cyclins. Cell 70 993 1006
69. ChangY
MoorePS
TalbotSJ
BoshoffCH
ZarkowskaT
1996 Cyclin encoded by KS herpesvirus. Nature 382 410
70. KnudsenES
WangJY
1996 Differential regulation of retinoblastoma protein function by specific Cdk phosphorylation sites. J Biol Chem 271 8313 8320
71. KnudsenES
WangJY
1997 Dual mechanisms for the inhibition of E2F binding to RB by cyclin-dependent kinase-mediated RB phosphorylation. Mol Cell Biol 17 5771 5783
72. LeeCP
HuangYH
LinSF
ChangY
ChangYH
2008 Epstein-Barr virus BGLF4 kinase induces disassembly of the nuclear lamina to facilitate virion production. J Virol 82 11913 11926
73. DechatT
PfleghaarK
SenguptaK
ShimiT
ShumakerDK
2008 Nuclear lamins: major factors in the structural organization and function of the nucleus and chromatin. Genes Dev 22 832 853
74. MettenleiterTC
2002 Herpesvirus assembly and egress. J Virol 76 1537 1547
75. HealdR
McKeonF
1990 Mutations of phosphorylation sites in lamin A that prevent nuclear lamina disassembly in mitosis. Cell 61 579 589
76. Cano-MonrealGL
WylieKM
CaoF
TavisJE
MorrisonLA
2009 Herpes simplex virus 2 UL13 protein kinase disrupts nuclear lamins. Virology 392 137 147
77. LeeMG
NurseP
1987 Complementation used to clone a human homologue of the fission yeast cell cycle control gene cdc2. Nature 327 31 35
78. ReedSI
WittenbergC
1990 Mitotic role for the Cdc28 protein kinase of Saccharomyces cerevisiae. Proc Natl Acad Sci U S A 87 5697 5701
79. Ninomiya-TsujiJ
NomotoS
YasudaH
ReedSI
MatsumotoK
1991 Cloning of a human cDNA encoding a CDC2-related kinase by complementation of a budding yeast cdc28 mutation. Proc Natl Acad Sci U S A 88 9006 9010
80. VerschurenEW
JonesN
EvanGI
2004 The cell cycle and how it is steered by Kaposi's sarcoma-associated herpesvirus cyclin. J Gen Virol 85 1347 1361
81. BernardiR
PandolfiPP
2007 Structure, dynamics and functions of promyelocytic leukaemia nuclear bodies. Nat Rev Mol Cell Biol 8 1006 1016
82. EverettRD
MurrayJ
2005 ND10 components relocate to sites associated with herpes simplex virus type 1 nucleoprotein complexes during virus infection. J Virol 79 5078 5089
83. IshovAM
StenbergRM
MaulGG
1997 Human cytomegalovirus immediate early interaction with host nuclear structures: definition of an immediate transcript environment. J Cell Biol 138 5 16
84. SaffertRT
KalejtaRF
2008 Promyelocytic leukemia-nuclear body proteins: herpesvirus enemies, accomplices, or both? Future Virology 3 265 277
85. TavalaiN
StammingerT
2008 New insights into the role of the subnuclear structure ND10 for viral infection. Biochim Biophys Acta 1783 2207 2221
86. KoriothF
MaulGG
PlachterB
StammingerT
FreyJ
1996 The nuclear domain 10 (ND10) is disrupted by the human cytomegalovirus gene product IE1. Exp Cell Res 229 155 158
87. SaffertRT
KalejtaRF
2006 Inactivating a cellular intrinsic immune defense mediated by Daxx is the mechanism through which the human cytomegalovirus pp71 protein stimulates viral immediate-early gene expression. J Virol 80 3863 3871
88. MaulGG
GuldnerHH
SpivackJG
1993 Modification of discrete nuclear domains induced by herpes simplex virus type 1 immediate early gene 1 product (ICP0). J Gen Virol 74 ( Pt 12) 2679 2690
89. LivingstonCM
IfrimMF
CowanAE
WellerSK
2009 Virus-Induced Chaperone-Enriched (VICE) domains function as nuclear protein quality control centers during HSV-1 infection. PLoS Pathog 5 e1000619
90. KlementIA
SkinnerPJ
KaytorMD
YiH
HerschSM
1998 Ataxin-1 nuclear localization and aggregation: role in polyglutamine-induced disease in SCA1 transgenic mice. Cell 95 41 53
91. BaekMC
KroskyPM
PearsonA
CoenDM
2004 Phosphorylation of the RNA polymerase II carboxyl-terminal domain in human cytomegalovirus-infected cells and in vitro by the viral UL97 protein kinase. Virology 324 184 193
92. KudohA
DaikokuT
IshimiY
KawaguchiY
ShirataN
2006 Phosphorylation of MCM4 at sites inactivating DNA helicase activity of the MCM4-MCM6-MCM7 complex during Epstein-Barr virus productive replication. J Virol 80 10064 10072
93. LeeCP
ChenJY
WangJT
KimuraK
TakemotoA
2007 Epstein-Barr virus BGLF4 kinase induces premature chromosome condensation through activation of condensin and topoisomerase II. J Virol 81 5166 5180
94. KudohA
FujitaM
KiyonoT
KuzushimaK
SugayaY
2003 Reactivation of lytic replication from B cells latently infected with Epstein-Barr virus occurs with high S-phase cyclin-dependent kinase activity while inhibiting cellular DNA replication. J Virol 77 851 861
95. JaultFM
JaultJM
RuchtiF
FortunatoEA
ClarkC
1995 Cytomegalovirus infection induces high levels of cyclins, phosphorylated Rb, and p53, leading to cell cycle arrest. J Virol 69 6697 6704
96. MarschallM
MarziA
aus dem SiepenP
JochmannR
KalmerM
2005 Cellular p32 recruits cytomegalovirus kinase pUL97 to redistribute the nuclear lamina. J Biol Chem 280 33357 33367
97. McLaughlin-DrubinME
MungerK
2009 The human papillomavirus E7 oncoprotein. Virology 384 335 344
98. MouF
WillsEG
ParkR
BainesJD
2008 Effects of lamin A/C, lamin B1, and viral US3 kinase activity on viral infectivity, virion egress, and the targeting of herpes simplex virus U(L)34-encoded protein to the inner nuclear membrane. J Virol 82 8094 8104
99. MalumbresM
BarbacidM
2005 Mammalian cyclin-dependent kinases. Trends Biochem Sci 30 630 641
100. MalumbresM
HarlowE
HuntT
HunterT
LahtiJM
2009 Cyclin-dependent kinases: a family portrait. Nat Cell Biol 11 1275 1276
101. AhnJH
HaywardGS
1997 The major immediate-early proteins IE1 and IE2 of human cytomegalovirus colocalize with and disrupt PML-associated nuclear bodies at very early times in infected permissive cells. J Virol 71 4599 4613
102. AdamsonAL
KenneyS
2001 Epstein-barr virus immediate-early protein BZLF1 is SUMO-1 modified and disrupts promyelocytic leukemia bodies. J Virol 75 2388 2399
103. WilemanT
2007 Aggresomes and pericentriolar sites of virus assembly: cellular defense or viral design? Annu Rev Microbiol 61 149 167
104. BurchAD
WellerSK
2004 Nuclear sequestration of cellular chaperone and proteasomal machinery during herpes simplex virus type 1 infection. J Virol 78 7175 7185
105. BurchAD
WellerSK
2005 Herpes simplex virus type 1 DNA polymerase requires the mammalian chaperone hsp90 for proper localization to the nucleus. J Virol 79 10740 10749
106. NozawaN
YamauchiY
OhtsukaK
KawaguchiY
NishiyamaY
2004 Formation of aggresome-like structures in herpes simplex virus type 2-infected cells and a potential role in virus assembly. Exp Cell Res 299 486 497
107. NgTI
TalaricoC
BurnetteTC
BironK
RoizmanB
1996 Partial substitution of the functions of the herpes simplex virus 1 U(L)13 gene by the human cytomegalovirus U(L)97 gene. Virology 225 347 358
108. RomakerD
SchregelV
MaurerK
AuerochsS
MarziA
2006 Analysis of the structure-activity relationship of four herpesviral UL97 subfamily protein kinases reveals partial but not full functional conservation. J Med Chem 49 7044 7053
109. DavisonAJ
2007 Comparative Analysis of the Genomes.
Ann ArvinGC-F
MocarskiEdward
MoorePatrickS
RoizmanBernard
WhitleyRichard
YamanishiKoichi
Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis Cambridge Cambridge University Press 10 26
110. PelletPE
RoizmanB
2007 The Family Herpesviridae: A Brief Introduction.
DavidM
KnipePMH
Fields' Virology Philadelphia Lipincott, Williams, and Wilkins 2479 2499
111. MillerME
EngelDA
SmithMM
1995 Cyclic AMP signaling is required for function of the N-terminal and CR1 domains of adenovirus E1A in Saccharomyces cerevisiae. Oncogene 11 1623 1630
112. MarschallM
Stein-GerlachM
FreitagM
KupferR
van den BogaardM
2002 Direct targeting of human cytomegalovirus protein kinase pUL97 by kinase inhibitors is a novel principle for antiviral therapy. J Gen Virol 83 1013 1023
113. NeipelF
EllingerK
FleckensteinB
1991 The unique region of the human herpesvirus 6 genome is essentially collinear with the UL segment of human cytomegalovirus. J Gen Virol 72 ( Pt 9) 2293 2297
114. TamuraK
DudleyJ
NeiM
KumarS
2007 MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol 24 1596 1599
115. KalejtaRF
BechtelJT
ShenkT
2003 Human cytomegalovirus pp71 stimulates cell cycle progression by inducing the proteasome-dependent degradation of the retinoblastoma family of tumor suppressors. Mol Cell Biol 23 1885 1895
116. SaffertRT
KalejtaRF
2007 Human cytomegalovirus gene expression is silenced by Daxx-mediated intrinsic immune defense in model latent infections established in vitro. J Virol 81 9109 9120
117. ZhuH
ShenY
ShenkT
1995 Human cytomegalovirus IE1 and IE2 proteins block apoptosis. J Virol 69 7960 7970
118. HanksSK
QuinnAM
HunterT
1988 The protein kinase family: conserved features and deduced phylogeny of the catalytic domains. Science 241 42 52
119. RussoAA
JeffreyPD
PattenAK
MassagueJ
PavletichNP
1996 Crystal structure of the p27Kip1 cyclin-dependent-kinase inhibitor bound to the cyclin A-Cdk2 complex. Nature 382 325 331
120. RussoAA
TongL
LeeJO
JeffreyPD
PavletichNP
1998 Structural basis for inhibition of the cyclin-dependent kinase Cdk6 by the tumour suppressor p16INK4a. Nature 395 237 243
121. JeffreyPD
RussoAA
PolyakK
GibbsE
HurwitzJ
1995 Mechanism of CDK activation revealed by the structure of a cyclinA-CDK2 complex. Nature 376 313 320
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2010 Číslo 9
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Měli bychom postcovidový syndrom léčit antidepresivy?
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- Structure of the Extracellular Portion of CD46 Provides Insights into Its Interactions with Complement Proteins and Pathogens
- The Length of Vesicular Stomatitis Virus Particles Dictates a Need for Actin Assembly during Clathrin-Dependent Endocytosis
- Inhibition of TIR Domain Signaling by TcpC: MyD88-Dependent and Independent Effects on Virulence
- The Coevolution of Virulence: Tolerance in Perspective