
Chemokine Receptor Ccr1 Drives Neutrophil-Mediated Kidney Immunopathology and Mortality in Invasive Candidiasis
Invasive candidiasis is the 4th leading cause of nosocomial bloodstream infection in the US with mortality that exceeds 40% despite administration of antifungal therapy; neutropenia is a major risk factor for poor outcome after invasive candidiasis. In a fatal mouse model of invasive candidiasis that mimics human bloodstream-derived invasive candidiasis, the most highly infected organ is the kidney and neutrophils are the major cellular mediators of host defense; however, factors regulating neutrophil recruitment have not been previously defined. Here we show that mice lacking chemokine receptor Ccr1, which is widely expressed on leukocytes, had selectively impaired accumulation of neutrophils in the kidney limited to the late phase of the time course of the model; surprisingly, this was associated with improved renal function and survival without affecting tissue fungal burden. Consistent with this, neutrophils from wild-type mice in blood and kidney switched from Ccr1lo to Ccr1high at late time-points post-infection, when Ccr1 ligands were produced at high levels in the kidney and were chemotactic for kidney neutrophils ex vivo. Further, when a 1∶1 mixture of Ccr1+/+ and Ccr1−/− donor neutrophils was adoptively transferred intravenously into Candida-infected Ccr1+/+ recipient mice, neutrophil trafficking into the kidney was significantly skewed toward Ccr1+/+ cells. Thus, neutrophil Ccr1 amplifies late renal immunopathology and increases mortality in invasive candidiasis by mediating excessive recruitment of neutrophils from the blood to the target organ.
Published in the journal:
. PLoS Pathog 8(8): e32767. doi:10.1371/journal.ppat.1002865
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1002865
Summary
Invasive candidiasis is the 4th leading cause of nosocomial bloodstream infection in the US with mortality that exceeds 40% despite administration of antifungal therapy; neutropenia is a major risk factor for poor outcome after invasive candidiasis. In a fatal mouse model of invasive candidiasis that mimics human bloodstream-derived invasive candidiasis, the most highly infected organ is the kidney and neutrophils are the major cellular mediators of host defense; however, factors regulating neutrophil recruitment have not been previously defined. Here we show that mice lacking chemokine receptor Ccr1, which is widely expressed on leukocytes, had selectively impaired accumulation of neutrophils in the kidney limited to the late phase of the time course of the model; surprisingly, this was associated with improved renal function and survival without affecting tissue fungal burden. Consistent with this, neutrophils from wild-type mice in blood and kidney switched from Ccr1lo to Ccr1high at late time-points post-infection, when Ccr1 ligands were produced at high levels in the kidney and were chemotactic for kidney neutrophils ex vivo. Further, when a 1∶1 mixture of Ccr1+/+ and Ccr1−/− donor neutrophils was adoptively transferred intravenously into Candida-infected Ccr1+/+ recipient mice, neutrophil trafficking into the kidney was significantly skewed toward Ccr1+/+ cells. Thus, neutrophil Ccr1 amplifies late renal immunopathology and increases mortality in invasive candidiasis by mediating excessive recruitment of neutrophils from the blood to the target organ.
Introduction
Invasive candidiasis, mainly caused by the opportunistic yeast Candida albicans, is the fourth leading cause of nosocomial bloodstream infection in the US and worldwide [1]. The annual incidence of invasive candidiasis has dramatically increased over the past decades and is now estimated at >20 cases per 100,000 population (>60,000 cases/year in the US) with an associated annual cost of two billion dollars in the US [1]–[3]. Vaccines are not available and despite administration of potent antifungal treatment, mortality of invasive candidiasis patients exceeds 40% [1], [4]. Thus, identification of novel therapeutic targets based on better understanding of its molecular pathogenesis is needed.
A mouse model has been used extensively to study invasive candidiasis. In the model, mice develop renal failure and septic shock [5], similar to humans with invasive candidiasis [4], and the kidney is the primary target organ [5], [6]. Several studies in this model have established the importance of innate immunity in anti-Candida host defense. Neutrophils and monocytes/macrophages are the predominant leukocytes accumulating in Candida-infected tissues in both mice and humans [7], [8]. In vivo depletion of neutrophils or macrophages using anti-Gr1 monoclonal antibody or clodronate-loaded liposomes respectively, results in increased tissue fungal burden and accelerated mortality [9], [10]. In contrast, mice with severe combined immunodeficiency that lack functional lymphocytes do not have increased mortality after invasive candidiasis [11]. In agreement with the requirement of innate over adaptive immunity for host defense against invasive candidiasis, patients with chronic granulomatous disease but not those with HIV infection are at heightened risk for invasive candidiasis [12].
At the molecular level, little information is available regarding the factors regulating phagocyte recruitment to Candida-infected organs. Here, we have defined the profile of chemoattractants and chemoattractant receptors induced by Candida in infected tissues of mice. We have used this profile to identify the chemokine receptor Ccr1 as a candidate, and have tested its functional importance genetically using Ccr1-deficient mice. Our data show that Ccr1 plays a pathogenic role in invasive candidiasis by mediating renal immunopathology via excessive neutrophil recruitment from the blood into the kidney.
Results
Invasive candidiasis induces expression of leukocyte chemotactic factors in infected organs
To identify molecules that might be important in the pathogenesis of invasive candidiasis, we screened 72 immunomodulatory factors (22 chemokine receptors, 38 chemokines, 12 cytokines) over time for differential mRNA expression in the kidney, spleen, liver and brain of uninfected mice versus in mice injected with 2.5×105 CFU, an inoculum that results in 100% mortality by day 8 post-infection (Figure 1A). Twelve of the 22 chemokine receptors, 31 of the 38 chemokines, and 11 of the 12 cytokines were induced ≥2-fold in at least two different organs of all mice examined (Figure 1B, 1C and 1D). Of the cytokines induced (Figure 1B), IL-1α, IL-1β, IL-6, IL-10, IL-17A, IL-18, IFN-γ, and TNF-α have been previously shown to play an important role in the pathogenesis of invasive candidiasis in vivo [13]–[21].

Of the chemoattractant mediators, several chemokines (Ccl25, Ccl27, Ccl28) and chemokine receptors (Ccr9, Ccr10, Cxcr5, Cxcr6, Cxcr7) associated with adaptive immunity were not induced after Candida infection (Figure 1C and 1D). In contrast, several chemoattractants associated with the innate immune response were significantly induced. Specifically, with regard to candidate chemotactic systems that might guide leukocyte accumulation in Candida-infected organs our data point to the phagocyte-targeted CC chemokines Ccl2, Ccl3, Ccl4, Ccl5 and Ccl7 and their receptors Ccr1, Ccr2 and Ccr5, and to the ELR CXC chemokines Cxcl1 (KC), Cxcl2 (MIP-2), Cxcl5 and Cxcl7 and their receptors Cxcr1 and Cxcr2 (Figure 1C and 1D). For the remainder of this paper, we focus our analysis on the role of Ccr1 in the pathogenesis of invasive candidiasis, as Ccr1 and its ligands were among the phagocyte-targeted chemotactic factors that were most highly induced in the model.
Lack of Ccr1 results in increased survival without impairing anti-Candida control
In separate experiments, induction of Ccr1 and its ligands Ccl3, Ccl5, Ccl6, Ccl7, Ccl8 and Ccl9 by invasive candidiasis was examined in greater detail in mice injected with 1.25×105 CFU of C. albicans, which causes ∼80% mortality by day 14 post-infection (Figure 1A). As shown in Figure 1E, Ccr1 induction was greatest in the kidney, peaking at a >100-fold increase over the uninfected state on day 9 post-infection. Ccr1 was induced to a lesser extent in the liver throughout the course of the infection and in the brain more transiently peaking on day 3. In contrast, weak induction was observed in the spleen. The Ccr1 ligands were all induced with variable dynamics; Ccl5 and Ccl8 were induced most prominently in the brain, the others most predominantly in the kidney (Figure 1F).
Because Ccr1 is expressed on neutrophils and monocytes/macrophages [22], which are critical for survival after invasive candidiasis, and since Ccr1 and its ligands were induced in the model, we investigated the in vivo role of Ccr1 in the pathogenesis of invasive candidiasis using Ccr1−/− mice. Surprisingly, Ccr1−/− mice infected with Candida had 3-fold better survival than Ccr1+/+ mice at day 14 post-infection (18% versus 56%; Figure 2A; P<0.001), an effect maintained through day 28 post-infection (data not shown; P = 0.001), and exhibited less pronounced weight loss (Figure 2B; P<0.05). Mortality with this inoculum began at day 6 post-infection and the greatest divergence in infection susceptibility between Ccr1+/+ and Ccr1−/− mice was seen around day 9 (Figure 2A). Since fungal burden in the kidney has been shown to correlate strongly with mortality in this model [5], [6], we examined tissue fungal burden in Ccr1+/+ and Ccr1−/− mice at different time-points after infection to determine whether fungal burden differences might account for the difference in survival. As shown in Figure 2C, 2D, 2E and 2F, Ccr1 deficiency did not affect Candida tissue burden in the kidney, spleen, liver or brain. Moreover, no differences were observed in the distribution or morphogenic states of Candida infiltration between Ccr1+/+ and Ccr1−/− mice in the kidney, spleen, liver or brain as determined by Periodic-acid Schiff staining (Figure S1A and data not shown). These data demonstrate that Ccr1 deficiency leads to improved survival after invasive candidiasis, which is not related to a differential ability of Ccr1+/+ and Ccr1−/− mice to control the pathogen burden.

Ccr1 promotes kidney tissue injury, immunopathology and renal failure after Candida infection
We next examined whether the survival difference between infected Ccr1+/+ and Ccr1−/− mice was due to dysregulated immunopathology. Indeed, Ccr1+/+ kidneys had severe swelling and pallor in gross pathology (Figure 3A) and exhibited a more pronounced increase in weight post-infection compared to Ccr1−/− kidneys (Figure 3B; P = 0.01), a kidney-specific finding not observed in the spleen, liver or brain (Figure S1B). Histopathological analysis revealed that Ccr1+/+ kidneys sustained more extensive tissue damage with larger multifocal areas of abscess formation compared to Ccr1−/− kidneys (Figure 3C, top row; Figure S1C shows uninfected kidney for comparison). This difference was particularly prominent in the renal cortex; >80% of the surface area was affected by abscess formation in Ccr1+/+ mice, whereas <20% of cortical involvement was observed in Ccr1−/− mice (Figure 3C, bottom row & Figure S1D; Figure S1E shows uninfected renal cortex for comparison). Of note, no difference in histopathology was observed between Candida-infected Ccr1+/+ and Ccr1−/− mice in the spleen, liver or brain (Figure S1F).
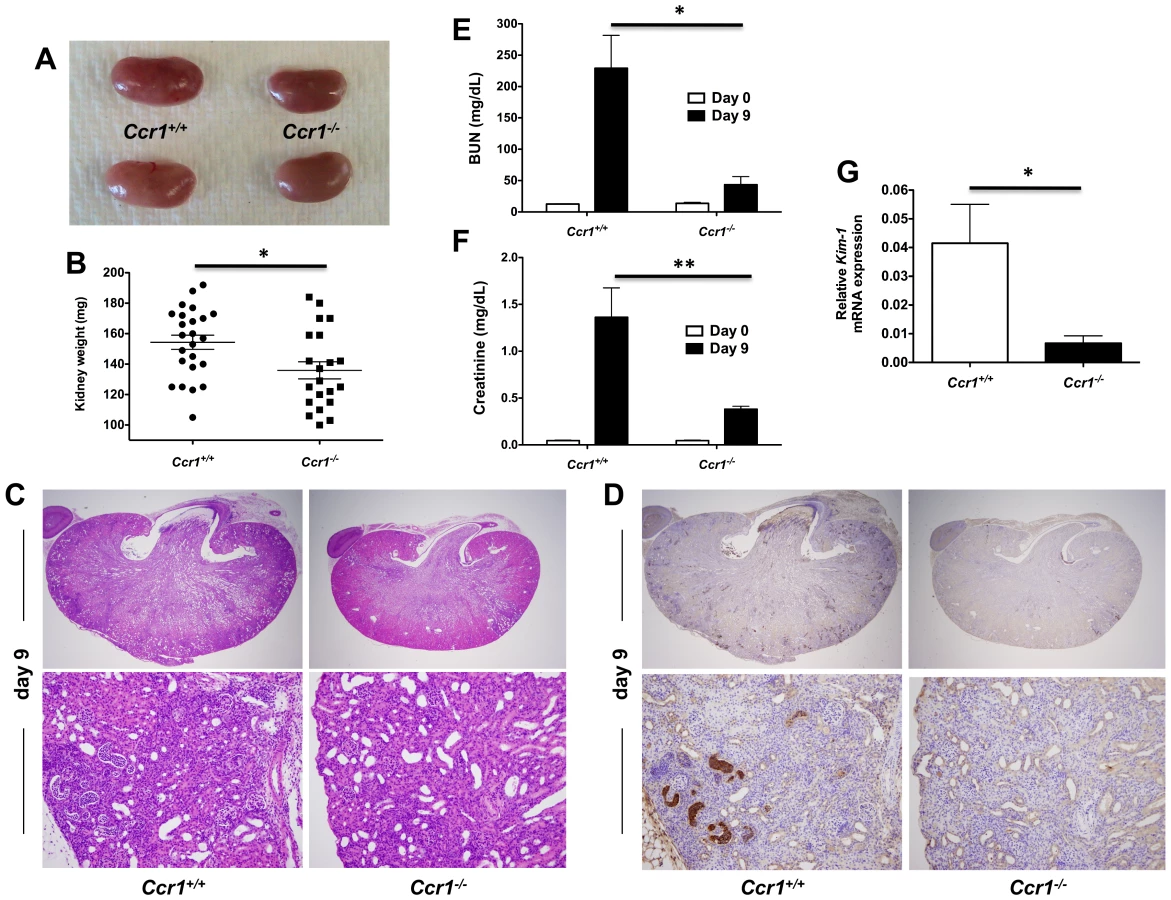
To characterize the cellular composition of the abscesses seen in Ccr1+/+ and Ccr1−/− kidneys post-infection, we performed immunohistochemistry (IHC) using 7/4, a marker on the surface of mouse neutrophils and monocytes/macrophages [23]. 7/4+ cells were more frequent in Ccr1+/+ kidneys than in Ccr1−/− kidneys post-infection (Figure 3D). Consistent with this, Ccr1+/+ mice developed more severe renal failure after infection (Figure 3E and 3F; P<0.01), and Ccr1+/+ kidneys displayed significantly greater expression of kidney injury molecule-1 (KIM-1; Figure 3G; P = 0.01), a marker of tubular epithelial damage [24] currently used for diagnosis of acute kidney injury in humans [25]. These data show that Ccr1 deficiency results in attenuated kidney tissue injury, less severe renal failure and decreased phagocyte accumulation and abscess formation in the kidney after Candida infection.
Ccr1 mediates neutrophil-specific accumulation in the kidney only late in the course of invasive candidiasis
To assess quantitatively and temporally the effect of Ccr1 on specific leukocyte accumulation in the model, we harvested leukocytes from the organs and defined their immunophenotypes by FACS on days 3, 6, 9 and 12 post-infection. As previously shown [8], neutrophils were the predominant leukocytes accumulating in the kidney after Candida infection, followed by macrophages and dendritic cells. Of interest, whereas no difference in accumulation of CD45+ leukocytes was seen up until day 6, Ccr1−/− kidneys accumulated ∼60% fewer leukocytes compared to Ccr1+/+ kidneys late at days 9 and 12 post-infection (Figure 4A; P≤0.01). This time-dependent difference in Ccr1-mediated leukocyte accumulation in the kidney was exclusively accounted for by Ly6cintLy6G+CD11b+ neutrophils, which accumulated in a Ccr1-independent manner until day 6 post-infection, but thereafter were ∼60% decreased in Ccr1−/− kidneys (Figure 4B, 4C and 4D; P≤0.01). In contrast, neutrophil accumulation was Ccr1-independent throughout the entire course of invasive candidiasis in the spleen, liver and brain (Figure S2A, S2B and S2C).

No differences were observed between Ccr1+/+ and Ccr1−/− kidneys post-infection in the accumulation of Ly6chiCD11b+ inflammatory monocytes, NK1.1+ cells, MHCII+F4/80+CD11c− macrophages, MHCII+CD11c+ dendritic cells, CD4+ and CD8+ T lymphocytes or CD19+ B cells (Figure S3A, S3B, S3C, S3D, S3E, S3F and S3G), although there was a trend towards lower B cell numbers in Ccr1−/− kidneys at days 9 and 12 post-infection (P = 0.07). Similarly, Ccr1 deficiency did not significantly affect accumulation of these immune cell types in the spleen, liver or brain (data not shown). These data indicate that Ccr1 is important for neutrophil accumulation in the kidney late but not early in the course of invasive candidiasis, while it is dispensable for neutrophil accumulation in the other organs tested, and for accumulation of other leukocytes in all the organs examined.
Decreased neutrophil accumulation in Ccr1−/− kidneys late after invasive candidiasis is not caused by reduced neutrophil survival or increased neutrophil egress into the urine
Because both histopathological and immunophenotypic analyses revealed differences between Ccr1+/+ and Ccr1−/− mice only in the kidney, in the remainder of our studies we focused on that organ. Thus, we next investigated the mechanism(s) underlying the late Ccr1-dependent phase of neutrophil accumulation in the kidney after invasive candidiasis. Decreased neutrophil accumulation in Ccr1−/− kidneys could be explained by the net effect of (1) decreased trafficking of neutrophils from the blood into the kidney, and/or (2) decreased survival of neutrophils in Ccr1−/− mice after entry into the kidney, and/or (3) increased egress of neutrophils from the kidney into the urine. First, we examined the effect of Ccr1 deficiency on neutrophil survival. Chemokine receptors have previously been reported to mediate cell survival signals [26]. However, Ccr1 deficiency did not appear to affect neutrophil survival in the model since Ccr1+/+ and Ccr1−/− neutrophils had similar levels by FACS of annexin V− 7-AAD− cells (as a measure of viability), annexin V+ 7-AAD− cells (as a measure of apoptosis) and annexin V+ 7-AAD+ cells (as a measure of death) in both the kidney (Figure 5A, 5B, 5C and 5D) and spleen (Figure S4). With regard to egress, it is conceivable that a critical threshold may exist for neutrophils in the kidney beyond which the collecting system and renal pelvis becomes obstructed and neutrophils no longer flow out of the kidney in the urine. This would plausibly be a time-dependent process and could explain the delayed Ccr1 dependence of neutrophil accumulation in the model. However, we observed no difference in the number of neutrophils in the urine of Ccr1+/+ and Ccr1−/− mice post-infection (Figure 5E). Hence, we found no evidence that decreased neutrophil accumulation in Ccr1−/− kidneys late after infection was caused by decreased neutrophil survival or increased neutrophil egress into the urine.

Ccr1 is expressed on blood and kidney neutrophils late in the course of invasive candidiasis
Based on the above findings, we focused next on the third possibility, that Ccr1 mediates neutrophil trafficking from the blood into the kidney, but only in the late phase of the time course of the model. For this to be plausible, (a) the receptor should be expressed on the surface of blood neutrophils (before the cells enter into the kidney) late after infection, and (b) the Ccr1-targeting chemokines should be induced in the kidney at the same time in order to generate a chemokine concentration gradient capable of recruiting Ccr1+ blood neutrophils into the kidney via mediating activation, firm adhesion and transmigration of neutrophils on the endothelial surface together with adhesion molecules [22]. Hence, we first looked at the expression of Ccr1 in blood neutrophils of Ccr1+/+ mice during invasive candidiasis. We found that Ccr1 was not detected on blood neutrophils in uninfected mice (data not shown), and was not detected until day 9 post-infection, when ∼20% of neutrophils stained positive (Figure 6A and 6B; P<0.05). Further, when MACS-sorted neutrophils from Ccr1+/+ kidneys were tested, Ccr1 mRNA was significantly greater in the late phase (days 6 and 9 post-infection) relative to the early phase (day 3 post-infection) (Figure 6C; P<0.05). This was confirmed at the protein level by FACS. In particular, the percentage of Ccr1+ kidney neutrophils increased from ∼20–30% on day 3 post-infection to ≥60% on day 9 post-infection (Figure 6D and 6E; P<0.0001). In addition to neutrophils, Ccr1 was expressed on other myeloid cells in the kidney (monocytes, macrophages) but not on T or B lymphocytes (Figure S5A, S5B, S5C, S5D and S5E). However, in contrast to neutrophils, Ccr1 expression on the other myeloid cells was constant throughout the course of invasive candidiasis (data not shown). Hence, these data show that Ccr1 expression increases on the surface of neutrophils in the blood and kidney late in the course of invasive candidiasis, when neutrophil accumulation is decreased in Ccr1−/− kidneys.

Ccr1 agonists are induced in the kidney after Candida infection and are chemotactic for neutrophils ex vivo
We next determined the levels of the Ccr1 agonists Ccl3, Ccl5, Ccl6, Ccl7, Ccl8 and Ccl9 in Ccr1+/+ and Ccr1−/− kidneys after infection. Although differential dynamics in induction were observed among them, all Ccr1 ligands were significantly induced at the mRNA (Figure 7A) and protein (Figure 7B) levels in both Ccr1+/+ and Ccr1−/− kidneys (but not spleen; Figure S6) both early and late after invasive candidiasis. In addition, in order to determine whether neutrophils obtained from Candida-infected mice respond to Ccr1 ligands, we sorted neutrophils from kidneys of Ccr1+/+ mice at day 9 post-infection and performed chemotaxis assays with recombinant Ccr1 agonists ex vivo. As shown in Figure 7C, five agonists were active with the rank order CCL9>CCL6>CCL3>CCL5>CCL8, whereas CCL7 was inactive. CCL3 has previously been reported to be chemotactic for mouse neutrophils [27], whereas the chemotactic potential of the other Ccr1 agonists has not been previously tested directly for mouse neutrophils.

Next, because both hematopoietic and non-hematopoietic cells have chemokine secreting potential [22], we attempted to define the source of the Ccr1 ligands in the kidney. Due to lack of reliable commercially available antibodies for these chemokines, an IHC approach was not feasible. Instead, we sorted kidney neutrophils during invasive candidiasis in Ccr1+/+ mice and determined the relative mRNA expression for the Ccr1 ligands. As shown in Figure 7D, three were expressed by kidney neutrophils with the rank order Ccl3>Ccl6>Ccl9, whereas Ccl5, Ccl7 and Ccl8 were barely detected or not detected (data not shown). When comparing the relative expression of these chemokines in sorted neutrophils versus whole kidney homogenates, neutrophils appear to be a significant source for Ccl3 and perhaps for Ccl6 and Ccl9 in the kidney after infection, suggestive of a positive feedback loop of neutrophil chemoattraction. These data collectively show that the Ccr1 agonists are induced in the kidney after invasive candidiasis and are chemotactic for kidney neutrophils ex vivo.
Ccr1 mediates neutrophil trafficking from the blood into the kidney after Candida infection
To test the role of Ccr1 in neutrophil trafficking from the blood to the kidney directly, we conducted a competitive repopulation study where equal numbers of Ccr1+/+ and Ccr1−/− Ly6cintLy6G+CD11b+ neutrophils were isolated by gradient centrifugation from the bone marrows of Ccr1+/+ and Ccr1−/− Candida-infected mice at day 9 post-infection, then differentially labeled, mixed 1∶1 and injected into Candida-infected Ccr1+/+ recipient mice on day 9 post infection. This time point was chosen because it is the time when (1) neutrophil accumulation in the kidney is Ccr1-dependent in the model (Figure 4B, 4C and 4D), (2) Ccr1 is expressed on the surface of neutrophils in the blood (Figure 6A and 6B), kidney (Figure 6C, 6D and 6E) and bone marrow (data not shown), and (3) Ccr1 ligands are produced in the kidney and are chemotactic for neutrophils (Figure 7A, 7B and 7C). Four hours after neutrophil transfer, mice were euthanized and kidneys were analyzed for the presence of labeled neutrophils. As shown in Figure 8A and 8B, the ratio of labeled Ccr1+/+:Ccr1−/− neutrophils prior to injection was approximately 1∶1, as intended. Four hours post-injection, the ratio of Ccr1+/+:Ccr1−/− neutrophils in the kidney was strongly skewed towards Ccr1+/+ neutrophils at approximately a 2.5∶1 ratio; this skewing was seen in all 16 mice tested (range: 1.7–4.1). The possibility that the labeling dyes had a differential effect on the survival of Ccr1+/+ versus Ccr1−/− neutrophils was excluded by FACS, which showed that the proportion of differentially labeled dead Ccr1+/+ and Ccr1−/− neutrophils was similar (Figure 8C). Therefore, these data indicate that Ccr1 on neutrophils directly mediates neutrophil trafficking from the blood into the kidney late in the course of invasive candidiasis.

Ccr1 deficiency does not affect the expression of other neutrophil-targeted chemotactic factors or adhesion molecules in the kidney after Candida infection
Next, we aimed to exclude the possibility that decreased accumulation of neutrophils in Ccr1−/− kidneys late after infection was caused by decreased expression of other neutrophil-targeted chemoattractant receptors and ligands and/or adhesion molecules in Ccr1−/− kidneys. For that, we determined the expression level of five major neutrophil-targeted chemoattractant receptors (i.e., the chemokine receptors Cxcr1 and Cxcr2, the complement C5a receptor [C5aR], the leukotriene B4 receptor Blt1, and the platelet activating factor receptor [Pafr]), two major neutrophil-targeted chemokines (i.e., the ELR CXC chemokines Cxcl1 [KC] and Cxcl2 [MIP-2]), and five adhesion molecules (i.e., α-integrin X, α-integrin L, β2-integrin, L-selectin and intercellular adhesion molecule 1 [ICAM-1]) in Ccr1+/+ and Ccr1−/− kidneys at days 3, 6 and 9 post-infection. We found no significant differences in the expression of these factors between Ccr1+/+ and Ccr1−/− kidneys at day 6 (Figure S7D, S7E and S7F) or 9 (Figure 9A, 9B and 9C) post-infection. Of the 12 factors examined, a difference in mRNA expression between the two strains was observed at day 3 post-infection only for two: β2-integrin and L-selectin. For both results, mRNA was increased slightly in the Ccr1−/− kidneys (Figure S7A, S7B and S7C). These data do not provide clear evidence that neutrophil accumulation in the model late after infection is associated with Ccr1-dependent expression of neutrophil-targeted chemotactic factors or adhesion molecules. The differences observed on day 3 post-infection could have a long-term effect but will require additional experimentation to establish any functional significance.

Ccr1 expression on kidney neutrophils does not affect their immunopathogenic potential or anti-Candida effector function
Last, we sought to determine whether the higher Ccr1 expression seen on kidney neutrophils late after infection is associated with an increased ability of neutrophils to cause tissue injury. For that, we sorted Ccr1lo kidney neutrophils at day 3 and Ccr1high kidney neutrophils at day 9 after infection from Ccr1+/+ Candida-infected mice and compared their potential for degranulation (beta-glucuronidase assay) and oxidative burst (dihydrorhodamine 123 [DHR] assay) ex vivo. As shown in Figure S8A, Ccr1lo and Ccr1high kidney neutrophils exhibited no difference in degranulation measured as beta-glucuronidase release. Similarly, we found no difference between Ccr1lo and Ccr1high kidney neutrophils in oxidative burst, both at rest and following stimulation with phorbol myristate acetate (PMA; Figure S8B). Last, Ccr1lo and Ccr1high kidney neutrophils did not exhibit significant differences in anti-Candida effector function as demonstrated by their similar capacity for phagocytosis (Figure S8C) and killing (Figure S8D) against Candida ex vivo. These data show that the magnitude of Ccr1 expression on kidney neutrophils after Candida infection does not appear to directly influence the immunopathogenic potential of neutrophils or their anti-Candida effector function in the kidney.
Discussion
In the present study we demonstrate that the chemokine receptor Ccr1 is associated with fatal neutrophil-mediated immunopathology selectively in the kidney in a mouse model of systemic Candida albicans infection. The receptor appears to act directly by promoting neutrophil trafficking from the blood into the kidney, but not until the late phase of the infection when it is turned on selectively by blood neutrophils and when most of its ligands Ccl3, Ccl5, Ccl6, Ccl7, Ccl8 and Ccl9 are expressed at high levels in the kidney. Our conclusions are based on detailed analysis of differences in clinical, pathological, microbiological, immunological and molecular parameters in the model between Ccr1+/+ and Ccr1−/− mice. Our study is the first identification of a specific chemoattractant receptor that mediates neutrophil trafficking into the kidney and drives neutrophil-mediated immunopathology and mortality in the mouse model of invasive candidiasis. Moreover, the conclusions regarding mechanism are based on a formal in vivo neutrophil trafficking study.
We focused on Ccr1 because phagocytes are known to be the main cellular mediators of the immune response in invasive candidiasis [9], [10], and because our broad survey of the chemokine system in the model showed that phagocyte-targeted chemokines and their receptors, including Ccr1 and its ligands, were among the most highly up-regulated. Ccr1 is a chemokine receptor whose functional importance for neutrophils has been more apparent in mice than in humans [27]–[30], and it is also expressed on other hematopoietic and some non-hematopoietic cell types [22].
The outcome in any infectious disease is determined by a balance between host and pathogen factors. In our study, we found several lines of evidence supporting excessive neutrophil accumulation in the kidney resulting in neutrophil-mediated immunopathology as the underlying mechanism for Ccr1-mediated decreased survival in the model. First, Ccr1 deficiency did not affect tissue Candida proliferation, an established correlate of survival in previous studies in the model of invasive candidiasis [5], [6], [8]. Second, Ccr1 deficiency was associated with markedly decreased destructive inflammatory changes and tissue injury in the kidney; this was accompanied by less severe kidney failure in Ccr1−/− mice, which is a major determinant of survival in the model [5]. Third, the decreased kidney tissue injury was associated with a large and selective reduction in neutrophil accumulation in Ccr1−/− kidneys after infection.
Neutrophils are key innate immune effector cells that play a critical role in phagocytosis and killing of Candida albicans [31]. In agreement, neutropenia is a major risk factor for mortality after invasive candidiasis in both mice and humans [9], [32]. Early neutrophil availability in particular has been shown to be critical for protection against invasive candidiasis in mice, as neutrophil depletion within the first 24 hours, but not at later time points post-infection, led to accelerated Candida growth and mortality [33]. On the other hand, neutrophils may exert detrimental effects on the host by mediating tissue injury. For example, patients with invasive candidiasis often require administration of corticosteroid therapy after recovery from neutropenia for amelioration of clinical symptoms and attenuation of exuberant inflammation [7]. A similar immune reconstitution inflammatory syndrome has also been described following neutrophil recovery in patients with invasive aspergillosis [34]. In mice, the pathogenic role of neutrophils in invasive candidiasis has been previously suggested by neutrophil depletion studies [33], in which rendering mice neutropenic at late time points after infection when inflammation is pronounced [8], markedly improved survival, presumably due to attenuated tissue injury. Our study is the first to identify a single molecular factor, Ccr1, as one of the mediators of immunopathology in the mouse model of invasive candidiasis.
Ccr1 has been shown to exert both pathogenic and protective effects depending on the inflammatory milieu. For example, Ccr1 was critical for effective host defense against Toxoplasma gondii, pneumovirus of mice and Aspergillus fumigatus by mediating neutrophil accumulation in the target infection organs [27], [35], [36]. Conversely, Ccr1-mediated neutrophil accumulation was pathogenic in a model of acute respiratory distress syndrome following pancreatitis and a model of sepsis following bacterial peritonitis [28], [29]. The role of Ccr1 has also been studied in the context of renal inflammation. Specifically, Ccr1 deficiency decreased neutrophil and macrophage accumulation in the kidney after ischemia-reperfusion injury, but it had no effect on kidney function or survival [37]. Moreover, Ccr1 deficiency decreased kidney injury in models of Alport disease, focal segmental glomerulosclerosis, unilateral ureteral obstruction and toxic nephropathy; however, the attenuated tissue damage was associated with decreased infiltration of lymphocytes and macrophages but not neutrophils in the kidney [38]–[42].
The mechanism of Ccr1-mediated neutrophil accumulation and immunopathology in the kidney in invasive candidiasis appears to lie at the level of neutrophil trafficking from the blood into the kidney, as shown by our competitive repopulation study using Ccr1+/+ and Ccr1−/− neutrophils. Additional work using gene-deficient mice and/or antibody depleting strategies will be needed to define which of the Ccr1 ligands, either alone or in synergy, mediate neutrophil trafficking into Candida-infected kidneys. Also, because Ccr1 is expressed on neutrophil precursors and mature neutrophils in the bone marrow [27], the impact of Ccr1 deficiency in neutrophil precursor production and egress of neutrophils from the bone marrow to the blood after Candida infection merits further investigation. Of note, although Ccr1 is expressed on several immune cell types [22], the reduction in leukocyte accumulation in Ccr1−/− kidneys was specific to neutrophils. In addition, neutrophil accumulation was Ccr1-dependent only in the kidney but not in the other organs examined, further attesting to the organ-specific nature of the innate immune response in invasive candidiasis [8].
Of interest, Ccr1 mediated neutrophil accumulation in the kidney only late in the course of invasive candidiasis. Three important new questions arise from this finding: the first relates to the signals responsible for Ccr1 induction on neutrophils late but not early in the course of invasive candidiasis. Factors that have been reported to induce Ccr1 on the surface of mouse and human neutrophils include TNF-α, GM-CSF and IFN-γ [43], [44], which were not found to be different between days 3 and 9 in blood or kidney in our model (data not shown). Candida albicans grows both in the yeast and hyphal forms in the kidney starting immediately post-infection [8], but unmasking of β-glucan, the pathogen-associated molecular pattern for dectin-1 [45], does not occur until late in the course of invasive candidiasis [46]. Hence, it is plausible that availability of soluble or membrane-bound β-glucan late after invasive candidiasis may contribute to Ccr1 induction on neutrophils.
The second important new question that arises from our study pertains to the chemotactic factors responsible for neutrophil recruitment in the kidney during the first 6 days after infection, when Ccr1 is dispensable for neutrophil accumulation in the kidney. Other neutrophil chemokine receptors such as Cxcr1 and Cxcr2, which were also induced in the model, may play such a role. In this regard, a sequential role for the leukotriene B4 receptor Blt1, Ccr1 and Cxcr2 has previously been reported for neutrophil recruitment in a mouse model of arthritis [47]. Because neutrophil availability during the early phase after infection is critical for survival [33], identifying the chemoattractant receptors that mediate trafficking into the kidney early in the course of the infection may reveal novel protective molecular factors against invasive candidiasis in the model. In fact, Cxcr2-deficient mice were previously reported to have impaired ability to control Candida proliferation in the kidney and other organs after invasive candidiasis, but accumulation of neutrophils in these tissues was not defined in that study [48].
The third important new question arises from our data that neutrophil accumulation was decreased but not entirely eliminated in Ccr1−/− kidneys late after infection (Figure 4), and from our competitive repopulation study, which showed that, despite the significant skewing toward Ccr1+/+ neutrophils, Ccr1−/− neutrophils also trafficked from the blood into the kidney at day 9 post-infection (Figure 8). Thus, future studies should aim to determine which chemotactic factors, in addition to Ccr1, also mediate neutrophil trafficking from the blood into the kidney late after infection. Identification of such factors may reveal other novel mediators of immunopathology in the model and could provide further insight into the balance between effective host defense and immunopathology in invasive candidiasis.
In contrast to the mouse model of the infection where kidney is the primary target organ [5], kidney abscesses are rare in patients with bloodstream-derived invasive candidiasis with the exception of neonates [49]; hence, whether CCR1 mediates immunopathology in human invasive candidiasis remains to be elucidated. Therefore, future studies should examine whether CCR1 is expressed on human neutrophils in Candida kidney abscesses. In addition, because neutropenic patients with gastrointestinal tract-derived hepatosplenic candidiasis often develop worsening liver and splenic abscesses immediately after neutrophil recovery [7], the role of Ccr1 in mediating immunopathology in that setting should be investigated using a neutropenic mouse model of gastrointestinal tract-derived invasive candidiasis [50], in which it would be important to determine whether neutrophils that repopulate the bloodstream after recovery from neutropenia express Ccr1.
In conclusion, our data identify Ccr1 as a critical pathogenic factor in systemic infection caused by Candida albicans in a mouse model. Our study shows for the first time that neutrophil-mediated immunopathology may be detrimental in an invasive fungal infection and that chemokine receptors regulate such a process. Further studies will be needed to investigate the role of Ccr1 ligands and to further understand the factors that shape and modulate protective versus pathogenic roles of neutrophils in invasive candidiasis.
Materials and Methods
Ethics statement
The mouse studies were performed in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health under the auspices of a protocol approved by the Animal Care and Use Committee of the National Institute of Allergy and Infectious Diseases. Every effort was made to minimize animal suffering.
Mouse model of invasive candidiasis
Female Ccr1+/+ WT and Ccr1−/− C57BL/6 mice were obtained from Taconic Farms (Germantown, NY) and were used at 8–10 weeks of age. Ccr1−/− mice, generated as previously described [27], were backcrossed onto the C57BL/6 background for 10 generations and maintained under specific pathogen-free housing conditions. Mice were injected via the lateral tail vein with 1.25 or 2.5×105 blastospores of C. albicans SC5314 (kind gift of Dr. John Bennett, NIAID, NIH). Candida was grown at 30°C in yeast extract, peptone, and dextrose medium containing penicillin and streptomycin (Mediatech Inc., Herndon, VA). Cells were centrifuged, washed in PBS, and counted using a hemocytometer.
Fungal burden determination
Infected mice were euthanized at 1, 6 and 9 days post-infection to determine the fungal burden in the kidney, brain, spleen and liver. The organs were aseptically removed, weighed, homogenized in PBS, serially diluted, and plated in duplicate on yeast extract, peptone, and dextrose agar plates containing penicillin and streptomycin. CFUs were determined after 48 hrs of incubation at 37°C and results were expressed as CFUs/gram of tissue.
Mouse histology and immunohistochemistry (IHC)
Mice were euthanized on day 9 post-infection and the kidney, brain, spleen and liver were removed, fixed with 10% formalin and embedded in paraffin wax. Tissue sections were processed for hematoxylin and eosin staining or Periodic-acid Schiff or were placed on poly-L-lysine coated glass slides, deparaffinized in xylene and rehydrated in a graded series of alcohol. After antigen retrieval for 20 min at 85°C, endogenous peroxidase was blocked by alcohol containing 0.3% hydrogen peroxide for 10 min at room temperature (RT). The slides were incubated with 5% BSA (Sigma-Aldrich, St. Louis, MO) for 20 min to block nonspecific protein binding and washed with Tris-buffered saline containing 0.05% Tween 20. Then, rat anti-mouse 7/4 (1∶10; Cedarlane Laboratories, Burlington, NC) was added in 1% BSA and slides were incubated overnight at RT. 1% BSA without primary antibody was used as negative control. The slides were washed twice with Tris-buffered saline containing 0.05% Tween 20 and detection of immunoreaction was achieved using the streptavidin-horse radish peroxidase system (Dako, Carpinteria, CA) containing streptavidin-peroxidase complex after a 30 min incubation at RT. Color was developed with 3,3′-diaminobenzidine (Dako) and hydrogen peroxide (Sigma), and slides were subsequently counterstained with hematoxylin, dehydrated, and mounted. At least three different sections of each organ per time-point were tested per experiment.
Determination of serum blood urea nitrogen (BUN) and creatinine
Serum BUN and creatinine were measured as markers of renal function at day 9 post-infection in Ccr1+/+ and Ccr1−/− mice. Blood was collected by cardiac puncture from each mouse at the time of sacrifice, and stored at −80°C until use. BUN and creatinine concentrations were determined at the NIH Clinical Chemistry Laboratory.
Chemoattractant receptor, chemokine and adhesion molecule gene expression analysis
In the initial mRNA analysis of the chemokine system in Ccr1+/+ mice, control uninfected mice and mice infected with 2.5×105 CFUs of Candida were euthanized on day 1, 4, and 7 post-infection (n = 3 mice per time point) and mRNA was extracted from the kidney, brain, liver and spleen using Trizol (Invitrogen, Carlsbad, CA) and the RNeasy kit (Qiagen, Valencia, CA) according to the manufacturer's instructions. cDNA was generated using the Superscript III First-Strand Synthesis SuperMix kit with random hexamers (Invitrogen) and real-time quantitative PCR (qPCR) for chemoattractant receptors and ligands was performed by SYBR Green or Taqman detection (Applied Biosystems, Foster City, CA) with the 7900HT Fast Real-Time PCR System in a total reaction volume of 22 µl using 2 µl cDNA, 11 µl 2× SYBR Green or Taqman PCR Master mix, and 1.1 µl of forward primer/1.1 µl of reverse primer (SYBR Green) or 1.1 µl primer–probe mix (Taqman). All qPCR assays were performed in duplicate and the relative gene expression of each molecule compared to uninfected mice was determined after normalization with GAPDH transcript levels using the ΔΔCT method, displayed as either gene amplicons or mRNA fold change. The primers using SYBR Green detection were designed from GenBank sequences using software Primer3 (http://frodo.wi.mit.edu/primer3/) and purchased from Invitrogen (sequences shown in Table S1), whereas Taqman primers/probes were predesigned by Applied Biosystems.
To determine the relative gene expression of chemoattractant receptors, chemokines and adhesion molecules in Ccr1+/+ versus Ccr1−/− mice, uninfected mice and mice infected with 1.25×105 CFUs of Candida were euthanized on day 3, 6, and 9 post-infection (n = 3–5 mice per time point) and qPCR was performed as mentioned above in two independent experiments. Moreover, to determine the extent of kidney epithelial cell injury in Ccr1+/+ versus Ccr1−/− mice, the relative expression of KIM-1 was determined at day 9 post-infection using the previously published Kim-1 primer pairs: 5′-ATGAATCAGATTCAAGTCTTC-3′, 5′-TCTGGTTTGTGAGTCCATGTG-3′ [51].
Chemokine quantification
Kidneys were homogenized into PBS with 0.5% Tween 20 (Bio-Rad, Hercules, CA) and a protease inhibitor cocktail (Roche, San Francisco, CA) and centrifuged at 13,000× rpm for 10 min at 4°C. The supernatants were aliquoted and frozen at −80°C until use. Mouse CCL3, CCL5, CCL8 and CCL9 were quantified using Duoset kits (R&D Systems, Minneapolis, MN). Mouse CCL6 was quantified by sandwich ELISA using a kit according to the manufacturer's instructions (Antigenix America, Huntington Station, NY), and mouse CCL7 was measured using the CCL7 Instant ELISA kit (BenderMed Systems, Vienna, Austria).
Single cell suspension from blood, urine and organs
Mice were euthanized on day 3, 6, 9 and 12 post-infection (n = 3 mice/time-point). Urine was collected by applying external pressure over the urinary bladder and the volume obtained was recorded. Mice were anesthetized using ketamine/xylazine and 300 µl of blood were obtained via cardiac puncture in EDTA tubes. Then, the animals were perfused with normal saline and organs were harvested. Single cell suspensions were obtained using previously described methods [8]. In brief, anti-coagulated peripheral blood was treated with 1× PharmLyse buffer (BD Biosciences, San Jose, CA) according to the manufacturer's protocol to remove red blood cells. Then, leukocytes were washed three times with HBSS+2 mM EDTA+1% BSA, suspended in PBS, and passed though a 40-µm filter (BD Biosciences). Urine leukocytes were washed three times with RPMI+10% FBS, suspended in PBS, and passed through a 40-µm filter. Brains were collected in 7 ml of FACS buffer and homogenized using the back of a 6-cc syringe on ice in a tissue culture plate. The suspension was brought to 10 ml by adding 3 ml of 90% Percoll (GE Healthcare, Piscataway, NJ) in PBS. After thorough mixing, the solution was underlaid with 1 ml of 70% Percoll (in PBS), and was centrifuged at 2,450 rpm for 30 min at 4°C. The leukocytes at the interphase were isolated, washed three times in FACS buffer, suspended in PBS, and passed through a 40-µm filter. Spleens were finely minced and digested at 37°C in digestion solution (RPMI 1640 with 20 mM HEPES without serum) containing liberase TL (Roche Diagnostics, Chicago, IL) and grade II DNAse I (Roche) for 20 min with shaking. Digested tissue was passed through a 100-µm filter and washed, and the remaining red cells were lysed with ACK lysing buffer (Lonza, Walkersville, MD). Then, cells were passed through a 70-µm filter and washed three times with FACS buffer. Finally, splenocytes were suspended in PBS and passed through a 40-µm filter. To obtain kidney and liver single cell suspensions, the organs were finely minced and digested at 37°C in digestion solution containing liberase TL and grade II DNAse I for 20 min with shaking. Digested tissue was passed through a 70-µm filter, washed, and the remaining red cells were lysed with ACK lysing buffer for 30 seconds. Then, the cells were passed through a 40-µm filter, washed, and suspended in 40% Percoll. The suspension was overlaid on 70% Percoll, and centrifuged at 2,000 rpm for 30 min at RT. The leukocytes at the interphase were isolated, washed three times in FACS buffer, suspended in PBS, and passed though a 40-µm filter.
Flow cytometry
The blood, urine, kidney, brain, spleen and liver single cell suspensions were stained with a Live/Dead fluorescent dye (Invitrogen, Carlsbad, CA) for 10 min (1∶500) and incubated with rat anti-mouse CD16/32 (2.4G2; BD Biosciences) for 10 min (1∶100) at 4°C to block Fc receptors. Then, cells were incubated at 4°C for 30 min with the following anti-mouse antibodies: efluor450 CD45 (Ly-5; eBioscience, San Diego, CA); FITC-conjugated Ly6c (AL-21), CD3 (17A2; BD Biosciences) and MHC II (M5/114.15.2; eBioscience); PE-conjugated CD45 (Ly-5), Ly6G (1A8), NK1.1 (PK136; BD Biosciences); PE-Cy7-conjugated F4/80 (BM8) or CD8 (53-6.7; eBioscience); allophycocyanin-conjugated CD45 (Ly-5), CD11c (HL3) (BD Biosciences) or CD19 (1D3; eBioscience); allophycocyanin-Cy7-conjugated CD11b (M1/70; BD Biosciences) or APC-eFluor 780-conjugated CD4 (RM4-5; eBioscience). After three washes with FACS buffer, cells were fixed with 2% paraformaldehyde. FACS was performed on an LSRII (BD Biosciences) and data were analyzed using FlowJo (version 8.8.4; Treestar, Ashland, OR). Cell numbers were quantified using PE-conjugated fluorescent counting beads (Spherotech, Lake Forest, IL).
FACS analysis of neutrophils for annexin V and 7-AAD expression
To determine whether Ccr1 deficiency influenced neutrophil survival post-infection, we performed FACS analysis on kidney and spleen neutrophils from Ccr1+/+ and Ccr1−/− mice on days 6 and 9 post-infection. Cells were initially incubated for 10 min with rat anti-mouse CD16/32 and then stained for 30 min with the combination of PE-conjugated Ly6G, allophycocyanin-conjugated CD45 and allophycocyanin-Cy7-conjugated CD11b (BD Biosciences). Then, the cells were washed twice with PBS, suspended in 1× Annexin V binding buffer (BD Biosciences), and incubated for 15 min with 5 µl each of FITC-conjugated Annexin V and 7-AAD according to the manufacturer's instructions (BD Biosciences). The cells were washed and FACS was performed on an LSRII.
FACS analysis of neutrophils for Ccr1 expression
To determine the expression of Ccr1 on neutrophils and other leukocytes after infection, we performed FACS analysis on kidney and blood cells on days 3, 6 and 9 post-infection. Cells were initially incubated for 15 min with rat anti-mouse CD16/32 and then stained for 30 min with efluor450-conjugated CD45 or CD3, FITC-conjugated Ly6c or Ly6G, PE-Cy7-conjugated F4/80, allophycocyanin-conjugated CD19, Ly6G, CD45 or CD11c, allophycocyanin-Cy7-conjugated CD11b, and PE-conjugated rat monoclonal anti-mouse Ccr1 (FAB5986P, clone 643854; R&D Systems) or PE-conjugated isotype control (rat IgG2B; IC013P, clone 141945; R&D Systems) according to the manufacturer's protocol. The cells were washed and FACS was performed on an LSRII. The specificity of the monoclonal Ccr1 antibody was confirmed by demonstration of binding on Ccr1-transfected, but not untransfected control, HEK293 cells (data not shown).
Magnetic sorting of mouse kidney neutrophils
To sort kidney neutrophils the AutoMACS automatic magnetic cell-sorting system was used. Kidney single cell suspensions from Ccr1+/+ mice euthanized on days 3, 6 and 9 post-infection were prepared as described above, and the kidney cells were incubated with the anti-Ly6G MicroBead kit (Miltenyi Biotec, Auburn, CA) according to the manufacturer's instructions. Ly6G+ cell purification was conducted on an AutoMACS separator (Miltenyi Biotec) consistently achieving an enriched population of >90% Ly6cintLy6G+CD11b+ neutrophils with >95% viability (data not shown).
Gene expression of Ccr1 and its ligands on kidney neutrophils after infection
mRNA was extracted from the MACS-purified kidney neutrophils of Ccr1+/+ mice at days 3, 6 and 9 post-infection (n = 3–5 per time-point) using Trizol per the manufacturer's protocol. cDNA was generated, and qPCR was performed for Ccr1, Ccl3, Ccl5, Ccl6, Ccl7, Ccl8 and Ccl9 as described above. Relative gene expression was determined after normalization with GAPDH transcript levels using the ΔΔCT method and displayed as gene amplicons.
Chemotaxis assay
Chemotaxis was measured using a Boyden Chamber assay. Briefly, MACS-purified mouse neutrophils from 12 pooled kidneys of 6 Ccr1+/+ mice at day 9 post-infection (50 µl, 1×106 cells/ml) were added to the upper chamber and recombinant mouse CCL3, CCL5, CCL7, CCL8 (Peprotech Inc., Rocky Hill, NJ), CCL6 or CCL9 (R&D Systems) (concentration range: 0–500 nM) was added to the lower chamber separated by a membrane containing 3 µm diameter pores (Neuro Probe Inc., Gaithersburg, MD). Following a 1 hr incubation in a 37°C, 5% CO2 incubator, chemotaxis was assessed in triplicate by counting the number of migrated cells in three random fields per well.
In vivo neutrophil adoptive transfer
Marrow cavities of the tibias and femurs of Candida-infected Ccr1+/+ and Ccr1−/− mice at day 9 post-infection were flushed with RPMI+10% FBS+2 mM EDTA. Following hypotonic red blood cell lysis with 0.2% and 1.6% NaCl, mature neutrophils were isolated by gradient centrifugation over Histopaque 1119 (density, 1.119 g/mL) and Histopaque 1077 (density, 1.077 g/mL) according to the manufacturer's instructions at 2000 rpm for 30 min at 25°C. Neutrophils recovered at the interface of the Histopaque 1119 and Histopaque 1077 layers were 80–90% pure and >95% viable as determined by FACS (data not shown). Neutrophils were washed twice with RPMI+10% FBS+penicillin/streptomycin and were resuspended at 5×106 cells/ml in PBS. Ccr1+/+ and Ccr1−/− neutrophils were then incubated with CMFDA Cell Tracker Orange (Invitrogen) and CMTMR Cell Tracker Green (Invitrogen), respectively, in a water bath at 37°C for 10 min (final concentration, 5 µM). Cells were washed with cold RPMI+10% FBS+penicillin/streptomycin three times, resuspended in sterile PBS at a final concentration of 40×106 cells/ml, and mixed at a 1∶1 ratio. A total of 8×106 neutrophils (200 µl) were injected intravenously per animal in 16 Ccr1+/+ Candida-infected mice on day 9 post-infection and 4 hours later, the mice were euthanized, their kidneys were harvested and single cell suspensions were prepared as described above. After live/dead staining and Fc blockade, leukocytes were stained with efluor450-conjugated CD45, allophycocyanin-conjugated Ly6G and allophycocyanin-Cy7 conjugated CD11b. Neutrophil migration was calculated based on the ratio detected in the kidney compared with the initial input ratio of labeled cells at the time of cell transfer (Ccr1+/+:Ccr1−/− cells) after gating on CD45+Ly6G+CD11b+ cells.
Beta-glucuronidase assay
The degranulation potential of Candida-infected kidney neutrophils was assessed using the beta-glucuronidase assay as previously described [52]. Kidney neutrophils of Ccr1+/+ mice were magnetically sorted as mentioned above at days 3 (Ccr1lo) and 9 (Ccr1high) post-infection. Then, 5×104 neutrophils were lysed using 0.2% Triton-X-100 and incubated for 30 minutes in black 96-well plates with 4-methylumbelliferyl-β-D-glucuronide (MUB-glucuronide; Sigma) in a 37°C water bath. The reaction was terminated by adding ice-cold glycine (50 mM) and ammonium hydroxide (200 mM) buffer (pH, 10.5). The neutrophil β-glucuronidase activity was then determined by measuring the chromogenic substrate β-methylumbelliferone (MUB; Sigma) using a fluorescent plate reader with excitation wavelength of 360 nm and emission wavelength of 455 nm. Two independent experiments were performed using a total of 6–10 mice per time-point.
Neutrophil oxidative burst
The oxidative burst of Candida-infected kidney neutrophils was assessed using the DHR assay as previously described [53]. Kidney neutrophils of Ccr1+/+ mice were magnetically sorted as mentioned above at days 3 (Ccr1lo) and 9 (Ccr1high) post-infection. Then, 2.5×105 neutrophils were suspended in sterile HBSS buffer (i.e., HBSS without calcium/magnesium/phenol red, plus 0.5 g of BSA, plus 1 mM of EDTA) and 2 µl of a 25 mM DHR solution (Invitrogen) was added. After a 5-minute incubation at 37°C, PMA (Sigma) was then added (final concentration, 400 ng/ml) and the neutrophils were incubated in a 37°C shaking water bath for 15 minutes. Neutrophils not stimulated with PMA were also tested to determine the “resting” levels of rhodamine 123 fluorescence. The mean fluorescence intensity was then analyzed using flow cytometry in the FL-1 channel as a measure of the conversion of the non-fluorescent reactive oxygen species indicator DHR to the oxidized cationic rhodamine 123, which exhibits green fluorescence. Two independent experiments were performed using a total of 6–10 mice per time-point.
Neutrophil phagocytosis and killing assays
Kidney neutrophils of Ccr1+/+ mice were magnetically sorted as mentioned above at days 3 (Ccr1lo) and 9 (Ccr1high) post-infection. For the phagocytosis assays, 5×105 neutrophils were incubated for 30 min at 37°C with 5×105 yeast cells of opsonized GFP-Candida albicans strain SC5314 (kind gift from Dr. Robert Wheeler, University of Maine) at a neutrophil∶Candida ratio of 1∶1. The percent of Candida phagocytosis was then determined using flow cytometry by gating on the FITC-positive neutrophils. For the killing assays, 5×105 neutrophils were plated on 96-well flat bottom plates for 30 minutes to create a monolayer and then 105 yeast cells of opsonized Candida albicans strain SC5314 were added at a neutrophil∶Candida ratio of 5∶1. Following 30 minutes of phagocytosis at 37°C, the non-phagocytosed Candida cells were washed out of the wells. The neutrophil anti-Candida killing capacity was then determined by plating the number of viable Candida cells after an additional 90-minute incubation at 37°C as previously described [54]. Two independent experiments were performed using a total of 7–10 mice per time-point for the phagocytosis assays and a total of 8–10 mice per time-point for the killing assays.
Statistical analysis
The data were analyzed using the two-tailed unpaired t-test or the Mann-Whitney test where appropriate with Prism 5.0 software (GraphPad Software, San Diego, CA) and are presented as the mean ± SEM. The cutoff for statistical significance was defined as P<0.05.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. ZaoutisTE, ArgonJ, ChuJ, BerlinJA, WalshTJ, et al. (2005) The epidemiology and attributable outcomes of candidemia in adults and children hospitalized in the United States: a propensity analysis. Clin Infect Dis 41 : 1232–1239.
2. MillerLG, HajjehRA, EdwardsJEJr (2001) Estimating the cost of nosocomial candidemia in the united states. Clin Infect Dis 32 : 1110.
3. WilsonLS, ReyesCM, StolpmanM, SpeckmanJ, AllenK, et al. (2002) The direct cost and incidence of systemic fungal infections. Value Health 5 : 26–34.
4. PappasPG (2006) Invasive candidiasis. Infect Dis Clin North Am 20 : 485–506.
5. SpellbergB, IbrahimAS, EdwardsJEJr, FillerSG (2005) Mice with disseminated candidiasis die of progressive sepsis. J Infect Dis 192 : 336–343.
6. SzaboEK, MacCallumDM (2011) The contribution of mouse models to our understanding of systemic candidiasis. FEMS Microbiol Lett 320 : 1–8.
7. LegrandF, LecuitM, DupontB, BellatonE, HuerreM, et al. (2008) Adjuvant corticosteroid therapy for chronic disseminated candidiasis. Clin Infect Dis 46 : 696–702.
8. LionakisMS, LimJK, LeeCC, MurphyPM (2011) Organ-specific innate immune responses in a mouse model of invasive candidiasis. J Innate Immun 3 : 180–199.
9. FulurijaA, AshmanRB, PapadimitriouJM (1996) Neutrophil depletion increases susceptibility to systemic and vaginal candidiasis in mice, and reveals differences between brain and kidney in mechanisms of host resistance. Microbiology 142 : 3487–3496.
10. QianQ, JutilaMA, Van RooijenN, CutlerJE (1994) Elimination of mouse splenic macrophages correlates with increased susceptibility to experimental disseminated candidiasis. J Immunol 152 : 5000–5008.
11. MahantyS, GreenfieldRA, JoyceWA, KincadePW (1988) Inoculation candidiasis in a murine model of severe combined immunodeficiency syndrome. Infect Immun 56 : 3162–3166.
12. WinkelsteinJA, MarinoMC, JohnstonRBJr, BoyleJ, CurnutteJ, et al. (2000) Chronic granulomatous disease. Report on a national registry of 368 patients. Medicine (Baltimore) 79 : 155–169.
13. VonkAG, NeteaMG, van KriekenJH, IwakuraY, van der MeerJW, et al. (2006) Endogenous interleukin (IL)-1 alpha and IL-1 beta are crucial for host defense against disseminated candidiasis. J Infect Dis 193 : 1419–1426.
14. StuytRJ, NeteaMG, VerschuerenI, FantuzziG, DinarelloCA, et al. (2002) Role of interleukin-18 in host defense against disseminated Candida albicans infection. Infect Immun 70 : 3284–3286.
15. NeteaMG, van TitsLJ, CurfsJH, AmiotF, MeisJF, et al. (1999) Increased susceptibility of TNF-alpha lymphotoxin-alpha double knockout mice to systemic candidiasis through impaired recruitment of neutrophils and phagocytosis of Candida albicans. J Immunol 163 : 1498–1505.
16. van EnckevortFH, NeteaMG, HermusAR, SweepCG, MeisJF, et al. (1999) Increased susceptibility to systemic candidiasis in interleukin-6 deficient mice. Med Mycol 37 : 419–426.
17. MencacciA, CenciE, Del SeroG, Fé d'OstianiC, MosciP, et al. (1998) IL-10 is required for development of protective Th1 responses in IL-12-deficient mice upon Candida albicans infection. J Immunol 161 : 6228–6237.
18. LavigneLM, SchopfLR, ChungCL, MaylorR, SypekJP (1998) The role of recombinant murine IL-12 and IFN-gamma in the pathogenesis of a murine systemic Candida albicans infection. J Immunol 160 : 284–292.
19. Vazquez-TorresA, Jones-CarsonJ, WagnerRD, WarnerT, BalishE (1999) Early resistance of interleukin-10 knockout mice to acute systemic candidiasis. Infect Immun 67 : 670–674.
20. BalishE, WarnerTF, NicholasPJ, PaullingEE, WestwaterC, et al. (2005) Susceptibility of germfree phagocyte oxidase - and nitric oxide synthase 2-deficient mice, defective in the production of reactive metabolites of both oxygen and nitrogen, to mucosal and systemic candidiasis of endogenous origin. Infect Immun 73 : 1313–1320.
21. HuangW, NaL, FidelPL, SchwarzenbergerP (2004) Requirement of interleukin-17A for systemic anti-Candida albicans host defense in mice. J Infect Dis 190 : 624–631.
22. MurphyPM, BaggioliniM, CharoIF, HébertCA, HorukR, et al. (2000) International union of pharmacology. XXII. Nomenclature for chemokine receptors. Pharmacol Rev 52 : 145–176.
23. TsouCL, PetersW, SiY, SlaymakerS, SlaymakerS, et al. (2007) Critical roles for CCR2 and MCP-3 in monocyte mobilization from bone marrow and recruitment to inflammatory sites. J Clin Invest 117 : 902–909.
24. IchimuraT, BonventreJV, BaillyV, WeiH, HessionCA, et al. (1998) Kidney injury molecule-1 (KIM-1), a putative epithelial cell adhesion molecule containing a novel immunoglobulin domain, is up-regulated in renal cells after injury. J Biol Chem 273 : 4135–4142.
25. VaidyaVS, OzerJS, DieterleF, CollingsFB, RamirezV, et al. (2010) Kidney injury molecule-1 outperforms traditional biomarkers of kidney injury in preclinical biomarker qualification studies. Nat Biotechnol 28 : 478–485.
26. KarlmarkKR, ZimmermannHW, RoderburgC, GasslerN, WasmuthHE, et al. (2010) The fractalkine receptor CX3CR1 protects against liver fibrosis by controlling differentiation and survival of infiltrating hepatic monocytes. Hepatology 52 : 1769–1782.
27. GaoJL, WynnTA, ChangY, LeeEJ, BroxmeyerHE, et al. (1997) Impaired host defense, hematopoiesis, granulomatous inflammation and type 1-type 2 cytokine balance in mice lacking CC chemokine receptor 1. J Exp Med 185 : 1959–1968.
28. GerardC, FrossardJL, BhatiaM, SalujaA, GerardNP, et al. (1997) Targeted disruption of the beta-chemokine receptor CCR1 protects against pancreatitis-associated lung injury. J Clin Invest 100 : 2022–2027.
29. HeM, HorukR, MoochhalaSM, BhatiaM (2007) Treatment with BX471, a CC chemokine receptor 1 antagonist, attenuates systemic inflammatory response during sepsis. Am J Physiol Gastrointest Liver Physiol 292 : 1173–1180.
30. ZhangS, YounBS, GaoJL, MurphyPM, KwonBS (1999) Differential effects of leukotactin-1 and macrophage inflammatory protein-1 alpha on neutrophils mediated by CCR1. J Immunol 162 : 4938–4942.
31. SchuitKE (1979) Phagocytosis and intracellular killing of pathogenic yeasts by human monocytes and neutrophils. Infect Immun 24 : 932–938.
32. UzunO, AsciogluS, AnaissieEJ, RexJH (2001) Risk factors and predictors of outcome in patients with cancer and breakthrough candidemia. Clin Infect Dis 32 : 1713–1717.
33. RomaniL, MencacciA, CenciE, Del SeroG, BistoniF, et al. (1997) An immunoregulatory role for neutrophils in CD4+ T helper subset selection in mice with candidiasis. J Immunol 158 : 2356–2362.
34. MiceliMH, MaertensJ, BuvéK, GrazziuttiM, WoodsG, et al. (2007) Immune reconstitution inflammatory syndrome in cancer patients with pulmonary aspergillosis recovering from neutropenia: Proof of principle, description, and clinical and research implications. Cancer 110 : 112–120.
35. KhanIA, MurphyPM, CasciottiL, SchwartzmanJD, CollinsJ, et al. (2001) Mice lacking the chemokine receptor CCR1 show increased susceptibility to Toxoplasma gondii infection. J Immunol 166 : 1930–1937.
36. DomachowskeJB, BonvilleCA, GaoJL, MurphyPM, EastonAJ, et al. (2000) The chemokine macrophage-inflammatory protein-1 alpha and its receptor CCR1 control pulmonary inflammation and antiviral host defense in paramyxovirus infection. J Immunol 165 : 2677–2682.
37. FuruichiK, GaoJL, HorukR, WadaT, KanekoS, et al. (2008) Chemokine receptor CCR1 regulates inflammatory cell infiltration after renal ischemia-reperfusion injury. J Immunol 181 : 8670–8676.
38. VielhauerV, BerningE, EisV, KretzlerM, SegererS, et al. (2004) CCR1 blockade reduces interstitial inflammation and fibrosis in mice with glomerulosclerosis and nephrotic syndrome. Kidney Int 66 : 2264–2278.
39. NinichukV, GrossO, ReichelC, KhandogaA, PawarRD, et al. (2005) Delayed chemokine receptor 1 blockade prolongs survival in collagen 4A3-deficient mice with Alport disease. J Am Soc Nephrol 16 : 977–985.
40. EisV, LuckowB, VielhauerV, SivekeJT, LindeY, et al. (2004) Chemokine receptor CCR1 but not CCR5 mediates leukocyte recruitment and subsequent renal fibrosis after unilateral ureteral obstruction. J Am Soc Nephrol 15 : 337–347.
41. AndersHJ, VielhauerV, FrinkM, LindeY, CohenCD, et al. (2002) A chemokine receptor CCR-1 antagonist reduces renal fibrosis after unilateral ureter ligation. J Clin Invest 109 : 251–259.
42. TophamPS, CsizmadiaV, SolerD, HinesD, GerardCJ, et al. (1999) Lack of chemokine receptor CCR1 enhances Th1 responses and glomerular injury during nephrotoxic nephritis. J Clin Invest 104 : 1549–1557.
43. BonecchiR, PolentaruttiN, LuiniW, BorsattiA, BernasconiS, et al. (1999) Up-regulation of CCR1 and CCR3 and induction of chemotaxis to CC chemokines by IFN-gamma in human neutrophils. J Immunol 162 : 474–479.
44. ChengSS, LaiJJ, LukacsNW, KunkelSL (2001) Granulocyte-macrophage colony stimulating factor up-regulates CCR1 in human neutrophils. J Immunol 166 : 1178–1184.
45. van de VeerdonkFL, KullbergBJ, van der MeerJW, GowNA, NeteaMG (2008) Host-microbe interactions: innate pattern recognition of fungal pathogens. Curr Opin Microbiol 11 : 305–312.
46. WheelerRT, KombeD, AgarwalaSD, FinkGR (2008) Dynamic, morphotype-specific Candida albicans beta-glucan exposure during infection and drug treatment. PLoS Pathog 4: e1000227.
47. ChouRC, KimND, SadikCD, SeungE, LanY, et al. (2010) Lipid-cytokine-chemokine cascade drives neutrophil recruitment in a murine model of inflammatory arthritis. Immunity 33 : 266–278.
48. BalishE, WagnerRD, Vazquez-TorresA, Jones-CarsonJ, PiersonC, et al. (1999) Mucosal and systemic candidiasis in IL-8Rh−/ − BALB/c mice. J Leukoc Biol 66 : 144–150.
49. BryantK, MaxfieldC, RabalaisG (1999) Renal candidiasis in neonates with candiduria. Pediatr Infect Dis J 18 : 959–963.
50. KohAY, KöhlerJR, CoggshallKT, Van RooijenN, PierGB (2008) Mucosal damage and neutropenia are required for Candida albicans dissemination. PLoS Pathog 4: e35.
51. IchimuraT, AsseldonkEJ, HumphreysBD, GunaratnamL, DuffieldJS, et al. (2008) Kidney injury molecule-1 is a phosphatidylserine receptor that confers a phagocytic phenotype on epithelial cells. J Clin Invest 118 : 1657–1668.
52. KolotilaMP, DiamondRD (1988) Stimulation of neutrophil actin polymerization and degranulation by opsonized and unopsonized Candida albicans hyphae and zymosan. Infect Immun 56 : 2016–2022.
53. JirapongsananurukO, MalechHL, KuhnsDB, NiemelaJE, BrownMR, et al. (2003) Diagnostic paradigm for evaluation of male patients with chronic granulomatous disease, based on the dihydrorhodamine 123 assay. J Allergy Clin Immunol 111 : 374–379.
54. VonkAG, NeteaMG, KullbergBJ (2012) Phagocytosis and intracellular killing of Candida albicans by murine polymorphonuclear neutrophils. Methods Mol Biol 845 : 277–287.
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2012 Číslo 8
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Měli bychom postcovidový syndrom léčit antidepresivy?
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- Invariant NKT Cells: Regulation and Function during Viral Infection
- Host Defense and Tolerance: Unique Challenges in the Placenta
- Nonhuman Primate Models for HIV Cure Research
- Exon Level Transcriptomic Profiling of HIV-1-Infected CD4 T Cells Reveals Virus-Induced Genes and Host Environment Favorable for Viral Replication