
Human Cytomegalovirus IE1 Protein Elicits a Type II Interferon-Like
Host Cell Response That Depends on Activated STAT1 but Not
Interferon-γ
Human cytomegalovirus (hCMV) is a highly prevalent pathogen that, upon primary
infection, establishes life-long persistence in all infected individuals. Acute
hCMV infections cause a variety of diseases in humans with developmental or
acquired immune deficits. In addition, persistent hCMV infection may contribute
to various chronic disease conditions even in immunologically normal people. The
pathogenesis of hCMV disease has been frequently linked to inflammatory host
immune responses triggered by virus-infected cells. Moreover, hCMV infection
activates numerous host genes many of which encode pro-inflammatory proteins.
However, little is known about the relative contributions of individual viral
gene products to these changes in cellular transcription. We systematically
analyzed the effects of the hCMV 72-kDa immediate-early 1 (IE1) protein, a major
transcriptional activator and antagonist of type I interferon (IFN) signaling,
on the human transcriptome. Following expression under conditions closely
mimicking the situation during productive infection, IE1 elicits a global type
II IFN-like host cell response. This response is dominated by the selective
up-regulation of immune stimulatory genes normally controlled by IFN-γ and
includes the synthesis and secretion of pro-inflammatory chemokines.
IE1-mediated induction of IFN-stimulated genes strictly depends on
tyrosine-phosphorylated signal transducer and activator of transcription 1
(STAT1) and correlates with the nuclear accumulation and sequence-specific
binding of STAT1 to IFN-γ-responsive promoters. However, neither synthesis
nor secretion of IFN-γ or other IFNs seems to be required for the
IE1-dependent effects on cellular gene expression. Our results demonstrate that
a single hCMV protein can trigger a pro-inflammatory host transcriptional
response via an unexpected STAT1-dependent but IFN-independent mechanism and
identify IE1 as a candidate determinant of hCMV pathogenicity.
Published in the journal:
. PLoS Pathog 7(4): e32767. doi:10.1371/journal.ppat.1002016
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1002016
Summary
Human cytomegalovirus (hCMV) is a highly prevalent pathogen that, upon primary
infection, establishes life-long persistence in all infected individuals. Acute
hCMV infections cause a variety of diseases in humans with developmental or
acquired immune deficits. In addition, persistent hCMV infection may contribute
to various chronic disease conditions even in immunologically normal people. The
pathogenesis of hCMV disease has been frequently linked to inflammatory host
immune responses triggered by virus-infected cells. Moreover, hCMV infection
activates numerous host genes many of which encode pro-inflammatory proteins.
However, little is known about the relative contributions of individual viral
gene products to these changes in cellular transcription. We systematically
analyzed the effects of the hCMV 72-kDa immediate-early 1 (IE1) protein, a major
transcriptional activator and antagonist of type I interferon (IFN) signaling,
on the human transcriptome. Following expression under conditions closely
mimicking the situation during productive infection, IE1 elicits a global type
II IFN-like host cell response. This response is dominated by the selective
up-regulation of immune stimulatory genes normally controlled by IFN-γ and
includes the synthesis and secretion of pro-inflammatory chemokines.
IE1-mediated induction of IFN-stimulated genes strictly depends on
tyrosine-phosphorylated signal transducer and activator of transcription 1
(STAT1) and correlates with the nuclear accumulation and sequence-specific
binding of STAT1 to IFN-γ-responsive promoters. However, neither synthesis
nor secretion of IFN-γ or other IFNs seems to be required for the
IE1-dependent effects on cellular gene expression. Our results demonstrate that
a single hCMV protein can trigger a pro-inflammatory host transcriptional
response via an unexpected STAT1-dependent but IFN-independent mechanism and
identify IE1 as a candidate determinant of hCMV pathogenicity.
Introduction
Human cytomegalovirus (hCMV), the prototypical β-herpesvirus, is an extremely widespread pathogen (reviewed in [1]). Primary hCMV infection is invariably followed by life-long viral persistence in all infected individuals. The groups most evidently affected by hCMV disease are humans with acquired or developmental immune deficits including allograft recipients receiving immunosuppressive drugs, human immunodeficiency virus-infected individuals, cancer patients undergoing intensive chemotherapy, and infants infected in utero (reviewed in [2]). In immunologically normal hosts, clinically relevant symptoms rarely accompany acute infections (reviewed in [3]), but viral persistence may contribute to chronic disease conditions including atherosclerosis, cardiovascular disease, inflammatory bowel disease, immune senescence, and certain malignancies (reviewed in [4], [5], [6], [7], [8]).
The pathogenesis of disease (e.g., pneumonitis, retinitis, hepatitis, enterocolitis, and encephalitis) associated with acute hCMV infection in immunocompromised people is most readily attributable to end organ damage either directly caused by cytopathic viral replication or by host immunological responses that target virus-infected cells. In contrast, chronic disease associated with persistent hCMV infection in immunocompetent individuals as well as in the allografts of transplant recipients is most likely related to prolonged inflammation (reviewed in [9]). In fact, hCMV has been frequently detected in the midst of intense inflammation, and a myriad of studies from transplant recipients and normal hosts have presented a strong case for this virus as an etiologic agent in chronic inflammatory processes, particularly those resulting in vascular disease (reviewed in [4]). At the molecular level, this is reflected by the fact that, in both human cells and animal models, cytomegalovirus infections activate numerous host genes many of which encode growth factors, cytokines, chemokines, and adhesion molecules with pro-inflammatory and immune stimulatory activities [10], [11], [12], [13], [14], [15], [16], [17], [18], [19], [20], [21], [22], [23]. A number of these virus-induced proteins are released from infected cells forming the viral “secretome” [4], [24], [25].
A large proportion of human genes that undergo activation during hCMV infection are normally controlled by interferons (IFNs) (reviewed in [26], [27]). The IFNs constitute a distinct group of cytokines synthesized and released by most vertebrate cells in response to the presence of many different pathogens including hCMV. They are divided among three classes: type I IFNs (primarily IFN-α and IFN-β), type II IFN (IFN-γ), and type III IFNs (IFN-λ or interleukin 28/29). The type I IFNs share many biological activities with type III IFNs, especially in host protection against viruses. IFN-γ, the sole type II IFN, is one of the most important mediators of inflammation and immunity exerting pleiotropic effects on activation, differentiation, expansion and/or survival of virtually any cell type of the immune system (reviewed in [28]). A significant body of research has identified the primary IFN pathway components and has characterized their roles in “canonical” signaling (reviewed in [29], [30]). In this pathway, IFNs bind to their cognate cell surface receptors to induce conformational changes that activate the receptor-associated enzymes of the Janus kinase (JAK) family. The post-translational modifications that follow this activation create docking sites for proteins of the signal transducer and activator of transcription (STAT) family with seven human members. In turn, the STAT proteins undergo JAK-mediated phosphorylation at a single tyrosine residue (Y701 in STAT1), which triggers their transition to an active dimer conformation. The STAT dimers accumulate in the nucleus where they may recruit additional proteins, and these complexes then bind sequence-specifically to short DNA motifs termed IFN-stimulated response element (ISRE) or gamma-activated sequence (GAS). ISREs are usually bound by a ternary complex composed of a STAT1-STAT2 heterodimer and IFN regulatory factor (IRF) 9, which forms upon induction by type I and type III IFNs and is referred to as IFN-stimulated gene factor 3 (ISGF3). In contrast, type II IFN typically signals via STAT1 homodimers that associate with GAS elements. Finally, promoter-associated STAT proteins stimulate transcription of numerous IFN-stimulated genes (ISGs) via their carboxy-terminal transcriptional activation domain. Within this domain, phosphorylation of a serine residue (S727 in STAT1) can augment STAT transcriptional activity. To some extent, the complex responses elicited by type I, type II, and type III IFNs are redundant as a consequence of partly overlapping ISGs.
Since many ISGs, especially those induced by type I IFNs, exhibit potent anti-viral activities most viruses have evolved escape mechanisms that mitigate IFN responses. In fact, both hCMV and murine cytomegalovirus (mCMV) are known to disrupt IFN pathways at multiple points (reviewed in [26], [27]). For example, JAK-STAT signaling is inhibited by the hCMV 72-kDa immediate-early 1 (IE1) gene product [31], [32], [33], a key regulatory nuclear protein required for viral early gene expression and replication in fibroblasts infected at low input multiplicities [34], [35], [36]. IE1 orthologs of mCMV and rat cytomegalovirus (rCMV) also contribute to replication and virulence in the respective animals [37], [38]. The hCMV IE1 protein counteracts virus - or type I IFN-induced ISG activation via complex formation with STAT1 and STAT2 resulting in reduced binding of ISGF3 to ISREs [31], [32], [33], [39]. STAT2 interaction contributes to hCMV type I IFN resistance and to IE1 function during productive infection [33], but the viral protein undergoes many additional host cell interactions (reviewed in [2], [40], [41]). For example, IE1 targets subnuclear structures known as promyelocytic leukemia (PML) bodies or nuclear domain 10 (ND10) ([42], [43], [44]; reviewed in [45], [46], [47], [48]). In addition, IE1 associates with chromatin [49] and interacts with a variety of transcription regulatory proteins [50], [51], [52], [53], [54], [55], [56], [57]. Consequently, IE1 stimulates expression from a broad range of viral and cellular promoters in transient transfection assays. However, IE1-mediated activation or repression of merely a few single endogenous human genes has been demonstrated so far [58], [59], [60], [61], [62], [63], [64].
Here we present the results of the first systematic human transcriptome analysis following expression of the hCMV IE1 protein. Surprisingly, the predominant response to IE1 was characterized by activation of pro-inflammatory and immune stimulatory genes normally controlled by IFN-γ. We further demonstrate that IE1 employs an unusual mechanism, which does not require induction of IFNs but nonetheless depends on activated (Y701-phosphorylated) STAT1, to up-regulate a subset of ISGs.
Results
Construction and characterization of human primary cells with inducible IE1 expression
The hCMV IE1 protein exhibits complex activities, and results obtained from experiments with IE1 mutant virus strains are inherently difficult to interpret. In fact, regarding the phenotype of IE1-deficient viruses at low input multiplicities, it seems almost impossible to discriminate between effects directly linked to any of the IE1 activities and indirect effects caused by delays in downstream viral gene expression and replication. On the other hand, following infection at high multiplicity, many consequences of absent IE1 expression are compensated for by excess viral structural components, such as tegument proteins and/or DNA, and therefore undetectable ([35], [36]; reviewed in [2], [40], [41]). Thus, it is apparent that cells with inducible expression of functional IE1 at physiological levels would be highly useful by allowing a definite assessment of the viral protein's activities outside the confounding context of infection. Furthermore, such cells would avoid potential difficulties typically associated with transient transfection, including variable frequency of positive cells and protein accumulation to non-physiologically high levels. Importantly, an inducible expression system would also preclude cells from adapting to long-term IE1 expression. In fact, the continued presence of IE1 is reportedly incompatible with genomic integrity and normal cell proliferation [65], [66], [67].
We used a tetracycline-dependent induction (Tet-on) system built into lentivirus vectors to generate cells in which IE1 expression can be synchronously induced and compared to cells not expressing the viral protein. The first component of this system is a lentiviral vector (pLKOneo.CMV.EGFPnlsTetR; [68], [69], [70]) that includes a hybrid gene encoding the tetracycline repressor (TetR) linked to a nuclear localization signal (NLS) derived from the SV40 large T antigen and the enhanced green fluorescent protein (EGFP) to produce an EGFPnlsTetR fusion protein [68]. In addition, this vector encodes neomycin resistance. The second component is a lentivirus vector (pLKO.DCMV.TetO.cIE1) conferring puromycin resistance, in which a fragment of the hCMV promoter-enhancer drives expression of the IE1 (Towne strain) cDNA. In this vector, tandem tetracycline operator (TetO) sequences are present immediately downstream of the TATA box. For the lentivirus transductions, we chose MRC-5 primary human embryonic lung fibroblasts, because they support robust wild-type hCMV replication, whereas IE1-deficient virus strains exhibit a severe growth defect after low multiplicity infection of these cells ([31], [33] and Figure 1 C). Initially, low passage MRC-5 cells were transduced with lentivirus prepared from plasmid pLKOneo.CMV.EGFPnlsTetR, and a neomycin-resistant polyclonal cell population (named TetR) was isolated in which almost all cells expressed the EGFP fusion protein located in the nucleus (data not shown). Next, TetR cells were transduced with lentivirus prepared from pLKO.DCMV.TetO.cIE1 and a mixed cell population (named TetR-IE1) exhibiting both neomycin and puromycin resistance was selected. Finally, fluorescence-activated cell sorting was performed to collect cells with high levels of EGFPnlsTetR and, consequently, low levels of IE1 in the absence of inductor.

To characterize the newly generated cells, TetR-IE1 cells were treated with doxycycline for 24 or 72 h and examined for IE1 expression by indirect immunofluorescence microscopy (Figure 1 A). Before induction, the majority (67.0%) of cells was IE1 negative, and most other cells expressed barely detectably levels of the viral protein. Interestingly, in the latter proportion of cells IE1 was present in a predominantly punctate nuclear pattern. This likely reflects stable co-localization between IE1 and ND10 due to viral protein levels insufficient to disrupt the nuclear structures. At 24 h following induction only 2.8% of cells were negative for IE1 expression and >97% stained positive for the viral protein. In almost all positive cells IE1 exhibited a largely diffuse nuclear staining indicating complete disruption of ND10. Very similar results were obtained for IE1 expression and localization 72 h post induction. Importantly, the observed temporal and spatial pattern of IE1 subnuclear localization in TetR-IE1 cells closely resembles that observed during productive hCMV infection in fibroblasts where initial colocalization between IE1 and ND10 is succeeded by ND10 disruption and diffuse nuclear distribution of the viral protein [43], [44], [71].
To compare the relative levels of IE1 expressed during hCMV infection and after induction of TetR-IE1 cells, TetR cells were infected with the hCMV Towne strain, and samples collected before or 3 h, 6 h, 12 h, 24 h, 48 h and 72 h after infection were analyzed for IE1 steady-state protein levels in comparison with samples of TetR-IE1 cells that had been treated with doxycycline (Figure 1 B). The timing of IE1 induction in TetR-IE1 cells was remarkably similar to the kinetics of IE1 accumulation in hCMV-infected cells. In addition, the IE1 levels detected at 24 to 72 h post induction were comparable to the protein amounts that had accumulated by 24 h post hCMV infection.
To confirm that TetR-IE1 cells express fully active IE1, replication of wild-type and IE1-deficient hCMV strains was compared by multi-step analyses conducted in doxycycline-treated TetR and TetR-IE1 cells (Figure 1 C). To this end, we employed a bacterial artificial chromosome (BAC)-based recombination approach to generate a “markerless” mutant virus strain (TNdlIE1) lacking the entire IE1-specific coding sequence. For details on the construction of TNdlIE1 and a revertant virus (TNrvIE1) see Materials and Methods. As expected, the replication of two independent TNdlIE1 clones was strongly attenuated in TetR cells, with a ∼2 to >3 log difference in titers between mutant and revertant virus strains. It is important to note that our previous work has shown that TNrvIE1 and the parental wild-type strain (TNwt) exhibit identical replication kinetics [33]. However, induced TetR-IE1 cells were able to support wild-type-like replication of the TNdlIE1 viruses demonstrating that the viral protein provided in trans can fully compensate for the lack of IE1 expression from the hCMV genome during productive infection. Interestingly, even the titers of TNrvIE1 were reproducibly up to ∼20-fold higher in TetR-IE1 as compared to IE1-negative cells between 3 and 12 days post infection.
Taken together, these results show that in TetR-IE1 cells expression of IE1 can be synchronously induced from the autologous hCMV major IE (MIE) promoter resulting in fully functional protein at levels present during the early stages of hCMV infection. Thus, TetR/TetR-IE1 cells present an ideal model to study the activities of the IE1 protein outside the complexity of infection, yet under physiological conditions.
IE1 triggers a pro-inflammatory and immune stimulatory human transcriptome response
The capacity of hCMV IE1 to activate transcription from both viral and cellular promoters has long been appreciated ([72]; reviewed in [2], [40], [41]). However, most reports on IE1-regulated host gene transcription have relied on transient transfections and promoter-reporter assays. To our knowledge, regulation of endogenous cellular transcription by IE1 has so far only been studied sporadically and at the level of single genes.
To comprehensively assess the impact of IE1 on the human transcriptome, we performed a systematic gene expression analysis using our TetR/TetR-IE1 cell model and Affymetrix GeneChip Human Gene 1.0 ST Arrays covering 28,869 genes (>99% of sequences currently present in the RefSeq database, National Center for Biotechnology Information). We compared the gene expression profiles at 24 h and 72 h post induction in induced versus non-induced TetR-IE1 cells and in induced TetR-IE1 versus induced TetR cells. Expression from the vast majority (99.9%) of genes represented on the arrays was not significantly affected by IE1. However, mRNA levels of 38 human genes differed by a factor of two or more (p>0.01) in both the induced TetR-IE1/non-induced TetR-IE1 and the induced TetR-IE1/induced TetR comparisons. For 32 (84%) of the 38 genes, changes in mRNA levels were only observed after 72 h (but not 24 h) of IE1 expression, and only six (16%) were differentially expressed at both 24 h and 72 h. Moreover, 13 (34%) of these genes were down-regulated by a factor between 2.0 and 5.5 (data not shown) and 25 (66%) were up-regulated by a factor between 2.0 and 41.9 (Table 1). For the present work, we concentrated on the set of genes that was found to be up-regulated by expression of IE1.

We utilized the Gene Ontology (GO) classification system (http://www.geneontology.org) to identify attributes which predominate among IE1-activated gene products regarding the three GO domains “biological process”, “molecular function”, and “cellular component”. Furthermore, we employed a set of analysis tools to construct maps that visualize overrepresented attributes on the GO hierarchy (Figure 2). According to GO, the most significantly enriched “biological process” terms with respect to the 25 IE1-activated genes are: “immune system process”, “immune response”, “inflammatory response”, “response to wounding”, “response to stimulus”, “defense response”, “chemotaxis”, “taxis”, and “regulation of cell proliferation” (Figure 2 A). In fact, virtually all IE1-induced genes with assigned functions have been implicated in adaptive or innate immune processes including inflammation. Moreover, 7 (28%) of the 25 genes encode known cytokines or other soluble mediators, namely the chemokine (C-X-C motif) ligands CXCL9, CXCL10 and CXCL11, the chemokine (C-C motif) ligand CCL11, endothelin 1 (encoded by EDN1), and the tumor necrosis factor (TNF) superfamily members 4 (TNFSF4, also known as OX40 ligand) and 18 (TNFSF18, also known as GITR ligand). This observation is also illustrated by the fact that, according to GO, the most significantly enriched “molecular function” terms in the IE1-activated transcriptome are: “cytokine receptor binding”, “cytokine activity”, “chemokine activity”, “chemokine receptor binding”, and “G-protein-coupled receptor binding” (Figure 2 B). Furthermore, the top “cellular component” category is “extracellular space” (Figure 2 C). For a more thorough assessment of overrepresented GO terms among IE1-induced genes, see Supporting Tables S1, S2 and S3.

Surprisingly, the genes induced by IE1 are generally associated with stimulatory rather than inhibitory effects on immune function including inflammation (Figure 2 A and Supporting Table S1). For example, some of the gene products are involved in the proteolysis (cathepsin S encoded by CTSS), intracellular transport (TAP1 transporter) or cell surface presentation (HLA-DRA) of antigens (reviewed in [73]). The chemokines CXCL9, CXCL10, and CXCL11 mediate leukocyte migration (see Discussion; reviewed in [73], [74], [75]). CD274 (also known as PDL1), TNFSF4, and TNFSF18 are co-stimulatory molecules which promote leukocyte (including T and B lymphocyte) activation, proliferation and/or survival (reviewed in [73], [76], [77], [78], [79]). Indoleamine 2,3-dioxygenase 1 (IDO1) and IRF1 have also been linked to T lymphocyte regulation, but they have additional functions in innate immune control of viral infection (reviewed in [73], [80], [81], [82], [83], [84], [85]. Likewise, GBP1 and murine GBP2 exhibit antiviral activity [86], [87], [88], [89].
Out of the 25 IE1-activated genes, 14 were selected for validation by qRT-PCR. The selected genes were representative of the entire range of expression kinetics and induction magnitudes measured by microarray analysis. The PCR approach confirmed expression of all tested genes typically reporting similar or larger fold increases compared to the array data (Figure 3 A–B and Figure 4 A). For example, in induced (72 h) versus non-induced TetR-IE1 cells the CXCL10 mRNA was found to be increased 24.6-fold by array analysis (Table 1) and 68.0-fold by PCR (Figure 3 A). Under the same conditions, the GBP4 transcript was induced 13.5-fold by array analysis (Table 1) as compared to 19.1-fold by PCR (Figure 3 A). The corresponding data for TAP1 were 2.1-fold (array analysis; Table 1) and 2.3-fold (PCR; Figure 3 A). Largely concordant results regarding induction magnitudes between array and PCR analyses were also obtained for CCDC3, CCL11, HES1, SERTAD4, TNFSF4, and TNFSF18 (Figure 3 B) as well as for CXCL9, CXCL11, IDO1, IFIT2, and IRF1 (Figure 4 A). In addition to the extent of gene activation, the precise timing of induction was exemplary investigated for CXCL10, GBP4 and TAP1 (Figure 3 A). A substantial increase in mRNA production from all three genes was evident at 72 h (and to a lesser extent at 48 h) but only minor effects were detected between 6 h and 24 h post IE1 induction consistent with the array data (Table 1). Tubulin-β (TUBB) gene expression, which is not affected by IE1, served as a negative control for the PCR experiments. Finally, the chemokines CXCL9 and CXCL11 were exclusively detected in supernatants from TetR-IE1 but not TetR cells (Figure 3 C). Moreover, the levels of CXCL10 protein were drastically increased in TetR-IE1 compared to TetR cells. This demonstrates that for these genes elevated mRNA levels also translate into enhanced protein synthesis and secretion.


The fact that increased expression of all tested IE1-activated genes was detectable with two or three alternative approaches strongly suggests that essentially all genes identified within the given experimental framework and data analysis settings are truly differentially expressed upon induction of IE1. Moreover, the activation of at least a subset of IE1-responsive genes appears to be temporally coupled.
Most IE1-activated genes are ISGs normally controlled by IFN-γ
A plethora of past studies has established that immune regulatory genes are preferential targets of IFN-based regulation [28], [29], [30]. Intriguingly, at least 21 (84%) of the 25 IE1-activated human genes identified by microarray analysis turned out to be bona fide ISGs (Table 2) according to informations retrieved from the Interferome database (http://www.interferome.org [90]) and other sources including our own qRT-PCR analyses (Figure 4 A and Supporting Table S4). Several of these ISGs cluster in certain chromosomal locations (e.g., 1p22, 4q21, and 10q23-q25; Table 2) apparently reflective of their co-regulation.
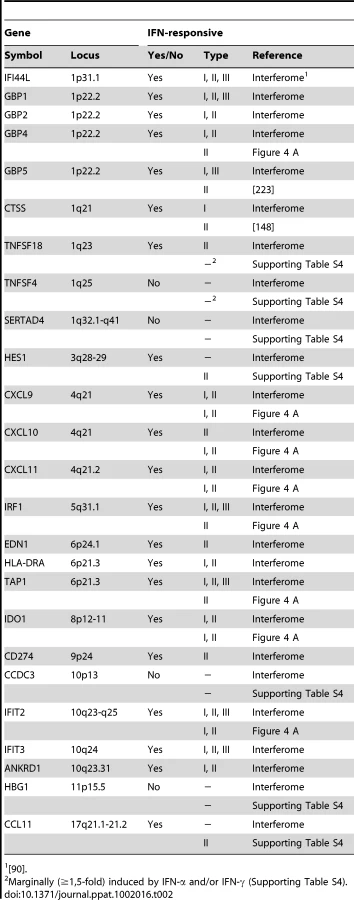
An initial assessment mainly based on the Interferome data revealed that IE1-activated ISGs are normally induced by either only IFN-γ or by both type II and type I IFNs (Table 2). To confirm this assignment and to further discriminate between type I and type II ISGs, we treated TetR and TetR-IE1 cells with exogenous IFN-α or IFN-γ and analyzed the effects on mRNA accumulation from a select subset of IE1-responsive ISGs. The transcript levels of all tested ISGs, namely CXCL9–11, GBP4, IDO1, IFIT2, IRF1, and TAP1 (Figure 4 A) as well as CCL11 (Supporting Table S4) were not only increased by IE1 expression (TetR-IE1 relative to TetR cells) but also by IFN-γ treatment of TetR cells, although to varying degrees (∼2 to >30,000-fold; Figure 4 A). Notably, there was a significant positive correlation (Pearson's correlation coefficient = 0.81) between the magnitudes of IE1 - and IFN-γ-mediated ISG induction. In contrast, the same genes were substantially less susceptible (CXCL9–11, GBP4, IDO1, and IFIT2) or entirely unresponsive (CCL11, IRF1, and TAP1) to IFN-α (Figure 4 A), and there was no correlation (Pearson's correlation coefficient = −0.04) between IE1 and IFN-α responsiveness. For comparison, three typical type I ISGs, the genes encoding eukaryotic translation initiation factor 2α kinase 2 (EIF2AK2, also known as PKR), myxovirus (influenza virus) resistance 1 (Mx1, also known as MxA), and 2′,5′-oligoadenylate synthetase (OAS1), were strongly induced by IFN-α but barely by IFN-γ or IE1 (Figure 4 B). Although no obvious synergistic or additive effects between IE1 expression and IFN-γ treatment were observed in these assays (Figure 4 A–B), IFN-α induction of type I ISGs was severely compromised in TetR-IE1 as compared to TetR cells (Figure 4 B). The latter observation is consistent with our previous work which has demonstrated that IE1 blocks STAT2-dependent signaling resulting in inhibition of type I ISG activation [31], [33].
Hence, it appears that expression of IE1 selectively activates a subset of ISGs and ISG gene clusters which are primarily responsive to IFN-γ indicating that the viral protein elicits a type II IFN-like transcriptional response.
IE1-mediated ISG activation is independent of IFNs
ISG activation typically requires synthesis, secretion and receptor binding of IFNs (reviewed in [26], [27], [29], [30]). IFN-α is encoded by a multi-gene family and is mainly expressed in leukocytes although some members are stimulated by IFN-β in fibroblasts [91]. However, neither of 12 IFN-α (IFNA) and three alternative type I IFN coding genes (IFNE, IFNK, and IFNW1 encoding IFN-ε, IFN-κ, and IFN-ω, respectively) was noticeably induced by IE1 as judged by our microarray results (Supporting Table S5). In contrast to IFN-α, IFN-β is encoded by a single gene (IFNB) and is produced by most cell types, especially by fibroblasts (IFN-β is also known as “fibroblast IFN”). However, previous work has shown that IE1 expression does not induce transcription from the IFN-β gene in fibroblasts [31], [32], [92]. Consistently, our microarray data did not reveal appreciable differences in IFNB1 mRNA levels between TetR and TetR-IE1 cells (Supporting Table S5). The single human IFN-γ gene (IFNG) is expressed upon stimulation of many immune cell types but not usually in fibroblasts, and our microarray results indicate that IE1 does not activate expression from this gene. Likewise, none of the known type III IFN genes (IL28A, IL28B, and IL29 encoding IFN-λ2/IL-28A, IFN-λ3/IL-28B, and IFN-λ1/IL-29, respectively) was significantly responsive to IE1 expression in this system (Supporting Table S5). For the IFN-β and IFN-γ transcripts, these results were confirmed by highly sensitive qRT-PCR from doxycycline-treated TetR-IE1 and TetR cells. Levels of the two IFN mRNAs did not significantly differ between TetR-IE1 and TetR cells at any of ten post induction time points (0 h–96 h) under investigation (Supporting Figure S1 and Supporting Table S6). Thus, IE1 does not seem to induce expression from the IFN-γ or any other human IFN gene.
To further rule out the possibility that ISG activation is a result of low level IFN production or secretion of any other soluble mediator from IE1 expressing cells, culture supernatants from TetR-IE1 cells induced with doxycycline for 24 h or 72 h were transferred to MRC-5 cells. As expected, MRC-5 cells did not undergo ISG induction 3 h to 72 h following media transfer (data not shown). Furthermore, we set up a transwell system with TetR cells in the top and TetR-IE1 cells in the bottom chamber (Figure 5). Following addition of IFN-γ to the lower chamber, we observed substantially increased mRNA levels of three IE1-responsive indicator ISGs (CXCL9, CXCL11, and GBP4) in both TetR and TetR-IE1 cells (Figure 5 A). In contrast, addition of doxycycline caused up-regulation of ISG mRNA levels in TetR-IE1 but not TetR cells (Figure 5 B). These results indicate that ISG induction is restricted to IE1 expressing cells and that a diffusible factor is not sufficient to mediate gene activation by the viral protein.

Finally, we performed experiments adding neutralizing antibodies directed against IFN-β and IFN-γ to the cell culture media (Figure 6). ISG-specific qRT-PCRs from TetR cells treated with a combination of antibodies and high doses of the respective exogenous IFN confirmed that cytokine neutralization was both highly effective and specific. At the same time, neither the IFN-β - nor the IFN-γ-specific neutralizing antibodies had any significant negative effect on IE1-mediated ISG induction. These results strongly support the view that ISG activation by IE1 is independent of IFN-β, IFN-γ, and likely other IFNs.

IE1-mediated ISG activation depends on STAT1 but not STAT2
Homodimeric STAT1 complexes are the central intracellular mediators of canonical IFN-γ signaling (reviewed in [26], [27], [28], [29], [30]). Interestingly, previous work has shown that the IE1 protein interacts with both STAT1 and STAT2, although STAT2 binding appeared to be more efficient [31], [32], [33], [39]. STAT2 has also been implicated in certain IFN-γ responses ([93], [94]; reviewed in [95]), although some (hCMV-mediated) activation of ISG transcription appears to occur entirely independent of STAT proteins ([96]; reviewed in [26], [27]).
To investigate whether ISG activation by IE1 requires the presence of STAT1 and/or STAT2, we employed siRNA-based gene silencing individually targeting the two STAT transcripts. Following transfection of MRC-5, TetR and/or TetR-IE1 cells with two different siRNA duplexes each for STAT1 and STAT2, we monitored endogenous STAT expression by immunoblotting (Figure 7 A) and qRT-PCR (Figure 7 B). An estimated ≥80% selective reduction in STAT1 and STAT2 protein accumulation was observed 2 days following siRNA transfection, and even after 5 days significantly lower protein levels were detected compared to cells transfected with a non-specific control siRNA (Figure 7 A). The knock-down of STAT1 and STAT2 was also evident at the level of mRNA accumulation (86 to 95% for STAT1 and 51 to 95% for STAT2 at day 5 post transfection; Figure 7 B). The knock-down specificity was verified by confirming that STAT1 siRNAs do not significantly reduce STAT2 mRNA levels and vice versa. Moreover, none of the STAT-directed siRNAs had any appreciable effect on IE1 expression (Figure 7 B). Again, expression from the CXCL10 and GBP4 genes was strongly up-regulated in doxycycline-treated TetR-IE1 versus TetR cells. However, STAT1 knock-down caused the CXCL10 and GBP4 genes to become almost entirely resistant to IE1-mediated activation in induced TetR-IE1 cells. In contrast, depletion of STAT2 had no negative effect on IE1-dependent ISG induction (Figure 7 B) although it diminished basal and IFN-α-induced type I ISG (OAS1) expression (Supporting Figure S2). These results demonstrate that STAT1, but not STAT2, is an essential mediator of the cellular transcriptional response to IE1 expression and suggest that the viral protein might mediate ISG activation via activation of JAK-STAT signaling.

IE1-mediated ISG activation requires STAT1 tyrosine phosphorylation
The activation-inactivation cycle of STAT transcription factors entails their transition between different dimer conformations. Unphosphorylated STATs can dimerize in an anti-parallel conformation, whereas tyrosine (Y701) phosphorylation triggers transition to a parallel dimer conformation resulting in increased DNA binding and nuclear retention of STAT1 (reviewed in [29], [30], [97]). In addition, serine (S727) phosphorylation is required for the full transcriptional and biological activity of STAT1 [98]. In order to investigate whether IE1 promotes STAT1 activation, we compared the levels of Y701 - and S727-phosphorylated STAT1 in doxycyline-induced TetR and TetR-IE1 cells (Figure 8 A). Total STAT1 steady-state protein levels were very similar in TetR and TetR-IE1 cells. In contrast, Y701-phosphorylated forms of STAT1 were only detectable in the presence of IE1 unless cells were treated with IFN-γ. In addition, IE1 was almost as efficient as IFN-γ in inducing STAT1 S727 phosphorylation. These results strongly suggest that IE1 expression triggers the formation of Y701 - and S727-phosphorylated, transcriptionally fully active STAT1 dimers.

To examine whether STAT1 Y701 and/or S727 phosphorylation is an essential step in IE1-mediated ISG activation, we set up a “knock-down/knock-in” system designed to study mutant STAT1 proteins in a context of diminished endogenous wild-type protein levels. We constructed an “siRNA-resistant” STAT1 coding sequence, termed STAT1*, containing two silent nucleotide exchanges in the sequence corresponding to siRNA STAT1 #146 (Figure 7 A). The STAT1* sequence was used as a substrate for further mutagenesis to generate siRNA-resistant constructs encoding mutant STAT1 proteins with conservative amino acid substitutions that preclude tyrosine or serine phosphorylation (Y701F or S727A, respectively; reviewed in [99], [100]). A retroviral gene transfer system based on vector pLHCX was utilized to efficiently express the different STAT1 proteins in TetR-IE1 cells. All STAT1 variants (STAT1*, STAT1*Y701F, and STAT1*S727A) were overexpressed to levels undiscernible from the wild-type protein and mRNA (Figure 8 B–C). In comparison to transfections with a non-specific control siRNA (#149), siRNA #146 severely reduced the levels of endogenous and overexpressed wild-type STAT1 without negatively affecting expression of the siRNA-resistant STAT1 variants or IE1 (Figure 8 B–C). As expected, the Y701F and S727A mutant STAT1 proteins did not undergo tyrosine or serine phosphorylation, respectively, upon stimulation by IFN-γ. Interestingly, while the S727A protein could still be tyrosine-phosphorylated, the Y701F mutant was defective for both tyrosine and serine phosphorylation (Figure 8 B). This observation is in agreement with previous findings showing that IFN-γ-dependent S727 phosphorylation occurs exclusively on Y701-phosphorylated STAT1 [101]. Ectopic expression of wild-type STAT1, STAT1*, and STAT1*S727A but not STAT1*Y701F in addition to the endogenous protein enhanced IE1-mediated activation of CXCL10 and GBP4 transcription. Conversely, siRNA-mediated depletion of endogenous STAT1 strongly reduced this response. Importantly, expression of STAT1* in cells depleted of endogenous STAT1 rescued ISG induction by IE1 almost completely. STAT1*S727A expression also compensated for the lack of endogenous STAT1, although slightly less efficiently compared to STAT1*, whereas STAT1*Y701F was unable to rescue IE1-mediated ISG activation (Figure 8 C).
Thus, although IE1 appears to trigger phosphorylation of STAT1 at both Y701 and S727, only the former modification is required for ISG activation. Nonetheless, STAT1 S727 phosphorylation may augment IE1-dependent gene activation.
IE1 facilitates STAT1 nuclear accumulation and promoter binding
Y701 phosphorylation usually causes a cytoplasmic to nuclear shift in steady-state localization and efficient sequence-specific DNA binding of STAT1 dimers (reviewed in [29], [30], [97]). Accordingly, immunofluorescence microscopy revealed that the presence of IE1 strongly promotes nuclear accumulation of STAT1, very similar to what was observed following addition of IFN-γ (Figure 9 A). In contrast, significant amounts of nuclear STAT2 were only detected after treatment of cells with IFN-α but not upon IE1 expression. These results were confirmed by nucleo-cytoplasmic cell fractionation (Figure 9 B). In these assays, IE1 induction for 72 h was as efficient in promoting STAT1 nuclear accumulation as treatment with type I or type II IFNs for 1 h. IFN treatment also strongly induced the nuclear accumulation of STAT2. However, the levels of nuclear STAT2 increased only marginally upon expression of IE1.

Finally, we asked whether IE1 may direct STAT1 to promoters of type II ISGs. Chromatin immunoprecipitation (ChIP) analyses demonstrated that the viral protein potentiates the recruitment of STAT1 to certain IFN-γ - and IE1-responsive ISG promoters (e.g., TAP1) but not to promoters of several non-ISGs (e.g., GAPDH; Figure 10 A). Moreover, there was a positive correlation between the magnitude of STAT1 chromatin association induced by IE1 and IFN-γ. At the same time, IE1 had no effect on association of STAT2 with these promoters (Figure 10 B). These results are in agreement with the fact that a previous global ChIP-sequencing study has experimentally demonstrated STAT1 association with 14 (56%) out of the 25 IE1-responsive gene promoters identified in this study ([102] and Supporting Table S7). In addition, 22 (88%) of these promoter sequences (all except EDN1, HBG1, and HLA-DRA) carry one or more (up to six) predicted STAT1β binding sites (GAS elements) according to the PROMO tool (version 3.0.2, default settings with 15% maximum matrix dissimilarity rate, http://alggen.lsi.upc.es), which predicts transcription factor binding sites as defined by position weight matrices derived from the TRANSFAC (version 8.3) database [103], [104]. Similar results were obtained with other in silico promoter analysis tools (data not shown).
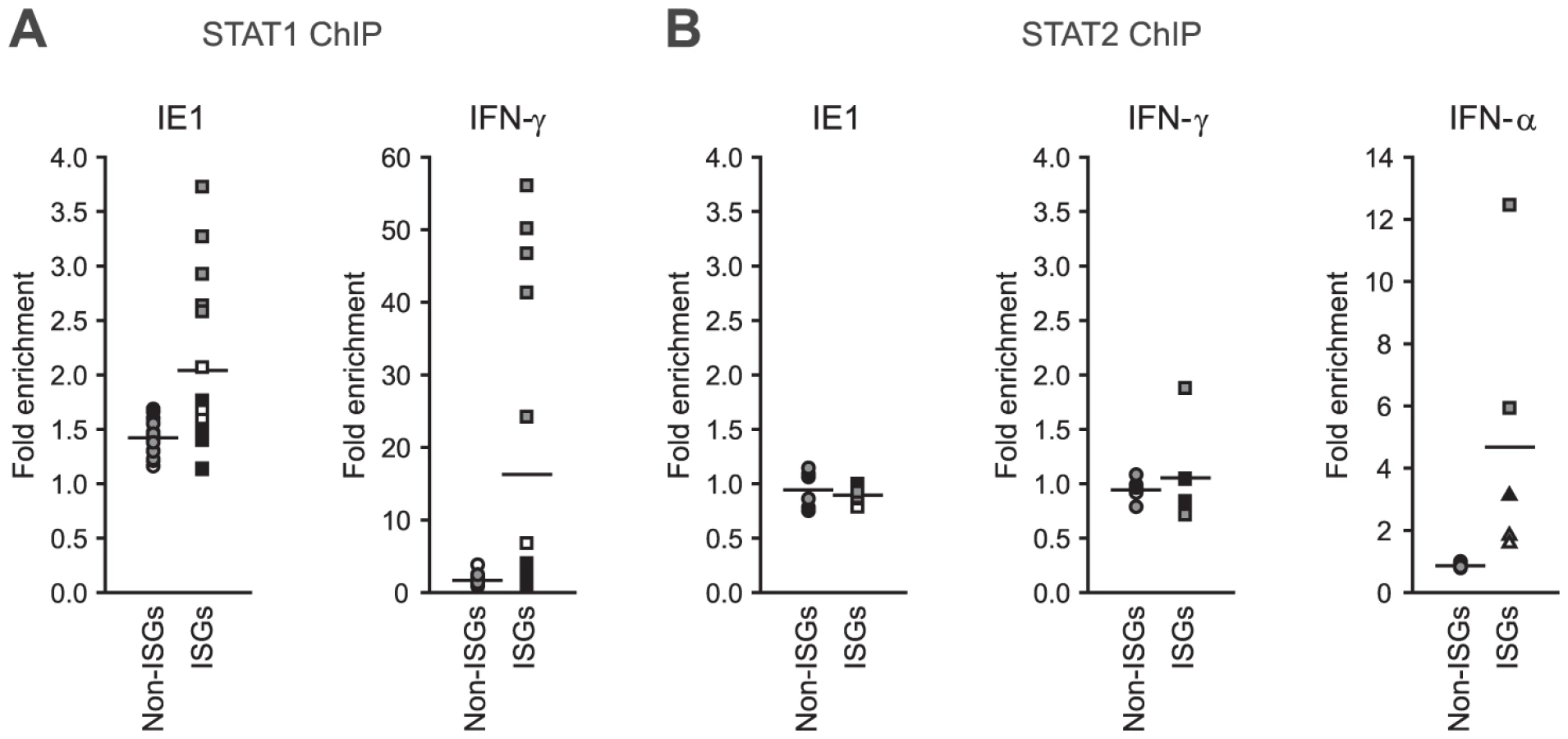
Based on these findings we propose that IE1 activates a subset of ISGs at least in part through increasing the nuclear concentration and sequence-specific DNA binding of phosphorylated STAT1 thereby modulating host gene expression in an unanticipated fashion.
Discussion
The transcriptional transactivation capacity of the hCMV MIE proteins has been recognized for decades ([72]; reviewed in [2], [40], [41]). For example, it has long been established that the 72-kDa IE1 protein can stimulate transcription from its own promoter-enhancer [36], [105], [106]. IE1 also activates at least a subset of hCMV early promoters therein collaborating with the viral 86-kDa IE2 protein [34], [35], [53], [71], [72], [107], [108], [109]. Furthermore, IE1 or combinations of IE1 and IE2 can stimulate expression from a variety of non-hCMV promoters. In fact, numerous heterologous viral and cellular promoters are responsive to IE1 or combinations of IE1 and IE2 [50], [51], [52], [57], [60], [61], [71], [72], [110], [111], [112], [113], [114], [115], [116], [117]. IE1 may accomplish transcriptional activation via interactions with a diverse set of cellular transcription regulatory proteins thereby acting through multiple DNA elements [50], [51], [52], [54], [55], [56], [57], [58], [59], [105], [106], [109], [110], [111], [112], [113], [117], [118], [119], [120], [121], [122], [123], [124], [125], [126] as well as epigenetic mechanisms including histone acetylation [53], [59], [127]. More recently, IE1 has also been implicated in transcriptional repression [31], [32], [33], [57], [62], [63], [64]. Our own work ([31] and this study, Figure 4 B) and a report by Huh et al. (2008) has demonstrated that IE1 can inhibit the hCMV - or IFN-α/β-dependent activation of human ISGs including ISG54, MxA, PKR, and CXCL10. The mechanism of inhibition appears to involve physical interactions of IE1 with the cellular STAT1 and STAT2 proteins that result in diminished DNA binding of the ternary ISGF3 complex to promoters of type I ISGs ultimately interfering with transcriptional activation [31], [32], [33]. Despite this plethora of studies, our understanding of the true transcriptional regulatory capacity of IE1 is still limited. This is mainly due to the fact that IE1-regulated transcription has almost exclusively been studied at the single gene level. Moreover, much of the past work has relied on transfection-based promoter-reporter assays, and IE1-dependent up - or down-regulation of only very few endogenous human genes has been demonstrated so far.
The present work constitutes the first systematic analysis of IE1-specific changes to transcription from the human genome. Importantly, to minimize cellular compensatory effects and to closely mimic the situation during hCMV infection, all experiments were based on short-term (up to 72 h) induction of IE1 expression from its autologous promoter (Figure 1 A–B). Just over 0.1% (25 out of 28,869) of all human transcripts under examination were found to be significantly up-regulated by IE1 under stringent analysis conditions (Table 1). This figure may be unexpected in the light of the reported interactions of IE1 with several ubiquitous transcription factors and its reputation as a “promiscuous” transactivator. However, rather than causing a broad transcriptional host response, IE1-specific gene activation was largely restricted to a subset of ISGs that are primarily responsive to IFN-γ (Table 2, Figure 4 and Supporting Table S4). Thus, IE1 appears to activate certain ISGs (typically type II ISGs) while simultaneously inhibiting the activation of other ISGs (typically type I ISGs). Importantly, more than half (at least 14 out of the 25) IE1-activated genes identified in this study were previously shown to be induced during hCMV infection of fibroblasts and/or other human cell types (Table 3). This strongly suggests that many if not all IE1-specific transcriptional changes observed in our expression model may be relevant to viral infection. On the other hand, our preliminary results indicate that the conditional replication defect of IE1 knock-out viruses in human fibroblasts [35], [36] may not result from an inability to initiate an IFN-γ-like response (data not shown). In fact, additional viral gene products are known or expected to contribute to ISG activation during hCMV infection (reviewed in [26], [27]) and may compensate for IE1 in this respect, at least during productive infection of fibroblasts.

In addition to being distinctively responsive to IFN-γ, most IE1-activated genes appear to share similar kinetics of induction (Table 1 and Figure 3), and many cluster in certain genomic locations (Table 2) suggesting a common underlying mechanism of activation. Specific siRNA-mediated STAT1 (but not STAT2) knock-down inhibited IE1-dependent activation of several target ISGs almost completely (Figure 7 A). Conversely, STAT1 overexpression proved to enhance ISG activation in IE1 expressing cells (Figure 8 C). Moreover, defective IE1-activated ISG transcription in cells depleted of endogenous STAT1 was efficiently rescued by ectopic STAT1 expression (Figure 8 C). These results demonstrate that the STAT1 protein is a critical mediator of the cellular transcriptional response to IE1. Moreover, this response appears to strictly depend on the Y701-phosphorylated form of STAT1 which is induced by IE1 expression (Figure 8). Although recent work has shown that some STAT1 functions are executed by the non-phosphorylated protein (reviewed in [97], [99], [100]), it is the Y701-phosphorylated form that preferentially accumulates in the nucleus and binds to DNA with high affinity (reviewed in [29], [30]) providing a mechanism for IE1-dependent ISG activation. IE1 also induces S727 phosphorylation of STAT1 (Figure 8 A), but this modification is dispensable merely serving an augmenting function in ISG activation triggered by the viral protein (Figure 8 C). Phosphorylation of S727 is thought to be required for the full transcriptional activity of STAT1 by recruiting histone acetyltransferase activity [98], [128], [129]. Interestingly, the hCMV IE1 protein can promote histone acetylation [53] suggesting it might compensate for S727 phosphorylation by binding to DNA-associated STAT1.
Our prior work has shown that IE1 physically interacts with STAT1 during hCMV infection and in vitro, and the two proteins co-localize in the nuclei of transfected cells treated with IFN-α [31]. The results of Figure 9 extend these observations by demonstrating that the viral protein facilitates nuclear accumulation and DNA binding of STAT1 in the absence of IFNs. The STATs were initially described as cytoplasmic proteins that enter the nucleus only in the presence of cytokines. However, it has now been established that STATs constantly shuttle between nucleus and cytoplasm irrespective of cytokine stimulation (reviewed in [97], [130], [131]). Thus, complex formation between nuclear resident IE1 and STAT1 passing through the nucleus may be sufficient to impair STAT1 export to the cytoplasm resulting in nuclear retention and increased DNA binding of the cellular protein. In this scenario, IE1 may increase the levels of Y701-phosphorylated STAT1 by interfering with nuclear dephosphorylation of the cellular protein. In fact, DNA binding was shown to protect STAT1 from dephosphorylation, which normally occurs at a step preceding export to the cytoplasm [132], [133]. This one-step “nuclear shortcut” model assumes that small amounts of Y701-phosphorylated STAT1 enter the nucleus in the absence of IFNs and any potential IE1-induced mediators of STAT1 activation. Conceivably, human fibroblasts (TetR cells) may constitutively release small amounts of soluble inducers (e.g., certain growth factors; see below) that maintain residual levels of activated STAT1 undetectable by immunoblotting (Figure 8 A). Moreover, we cannot rule out that the fetal calf serum used for cell culture media may contain factors causing a limited number of STAT1 molecules to undergo Y701 phosphorylation. In contrast, increased S727 phosphorylation in the presence of IE1 may result from higher levels of DNA-targeted STAT1, as this modification is preferentially or exclusively incorporated into the nuclear chromatin-associated cellular protein, at least during the normal IFN-γ response [101].
Alternatively, IE1 may actively induce STAT1 Y701 phosphorylation thereby promoting nuclear import of STAT1 dimers. This phosphorylation event is typically mediated by cytoplasmic JAK family kinases upon ligand-mediated activation of IFN receptors. However, our results demonstrate that IE1 does not induce the expression of human IFN genes, and we found no evidence for IFN-γ or IFN-β secretion from IE1 expressing cells (Supporting Table S5, Figure 6 and data not shown). Moreover, our transwell and media transfer experiments indicate that cytokines or other soluble mediators that may constitute a hypothetical IE1 “secretome” are not sufficient to stimulate ISG expression (Figure 5 and data not shown). However, this does not rule out the possibility that IE1 may cooperate with one or more soluble factors to trigger the observed transcriptional response. In fact, 80% of all IE1 target genes were not found activated within the first 24 h after induction of IE1 expression despite the fact that the viral protein had reached almost peak levels by this time (Figure 1 B and Table 1). Instead, up-regulation typically started at 48 h and increased until at least 72 h following IE1 expression (Table 1 and Figure 3 A). This timing of induction is compatible with a two-step model in which IE1 first initiates de novo synthesis and secretion of an unidentified cellular gene product required to trigger STAT1 Y701 phosphorylation (step 1). Besides IFNs, STAT1 signaling can be induced by several interleukins (e.g., IL-6) some of which are known to be up-regulated by IE1 [58], [60], [61], [110]. However, STAT1 Y701 phosphorylation can also occur independently of cytokines (reviewed in [134]). In fact, growth factors including the epidermal growth factor and certain hormones are also able to induce STAT1 Y701 phosphorylation [135], [136], [137], [138], [139]. In addition, tumor necrosis factor (TNF) has been shown to signal through activated STAT1 [140] raising the intriguing possibility that the soluble protein products of TNFSF4 and/or TNFSF18, two TNF family members belonging to the few genes already activated by 24 h following IE1 induction (Table 1), may be involved in IE1-mediated Y701 phosphorylation of STAT1. However, activation of one or more of these IFN-independent pathways may not produce enough activated nuclear STAT1 to trigger efficient ISG expression and may therefore be required but not sufficient for IE1-mediated gene induction. In accordance with this possibility, the levels of Y701-phosphorylated STAT1 were much higher in IFN-γ-treated as compared to IE1 expressing cells (Figure 8 A). Thus, on top of low level Y701 phosphorylation, IE1-dependent nuclear retention of STAT1 through complex formation between the viral and cellular protein (as outlined for the one-step model; see above) may be necessary in order to elicit a significant transcriptional response (step 2).
Although activated STAT1 is clearly a key mediator of IE1-dependent ISG induction, additional factors may be involved. In fact, not all known STAT1-activated human genes seem to be included in the IE1-specific transcriptome implying that additional gene products likely contribute to target specificity. One of the candidate co-factors that has been repeatedly linked to IE1 function is NFκB. In fact, IE1 was shown to activate the NFκB p65 (RelA) and RelB promoters [55], [112], [113], [121], to facilitate expression of the NFκB RelB subunit and/or NFκB post-translational activation [58], [113], [119], [121], and to activate transcription through NFκB binding sites [58], [105], [106], [113], [119], [126]. At the same time, NFκB has been implicated in IFN-γ-induced activation of a subset of ISGs including CXCL10 and GBP2 ([141], [142], [143], [144], [145]; reviewed in [146], [147]). However, we did not observe nuclear translocation of NFκB following induction of IE1 in TetR-IE1 cells. Moreover, siRNA-mediated knock-down of NFκB p65 had no significant impact on IE1-activated CXCL10 and GBP4 expression in these cells (data not shown). These observations indicate that the transcriptional response to IE1 is largely independent of NFκB, at least within our experimental setup. IRF1 is another transcription factor that contributes to the activation of certain ISGs including CTSS, GBP2, and TAP1 ([128], [148], [149], [150]; reviewed in [80], [81], [82]). IRF1 might enhance IE1-mediated ISG activation, especially since its mRNA is up-regulated by expression of the viral protein (Table 1 and Figure 4 A).
A key feature of the IE1 protein appears to be its ability to target to and disrupt subnuclear multi-protein structures known as PML bodies or ND10 during the early phase of hCMV infection and upon ectopic expression [42], [43], [44]. The mechanism of IE1-dependent ND10 disruption most likely involves binding to the PML protein, a major constituent of ND10 [54]. We have not specifically investigated the role of PML in IE1-mediated gene induction. Nonetheless, our results are compatible with the possibility that ND10 disruption is required for the transcriptional response to IE1 since the nuclear structures were confirmed to be disintegrated at both post-induction time points (24 h and 72 h) of our microarray analysis (data not shown). Although the exact function of ND10 remains unclear, the structures have been implicated in a variety of processes including inflammation [151] and anti-viral defense (reviewed in [45], [46], [47], [48]). Besides a proposed role of ND10 in viral gene expression, they may also function in transcriptional regulation of certain cellular genes. Several examples of selective associations between ND10 and genes or chromosomal loci, especially regions of high transcription activity and/or gene density, have been reported (reviewed in [152]). For example, immunofluorescent in situ hybridization analyses demonstrated that the major histocompatibility (MHC) class I gene cluster on chromosome 6 (6p21) is non-randomly associated with ND10 in human fibroblasts [153]. Transcriptional activation in the presence of IFN-γ correlates with the relocalization of this locus to the exterior of the chromosome 6 territory in a process that appears to involve DNA binding of Y701-phosphorylated STAT1, changes in chromatin loop architecture, and histone hyperacetylation [154], [155], [156]. Interestingly, many IE1-activated genes cluster in certain genomic locations (Table 2). This includes the HLA-DRA and TAP1 genes located within the ND10-associated MHC locus at 6p21. Together these observations raise the intriguing possibility that, through a combination of PML disruption and STAT1 activation, IE1 might cause higher order chromatin remodeling of entire chromosomal loci resulting in transcriptional activation.
One of the most surprising findings of the present study concerns the fact that most IE1-induced cellular genes are generally associated with stimulatory rather than inhibitory effects on immune function and inflammation (Table 1, Figure 2 and Supporting Tables S1, S2). It has been proposed that certain inflammatory and innate defense mechanisms launched by the host to limit hCMV replication may actually facilitate viral dissemination, for example by increasing target cell availability and/or by creating an environment conducive to virus reactivation (coined “no pain, no gain” by Mocarski [157]). Thus, it is plausible that hCMV not just attenuates host immunity through the numerous immune evasion mechanisms ascribed to this virus (reviewed in [158]), but rather aims at counterbalancing the effects of the innate and inflammatory response in restricting and facilitating viral replication. This strategy may be crucial in allowing for what has been termed “mutually assured survival” of both virus and host [159].
The functional group of IE1-induced pro-inflammatory proteins potentially involved in viral target cell recruitment is best represented by the chemokines CXCL9, CXCL10, and CXCL11. All three proteins are not only induced by IE1 (Table 1 and Figures 3–7) but also during hCMV infection of various cell types, and they represent major constituents of the viral secretome ([4], [18], [24], [160], [161], [162], [163], [164], [165] and Table 3). By binding to a common receptor, termed CXCR3, the three chemokines have the ability to attract subsets of circulating leukocytes to sites of infection and/or inflammation (reviewed in [74], [75]). Although CXCR3 is preferentially expressed on activated T helper 1 cells, the receptor protein is also present on many other cell types including CD34+ hematopoietic progenitors [166] which are preferential sites of hCMV latency [167], [168], [169], [170], [171], [172]. CXCR3 and its ligands have been implicated in a large variety of inflammatory and immune disorders (reviewed in [74], [75]). For example, cells expressing CXCR3 are found at high numbers in biopsies taken from patients experiencing organ transplant dysfunction and/or rejection [173], [174], [175], [176], [177], [178], [179], [180], [181]. Moreover, CXCL9 [175], [176], [177], [179], [180], CXCL10 [173], [174], [175], [176], [177], [179], [180], and CXCL11 [175], [176], [177], [178], [179], [180], [181] mRNA and protein levels are increased in tissues of organs undergoing rejection. Importantly, the levels of CXCR3-positive cells and CXCR3 ligand mRNA in the biopsy samples frequently correlate with the grade of graft rejection [174], [176], [177], [178], [180] suggesting a causative role of this pathway. Up-regulation of CXCL10 and other chemokines also correlated with transplant vascular sclerosis and chronic rejection in an rCMV cardiac allograft infection model [4], [182], [183]. In addition to CXCL9, CXCL10, and CXCL11, IE1 also up-regulates expression of CCL11 (Table 1), another CXCR3-interacting chemokine [184]. Through activation of the CXCR3 axis, IE1 might contribute to hCMV dissemination and pathogenesis in unexpected ways.
The IE1 protein has long been suspected to be a key player in the events leading to reactivation from hCMV latency although this view has recently been challenged by functional analysis of the mCMV and rCMV IE1 orthologs in mouse and rat models of infection, respectively [37], [185]. Nonetheless, inflammatory (including allogeneic) immune responses are believed to be efficient stimuli for hCMV reactivation. In fact, stimulation of latently infected monocytes or myeloid progenitor cells with pro-inflammatory cytokines including IFN-γ can reactivate viral replication ([186], [187], [188], [189]; reviewed in [190], [191], [192]). IFN-γ may aid hCMV reactivation by affecting cellular differentiation ([193]; reviewed in [28], [190], [191], [192]) and/or by activating transcription through GAS-like elements present in the viral MIE promoter-enhancer [194]. These GAS-like elements were shown to be required for efficient hCMV transcription and replication, at least after low multiplicity infection, and IFNs enhanced MIE gene expression [194]. Conceivably, the IE1 protein may phenocopy the effect of IFN-γ in activating both cellular ISGs and the viral MIE promoter thereby facilitating viral reactivation. Conversely, along the lines of the “immune sensing hypothesis of latency control” proposed by Reddehase and colleagues [195], episodes of IE1 expression may promote maintenance of viral latency not only through providing antigenic peptides (reviewed in [196]) but also by concomitantly activating critical immune effector functions including antigen transport (TAP1), processing (CTSS) and presentation (HLA-DRA) as well as immune cell recruitment (CXCL9, CXCL10, CXCL11, CCL11; see above) and co-stimulation (TNFSF4, TNFSF18 and CD274).
Current anti-hCMV strategies are directed against viral DNA replication, but sometimes fail to halt disease. This may be due to virus-induced “side effects” that are not correlated to production of virus particles and lysis of host cells. In fact, in hCMV pneumonitis and retinitis, disease symptoms were repeatedly found in the absence of replicating virus or viral cytopathogenicity [197], [198]. Similarly, in mouse models of viral pneumonitis mCMV replication per se was not sufficient to cause disease [197], [199], [200]. Conversely, mCMV disease could be triggered immunologically without inducing viral replication [201]. Here we have shown that out of >160 different hCMV gene products, a single protein (IE1) is sufficient to alter the expression of human genes with strong pro-inflammatory and immune stimulatory potential without the requirement for virus replication. The present work supports the idea that the hCMV MIE gene and specifically the IE1 protein may play a direct and predominant role in viral immunopathogenesis and inflammatory disease [202], [203], [204], [205]. Thus, the IE1 protein should be considered a prime target for the development of improved prevention and treatment options directed against hCMV.
Materials and Methods
Plasmids
The pMD2.G and psPAX2 packaging vectors for recombinant lentivirus production were obtained from Addgene (http://www.addgene.org; plasmids 12259 and 12260, respectively). Plasmids pLKOneo.CMV.EGFPnlsTetR, pLKO.DCMV.TetO.cICP0, and pCMV.TetO.cICP0 were kindly provided by Roger Everett (Glasgow, UK). pLKOneo.CMV.EGFPnlsTetR contains the complete hCMV MIE promoter upstream of a sequence encoding EGFP fused to an NLS and TetR [68], [69], [70]. In the pLKO.1puro derivative pLKO.DCMV.TetO.cICP0, expression of the herpes simplex virus type 1 infected cell protein 0 cDNA (cICP0) is under the control of a tandem TetO sequence located downstream of a truncated version of the hCMV MIE promoter (DCMV) [69], [70]. To generate pLKO.DCMV.TetO.cIE1, the IE1 cDNA of the hCMV Towne strain was PCR-amplified from pEGFP-IE1 [71] with upstream primer #483 containing a HindIII site and downstream primer #484 containing an EcoRI site (the sequences of all primers used in this study are listed in Supporting Table S8). The IE1 sequence was subcloned into the HindIII and EcoRI sites of pCMV.TetO.cICP0. The NdeI-EcoRI fragment of the resulting plasmid pCMV.TetO.IE1 was verified by sequencing and used to replace the ICP0 cDNA in pLKO.DCMV.TetO.cICP0 thereby generating plasmid pLKO.DCMV.TetO.cIE1.
QuikChange site-directed mutagenesis of plasmid pRc/CMV-hSTAT1p91 (kindly provided by Christian Schindler, New York, USA) with oligonucleotides #660 and #661 resulted in pCMV-STAT1* encoding a STAT1 variant mRNA resistant to silencing by the STAT1-specific siRNA duplex #146 (the sequences of all siRNAs used in this study are listed in Supporting Table S9). The plasmids pCMV-STAT1*Y701F and pCMV-STAT1*S727A were generated by QuikChange mutagenesis of pCMV-STAT1* with primer pairs #662/#663 and #664/#665, respectively. BamHI-EcoRV fragments of pRc/CMV-hSTAT1p91, pCMV-STAT1*, pCMV-STAT1*Y701F, and pCMV-STAT1*S727A were treated with Klenow fragment and ligated to the HpaI-digested, dephosphorylated retroviral vector pLHCX (Clontech, no. 631511) resulting in plasmids pLHCX-STAT1, pLHCX-STAT1*, pLHCX-STAT1*Y701F, and pLHCX-STAT1*S727A, respectively. The correct orientations and nucleotide sequences of the inserted STAT1 cDNAs were verified by sequencing.
Cells and retroviruses
Human MRC-5 embryonic lung fibroblasts (Sigma-Aldrich, no. 05011802), the human p53-negative non-small cell lung carcinoma cell line H1299 (ATCC, no. CRL-5803 [206]), and Phoenix-Ampho retrovirus packaging cells (from Garry Nolan, Stanford, USA [207]) were maintained in Dulbecco's Modified Eagle's Medium supplemented with 10% fetal calf serum, 100 units/ml penicillin, and 100 µg/ml streptomycin. All cultures were regularly screened for mycoplasma contamination using the PCR Mycoplasma Test Kit II from PromoKine. Where applicable, cells were treated with 1,000 U/ml recombinant human IFN-α A/D (R&D Systems, no. 11200), 10 ng/ml recombinant human IFN-β 1a (Biomol, no. 86421), or 10 ng/ml recombinant human IFN-γ (R&D Systems, no. 285-IF) for various durations. Neutralizing goat antibodies to human IFN-β (no. AF814) or IFN-γ (no. AF-285-NA) and normal goat IgG (no. AB-108-C) were purchased from R&D Systems and used at concentrations of 1 µg/ml (anti-IFN-β) or 2 µg/ml (anti-IFN-γ, normal IgG). Transwell assays were performed in tissue-culture-treated 100-mm plates with polycarbonate membrane and 0.4 µm pore size (Corning, no. 3419).
During the week prior to transfection, Phoenix-Ampho cells were grown in medium containing hygromycin (300 µg/ml) and diphtheria toxin (1 µg/ml). Production of replication-deficient retroviral particles, retrovirus infections, and selection of stable cell lines were performed according to the pLKO.1 protocol available on the Addgene website (http://www.addgene.org/pgvec1?f=c&cmd=showcol&colid=170&page=2) with minor modifications. Retroviral particles were generated by transient transfection of H1299 cells (pLKO-based vectors) or Phoenix-Ampho cells (pLHCX-based vectors) using the calcium phosphate co-precipitation technique [208]. Recombinant viruses were collected 36 h and 60 h after transfection, and were used for transduction of target cells by two subsequent 16 h incubations. To generate TetR cells, MRC-5 fibroblasts at population doubling 19 were infected with pLKOneo.CMV.EGFPnlsTetR-derived lentiviruses and selected with G418 (0.2 mg/ml). To generate TetR-IE1 cells, TetR cells were transduced by pLKO.DCMV.TetO.cIE1-derived lentiviruses and selected with puromycin (1 µg/ml). Cells with high level EGFPnlsTetR expression (and low IE1 background) were enriched by fluorescence-activated cell sorting in a FACSCanto II flow cytometer (BD Biosciences). TetR cells were maintained in medium containing G418 (0.1 mg/ml), while TetR-IE1 cells were cultured in the presence of both G418 (0.1 mg/ml) and puromycin (0.5 µg/ml). To induce IE1 expression, cells were treated with doxycycline (Clontech, no. 631311) at a final concentration of 1 µg/ml. To generate TetR-IE1 cells with stable expression of ectopic STAT1 proteins, uninduced TetR-IE1 cells were transduced with pLHCX-derived retroviruses encoding STAT1, STAT1*, STAT1*Y701F, or STAT1*S727A.
hCMV mutagenesis and infection
The EGFP-expressing wild-type Towne strain (TNwt) of hCMV was derived from an infectious BAC clone (T-BACwt [209]) of the viral genome. Allelic exchange to generate IE1-deficient viruses (TNdlIE1) and corresponding “revertants” (TNrvIE1) utilized the following derivatives of transfer plasmid pGS284 [210]: pGS284-TNIE1kanlacZ, pGS284-TNMIEdlIE1, pGS248-TNMIE, and pGS284-TNMIErvIE1. Plasmid pGS284-TNIE1kanlacZ contains the kanamycin resistance gene (kan) and the lacZ gene cloned between sequences flanking the IE1-specific exon four of the hCMV TN MIE transcription unit. The ∼1000-bp flanking sequences were obtained by PCR amplification using primers #136 and #137 (downstream flanking sequence) or #139 and #140 (upstream flanking sequence; for PCR primer sequences, see Supporting Table S8) and T-BACwt as template. The amplified downstream flanking sequence was cloned into pGS284 via BglII and NotI sites present in both the PCR primers and target vector sequences. Following addition of adenosine nucleotide overhangs to the 3′-ends of the PCR product, the upstream flanking sequence was first subcloned into vector pCR4-TOPO (Invitrogen) and subsequently inserted via NotI sites into pGS284 carrying the downstream flanking sequence. The kanlacZ expression cassette was released from plasmid YD-C54 [211] and cloned into the PacI sites (introduced through PCR primers #137 and #139) located between the hCMV flanking sequences in the pGS284 derivative described above. Plasmid pGS284-TNMIEdlIE1 contains an MIE fragment lacking 1,413 bp between the AccI sites upstream and downstream of exon four. The exon four-deleted MIE fragment was obtained from T-BACwt by overlap extension PCR as previously described [212]. The primer pairs used for PCR mutagenesis were #348/#349 (upstream fragment), #350/#351 (downstream fragment), and #348/#351 (complete fragment). The final PCR product was cloned via BglII and NotI sites into pGS284. For the construction of pGS248-TNMIE (previously termed pGS248-MIE; [33]), a ∼3000-bp sequence of the MIE region was amplified by PCR using template T-BACwt and primers #155 and #156. After phosphorylation, the PCR product was first inserted into the SmaI site of pUC18 and then excised from this vector via FseI and NotI sites. The FseI-NotI fragment was subsequently cloned into the same sites of pGS284-TNMIEdlIE1 thereby repairing the exon four deletion in this plasmid to generate pGS284-TNMIErvIE1. DNA sequence analysis was completed on all hCMV-specific PCR amplification products to confirm their integrity. Allelic exchange was performed through homologous recombination in Escherichia coli strain GS500 as previously described [33], [210], [211]. First, the BAC pTNIE1kanlacZ was generated by recombination of T-BACwt with pGS284-TNIE1kanlacZ followed by selection for kanamycin resistance and LacZ expression. After that, the BACs pTNdlIE1 and pTNrvIE1 were made through recombination of pTNIE1kanlacZ with pGS284-TNMIEdlIE1 and pGS284-TNMIErvIE1, respectively, followed by selection for the loss of kanamycin resistance and LacZ expression. The BAC constructs were analyzed by EcoRI digestion. The BACs pTNdlIE1 and pTNrvIE1 were used for electroporation of MRC-5 cells to reconstitute viruses TNdlIE1 and TNrvIE1, respectively, as has been described previously [211]. Cell - and serum-free virus stocks were produced upon BAC transfection of MRC-5 fibroblasts (TNwt and TNrvIE1) or TetR-IE1 cells (TNdlIE1), and the titers of the wild-type TN and revertant preparations were determined by standard plaque assay on MRC-5 cells. Titration of TNdlIE1 stocks was performed by quantification of intracellular genome equivalents [33]. Multistep replication analysis of recombinant viruses on TetR and TetR-IE1 cells has been described previously [33].
GeneChip analysis
For global transcriptome analysis, 1.9×106 TetR or TetR-IE1 cells of the same passage number were seeded on 10-cm dishes. When cells reached confluency (three days after plating), the medium was replaced, and cells were growth-arrested by maintaining them in the same medium for seven days before they were collected for transcriptome analysis. During the last 72 h or 24 h prior to collection, cultures were treated with doxycycline at a final concentration of 1 µg/ml or were left untreated. Total RNA was isolated using TRIzol reagent (Invitrogen) and Phase Lock Gel Heavy (Eppendorf) according to the manufacturers' instructions. A second purification step with on-column DNase digestion was performed on the isolated RNA using the RNeasy Mini Kit from Qiagen. All subsequent steps were performed at the Kompetenzzentrum für Fluoreszente Bioanalytik (Regensburg, Germany). Total RNA (100 ng) was labeled using reagents and protocols specified in the Affymetrix GeneChip Whole Transcript (WT) Sense Target Labeling Assay Manual (P/N 701880 Rev. 4). Quantity and quality of starting total RNA, cRNA, and single-stranded cDNA were assessed in a NanoDrop spectrophotometer (Thermo Fisher Scientific) and a 2100 Bioanalyzer (Agilent Technologies), respectively. Samples were hybridized to Affymetrix Human Gene 1.0 ST Arrays which interrogate 28,869 well-annotated genes and cover >99% of sequences present in the RefSeq database (National Center for Biotechnology Information). We probed a total of 18 microarrays, which allowed us to monitor three biological replicates for each experimental condition (TetR and TetR-IE1 cells without and with 24 h and 72 h of doxycycline treatment). For creation of the summarized probe intensity signals, the Robust Multi-Array Average algorithm [213] was used. Files generated by the Affymetrix GeneChip Operating 1.4 and Expression Console 1.1 software have been deposited in Gene Expression Omnibus (GEO, National Center for Biotechnology Information [214]) and are accessible through GEO Series accession number GSE24434 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE24434).
qRT-PCR
In order to determine steady-state mRNA levels by qRT-PCR, total RNA was isolated from 3 to 4×105 fibroblasts using Qiagen's RNeasy Mini Kit and RNase-Free DNase Set according to the manufacturer's instructions. First-strand cDNA was synthesized using SuperScript III and Oligo(dT)20 primers (Invitrogen) starting from 2 µg of total RNA. Unless otherwise noted, first-strand cDNA was diluted 10-fold with sterile ultrapure water, and 5 µl were used to template 20-µl real-time PCRs performed in a Roche LightCycler 1.5 [33]. The instrument was operated with a ramp rate of 20°C per sec using the following protocol: pre-incubation cycle (95°C for 10 min, analysis mode: none), 40 to 50 amplification cycles with single fluorescence measurement at the end of the extension step (denaturation at 95°C for 10 sec, primer-dependent annealing at 66 to 56°C for 10 sec, primer-dependent extension at 72°C for 8 to 10 sec, analysis mode: quantification), melting curve cycle with continuous data acquisition during the melting step (denaturation at 95°C for 0 sec, annealing at 65°C for 60 sec, melting at 95°C for 0 sec with a ramp rate of 0.1°C/sec, analysis mode: melting curves), cooling cycle (40°C for 30 sec, analysis mode: none). The PCR mix was composed of 9 µl PCR grade water, 1 µl forward primer solution (10 µM), 1 µl reverse primer solution (10 µM), and 4 µl 5× concentrated Master Mix from the LightCycler FastStart DNA MasterPLUS SYBR Green I kit. The sequences of the high pressure liquid chromatography-purified PCR primers are listed in Supporting Table S8. All samples were quantified at least in duplicate, and each analysis included positive, minus-RT, and non-templated controls. The second derivate maximum method with arithmetic baseline adjustment (LightCycler Software 3.5) was used to determine quantification cycle (Cq) values. Cq values were further validated by ensuring they meet the following criteria: (i) corresponding melting peaks of the generated PCR products, calculated using the polynomial method with digital filters enabled, had to match the single peak of the positive control sample, (ii) standard deviations of Cq values from technical replicates had to be below 0.33, (iii) Cq values had to be significantly different from minus-RT controls (CqCq-RT-1), and (iv) Cq values had to be within the linear quantification range. The linear quantification range was individually determined for each primer pair by generating a standard curve with serial dilutions of first-strand cDNA from the sample with the highest expression level. PCR efficiency (E) was calculated from the slope of the standard curve according to equation (1):(1)The relative expression ratio (R) of the target (trgt) and reference (ref) gene in an experimental (eptl) versus control (ctrl) sample was calculated using the efficiency-corrected model shown in equation (2):(2)
Control samples of all experiments had reference and target gene expression levels well above the limits of detection. The tubulin-β gene (TUBB) was chosen as a reference, because (i) expression levels did not change upon IE1 induction, IFN treatment, siRNA transfection, or hCMV infection, (ii) it allowed for RNA-specific detection with no spurious product generation in minus-RT controls, and (iii) it exhibited similar expression levels compared to the target genes under investigation, which were generally expressed at levels lower than TUBB in the absence and at similar or higher levels relative to TUBB in the presence of IE1 expression, IFN treatment, or hCMV infection.
Chemokine quantification
CXCL9, CXCL10, and CXCL11 chemokine concentrations in cell culture supernatants were determined using commercially available colorimetric sandwich enzyme immunoassay kits (Quantikine Immunoassays no. DCX900, DIP100, and DCX110 from R&D Systems) following the manufacturer's instructions.
RNA interference
The sequences of siRNA duplexes used for mRNA knock-down experiments are listed in Supporting Table S9. They were introduced into cells at 30 nM final concentration using the Lipofectamine RNAiMAX Reagent (Invitrogen) following the manufacturer's instructions. Briefly, exponentially growing cells were seeded either in 12-well dishes at 2.5×105 cells/well for RNA analyses or in 6-well dishes at 5×105 cells/well for protein analyses. Transfections were performed in Opti-MEM I Reduced Serum Medium (Invitrogen) with 2 µl or 5 µl of RNAiMAX Reagent for 12 - or 6-wells, respectively.
Subcellular fractionation, immunoblotting, and microscopy
Cells (3.8×106) on 10-cm dishes were collected with trypsin/EDTA and then centrifuged for 5 min at 500× g and 4°C. Supernatants were removed and cells resuspended in 100 µl CSK buffer (10 mM PIPES [pH 6.8], 300 mM sucrose, 100 mM NaCl, 3 mM MgCl2, 1 mM EDTA, 0.1% (v/v) Igepal CA-630) with freshly added protease and phosphatase inhibitor cocktails. Lysates were centrifuged for 1 min at 1,300× g and 4°C, and the supernatants (cytoplasmic extracts) were transferred to clean pre-chilled tubes and combined with one volume of 2× protein sample buffer (100 mM Tris-HCl [pH 6.8], 4% (w/v) SDS, 20% (v/v) glycerol, 200 mM β-mercaptoethanol, 0.1% (w/v) bromophenol blue). The insoluble (pellet) fractions containing nuclei were washed once with 500 µl CSK buffer before they were suspended in 200 µl 2× protein sample buffer and sonified in a Bioruptor (Diagenode; “H” setting; 30 sec on-off interval) for 15 min. Samples were centrifuged for 10 min at 20,000× g and 4°C, and the supernatants (nuclear extracts) were transferred to clean pre-chilled tubes. Cytosolic and nuclear extracts were heated to 95°C for 5 min before immunoblot analysis. Generation of whole cell extracts, sodium dodecyl sulfate-polyacrylamide gel electrophoresis, immunoblotting, and (immuno)fluorescence microscopy were performed according to previously published protocols [33], [53], [215]. Immunodetection employed primary mono - or polyclonal antibodies directed against hCMV IE1 (1B12; [216]) or human GAPDH (Abcam, no. ab9485), histone H2A (Abcam, no. ab13923), STAT1 (no. sc-464 for immunoblotting and no. sc-346 for immunofluorescence, both from Santa Cruz), STAT1α (Santa Cruz, no. sc-345), STAT2 (Santa Cruz, no. sc-22816), and phosphorylated STAT1 (Y701-specific antibody no. 9171 and S727-specific antibody no. 9177, both from Cell Signaling Technologies). The secondary antibodies used were peroxidase-conjugated goat anti-mouse (no. 115-035-166) or goat anti-rabbit IgG (no. 111-035-144) from Dianova for immunoblotting, and highly cross-adsorbed Alexa Fluor 594 - or Alexa Fluor 633-conjugated goat anti-mouse (no. A-11032 or no. A-21052, respectively) and Alexa Fluor 546-conjugated goat anti-rabbit IgG (no. A-11035) from Invitrogen for immunofluorescence.
ChIP assay
ChIP was performed essentially as described by Nelson et al. [217], [218]. Resting cells on a 15-cm dish were cross-linked by treatment with 1% (v/v) formaldehyde for 10 min at 37°C. Isolated chromatin was sonified for 15 min in a Bioruptor (Diagenode; “H” setting, 30 sec on-off interval) and cleared by centrifugation for 20 min at 20,000× g and 4°C. Sheared chromatin from 7×106 cells (0.7 ml) was subjected to immunoprecipitation for 16 h at 4°C with gentle rotation using 10 µg of antibody. Two different polyclonal rabbit antibodies each against STAT1 (no. sc-3454 and sc-346 from Santa Cruz) and STAT2 (no. sc-476 and sc-839 from Santa Cruz) were used. After the antibody incubation step, insoluble material was removed by centrifugation (10 min at 20,000× g and 4°C) and 0.63 ml (90%) supernatant was transferred to a clean pre-chilled tube. Antibody-antigen complexes were isolated by sedimentation following incubation with 60 µl of Protein A Agarose/Salmon Sperm DNA slurry (Millipore) for 60 min at 4°C. PCR-ready DNA was prepared using Chelex-100 and duplicate samples of 5 µl (25% of the final reaction volume) each were used for DNA quantification by qPCR as described above and in recent publications [33], [215]. The PCR primer sequences are listed in Supporting Table S8.
Supporting Information
Zdroje
1. CannonMJSchmidDSHydeTB
2010
Review of cytomegalovirus seroprevalence and demographic
characteristics associated with infection.
Rev Med Virol
20
202
213
2. MocarskiESShenkTPassRF
2007
Cytomegaloviruses.
KnipeDMHowleyPMGriffinDELambRAMartinMA
Fields virology
Philadelphia
Lippincott Williams and Wilkins
2701
2773
3. RafailidisPIMourtzoukouEGVarbobitisICFalagasME
2008
Severe cytomegalovirus infection in apparently immunocompetent
patients: a systematic review.
Virol J
5
47
4. StreblowDNDumortierJMosesAVOrloffSLNelsonJA
2008
Mechanisms of cytomegalovirus-accelerated vascular disease:
induction of paracrine factors that promote angiogenesis and wound
healing.
Curr Top Microbiol Immunol
325
397
415
5. Soderberg-NauclerC
2008
HCMV microinfections in inflammatory diseases and
cancer.
J Clin Virol
41
218
223
6. MichaelisMDoerrHWCinatlJ
2009
The story of human cytomegalovirus and cancer: increasing
evidence and open questions.
Neoplasia
11
1
9
7. CrumpackerCS
2010
Invited commentary: human cytomegalovirus, inflammation,
cardiovascular disease, and mortality.
Am J Epidemiol
172
372
374
8. BrunnerSHerndler-BrandstetterDWeinbergerBGrubeck-LoebensteinB
2010
Persistent viral infections and immune aging.
Ageing Res Rev
In press
9. BrittW
2008
Manifestations of human cytomegalovirus infection: proposed
mechanisms of acute and chronic disease.
Curr Top Microbiol Immunol
325
417
470
10. CraigenJLYongKLJordanNJMacCormacLPWestwickJ
1997
Human cytomegalovirus infection up-regulates interleukin-8 gene
expression and stimulates neutrophil transendothelial
migration.
Immunology
92
138
145
11. GrundyJEDownesKL
1993
Up-regulation of LFA-3 and ICAM-1 on the surface of fibroblasts
infected with cytomegalovirus.
Immunology
78
405
412
12. ZhuHCongJPMamtoraGGingerasTShenkT
1998
Cellular gene expression altered by human cytomegalovirus: global
monitoring with oligonucleotide arrays.
Proc Natl Acad Sci U S A
95
14470
14475
13. BrowneEPWingBColemanDShenkT
2001
Altered cellular mRNA levels in human cytomegalovirus-infected
fibroblasts: viral block to the accumulation of antiviral
mRNAs.
J Virol
75
12319
12330
14. ChallacombeJFRechtsteinerAGottardoRRochaLMBrowneEP
2004
Evaluation of the host transcriptional response to human
cytomegalovirus infection.
Physiol Genomics
18
51
62
15. BrowneEPShenkT
2003
Human cytomegalovirus UL83-coded pp65 virion protein inhibits
antiviral gene expression in infected cells.
Proc Natl Acad Sci U S A
100
11439
11444
16. SimmenKASinghJLuukkonenBGLopperMBittnerA
2001
Global modulation of cellular transcription by human
cytomegalovirus is initiated by viral glycoprotein B.
Proc Natl Acad Sci U S A
98
7140
7145
17. HertelLMocarskiES
2004
Global analysis of host cell gene expression late during
cytomegalovirus infection reveals extensive dysregulation of cell cycle gene
expression and induction of pseudomitosis independent of US28
function.
J Virol
78
11988
12011
18. ChanGBivins-SmithERSmithMSSmithPMYurochkoAD
2008
Transcriptome analysis reveals human cytomegalovirus reprograms
monocyte differentiation toward an M1 macrophage.
J Immunol
181
698
711
19. Tang-FeldmanYJWojtowiczALochheadGRHaleMALiY
2006
Use of quantitative real-time PCR (qRT-PCR) to measure cytokine
transcription and viral load in murine cytomegalovirus
infection.
J Virol Methods
131
122
129
20. RottDZhuJZhouYFBurnettMSZalles-GanleyA
2003
IL-6 is produced by splenocytes derived from CMV-infected mice in
response to CMV antigens, and induces MCP-1 production by endothelial cells:
a new mechanistic paradigm for infection-induced
atherogenesis.
Atherosclerosis
170
223
228
21. DenglerTJRafteryMJWerleMZimmermannRSchonrichG
2000
Cytomegalovirus infection of vascular cells induces expression of
pro-inflammatory adhesion molecules by paracrine action of secreted
interleukin-1beta.
Transplantation
69
1160
1168
22. ChengJKeQJinZWangHKocherO
2009
Cytomegalovirus infection causes an increase of arterial blood
pressure.
PLoS Pathog
5
e1000427
23. ComptonTKurt-JonesEABoehmeKWBelkoJLatzE
2003
Human cytomegalovirus activates inflammatory cytokine responses
via CD14 and Toll-like receptor 2.
J Virol
77
4588
4596
24. DumortierJStreblowDNMosesAVJacobsJMKreklywichCN
2008
Human cytomegalovirus secretome contains factors that induce
angiogenesis and wound healing.
J Virol
82
6524
6535
25. GrundyJELawsonKMMacCormacLPFletcherJMYongKL
1998
Cytomegalovirus-infected endothelial cells recruit neutrophils by
the secretion of C-X-C chemokines and transmit virus by direct
neutrophil-endothelial cell contact and during neutrophil transendothelial
migration.
J Infect Dis
177
1465
1474
26. DeFilippisVR
2007
Induction and evasion of the type I interferon response by
cytomegaloviruses.
Adv Exp Med Biol
598
309
324
27. MarshallEEGeballeAP
2009
Multifaceted evasion of the interferon response by
cytomegalovirus.
J Interferon Cytokine Res
29
609
619
28. SahaBJyothi PrasannaSChandrasekarBNandiD
2010
Gene modulation and immunoregulatory roles of interferon
gamma.
Cytokine
50
1
14
29. SchindlerCPlumleeC
2008
Inteferons pen the JAK-STAT pathway.
Semin Cell Dev Biol
19
311
318
30. LiWX
2008
Canonical and non-canonical JAK-STAT signaling.
Trends Cell Biol
18
545
551
31. PaulusCKraussSNevelsM
2006
A human cytomegalovirus antagonist of type I IFN-dependent signal
transducer and activator of transcription signaling.
Proc Natl Acad Sci U S A
103
3840
3845
32. HuhYHKimYEKimETParkJJSongMJ
2008
Binding STAT2 by the acidic domain of human cytomegalovirus IE1
promotes viral growth and is negatively regulated by SUMO.
J Virol
82
10444
10454
33. KraussSKapsJCzechNPaulusCNevelsM
2009
Physical requirements and functional consequences of complex
formation between the cytomegalovirus IE1 protein and human
STAT2.
J Virol
83
12854
12870
34. GawnJMGreavesRF
2002
Absence of IE1 p72 protein function during low-multiplicity
infection by human cytomegalovirus results in a broad block to viral
delayed-early gene expression.
J Virol
76
4441
4455
35. GreavesRFMocarskiES
1998
Defective growth correlates with reduced accumulation of a viral
DNA replication protein after low-multiplicity infection by a human
cytomegalovirus IE1 mutant.
J Virol
72
366
379
36. MocarskiESKembleGWLyleJMGreavesRF
1996
A deletion mutant in the human cytomegalovirus gene encoding
IE1(491aa) is replication defective due to a failure in
autoregulation.
Proc Natl Acad Sci U S A
93
11321
11326
37. SandfordGRSchumacherUEttingerJBruneWHaywardGS
2010
Deletion of the rat cytomegalovirus immediate-early 1 gene
results in a virus capable of establishing latency, but with lower levels of
acute virus replication and latency that compromise reactivation
efficiency.
J Gen Virol
91
616
621
38. GhazalPVisserAEGustemsMGarciaRBorstEM
2005
Elimination of IE1 significantly attenuates murine
cytomegalovirus virulence but does not alter replicative capacity in cell
culture.
J Virol
79
7182
7194
39. DimitropoulouPCaswellRMcSharryBPGreavesRFSpandidosDA
2010
Differential relocation and stability of PML-body components
during productive human cytomegalovirus infection: Detailed characterization
by live-cell imaging.
Eur J Cell Biol
89
757
768
40. PaulusCNevelsM
2009
The human cytomegalovirus major immediate-early proteins as
antagonists of intrinsic and innate antiviral host reponses.
Viruses
1
760
779
41. CastilloJPKowalikTF
2002
Human cytomegalovirus immediate early proteins and cell growth
control.
Gene
290
19
34
42. WilkinsonGWKellyCSinclairJHRickardsC
1998
Disruption of PML-associated nuclear bodies mediated by the human
cytomegalovirus major immediate early gene product.
J Gen Virol
79
1233
1245
43. KoriothFMaulGGPlachterBStammingerTFreyJ
1996
The nuclear domain 10 (ND10) is disrupted by the human
cytomegalovirus gene product IE1.
Exp Cell Res
229
155
158
44. AhnJHHaywardGS
1997
The major immediate-early proteins IE1 and IE2 of human
cytomegalovirus colocalize with and disrupt PML-associated nuclear bodies at
very early times in infected permissive cells.
J Virol
71
4599
4613
45. TavalaiNStammingerT
2009
Interplay between herpesvirus infection and host defense by PML
nuclear bodies.
Viruses
1
1240
1264
46. MaulGG
2008
Initiation of cytomegalovirus infection at ND10.
Curr Top Microbiol Immunol
325
117
132
47. TavalaiNStammingerT
2008
New insights into the role of the subnuclear structure ND10 for
viral infection.
Biochim Biophys Acta
1783
2207
2221
48. SaffertRKalejtaR
2008
Promyelocytic leukemia-nuclear body proteins: herpesvirus
enemies, accomplices, or both?
Future Virology
3
265
277
49. LafeminaRLPizzornoMCMoscaJDHaywardGS
1989
Expression of the acidic nuclear immediate-early protein (IE1) of
human cytomegalovirus in stable cell lines and its preferential association
with metaphase chromosomes.
Virology
172
584
600
50. HayhurstGPBryantLACaswellRCWalkerSMSinclairJH
1995
CCAAT box-dependent activation of the TATA-less human DNA
polymerase alpha promoter by the human cytomegalovirus 72-kilodalton major
immediate-early protein.
J Virol
69
182
188
51. PomaEEKowalikTFZhuLSinclairJHHuangES
1996
The human cytomegalovirus IE1-72 protein interacts with the
cellular p107 protein and relieves p107-mediated transcriptional repression
of an E2F-responsive promoter.
J Virol
70
7867
7877
52. MargolisMJPajovicSWongELWadeMJuppR
1995
Interaction of the 72-kilodalton human cytomegalovirus IE1 gene
product with E2F1 coincides with E2F-dependent activation of dihydrofolate
reductase transcription.
J Virol
69
7759
7767
53. NevelsMPaulusCShenkT
2004
Human cytomegalovirus immediate-early 1 protein facilitates viral
replication by antagonizing histone deacetylation.
Proc Natl Acad Sci U S A
101
17234
17239
54. AhnJHBrignoleEJ3rdHaywardGS
1998
Disruption of PML subnuclear domains by the acidic IE1 protein of
human cytomegalovirus is mediated through interaction with PML and may
modulate a RING finger-dependent cryptic transactivator function of
PML.
Mol Cell Biol
18
4899
4913
55. YurochkoADMayoMWPomaEEBaldwinASJrHuangES
1997
Induction of the transcription factor Sp1 during human
cytomegalovirus infection mediates upregulation of the p65 and p105/p50
NF-kappaB promoters.
J Virol
71
4638
4648
56. LukacDMHarelNYTaneseNAlwineJC
1997
TAF-like functions of human cytomegalovirus immediate-early
proteins.
J Virol
71
7227
7239
57. HwangESZhangZCaiHHuangDYHuongSM
2009
Human cytomegalovirus IE1-72 protein interacts with p53 and
inhibits p53-dependent transactivation by a mechanism different from that of
IE2-86 protein.
J Virol
83
12388
12398
58. MurayamaTMukaidaNSadanariHYamaguchiNKhabarKS
2000
The immediate early gene 1 product of human cytomegalovirus is
sufficient for up-regulation of interleukin-8 gene
expression.
Biochem Biophys Res Commun
279
298
304
59. StraatKLiuCRahbarAZhuQLiuL
2009
Activation of telomerase by human
cytomegalovirus.
J Natl Cancer Inst
101
488
497
60. IwamotoGKMonickMMClarkBDAuronPEStinskiMF
1990
Modulation of interleukin 1 beta gene expression by the immediate
early genes of human cytomegalovirus.
J Clin Invest
85
1853
1857
61. IwamotoGKKonicekSA
1997
Cytomegalovirus immediate early genes upregulate interleukin-6
gene expression.
J Investig Med
45
175
182
62. LeeKJeonKKimJMKimVNChoiDH
2005
Downregulation of GFAP, TSP-1, and p53 in human glioblastoma cell
line, U373MG, by IE1 protein from human cytomegalovirus.
Glia
51
1
12
63. KohKLeeKAhnJHKimS
2009
Human cytomegalovirus infection downregulates the expression of
glial fibrillary acidic protein in human glioblastoma U373MG cells:
identification of viral genes and protein domains involved.
J Gen Virol
90
954
962
64. KlineJNGeistLJMonickMMStinskiMFHunninghakeGW
1994
Regulation of expression of the IL-1 receptor antagonist (IL-1ra)
gene by products of the human cytomegalovirus immediate early
genes.
J Immunol
152
2351
2357
65. CobbsCSSoroceanuLDenhamSZhangWKrausMH
2008
Modulation of oncogenic phenotype in human glioma cells by
cytomegalovirus IE1-mediated mitogenicity.
Cancer Res
68
724
730
66. CastilloJPFrameFMRogoffHAPickeringMTYurochkoAD
2005
Human cytomegalovirus IE1-72 activates ataxia telangiectasia
mutated kinase and a p53/p21-mediated growth arrest
response.
J Virol
79
11467
11475
67. ShenYZhuHShenkT
1997
Human cytomagalovirus IE1 and IE2 proteins are mutagenic and
mediate “hit-and-run” oncogenic transformation in cooperation
with the adenovirus E1A proteins.
Proc Natl Acad Sci U S A
94
3341
3345
68. SourvinosGEverettRD
2002
Visualization of parental HSV-1 genomes and replication
compartments in association with ND10 in live infected
cells.
EMBO J
21
4989
4997
69. EverettRDParsyMLOrrA
2009
Analysis of the functions of herpes simplex virus type 1
regulatory protein ICP0 that are critical for lytic infection and
derepression of quiescent viral genomes.
J Virol
83
4963
4977
70. EverettRDOrrA
2009
Herpes simplex virus type 1 regulatory protein ICP0 aids
infection in cells with a preinduced interferon response but does not impede
interferon-induced gene induction.
J Virol
83
4978
4983
71. NevelsMBruneWShenkT
2004
SUMOylation of the human cytomegalovirus major immediate-early
protein IE1-72kDa contributes to efficient viral replication by promoting
the accumulation of IE2-86kDa.
J Virol
78
7803
7812
72. EverettRD
1984
Transactivation of transcription by herpes virus products:
requirement for two HSV-1 immediate-early polypeptides for maximum
activity.
EMBO J
3
3135
3141
73. MurphyKTraversPWalportM
2008
Janeway's Immunobiology
New York
Garland Science
887
74. LacotteSBrunSMullerSDumortierH
2009
CXCR3, inflammation, and autoimmune diseases.
Ann N Y Acad Sci
1173
310
317
75. CollinsTLJohnsonMGMedinaJC
2007
Antagonists of CXCR3: a review of current
progress.
NeoteKLettsGLMoserB
Chemokine biology - basic research and clinical applications
Basel
Birkhauser
79
88
76. NocentiniGRiccardiC
2009
GITR: a modulator of immune response and
inflammation.
Adv Exp Med Biol
647
156
173
77. DuttaguptaPABoesteanuACKatsikisPD
2009
Costimulation signals for memory CD8+ T cells during viral
infections.
Crit Rev Immunol
29
469
486
78. CroftM
2010
Control of immunity by the TNFR-related molecule OX40
(CD134).
Annu Rev Immunol
28
57
78
79. IshiiNTakahashiTSorooshPSugamuraK
2010
OX40-OX40 ligand interaction in T-cell-mediated immunity and
immunopathology.
Adv Immunol
105
63
98
80. KrogerAKosterMSchroederKHauserHMuellerPP
2002
Activities of IRF-1.
J Interferon Cytokine Res
22
5
14
81. BattistiniA
2009
Interferon regulatory factors in hematopoietic cell
differentiation and immune regulation.
J Interferon Cytokine Res
29
765
780
82. SavitskyDTamuraTYanaiHTaniguchiT
2010
Regulation of immunity and oncogenesis by the IRF transcription
factor family.
Cancer Immunol Immunother
59
489
510
83. MacKenzieCRHeselerKMullerADaubenerW
2007
Role of indoleamine 2,3-dioxygenase in antimicrobial defence and
immuno-regulation: tryptophan depletion versus production of toxic
kynurenines.
Curr Drug Metab
8
237
244
84. CherayilBJ
2009
Indoleamine 2,3-dioxygenase in intestinal immunity and
inflammation.
Inflamm Bowel Dis
15
1391
1396
85. JiaLTianPDingC
2009
Immunoregulatory effects of indoleamine 2, 3-dioxygenase in
transplantation.
Transpl Immunol
21
18
22
86. ItsuiYSakamotoNKakinumaSNakagawaMSekine-OsajimaY
2009
Antiviral effects of the interferon-induced protein guanylate
binding protein 1 and its interaction with the hepatitis C virus NS5B
protein.
Hepatology
50
1727
1737
87. AndersonSLCartonJMLouJXingLRubinBY
1999
Interferon-induced guanylate binding protein-1 (GBP-1) mediates
an antiviral effect against vesicular stomatitis virus and
encephalomyocarditis virus.
Virology
256
8
14
88. ItsuiYSakamotoNKurosakiMKanazawaNTanabeY
2006
Expressional screening of interferon-stimulated genes for
antiviral activity against hepatitis C virus replication.
J Viral Hepat
13
690
700
89. CarterCCGorbachevaVYVestalDJ
2005
Inhibition of VSV and EMCV replication by the interferon-induced
GTPase, mGBP-2: differential requirement for wild-type GTP binding
domain.
Arch Virol
150
1213
1220
90. SamarajiwaSAForsterSAuchettlKHertzogPJ
2009
INTERFEROME: the database of interferon regulated
genes.
Nucleic Acids Res
37
D852
857
91. ErlandssonLBlumenthalRElorantaMLEngelHAlmG
1998
Interferon-beta is required for interferon-alpha production in
mouse fibroblasts.
Curr Biol
8
223
226
92. TaylorRTBresnahanWA
2005
Human cytomegalovirus immediate-early 2 gene expression blocks
virus-induced beta interferon production.
J Virol
79
3873
3877
93. MatsumotoMTanakaNHaradaHKimuraTYokochiT
1999
Activation of the transcription factor ISGF3 by
interferon-gamma.
Biol Chem
380
699
703
94. ZimmermannATrillingMWagnerMWilbornMBubicI
2005
A cytomegaloviral protein reveals a dual role for STAT2 in
IFN-γ signaling and antiviral responses.
J Exp Med
201
1543
1553
95. WesolyJSzweykowska-KulinskaZBluyssenHA
2007
STAT activation and differential complex formation dictate
selectivity of interferon responses.
Acta Biochim Pol
54
27
38
96. NavarroLMowenKRodemsSWeaverBReichN
1998
Cytomegalovirus activates interferon immediate-early response
gene expression and an interferon regulatory factor 3-containing
interferon-stimulated response element-binding complex.
Mol Cell Biol
18
3796
3802
97. SehgalPB
2008
Paradigm shifts in the cell biology of STAT
signaling.
Semin Cell Dev Biol
19
329
340
98. VarinouLRamsauerKKaraghiosoffMKolbeTPfefferK
2003
Phosphorylation of the Stat1 transactivation domain is required
for full-fledged IFN-gamma-dependent innate immunity.
Immunity
19
793
802
99. YangJStarkGR
2008
Roles of unphosphorylated STATs in signaling.
Cell Res
18
443
451
100. BrownSZeidlerMP
2008
Unphosphorylated STATs go nuclear.
Curr Opin Genet Dev
18
455
460
101. SadzakISchiffMGattermeierIGlinitzerRSauerI
2008
Recruitment of Stat1 to chromatin is required for
interferon-induced serine phosphorylation of Stat1 transactivation
domain.
Proc Natl Acad Sci U S A
105
8944
8949
102. RobertsonGHirstMBainbridgeMBilenkyMZhaoY
2007
Genome-wide profiles of STAT1 DNA association using chromatin
immunoprecipitation and massively parallel sequencing.
Nat Methods
4
651
657
103. MesseguerXEscuderoRFarreDNunezOMartinezJ
2002
PROMO: detection of known transcription regulatory elements using
species-tailored searches.
Bioinformatics
18
333
334
104. FarreDRosetRHuertaMAdsuaraJERoselloL
2003
Identification of patterns in biological sequences at the ALGGEN
server: PROMO and MALGEN.
Nucleic Acids Res
31
3651
3653
105. CherringtonJMMocarskiES
1989
Human cytomegalovirus ie1 transactivates the alpha
promoter-enhancer via an 18-base-pair repeat element.
J Virol
63
1435
1440
106. SambucettiLCCherringtonJMWilkinsonGWMocarskiES
1989
NF-kappa B activation of the cytomegalovirus enhancer is mediated
by a viral transactivator and by T cell stimulation.
EMBO J
8
4251
4258
107. LuuPFloresO
1997
Binding of SP1 to the immediate-early protein-responsive element
of the human cytomegalovirus DNA polymerase promoter.
J Virol
71
6683
6691
108. MaloneCLVesoleDHStinskiMF
1990
Transactivation of a human cytomegalovirus early promoter by gene
products from the immediate-early gene IE2 and augmentation by IE1:
mutational analysis of the viral proteins.
J Virol
64
1498
1506
109. ReevesMWoodhallDComptonTSinclairJ
2010
Human cytomegalovirus IE72 protein interacts with the
transcriptional repressor hDaxx to regulate LUNA gene expression during
lytic infection.
J Virol
84
7185
7194
110. GeistLJDaiLY
1996
Cytomegalovirus modulates interleukin-6 gene
expression.
Transplantation
62
653
658
111. WadeMKowalikTFMudryjMHuangESAzizkhanJC
1992
E2F mediates dihydrofolate reductase promoter activation and
multiprotein complex formation in human cytomegalovirus
infection.
Mol Cell Biol
12
4364
4374
112. YurochkoADKowalikTFHuongSMHuangES
1995
Human cytomegalovirus upregulates NF-kappa B activity by
transactivating the NF-kappa B p105/p50 and p65 promoters.
J Virol
69
5391
5400
113. JiangHYPetrovasCSonensheinGE
2002
RelB-p50 NF-kappa B complexes are selectively induced by
cytomegalovirus immediate-early protein 1: differential regulation of
Bcl-x(L) promoter activity by NF-kappa B family members.
J Virol
76
5737
5747
114. ShirakataMTerauchiMAblikimMImadomeKHiraiK
2002
Novel immediate-early protein IE19 of human cytomegalovirus
activates the origin recognition complex I promoter in a cooperative manner
with IE72.
J Virol
76
3158
3167
115. TevethiaMJSpectorDJLeisureKMStinskiMF
1987
Participation of two human cytomegalovirus immediate early gene
regions in transcriptional activation of adenovirus
promoters.
Virology
161
276
285
116. MonickMMGeistLJStinskiMFHunninghakeGW
1992
The immediate early genes of human cytomegalovirus upregulate
expression of the cellular genes myc and fos.
Am J Respir Cell Mol Biol
7
251
256
117. KimJMHongYKimSChoMHYoshidaM
1999
Sequences downstream of the RNA initiation site of the HTLV type
I long terminal repeat are sufficient for trans-activation by human
cytomegalovirus immediate-early proteins.
AIDS Res Hum Retroviruses
15
545
550
118. JohnsonRAYurochkoADPomaEEZhuLHuangES
1999
Domain mapping of the human cytomegalovirus IE1-72 and cellular
p107 protein-protein interaction and the possible functional
consequences.
J Gen Virol
80
1293
1303
119. KimSYuSSKimVN
1996
Essential role of NF-kappa B in transactivation of the human
immunodeficiency virus long terminal repeat by the human cytomegalovirus 1E1
protein.
J Gen Virol
77
83
91
120. WalkerSHagemeierCSissonsJGSinclairJH
1992
A 10-base-pair element of the human immunodeficiency virus type 1
long terminal repeat (LTR) is an absolute requirement for transactivation by
the human cytomegalovirus 72-kilodalton IE1 protein but can be compensated
for by other LTR regions in transactivation by the 80-kilodalton IE2
protein.
J Virol
66
1543
1550
121. WangXSonensheinGE
2005
Induction of the RelB NF-kappaB subunit by the cytomegalovirus
IE1 protein is mediated via Jun kinase and c-Jun/Fra-2 AP-1
complexes.
J Virol
79
95
105
122. CrumpJWGeistLJAuronPEWebbACStinskiMF
1992
The immediate early genes of human cytomegalovirus require only
proximal promoter elements to upregulate expression of interleukin-1
beta.
Am J Respir Cell Mol Biol
6
674
677
123. LukacDMManuppelloJRAlwineJC
1994
Transcriptional activation by the human cytomegalovirus
immediate-early proteins: requirements for simple promoter structures and
interactions with multiple components of the transcription
complex.
J Virol
68
5184
5193
124. KimJMHongYKimS
2000
Artificial recruitment of Sp1 or TBP can replace the role of IE1
in the synergistic transactivation by IE1 and IE2.
Biochem Biophys Res Commun
269
302
308
125. Dal MontePLandiniMPSinclairJVirelizierJLMichelsonS
1997
TAR and Sp1-independent transactivation of HIV long terminal
repeat by the Tat protein in the presence of human cytomegalovirus
IE1/IE2.
Aids
11
297
303
126. GeistLJHopkinsHADaiLYHeBMonickMM
1997
Cytomegalovirus modulates transcription factors necessary for the
activation of the tumor necrosis factor-alpha promoter.
Am J Respir Cell Mol Biol
16
31
37
127. CinatlJJrNevelsMPaulusCMichaelisM
2009
Activation of telomerase in glioma cells by human
cytomegalovirus: another piece of the puzzle.
J Natl Cancer Inst
101
441
443
128. RamsauerKFarlikMZupkovitzGSeiserCKrogerA
2007
Distinct modes of action applied by transcription factors STAT1
and IRF1 to initiate transcription of the IFN-gamma-inducible gbp2
gene.
Proc Natl Acad Sci U S A
104
2849
2854
129. StrassheimDRiddleSRBurkeDLGeraciMWStenmarkKR
2009
Prostacyclin inhibits IFN-gamma-stimulated cytokine expression by
reduced recruitment of CBP/p300 to STAT1 in a SOCS-1-independent
manner.
J Immunol
183
6981
6988
130. ReichNC
2007
STAT dynamics.
Cytokine Growth Factor Rev
18
511
518
131. MeyerTVinkemeierU
2004
Nucleocytoplasmic shuttling of STAT transcription
factors.
Eur J Biochem
271
4606
4612
132. HaspelRLDarnellJEJr
1999
A nuclear protein tyrosine phosphatase is required for the
inactivation of Stat1.
Proc Natl Acad Sci U S A
96
10188
10193
133. MeyerTMargALemkePWiesnerBVinkemeierU
2003
DNA binding controls inactivation and nuclear accumulation of the
transcription factor Stat1.
Genes Dev
17
1992
2005
134. SubramaniamPSTorresBAJohnsonHM
2001
So many ligands, so few transcription factors: a new paradigm for
signaling through the STAT transcription factors.
Cytokine
15
175
187
135. AndersenPPedersenMWWoetmannAVillingshojMStockhausenMT
2008
EGFR induces expression of IRF-1 via STAT1 and STAT3 activation
leading to growth arrest of human cancer cells.
Int J Cancer
122
342
349
136. GrudinkinPSZeninVVKropotovAVDoroshVNNikolskyNN
2007
EGF-induced apoptosis in A431 cells is dependent on STAT1, but
not on STAT3.
Eur J Cell Biol
86
591
603
137. KennedyAMShogrenKLZhangMTurnerRTSpelsbergTC
2005
17beta-estradiol-dependent activation of signal transducer and
activator of transcription-1 in human fetal osteoblasts is dependent on Src
kinase activity.
Endocrinology
146
201
207
138. SadowskiHBShuaiKDarnellJEJrGilmanMZ
1993
A common nuclear signal transduction pathway activated by growth
factor and cytokine receptors.
Science
261
1739
1744
139. FuXYZhangJJ
1993
Transcription factor p91 interacts with the epidermal growth
factor receptor and mediates activation of the c-fos gene
promoter.
Cell
74
1135
1145
140. GuoDDunbarJDYangCHPfefferLMDonnerDB
1998
Induction of Jak/STAT signaling by activation of the type 1 TNF
receptor.
J Immunol
160
2742
2750
141. SizemoreNAgarwalADasKLernerNSulakM
2004
Inhibitor of kappaB kinase is required to activate a subset of
interferon gamma-stimulated genes.
Proc Natl Acad Sci U S A
101
7994
7998
142. DebAHaqueSJMogensenTSilvermanRHWilliamsBR
2001
RNA-dependent protein kinase PKR is required for activation of
NF-kappa B by IFN-gamma in a STAT1-independent pathway.
J Immunol
166
6170
6180
143. ShultzDBFullerJDYangYSizemoreNRaniMR
2007
Activation of a subset of genes by IFN-gamma requires IKKbeta but
not interferon-dependent activation of NF-kappaB.
J Interferon Cytokine Res
27
875
884
144. ShultzDBRaniMRFullerJDRansohoffRMStarkGR
2009
Roles of IKK-beta, IRF1, and p65 in the activation of chemokine
genes by interferon-gamma.
J Interferon Cytokine Res
29
817
824
145. WeiLFanMXuLHeinrichKBerryMW
2008
Bioinformatic analysis reveals cRel as a regulator of a subset of
interferon-stimulated genes.
J Interferon Cytokine Res
28
541
551
146. DuZWeiLMurtiAPfefferSRFanM
2007
Non-conventional signal transduction by type 1 interferons: the
NF-kappaB pathway.
J Cell Biochem
102
1087
1094
147. GoughDJLevyDEJohnstoneRWClarkeCJ
2008
IFNgamma signaling-does it mean JAK-STAT?
Cytokine Growth Factor Rev
19
383
394
148. Storm van's GravesandeKLayneMDYeQLeLBaronRM
2002
IFN regulatory factor-1 regulates IFN-gamma-dependent cathepsin S
expression.
J Immunol
168
4488
4494
149. WhiteLCWrightKLFelixNJRuffnerHReisLF
1996
Regulation of LMP2 and TAP1 genes by IRF-1 explains the paucity
of CD8+ T cells in IRF-1−/ − mice.
Immunity
5
365
376
150. KimuraTKadokawaYHaradaHMatsumotoMSatoM
1996
Essential and non-redundant roles of p48 (ISGF3 gamma) and IRF-1
in both type I and type II interferon responses, as revealed by gene
targeting studies.
Genes Cells
1
115
124
151. TerrisBBaldinVDuboisSDegottCFlejouJF
1995
PML nuclear bodies are general targets for inflammation and cell
proliferation.
Cancer Res
55
1590
1597
152. ChingRWDellaireGEskiwCHBazett-JonesDP
2005
PML bodies: a meeting place for genomic loci?
J Cell Sci
118
847
854
153. ShielsCIslamSAVatchevaRSasieniPSternbergMJ
2001
PML bodies associate specifically with the MHC gene cluster in
interphase nuclei.
J Cell Sci
114
3705
3716
154. VolpiEVChevretEJonesTVatchevaRWilliamsonJ
2000
Large-scale chromatin organization of the major
histocompatibility complex and other regions of human chromosome 6 and its
response to interferon in interphase nuclei.
J Cell Sci
113
1565
1576
155. ChristovaRJonesTWuPJBolzerACosta-PereiraAP
2007
P-STAT1 mediates higher-order chromatin remodelling of the human
MHC in response to IFNgamma.
J Cell Sci
120
3262
3270
156. KumarPPBischofOPurbeyPKNotaniDUrlaubH
2007
Functional interaction between PML and SATB1 regulates
chromatin-loop architecture and transcription of the MHC class I
locus.
Nat Cell Biol
9
45
56
157. MocarskiESJr
2002
Virus self-improvement through inflammation: no pain, no
gain.
Proc Natl Acad Sci U S A
99
3362
3364
158. PowersCDeFilippisVMalouliDFruhK
2008
Cytomegalovirus immune evasion.
Curr Top Microbiol Immunol
325
333
359
159. Miller-KittrellMSparerTE
2009
Feeling manipulated: cytomegalovirus immune
manipulation.
Virol J
6
4
160. CheeranMCHuSShengWSPetersonPKLokensgardJR
2003
CXCL10 production from cytomegalovirus-stimulated microglia is
regulated by both human and viral interleukin-10.
J Virol
77
4502
4515
161. RennesonJDuttaBGorielySDanisBLecomteS
2009
IL-12 and type I IFN response of neonatal myeloid DC to human CMV
infection.
Eur J Immunol
39
2789
2799
162. MezgerMBoninMKesslerTGebhardtFEinseleH
2009
Toll-like receptor 3 has no critical role during early immune
response of human monocyte-derived dendritic cells after infection with the
human cytomegalovirus strain TB40E.
Viral Immunol
22
343
351
163. CaposioPMussoTLuganiniAInoueHGariglioM
2007
Targeting the NF-kappaB pathway through pharmacological
inhibition of IKK2 prevents human cytomegalovirus replication and
virus-induced inflammatory response in infected endothelial
cells.
Antiviral Res
73
175
184
164. GravelSPServantMJ
2005
Roles of an IkappaB kinase-related pathway in human
cytomegalovirus-infected vascular smooth muscle cells: a molecular link in
pathogen-induced proatherosclerotic conditions.
J Biol Chem
280
7477
7486
165. TaylorRTBresnahanWA
2006
Human cytomegalovirus immediate-early 2 protein IE86 blocks
virus-induced chemokine expression.
J Virol
80
920
928
166. JinquanTQuanSJacobiHHJingCMillnerA
2000
CXC chemokine receptor 3 expression on CD34(+) hematopoietic
progenitors from human cord blood induced by granulocyte-macrophage
colony-stimulating factor: chemotaxis and adhesion induced by its ligands,
interferon gamma-inducible protein 10 and monokine induced by interferon
gamma.
Blood
96
1230
1238
167. GoodrumFJordanCTTerhuneSSHighKShenkT
2004
Differential outcomes of human cytomegalovirus infection in
primitive hematopoietic cell subpopulations.
Blood
104
687
695
168. GoodrumFDJordanCTHighKShenkT
2002
Human cytomegalovirus gene expression during infection of primary
hematopoietic progenitor cells: a model for latency.
Proc Natl Acad Sci U S A
99
16255
16260
169. MintonEJTysoeCSinclairJHSissonsJG
1994
Human cytomegalovirus infection of the monocyte/macrophage
lineage in bone marrow.
J Virol
68
4017
4021
170. MaciejewskiJPBrueningEEDonahueREMocarskiESYoungNS
1992
Infection of hematopoietic progenitor cells by human
cytomegalovirus.
Blood
80
170
178
171. MendelsonMMonardSSissonsPSinclairJ
1996
Detection of endogenous human cytomegalovirus in CD34+ bone
marrow progenitors.
J Gen Virol
77
3099
3102
172. von LaerDMeyer-KoenigUSerrAFinkeJKanzL
1995
Detection of cytomegalovirus DNA in CD34+ cells from blood
and bone marrow.
Blood
86
4086
4090
173. AgostiniCCalabreseFReaFFaccoMTosoniA
2001
Cxcr3 and its ligand CXCL10 are expressed by inflammatory cells
infiltrating lung allografts and mediate chemotaxis of T cells at sites of
rejection.
Am J Pathol
158
1703
1711
174. MelterMExeniAReindersMEFangJCMcMahonG
2001
Expression of the chemokine receptor CXCR3 and its ligand IP-10
during human cardiac allograft rejection.
Circulation
104
2558
2564
175. GoddardSWilliamsAMorlandCQinSGladueR
2001
Differential expression of chemokines and chemokine receptors
shapes the inflammatory response in rejecting human liver
transplants.
Transplantation
72
1957
1967
176. ZhaoDXHuYMillerGGLusterADMitchellRN
2002
Differential expression of the IFN-gamma-inducible CXCR3-binding
chemokines, IFN-inducible protein 10, monokine induced by IFN, and
IFN-inducible T cell alpha chemoattractant in human cardiac allografts:
association with cardiac allograft vasculopathy and acute
rejection.
J Immunol
169
1556
1560
177. FahmyNMYamaniMHStarlingRCRatliffNBYoungJB
2003
Chemokine and receptor-gene expression during early and late
acute rejection episodes in human cardiac allografts.
Transplantation
75
2044
2047
178. KaoJKobashigawaJFishbeinMCMacLellanWRBurdickMD
2003
Elevated serum levels of the CXCR3 chemokine ITAC are associated
with the development of transplant coronary artery disease.
Circulation
107
1958
1961
179. FahmyNMYamaniMHStarlingRCRatliffNBYoungJB
2003
Chemokine and chemokine receptor gene expression indicates acute
rejection of human cardiac transplants.
Transplantation
75
72
78
180. HuHAizensteinBDPuchalskiABurmaniaJAHamawyMM
2004
Elevation of CXCR3-binding chemokines in urine indicates acute
renal-allograft dysfunction.
Am J Transplant
4
432
437
181. PanzerUReinkingRRSteinmetzOMZahnerGSudbeckU
2004
CXCR3 and CCR5 positive T-cell recruitment in acute human renal
allograft rejection.
Transplantation
78
1341
1350
182. StreblowDNKreklywichCNAndohTMosesAVDumortierJ
2008
The role of angiogenic and wound repair factors during
CMV-accelerated transplant vascular sclerosis in rat cardiac
transplants.
Am J Transplant
8
277
287
183. StreblowDNKreklywichCYinQDe La MelenaVTCorlessCL
2003
Cytomegalovirus-mediated upregulation of chemokine expression
correlates with the acceleration of chronic rejection in rat heart
transplants.
J Virol
77
2182
2194
184. XanthouGDuchesnesCEWilliamsTJPeaseJE
2003
CCR3 functional responses are regulated by both CXCR3 and its
ligands CXCL9, CXCL10 and CXCL11.
Eur J Immunol
33
2241
2250
185. BuscheAMarquardtABleichAGhazalPAnguloA
2009
The mouse cytomegalovirus immediate-early 1 gene is not required
for establishment of latency or for reactivation in the
lungs.
J Virol
83
4030
4038
186. Soderberg-NauclerCFishKNNelsonJA
1997
Interferon-gamma and tumor necrosis factor-alpha specifically
induce formation of cytomegalovirus-permissive monocyte-derived macrophages
that are refractory to the antiviral activity of these
cytokines.
J Clin Invest
100
3154
3163
187. Soderberg-NauclerCFishKNNelsonJA
1997
Reactivation of latent human cytomegalovirus by allogeneic
stimulation of blood cells from healthy donors.
Cell
91
119
126
188. Soderberg-NauclerCStreblowDNFishKNAllan-YorkeJSmithPP
2001
Reactivation of latent human cytomegalovirus in CD14(+)
monocytes is differentiation dependent.
J Virol
75
7543
7554
189. HahnGJoresRMocarskiES
1998
Cytomegalovirus remains latent in a common precursor of dendritic
and myeloid cells.
Proc Natl Acad Sci U S A
95
3937
3942
190. ReevesMSinclairJ
2008
Aspects of human cytomegalovirus latency and
reactivation.
Curr Top Microbiol Immunol
325
297
313
191. SinclairJ
2008
Human cytomegalovirus: Latency and reactivation in the myeloid
lineage.
J Clin Virol
41
180
185
192. HummelMAbecassisMM
2002
A model for reactivation of CMV from latency.
J Clin Virol
25
Suppl 2
S123
136
193. DelnesteYCharbonnierPHerbaultNMagistrelliGCaronG
2003
Interferon-gamma switches monocyte differentiation from dendritic
cells to macrophages.
Blood
101
143
150
194. NetterwaldJYangSWangWGhannySCodyM
2005
Two gamma interferon-activated site-like elements in the human
cytomegalovirus major immediate-early promoter/enhancer are important for
viral replication.
J Virol
79
5035
5046
195. ReddehaseMJSimonCOSeckertCKLemmermannNGrzimekNK
2008
Murine model of cytomegalovirus latency and
reactivation.
Curr Top Microbiol Immunol
325
315
331
196. ReddehaseMJ
2000
The immunogenicity of human and murine
cytomegaloviruses.
Curr Opin Immunol
12
390
396
197. GrundyJEShanleyJDGriffithsPD
1987
Is cytomegalovirus interstitial pneumonitis in transplant
recipients an immunopathological condition?
Lancet
2
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2011 Číslo 4
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Měli bychom postcovidový syndrom léčit antidepresivy?
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- NF-κB Hyper-Activation by HTLV-1 Tax Induces Cellular Senescence, but Can Be Alleviated by the Viral Anti-Sense Protein HBZ
- Bacterial and Host Determinants of MAL Activation upon EPEC Infection: The Roles of Tir, ABRA, and FLRT3
- : Reservoir Hosts and Tracking the Emergence in Humans and Macaques
- On Being the Right Size: The Impact of Population Size and Stochastic Effects on the Evolution of Drug Resistance in Hospitals and the Community