
Measles Immune Suppression: Lessons from the Macaque Model
Measles remains a significant childhood disease, and is associated with a transient immune suppression. Paradoxically, measles virus (MV) infection also induces robust MV-specific immune responses. Current hypotheses for the mechanism underlying measles immune suppression focus on functional impairment of lymphocytes or antigen-presenting cells, caused by infection with or exposure to MV. We have generated stable recombinant MVs that express enhanced green fluorescent protein, and remain virulent in non-human primates. By performing a comprehensive study of virological, immunological, hematological and histopathological observations made in animals euthanized at different time points after MV infection, we developed a model explaining measles immune suppression which fits with the “measles paradox”. Here we show that MV preferentially infects CD45RA− memory T-lymphocytes and follicular B-lymphocytes, resulting in high infection levels in these populations. After the peak of viremia MV-infected lymphocytes were cleared within days, followed by immune activation and lymph node enlargement. During this period tuberculin-specific T-lymphocyte responses disappeared, whilst strong MV-specific T-lymphocyte responses emerged. Histopathological analysis of lymphoid tissues showed lymphocyte depletion in the B - and T-cell areas in the absence of apoptotic cells, paralleled by infiltration of T-lymphocytes into B-cell follicles and reappearance of proliferating cells. Our findings indicate an immune-mediated clearance of MV-infected CD45RA− memory T-lymphocytes and follicular B-lymphocytes, which causes temporary immunological amnesia. The rapid oligoclonal expansion of MV-specific lymphocytes and bystander cells masks this depletion, explaining the short duration of measles lymphopenia yet long duration of immune suppression.
Published in the journal:
. PLoS Pathog 8(8): e32767. doi:10.1371/journal.ppat.1002885
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1002885
Summary
Measles remains a significant childhood disease, and is associated with a transient immune suppression. Paradoxically, measles virus (MV) infection also induces robust MV-specific immune responses. Current hypotheses for the mechanism underlying measles immune suppression focus on functional impairment of lymphocytes or antigen-presenting cells, caused by infection with or exposure to MV. We have generated stable recombinant MVs that express enhanced green fluorescent protein, and remain virulent in non-human primates. By performing a comprehensive study of virological, immunological, hematological and histopathological observations made in animals euthanized at different time points after MV infection, we developed a model explaining measles immune suppression which fits with the “measles paradox”. Here we show that MV preferentially infects CD45RA− memory T-lymphocytes and follicular B-lymphocytes, resulting in high infection levels in these populations. After the peak of viremia MV-infected lymphocytes were cleared within days, followed by immune activation and lymph node enlargement. During this period tuberculin-specific T-lymphocyte responses disappeared, whilst strong MV-specific T-lymphocyte responses emerged. Histopathological analysis of lymphoid tissues showed lymphocyte depletion in the B - and T-cell areas in the absence of apoptotic cells, paralleled by infiltration of T-lymphocytes into B-cell follicles and reappearance of proliferating cells. Our findings indicate an immune-mediated clearance of MV-infected CD45RA− memory T-lymphocytes and follicular B-lymphocytes, which causes temporary immunological amnesia. The rapid oligoclonal expansion of MV-specific lymphocytes and bystander cells masks this depletion, explaining the short duration of measles lymphopenia yet long duration of immune suppression.
Introduction
Measles is associated with a transient but profound immune suppression, which may last for several weeks to months after the acute stage of the disease. The clinical importance of this immune suppression is illustrated by the observation that measles mortality is typically caused by secondary infections in the respiratory or digestive tract [1]–[3]. However, the mechanism by which measles virus (MV) infection causes immune suppression is not completely understood. Multiple in vivo correlates of immune suppression have been described, including disappearance of Mantoux responses [4], [5], lymphopenia [6], [7] and impaired responses to vaccination [8], [9]. Decreased lymphoproliferative responses [10], [11], altered cytokine response profiles [12] and impairment of antigen-presenting cell function [13]–[15] have been described in vitro. The relevance of these observations to immune suppression and enhanced susceptibility to opportunistic infections remains unclear. The paradox of measles is that the acute phase of the disease is not only associated with immune suppression, but also with immune activation [16] and induction of robust MV-specific humoral and cellular immune responses that result in lifelong immunity.
MV infection is initiated in the respiratory tract. It has long been thought that the initial target cells of the virus were epithelial cells of the upper respiratory tract, but recent studies have demonstrated a major role for alveolar macrophages and dendritic cells (DC) in this process [17], [18]. These first MV-infected cells transmit the virus to the bronchus-associated lymphoid tissue (BALT) and/or the draining lymph nodes, where the infection is further amplified in lymphocytes and viremia is initiated [18], [19]. MV infects both T - and B-lymphocytes by binding of the MV-hemagglutinin (H) glycoprotein to the cellular receptor CD150 [20]. Recently, the adherens junction protein PVRL4 was identified as cellular receptor on epithelial cells [21], [22]. However, as this receptor is exclusively expressed on the basolateral surface of epithelial cells, it does not facilitate MV infection of epithelial cells during the early stages of the disease, but instead is thought to play a role in transmission during the late stages of measles pathogenesis [23].
It has been proposed that direct infection of lymphocytes and the subsequent lymphopenia could explain measles-associated immune suppression [24], [25]. However, this hypothesis has often been dismissed based on the observation that lymphopenia only lasts about a week, whilst immune suppression persists for several weeks to months. In addition, during the peak of viremia no more than 1 to 5% of the total lymphocyte population in peripheral blood is infected [26], [27]. Recent observations that MV infects high percentages of cells in lymphoid tissues [27] and preferentially targets CD45RA− or CD45R0+ memory T-lymphocytes [27], [28] led us to revisit the lymphocyte depletion hypothesis for immune suppression using the macaque model. Analysis of virological, immunological and histopathological parameters has demonstrated a remarkable similarity between measles in macaques and humans [29]. Here we present a comprehensive overview of a number of in vivo studies performed in macaques which provides a unifying model for the etiology of measles immune suppression that is both compatible with the measles paradox and with historical in vitro and in vivo correlates of measles immune suppression.
Results
MV targets lymphoid tissues and preferentially infects CD45RA− memory T-lymphocytes
We have analyzed data from rhesus or cynomolgus macaques (n = 40) infected with recombinant (r) MV strains (rMVIC323 or rMVKS) expressing enhanced green fluorescent protein (EGFP), spanning the early, intermediate and late stages of MV infection (Table S1). Data from previous studies [18], [27], [30] and from additional experimentally infected macaques (n = 14) were combined. The rMVs expressed EGFP from an additional transcription unit, and MV replication results in the host cell becoming EGFP+. At the peak of viremia, EGFP fluorescence was macroscopically detected in all lymphoid tissues (Figure 1A–D). The percentages MV-infected cells in lymphocyte subsets in PBMC or lymphoid tissues collected 9 or 11 days post-infection (d.p.i.) were determined by flow cytometry. Lymphocytes were subtyped as CD4+ or CD8+ naive (CD45RA+, Tn), central memory (CD45RA−CCR7+, TCM) or effector memory (CD45RA−CCR7−, TEM) T-lymphocytes or as naive (IgD+CD27−, Bn) or memory (IgD−CD27+, BM) CD20+HLA-DR+ B-lymphocytes (Figure S1). TCM and TEM were infected at a significantly higher level than Tn. In contrast, BM were not preferentially infected (Figure 1E–H).

To determine whether the increased susceptibility of CD45RA− memory T-lymphocytes (TM) to MV infection is an inherent property of these cells, and comparable between humans and macaques, we sorted naive and memory CD4+ and CD8+ T-lymphocyte populations from human or macaque PBMC on basis of CD45A expression. In vitro co-culture of these populations with MV-infected autologous cells showed that both human and macaque CD45RA− TM were preferentially infected by MV (Figure 2A). In an alternative approach, unsorted human or macaque PBMC were co-cultured with autologous MV-infected cells, and infection percentages in Tn, TCM and TEM were determined by flow cytometry, resulting in similar differences in the susceptibility of T-lymphocyte subpopulations (Figure 2B). This not only corroborated the in vivo results from the experimentally infected macaques, but also demonstrated a virtually identical trend for human and macaque subpopulations.

The MV receptor CD150 is expressed at high levels by activated human CD45ROhigh memory T-lymphocytes [31]. We determined the expression levels of CD150 on the different human and macaque T-lymphocyte subsets. This confirmed the higher level of CD150 expression by human CD45RA− TCM and TEM when compared to CD45RA+ Tn, and showed that the expression levels of CD150 on human and macaque T-lymphocyte subsets are comparable, which likely explains the increased susceptibility of CD45RA− TM to MV infection (Figure 2C–F). Although we were unsuccessful in further subtyping B-lymphocyte subsets on basis of expression patterns of different surface markers, the observed efficient infection of both Bn and BM lymphocytes seems in accordance with recent studies demonstrating CD150 expression on virtually all human B-lymphocyte subpopulations [32].
MV causes lymphocyte depletion in lymphoid tissues
To address the impact of MV infection in situ, immunohistochemical analyses of serial sections of lymphoid tissues collected at different d.p.i. were performed. This demonstrated that MV mainly replicated in B-cell follicles (Figure 3; 7 and 9 d.p.i.), as previously described [27], [33]. Multiple syncytia were observed 9 d.p.i., and dual immunofluorescence showed these were of B-lymphocyte origin (Figure S2). Strikingly similar to classic observations in humans [34], lymphoid exhaustion of the centers of the B-cell follicles was observed during and shortly after the peak of viremia (Figure 3; 9 and 11 d.p.i.).

MV infection suppresses tuberculin-specific T-lymphocyte responses
In vitro and in vivo recall T-lymphocyte responses to tuberculin were measured in Bacille Calmette-Guérin (BCG)-vaccinated macaques, prior to and 11 or 13 d.p.i. IFN-γ production of PBMC in response to purified-protein derivative (PPD) stimulation was reduced after MV infection. Notably, an MV-specific IFN-γ response was detected in PBMC collected from macaques sacrificed 13 d.p.i. (Figure 4A). The in vivo recall response was determined by Mantoux testing. Before MV infection a characteristic delayed-type hypersensitivity response developed on the site of intra-dermal tuberculin injection, characterized by a well-delineated soft swelling of the cutus, corresponding with an influx of CD3+ T-lymphocytes (Figure 4B). In line with classical observations [4], [5], Mantoux responses were suppressed after MV infection, with a much smaller and harder swelling in the cutus, potentially corresponding with epidermal repair, skin-infiltrating CD3+ T-lymphocytes were absent (Figure 4B). Interestingly, we also observed macroscopically detectable EGFP expression in the skin at the sites where the animals had been vaccinated intra-cutaneously with BCG three months prior to MV infection (Figure S3), suggesting infection of tissue-resident memory lymphocytes in the skin.

MV infection causes transient leukopenia followed by massive lymphocyte expansion
Analysis of macaque white blood cell (WBC) counts during the acute infection demonstrated a profound but transient leukopenia (Figure 5B, circles), which coincided with the peak of viremia. There was a relative decrease in size of the CD45RA− CD4+ and CD8+ TCM and TEM populations between 0 and 9 d.p.i. (Figure 5A), suggesting that leukopenia was related to depletion of MV-infected cells. WBC counts rapidly returned to pre-infection levels between 9 and 15 d.p.i. (Figure 5B), paralleled by a restoration of the relative CD45RA− TCM and TEM population sizes (Figure 5A). Lymph nodes were enlarged in all animals euthanized between 11 and 15 d.p.i., which was paralleled by the clearance of EGFP+ lymphocytes from lymphoid tissues. During this period large numbers of Ki67+ proliferating cells repopulated the B-cell follicles in the germinal centers of lymphoid tissues (Figure 3; 13 and 15 d.p.i.). Numbers of apoptotic lymphocytes, as detected by staining for cleaved caspase 3 (CC3), remained low at all time-points (Figure S4). Infiltration of CD3+ T-lymphocytes into the B-cell follicles suggests that MV-infected cells were cleared by cytotoxic T-lymphocyte-mediated killing, rather than undergoing apoptosis [35].
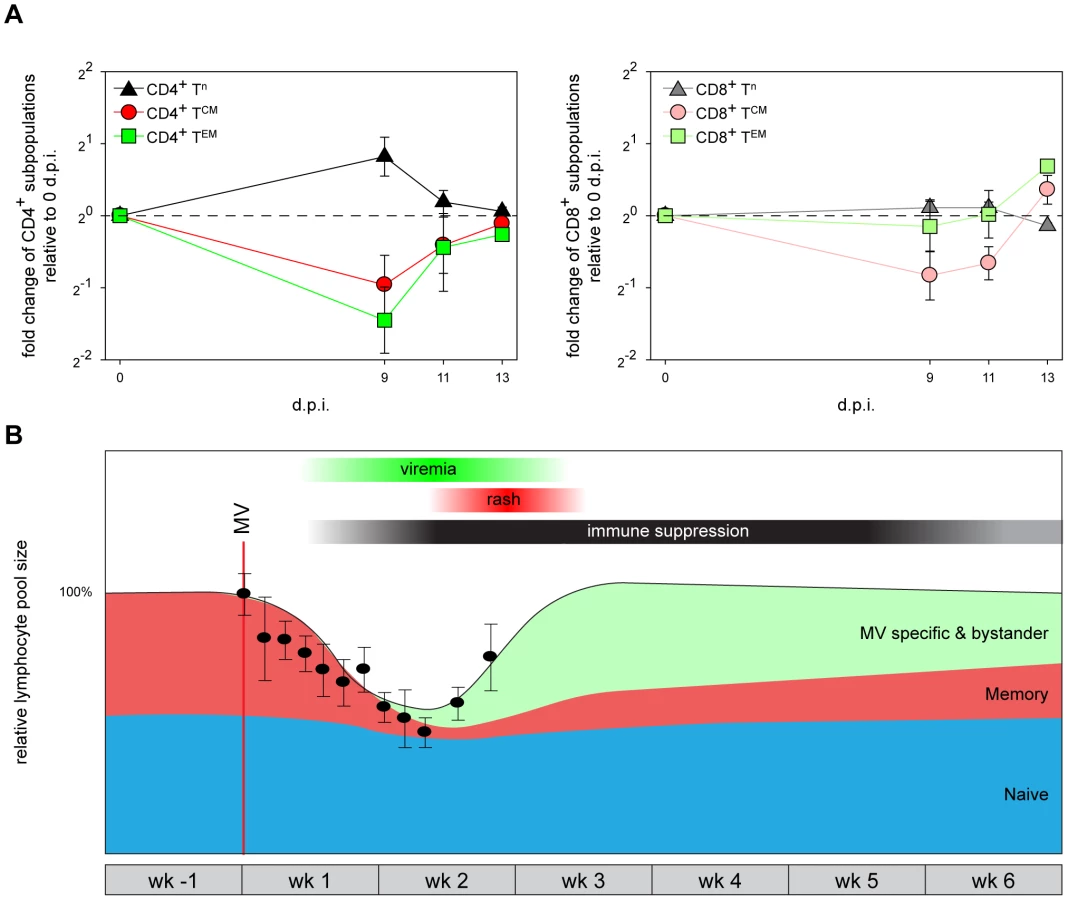
A unified model for measles immune suppression
Based on the observations described above we propose a model for the events leading to measles immune suppression. Infection and subsequent immune-mediated clearance of CD150+ lymphocytes results in specific depletion of memory T-lymphocytes and follicular B-lymphocytes, whilst the naive T-lymphocyte population remains relatively unaffected (Figure 5B, populations shown in red and blue, respectively). However, this leaves the question how such a short duration of measles lymphopenia can be reconciled with the long-lasting immune suppression. We hypothesize that the lymphocyte depletion is masked by the massive expansion of MV-specific and bystander lymphocytes (Figure 5B, population shown in green), which has also been observed in humans by a massive expansion of CD8+ T-lymphocytes [36] and by demonstrating skewing of the T-cell receptor repertoire after measles [37]. As a consequence, the qualitative composition of the lymphocyte population immediately after recovery from lymphopenia is dramatically different from that before MV infection, as pre-existing memory lymphocytes have been depleted and replaced by MV-specific and bystander lymphocytes. The net result is a temporary immunological amnesia, and restoration of immunological memory may take several weeks. Thus this model explains why measles-associated immune suppression extends well beyond the transient lymphopenia.
Discussion
Based on our observations, we conclude that measles immune suppression can, at least in part, be explained by massive infection and subsequent immune-mediated clearance of CD150+ memory T-lymphocytes and follicular B-lymphocytes. Depletion of T - and B-lymphocytes has also been described in the BALT of measles patients [38]. We show that MV preferentially infects CD45RA− TCM and TEM, which during secondary immune responses are the primary source of T-lymphocyte expansion or generation of effector T-lymphocytes, respectively [39]. Infection and subsequent immune-mediated depletion of memory T-lymphocyte subsets fits with the first description of measles-induced immune suppression, namely the disappearance of Mantoux responses in measles patients [4].
Several hypotheses for the underlying mechanism of measles immune suppression have been described previously. However, none of these adequately explain the “measles paradox”: the disease is associated with immune suppression but also with induction of strong MV-specific immune responses. Our observations not only explain the measles paradox, but also shed light on many of the in vivo and in vitro correlates of measles immune suppression described in literature [40]–[42]. Unresponsiveness of PBMC to mitogens and altered cytokine profiles during and after the acute phase of measles have been demonstrated in vitro in several studies [10], [11]. These observations are not disputed, but we consider it difficult to extrapolate unresponsiveness of PBMC to mitogens in vitro to immune suppression in vivo. In parallel with the clearance of MV-infected cells we observed high numbers of proliferating Ki67+ cells in B-cell follicles. This fits well with classical observations of immune activation following measles [16], suggesting that lymphoproliferation is not impaired in vivo during the convalescent phase. The observed in vitro suppression of mitogen-induced lymphoproliferation could also be explained by the altered qualitative composition of lymphocyte populations in convalescent measles patients, compared to healthy controls. Our model and previously published observations [16] show that a large proportion of the lymphocytes that circulate during the first weeks after measles infection have been recently activated in vivo, potentially making these cells less susceptible to re-stimulation in vitro.
We do not exclude that a functional impairment of lymphocytes or DC contributes to immune suppression and thus may augment the extent of immune suppression. Actually, it is likely that there are many factors that contribute to immune suppression in vivo. To date, direct evidence of DC infection by MV in humans has not been obtained. However, it has been demonstrated in vitro that MV is capable of infecting mature DC and Langerhans cells (LC) [13], [14], [43]–[47]. In vivo in experimentally MV-infected macaques there was strong evidence for infection of DC in the skin and secondary lymphoid tissues [27]. The role of DC in immune suppression has not been extensively studied in vivo, but it is possible that they play a role either by directly being targeted and depleted by MV or indirectly by interaction with and silencing of T-lymphocytes. Furthermore, the capacity to function as antigen presenting cells might be affected [40], [48].
Our study covers a time period of two weeks after MV infection, and as such does not provide experimental proof of what happens during resolution of measles immune suppression. The strength of our model using recombinant EGFP-expressing MV mainly lies in the sensitive detection of MV-infected cells, which is limited to the first two weeks after MV infection. Clearly, the memory lymphocyte populations specific for previously encountered pathogens are not completely depleted, and are largely restored during the weeks to months after measles. For instance, previously positive Mantoux responses disappear after onset of rash [4], [5], but eventually reappear. We have indicated this in our model (Figure 5B) by showing a gradual increase of the memory lymphocyte population after clearance of MV. Although we cannot fully explain the drivers of the resolution of immune suppression, it is possible that expansion of non-depleted TCM upon renewed antigen encounter may play an important role. However, homeostatic restoration by the immune system itself could be an alternative explanation.
Some of the individual observations described here have been reported earlier in relationship to animal morbillivirus-related immune suppression [49]–[51]. The novelty of our model lies in the immune-mediated lymphodepletion being masked by the massive expansion of MV-specific and bystander lymphocytes. Although effective MV-specific CD8+ T-lymphocyte responses as well as immune activation and lymph node enlargement have been described earlier [16], [36], [37], [51], they have not been associated with immune suppression in this way.
The most important consequence of our model is that the qualitative composition of lymphocyte populations changes dramatically upon MV infection. Although lymphocyte numbers in peripheral blood and lymphoid organs appear normal, depletion of pre-existing specific T - and B-lymphocytes subpopulations provides a direct explanation for the suppression of recall responses to other pathogens during and after measles. This allows such pathogens to cause severe disease and in the developing world leads to the high level of MV-associated mortality. In addition, MV efficiently replicates in B-lymphocytes, resulting in follicular exhaustion and disorganization of the germinal centers, which are essential in actively ongoing humoral immune responses.
We observed comparable levels of MV-infected cells in TCM, TEM and Bn, and in parallel observed lymphocyte depletion and disorganization in B-cell follicles during the acute phase of MV infection (Figure 3, 9–11 d.p.i.). However, we have also shown that proliferating cells can be detected in lymphoid tissues as early as 11 d.p.i., after which time the follicle structure is being restored. This matches the kinetics of antibody responses in the macaque model, in that MV-specific IgM and IgG responses are first detected around 11 d.p.i. and peak at 17 (IgM) and 24 (IgG) d.p.i. [27], [52]. These kinetics fit well with our conclusions from the data and map well onto the immune suppression model.
It has been described that MV infection can result in transient remissions of certain autoimmune diseases [53]–[55]. Our observations suggest that this can be explained by direct MV infection of CD150+ lymphocytes, followed by immune-mediated depletion. Similarly, this mechanism could also explain reductions in HIV-1 loads during acute measles [56], [57]: MV infection of memory CD4+ T-lymphocytes could result in depletion of HIV-1-infected cells. In certain auto-immune diseases, and in animal studies in which lymphocyte populations were experimentally depleted, commensal or opportunistic infectious agents that would normally be controlled by the immune system have been shown to cause severe disease [58]. The high incidence of respiratory and gastro-intestinal complications following measles [1]–[3] may therefore be directly related to the observed high percentages of MV-infection and subsequent lymphocyte depletion in the adenoids, tonsils and gut-associated lymphoid tissue, which form a first line of defense against inhaled or ingested pathogens. We conclude that MV infection wipes immunological memory, resulting in increased susceptibility to commensal or opportunistic infections.
Materials and Methods
Ethics statement
Animal experiments were conducted in compliance with European guidelines (EU directive on animal testing 86/609/EEC) and Dutch legislation (Experiments on Animals Act, 1997). The protocols were approved by the independent animal experimentation ethical review committee DCC in Driebergen, The Netherlands. Animal welfare was observed on daily basis, animal handling was performed under light anesthesia using ketamine and medetomidine. After handling atipamezole was administered to antagonize the effect of medetomidine. For experiments involving PBMC from human donors, written informed consent for research use was obtained by the Sanquin blood bank.
Animal study design
PBMC and tissues were collected from cynomolgus (Macaca fascicularis) (n = 35) or rhesus (Macaca mulatta) (n = 5) macaques included in previously published studies [18], [27], [30] (n = 26) or from unpublished infection experiments with rMVIC323EGFP or rMVKSEGFP (Table S1 and Dataset S1) (n = 14). Macaques were infected by intra-tracheal inoculation or aerosol inhalation and euthanized at 2 (n = 3), 3 (n = 3), 4 (n = 3), 5 (n = 4), 7 (n = 9), 9 (n = 8), 11 (n = 6), 13 (n = 2) or 15 (n = 2) d.p.i. Although some of the experiments had been designed to address different research questions, the accumulated samples effectively covered all stages of MV infection in macaques.
Mantoux tests
Four macaques received intra-dermal vaccinations with 4×0.1 ml of live BCG (NVI, Bilthoven, Netherlands). The animals received an intra-dermal Mantoux test with old tuberculin (0.1 ml, 25,000 IU/ml, Statens Serum Institut) [59] 3 months post-vaccination at 7 days pre-MV-infection, or 3 days prior to necropsy. Skin reactivity was assessed for three consecutive days. Skin samples from both Mantoux tests (pre - and post-MV infection) were collected into formalin.
Tuberculin - and MV-specific T-lymphocyte responses
PBMC obtained 7 days pre-infection and during necropsy were thawed and plated into 96-wells round-bottom plates at 2×105 cells per well. Cells were stimulated with either PPD (10 µg/ml), UV-inactivated MV (10 µg/ml) or live rMVrEdtEGFP [30] (5×104 CCID50 in the presence of 10 µg/ml infection-enhancing lipopeptide PHCSK4 [60]) for 48 hours in triplicate. IFN-γ concentrations were measured in supernatants by ELISA (U-CyTech Biosciences).
Leukopenia
After each blood collection, total WBC counts were obtained using an automated counter (Sysmex pocH-100iV). To address measles-induced leukopenia in these animals, mean WBC counts were determined on 0 (n = 31), 1 (n = 6), 2 (n = 20), 3 (n = 17), 4 (n = 18), 5 (n = 11), 6 (n = 20), 7 (n = 10), 8 (n = 6), 9 (n = 12), 11 (n = 8) and 13 (n = 2) d.p.i. Different animals were included at each time-point.
Necropsy
Animals were euthanized by exsanguination under ketamine anesthesia. For the purpose of detecting EGFP fluorescence, a lamp was custom-made containing six 5-volt LEDs (Luxeon Lumileds, lambertian, cyan, peak emission 490–495 nm) mounted with D480/40 bandpass filters (Chroma) in a frame that allowed decontamination with 70% (v/v) alcohol or fumigation with formaldehyde. Emitted fluorescence was visualized through the amber cover of a UV transilluminator normally used for screening DNA gels. Photographs were made using a Nikon D80 digital SLR camera. Lymphoid tissues were collected in buffered formalin for immunohistochemistry or PBS for preparation of single cell suspensions, which were used directly for flow cytometry.
Flow cytometry
T-lymphocytes were subdivided into Tn, TCM and TEM populations (Figure S1) by staining with CD3PerCP (BD Biosciences, clone SP34-2), CD4V450 (BD Biosciences, clone L200), CD8AmCyan (BD Biosciences, clone SK1), CD45RAPE-Cy7 (BD Biosciences, clone L48) and CCR7APC (R&D Systems, clone 150503). The APC signal was enhanced using an APC-FASER Kit (Miltenyi Biotec). B-lymphocytes were subdivided into Bn and BM populations (Figure S1) by staining with CD20PE-Cy7 (BD Biosciences, clone L27), HLA-DRPacific Blue (Biolegend, clone L243), CD27APC (eBioscience, clone O323) and a combination of IgDBiotin (Southern Biotech, goat polyclonal) and streptavidinPerCP (BD Biosciences). CD150 expression was determined by staining with CD150FITC (AbD Serotec, clone A12). The infection percentages within the populations were determined by detection of EGFP. All flow cytometry was performed on a FACS Canto II (BD Biosciences).
Histological and immunohistochemical analysis
H&E staining was performed to evaluate histological changes. Immunohistochemical staining was performed using a fully automated BondMax immunostainer with a polymer-based peroxidase detection system. MV-infected cells were detected using a polyclonal rabbit antibody to EGFP (Invitrogen). Similar stainings were performed with the following monoclonal antibodies: T-lymphocyte marker CD3 (DAKO, clone F7.2.38), B-lymphocyte marker CD20 (DAKO, clone L26), proliferation marker Ki67 (DAKO, clone MIB1) and apoptosis marker cleaved caspase 3 (Cell Signaling, clone 5A1E). Glass slides were scanned with a 40X/0.75 Olympus UPlan FLN objective on an Aperio Scanscope CS-O SS5200 equipped with Spectrum Plus. An Aperio Positive Pixel Count Algorithm was applies to quantify and therefore standardize the intensity of stains present to produce optimal discrimination between immunoperoxidase diaminobenzidine (DAB) tetrahydrochloride reactions and hematoxylin stained nuclei. Scanners are kept at ambient temperature in a temperature-controlled area to eliminate loss of performance due to overheating.
Susceptibility of lymphocyte subsets to infection
PBMC from healthy human or macaque donors were sorted into pure CD4+ or CD8+ naive (CD45RA+) and memory (CD45RA−) T-lymphocyte populations on basis of CD45RA expression. Unsorted or sorted PBMC from humans or macaques were infected by co-culture with low numbers (1∶1000) of autologous MV-infected B-LCL or BAL cells (infected with cell-free rMVKSEGFP at an MOI of 1 for 48 hours), respectively. After two days of co-culture infection percentages in the different subsets were determined by flow cytometry.
Statistical analysis
Differences between percentages infected cells or CD150 expression were tested by the non-parametric Wilcoxon rank test using SPSS software.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. BeckfordAP, KaschulaRO, StephenC (1985) Factors associated with fatal cases of measles. A retrospective autopsy study. S Afr Med J 68 : 858–863.
2. AkramuzzamanSM, CuttsFT, WheelerJG, HossainMJ (2000) Increased childhood morbidity after measles is short-term in urban Bangladesh. Am J Epidemiol 151 : 723–735.
3. ShanksGD, LeeSE, HowardA, BrundageJF (2011) Extreme mortality after first introduction of measles virus to the polynesian island of Rotuma, 1911. Am J Epidemiol 173 : 1211–1222.
4. Von PirquetCE (1908) Das Verhalten der kutanen Tuberkulin-reaktion während der Masern. Dtsch Med Wochenschr 34 : 1297–1300.
5. TamashiroVG, PerezHH, GriffinDE (1987) Prospective study of the magnitude and duration of changes in tuberculin reactivity during uncomplicated and complicated measles. Pediatr Infect Dis J 6 : 451–454.
6. LisseI, SambB, WhittleH, JensenH, SoumareM, et al. (1998) Acute and long-term changes in T-lymphocyte subsets in response to clinical and subclinical measles. A community study from rural Senegal. Scand J Infect Dis 30 : 17–21.
7. RyonJJ, MossWJ, MonzeM, GriffinDE (2002) Functional and phenotypic changes in circulating lymphocytes from hospitalized Zambian children with measles. Clin Diagn Lab Immunol 9 : 994–1003.
8. Premenko-LanierM, RotaPA, RhodesGH, BelliniWJ, McChesneyMB (2004) Protection against challenge with measles virus (MV) in infant macaques by an MV DNA vaccine administered in the presence of neutralizing antibody. J Infect Dis 189 : 2064–2071.
9. BankampB, HodgeG, McChesneyMB, BelliniWJ, RotaPA (2008) Genetic changes that affect the virulence of measles virus in a rhesus macaque model. Virology 373 : 39–50.
10. HirschRL, GriffinDE, JohnsonRT, CooperSJ, Lindo de SorianoI, et al. (1984) Cellular immune responses during complicated and uncomplicated measles virus infections of man. Clin Immunol Immunopathol 31 : 1–12.
11. WardBJ, JohnsonRT, VaisbergA, JaureguiE, GriffinDE (1991) Cytokine production in vitro and the lymphoproliferative defect of natural measles virus infection. Clin Immunol Immunopathol 61 : 236–248.
12. GriffinDE, WardBJ (1993) Differential CD4 T cell activation in measles. J Infect Dis 168 : 275–281.
13. Fugier-VivierI, Servet-DelpratC, RivaillerP, RissoanM-C, LiuY-J, et al. (1997) Measles virus suppresses cell-mediated immunity by interfering with the survival and functions of dendritic and T cells. J Exp Med 186 : 813–823.
14. GrosjeanI, CauxC, BellaC, BergerI, WildF, et al. (1997) Measles virus infects human dendritic cells and blocks their allostimulatory properties for CD4+ T cells. J Exp Med 186 : 801–812.
15. SchnorrJ-J, XanthakosS, KeikavoussiP, KampgenE, Ter MeulenV, et al. (1997) Induction of maturation of human blood dendritic cell precursors by measles virus is associated with immmunosuppression. Proc Natl Acad Sci U S A 94 : 5326–5331.
16. GriffinDE, WardBJ, JaureguiE, JohnsonRT, VaisbergA (1989) Immune activation in measles. N Engl J Med 320 : 1667–1672.
17. FerreiraCS, FrenzkeM, LeonardVH, WelsteadGG, RichardsonCD, et al. (2010) Measles virus infection of alveolar macrophages and dendritic cells precedes spread to lymphatic organs in transgenic mice expressing human signaling lymphocytic activation molecule (SLAM, CD150). J Virol 84 : 3033–3042.
18. LemonK, De VriesRD, MesmanAW, McQuaidS, Van AmerongenG, et al. (2011) Early target cells of measles virus after aerosol infection of non-human primates. PLoS Pathog 7: e1001263.
19. De VriesRD, MesmanAW, GeijtenbeekTBH, DuprexWP, De SwartRL (2012) The pathogenesis of measles. Curr Opin Virol In press: DOI 10.1016/j.coviro.2012.03.005..
20. TatsuoH, OnoN, YanagiY (2001) Morbilliviruses use signaling lymphocyte activation molecules (CD150) as cellular receptors. J Virol 75 : 5842–5850.
21. NoyceRS, BondreDG, HaMN, LinLT, SissonG, et al. (2011) Tumor cell marker PVRL4 (nectin 4) is an epithelial cell receptor for measles virus. PLoS Pathog 7: e1002240.
22. MühlebachMD, MateoM, SinnPL, PruferS, UhligKM, et al. (2011) Adherens junction protein nectin-4 is the epithelial receptor for measles virus. Nature 480 : 530–3.
23. RacanielloV (2011) An exit strategy for measles virus. Science 334 : 1650–1651.
24. SullivanJL, BarryDW, LucasSJ, AlbrechtP (1975) Measles infection of human mononuclear cells. I. Acute infection of peripheral blood lymphocytes and monocytes. J Exp Med 142 : 773–784.
25. HuddlestoneJR, LampertPW, OldstoneMBA (1980) Virus-lymphocyte interactions: infection of Tg and Tm subsets by measles virus. Clin Immunol Immunopathol 15 : 502–509.
26. ForthalDN, AarnaesS, BlandingJ, De la MazaL, TillesJG (1992) Degree and length of viremia in adults with measles. J Infect Dis 166 : 421–424.
27. De SwartRL, LudlowM, De WitteL, YanagiY, Van AmerongenG, et al. (2007) Predominant infection of CD150+ lymphocytes and dendritic cells during measles virus infection of macaques. PLoS Pathog 3: e178.
28. CondackC, GrivelJ-C, DevauxP, MargolisL, CattaneoR (2007) Measles virus vaccine attenuation: suboptimal infection of lymphatic tissue and tropism alteration. J Infect Dis 196 : 541–549.
29. De SwartRL (2009) Measles studies in the macaque model. Curr Top Microbiol Immunol 330 : 55–72.
30. De VriesRD, LemonK, LudlowM, McQuaidS, YükselS, et al. (2010) In vivo tropism of attenuated and pathogenic measles virus expressing green fluorescent protein in macaques. J Virol 84 : 4714–4724.
31. CocksBG, ChangCC, CarballidoJM, YsselH, De VriesJE, et al. (1995) A novel receptor involved in T-cell activation. Nature 376 : 260–263.
32. De SalortJ, SintesJ, LlinasL, Matesanz-IsabelJ, EngelP (2011) Expression of SLAM (CD150) cell-surface receptors on human B-cell subsets: from pro-B to plasma cells. Immunol Lett 134 : 129–136.
33. McChesneyMB, MillerCJ, RotaPA, ZhuY, AntipaL, et al. (1997) Experimental measles I. Pathogenesis in the normal and the immunized host. Virology 233 : 74–84.
34. WarthinAS (1931) Occurrence of numerous large giant cells in the tonsils and pharyngeal mucosa in the prodromal stage of measles. Arch Pathol 11 : 864–874.
35. De VriesRD, YükselS, OsterhausADME, De SwartRL (2010) Specific CD8+ T-lymphocytes control dissemination of measles virus. Eur J Immunol 40 : 388–395.
36. Van BinnendijkRS, PoelenMCM, KuijpersKC, OsterhausADME, UytdeHaagFGCM (1990) The predominance of CD8+ T cells after infection with measles virus suggests a role for CD8+ class I MHC-restricted cytotoxic T lymphocytes (CTL) in recovery from measles. J Immunol 144 : 2394–2399.
37. MongkolsapayaJ, JayeA, CallanMFC, MagnusenAF, McMichaelAJ, et al. (1999) Antigen-specific expansion of cytotoxic T lymphocytes in acute measles virus infection. J Virol 73 : 67–71.
38. MoussallemTM, GuedesF, FernandesER, PagliariC, LancellottiCLP, et al. (2007) Lung involvement in childhood measles: severe immune dysfunction revealed by quantitative immunohistochemistry. Hum Pathol 38 : 1239–1247.
39. SallustoF, LenigD, ForsterR, LippM, LanzavecchiaA (1999) Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature 401 : 708–712.
40. Schneider-SchauliesS, Schneider-SchauliesJ (2009) Measles virus-induced immunosuppression. Curr Top Microbiol Immunol 330 : 243–269.
41. HahmB (2009) Hostile communication of measles virus with host innate immunity and dendritic cells. Curr Top Microbiol Immunol 330 : 271–287.
42. GriffinDE (2010) Measles virus-induced suppression of immune responses. Immunol Rev 236 : 176–189.
43. SteineurMP, GrosjeanI, BellaC, KaiserlianD (1998) Langerhans cells are susceptible to measles virus infection and actively suppress T cell proliferation. Eur J Dermatol 8 : 413–420.
44. Servet-DelpratC, VidalainP-O, BausingerH, ManieS, Le DeistF, et al. (2000) Measles virus induces abnormal differentiation of CD40 ligand-activated human dendritic cells. J Immunol 164 : 1753–1760.
45. VidalainP-O, AzocarO, Rabourdin-CombeC, Servet-DelpratC (2001) Measles virus-infected dendritic cells develop immunosuppressive and cytotoxic activities. Immunobiol 204 : 629–638.
46. De WitteL, De VriesRD, Van der VlistM, YükselS, LitjensM, et al. (2008) DC-SIGN and CD150 have distinct roles in transmission of measles virus from dendritic cells to T-lymphocytes. PLoS Pathog 4: e1000049.
47. Van der VlistM, De WitteL, De VriesRD, LitjensM, De JongMAWP, et al. (2011) Human Langerhans cells capture measles virus through Langerin and present viral antigens to CD4(+) T-cells but are incapable of cross-presentation. Eur J Immunol 41 : 2619–2631.
48. AvotaE, AvotsA, NiewieskS, KaneLP, BommhardtU, et al. (2001) Disruption of Akt kinase activation is important for immunosuppression induced by measles virus. Nat Med 7 : 725–731.
49. McCulloughB, KrakowkaS, KoestnerA (1974) Experimental canine distemper virus-induced lymphoid depletion. Am J Pathol 74 : 155–170.
50. von MesslingV, SpringfeldC, DevauxP, CattaneoR (2003) A ferret model of canine distemper virus virulence and immunosuppression. J Virol 77 : 12579–12591.
51. BeinekeA, PuffC, SeehusenF, BaumgartnerW (2009) Pathogenesis and immunopathology of systemic and nervous canine distemper. Vet Immunol Immunopathol 127 : 1–18.
52. De SwartRL, VosHW, UytdeHaagFGCM, OsterhausADME, Van BinnendijkRS (1998) Measles virus fusion protein - and hemagglutinin-transfected cell lines are a sensitive tool for the detection of specific antibodies by a FACS-measured immunofluorescence assay. J Virol Methods 71 : 35–44.
53. SimpanenE, vanER, IsomakiH (1977) Remission of juvenile rheumatoid arthritis (Still's disease) after measles. Lancet 2 : 987–988.
54. YoshiokaK, MiyataH, MakiS (1981) Transient remission of juvenile rheumatoid arthritis after measles. Acta Paediatr Scand 70 : 419–420.
55. LinCY, LinMT, HsiehYL, TsaoLY (1988) Transient disappearance of immunologic disorders and remission after intercurrent measles infections in children with chronic idiopathic thrombocytopenic purpura. J Clin Immunol 8 : 207–213.
56. MossWJ, ScottS, NdhlovuZ, MonzeM, CuttsFT, et al. (2009) Suppression of human immunodeficiency virus type 1 viral load during acute measles. Pediatr Infect Dis J 28 : 63–65.
57. RuelTD, AchanJ, GasasiraAF, CharleboisED, MehbratuT, et al. (2008) HIV RNA suppression among HIV-infected Ugandan children with measles. J Acquir Immune Defic Syndr 48 : 225–227.
58. KretschmerR, JanewayCA, RosenFS (1968) Immunologic amnesia. Study of an 11-year-old girl with recurrent severe infections associated with dysgammaglobulinemia, lymphopenia and lymphocytotoxic antibody, resulting in loss of immunologic memory. Pediat Res 2 : 7–16.
59. LangermansJA, AndersenP, van SoolingenD, VervenneRA, FrostPA, et al. (2001) Divergent effect of bacillus Calmette-Guerin (BCG) vaccination on Mycobacterium tuberculosis infection in highly related macaque species: implications for primate models in tuberculosis vaccine research. Proc Natl Acad Sci U S A 98 : 11497–11502.
60. NguyenDT, De WitteL, LudlowM, YükselS, WiesmullerK-H, et al. (2010) The synthetic bacterial lipopeptide Pam3CSK4 modulates respiratory syncytial virus infection independent of TLR activation. PLoS Pathog 6: e1001049.
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2012 Číslo 8
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Měli bychom postcovidový syndrom léčit antidepresivy?
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- Invariant NKT Cells: Regulation and Function during Viral Infection
- Host Defense and Tolerance: Unique Challenges in the Placenta
- Nonhuman Primate Models for HIV Cure Research
- Exon Level Transcriptomic Profiling of HIV-1-Infected CD4 T Cells Reveals Virus-Induced Genes and Host Environment Favorable for Viral Replication