
B-Cyclin/CDKs Regulate Mitotic Spindle Assembly by Phosphorylating Kinesins-5 in Budding Yeast
Although it has been known for many years that B-cyclin/CDK complexes regulate the assembly of the mitotic spindle and entry into mitosis, the full complement of relevant CDK targets has not been identified. It has previously been shown in a variety of model systems that B-type cyclin/CDK complexes, kinesin-5 motors, and the SCFCdc4 ubiquitin ligase are required for the separation of spindle poles and assembly of a bipolar spindle. It has been suggested that, in budding yeast, B-type cyclin/CDK (Clb/Cdc28) complexes promote spindle pole separation by inhibiting the degradation of the kinesins-5 Kip1 and Cin8 by the anaphase-promoting complex (APCCdh1). We have determined, however, that the Kip1 and Cin8 proteins are present at wild-type levels in the absence of Clb/Cdc28 kinase activity. Here, we show that Kip1 and Cin8 are in vitro targets of Clb2/Cdc28 and that the mutation of conserved CDK phosphorylation sites on Kip1 inhibits spindle pole separation without affecting the protein's in vivo localization or abundance. Mass spectrometry analysis confirms that two CDK sites in the tail domain of Kip1 are phosphorylated in vivo. In addition, we have determined that Sic1, a Clb/Cdc28-specific inhibitor, is the SCFCdc4 target that inhibits spindle pole separation in cells lacking functional Cdc4. Based on these findings, we propose that Clb/Cdc28 drives spindle pole separation by direct phosphorylation of kinesin-5 motors.
Published in the journal:
. PLoS Genet 6(5): e32767. doi:10.1371/journal.pgen.1000935
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1000935
Summary
Although it has been known for many years that B-cyclin/CDK complexes regulate the assembly of the mitotic spindle and entry into mitosis, the full complement of relevant CDK targets has not been identified. It has previously been shown in a variety of model systems that B-type cyclin/CDK complexes, kinesin-5 motors, and the SCFCdc4 ubiquitin ligase are required for the separation of spindle poles and assembly of a bipolar spindle. It has been suggested that, in budding yeast, B-type cyclin/CDK (Clb/Cdc28) complexes promote spindle pole separation by inhibiting the degradation of the kinesins-5 Kip1 and Cin8 by the anaphase-promoting complex (APCCdh1). We have determined, however, that the Kip1 and Cin8 proteins are present at wild-type levels in the absence of Clb/Cdc28 kinase activity. Here, we show that Kip1 and Cin8 are in vitro targets of Clb2/Cdc28 and that the mutation of conserved CDK phosphorylation sites on Kip1 inhibits spindle pole separation without affecting the protein's in vivo localization or abundance. Mass spectrometry analysis confirms that two CDK sites in the tail domain of Kip1 are phosphorylated in vivo. In addition, we have determined that Sic1, a Clb/Cdc28-specific inhibitor, is the SCFCdc4 target that inhibits spindle pole separation in cells lacking functional Cdc4. Based on these findings, we propose that Clb/Cdc28 drives spindle pole separation by direct phosphorylation of kinesin-5 motors.
Introduction
Cyclin-dependent kinases (CDKs) complexed with various cyclins coordinate many duplication and segregation events during the eukaryotic cell division cycle [1], [2]. The duplication of the cell's microtubule organizing center, the centrosome, and the subsequent separation of the duplicated centrosomes is one such event [3], [4]. Timely separation of the duplicated centrosomes is required for the assembly of the bipolar spindle at metaphase which, in turn, is necessary for the equal segregation of sister chromatids during anaphase and the preservation of genome stability.
The budding yeast centrosome, called the spindle pole body (SPB), is functionally equivalent to the metazoan centrosome. Although structurally dissimilar [5], they appear to be regulated by similar mechanisms [3], [6]. Thus, the budding yeast SPB is a powerful model for understanding the metazoan centrosome, as demonstrated by genetic studies that have identified many components of the eukaryotic cellular machinery critical to both SPB and centrosome separation (reviewed in [5]–[9]).
Three classes of mutations that cause cells to arrest with duplicated but unseparated SPBs have been identified in Saccharomyces cerevisiae. The first class includes mutations in the genes encoding Cdc28, the yeast Cdk1, and the B-cyclins which bind to Cdc28. Cells lacking all six B-type cyclin genes (CLB1, CLB2, CLB3, CLB4, CLB5, and CLB6) are unable to separate SPBs [4]. The mutation of tyrosine 19 in Cdc28 to mimic an inhibitory phosphorylation (cdc28Y19E) [10], [11] has also been reported to result in a SPB separation defect [12]. This phosphomimetic mutation is thought to specifically inhibit Clb1,2,3,4/Cdc28 complexes, but likely not Clb5,6/Cdc28 [11]. Not surprisingly, Δclb1,2,3,4 mutants also appear to have a diminished capacity to separate SPBs [13], , although separation can occur after extended time periods [4], [13].
The second class of SPB separation mutations affects genes encoding components of the SCFCdc4 E3 ubiquitin ligase complex (CDC4, CDC53 and SKP1 [15]–[17]) as well as CDC34 [18], the E2 ubiquitin protein-conjugating enzyme that is associated with SCFCdc4. Temperature-sensitive cdc4, cdc53, and cdc34 mutants arrest with multiple elongated buds and unreplicated DNA, as well as duplicated but unseparated SPBs [15]–[19]. The arrest phenotype of these mutants is likely to be identical to that of Δclb1,2,3,4,5,6 [4], [20] mutants due to a buildup of Sic1 [15], [20]. Sic1 is a Clb/Cdc28-specific inhibitor whose degradation is normally triggered by the SCFCdc4 complex in G1 to allow entry into S phase [20]–[22]. However, it is possible that there is a SCFCdc4 target that is directly involved in maintaining cohesion between the duplicated SPBs and which must be destroyed before separation can occur. Such a protein could be a component of the proteinaceous bridge structure that physically joins newly duplicated SPBs and would need to be overcome for separation to occur [8], [9]. Direct phosphorylation by CDK complexes is generally required to trigger the ubiquitination of SCF targets [23]; so Clb/Cdc28 complexes might work in concert with the SCFCdc4 to destroy such a separation-inhibiting element.
The third class of mutations lies in the KIP1 and CIN8 genes [24], [25] which encode members of the kinesin-5 family of bipolar, microtubule-based motor proteins [26]. Kinesins-5 have been shown to be important in both the establishment and maintenance of the bipolar spindle in many fungal and metazoan systems [27]–[30]. It is thought that kinesin-5 motors crosslink and move spindle microtubules, which are also required for SPB separation [31], [32], in order to mechanically separate the spindle poles and establish the spindle (reviewed in [33]). Accordingly, cells lacking both functional Kip1 and Cin8, arrest with duplicated and unseparated SPBs when released from a G1 arrest [24], [25].
Together, these findings suggest that Clb/Cdc28 complexes promote the timely separation of SPBs, and that kinesin-5 motors may be subject to phosphoregulation by Clb/Cdc28 complexes [14], [34]. Although several of the genetic requirements for SPB separation are now known, the molecular mechanisms that regulate separation remain unclear. cyclin B/Cdk1 phosphorylation of the tail domain of the Homo sapiens [35], [36], Xenopus laevis [37], [38], and Drosophila melanogaster [39], [40] kinesin-5 orthologues (HsEg5 or Kif11, XlEg5, and KLP61F, respectively) has been shown to be required for their localization to the spindle. The BimC box motif [28] where this phosphorylation occurs is not found, however, in either Kip1 or Cin8, although other consensus CDK phosphorylation sites exist in both proteins.
Two recent studies [41], [42] suggest that Clb/Cdc28 complexes regulate kinesin-5 protein stability indirectly by phosphorylating Cdh1, a substrate-specific activator of the anaphase-promoting complex (APC) [43]. The APCCdh1 is active in G1, and is thought to be inactivated in S phase by B-cyclin/CDK-mediated phosphorylation of Cdh1 [44], [45]. The degradation of Kip1 and Cin8 is dependent on the APC in complex with Cdc20 [46] or with Cdh1 [47], respectively. Motor stability thus depends on APC activity. The studies by Crasta et al., however, relied heavily on two mutant alleles of CDC28, cdc28Y19E and cdc28-as1, which retain some CDK activity under the experimental conditions employed [41], [48]. They therefore do not rule out the possibility that CDKs also regulate kinesin-5 motors directly.
In this study, we asked if Clb/Cdc28 directly regulates kinesin-5 activity in order to trigger SPB separation and spindle assembly. We first determined that the only target of SCFCdc4 involved in regulating SPB separation is the Clb/Cdc28-specific inhibitor, Sic1. Thus, SCF-mediated destruction of a bridge component is likely not required for spindle assembly. We next determined that Clb2/Cdc28 phosphorylates Kip1 and Cin8 in vitro, and also that Clb/Cdc28 complexes do not regulate either the abundance or localization of these kinesins-5 in vivo. Moreover, by genetic mapping, we identified a CDK phosphorylation site in the motor domain of Kip1 that is critical to SPB separation. We also identified two non-conserved CDK sites in the tail domain of Kip1 that are important for timely SPB separation, and verified that they are phosphorylated in vivo by mass spectrometry. As the site in the motor domain is conserved across almost all of the kinesin-5 family, we propose that direct regulation of kinesin-5 motor functions by B-cyclin/CDK may not be exclusive to S. cerevisiae.
Results
Deleting SIC1 is sufficient to permit SPB separation in cells lacking functional Cdc4
Mutants lacking SCFCdc4 E3 ubiquitin ligase activity fail to separate duplicated SPBs. This observation suggests that there is a protein or proteins that must be ubiquitinated by the SCFCdc4 and subsequently degraded in order for the SPBs to separate. A likely candidate is the Clb/Cdc28-specific inhibitor Sic1, which accumulates at the restrictive temperature in mutants with temperature-sensitive alleles of the SCFCdc4 complex components such as Cdc4, Cdc34, Cdc53, and Skp1 [15], [20]. Sic1 inhibits Clb/Cdc28 kinases essential for S-phase entry [15], [20] as well as SPB separation [4], [13]. Moreover, cells that express a hyperstabilized allele of Sic1 arrest with a phenotype identical to that of cells which lack SCFCdc4 activity [49]. These findings do not, however, rule out the possibility that the destruction of additional SCFCdc4 targets, such as components of the bridge structure which physically joins newly duplicated SPBs, may be essential for SPB separation.
To determine if Sic1 is the only SCFCdc4 target important for SPB separation, we deleted SIC1 in a strain carrying a temperature-sensitive CDC4 allele, cdc4-3 [19], [50], [51], and expressing GFP-tagged Spc42, a SPB component. We then asked if the ability of these mutant cells to separate SPBs at the restrictive temperature was restored. In agreement with earlier studies [20], [51], [52], we observed that an asynchronous culture of cdc4-3 cells arrests almost uniformly at the G1/S border with elongated buds, unreplicated DNA, and duplicated but unseparated SPBs 2–4 hours after being shifted to 37°C (Figure 1). In contrast, an asynchronous culture of cdc4-3 sic1Δ arrests at 37°C with a majority of cells in G2/M with large, round buds and replicated DNA, as has been observed previously [20], [52]. Most importantly, for this study, duplicated and separated SPBs were observed in the majority (>90%) of cdc4-3 sic1Δ cells within 4 hours of the shift to the restrictive temperature (Figure 1). This finding indicates that the failure to separate SPBs in the absence of SCFCdc4 activity is due solely to the stabilization of Sic1 and the consequent inhibition of Clb/Cdc28 kinase activity. These results are consistent with observations made by Goh & Surana who observed the formation of short spindles by immunofluorescence in asynchronous GAL-CDC4 cdc4Δ sic1Δ cells shifted to glucose to inhibit Cdc4 expression [53]. Those results were, however, inconclusive because the control cells (PGAL1-CDC4 cdc4) used in that particular study did not arrest at the G1/S border, potentially due to the persistence of a low level of Cdc4.
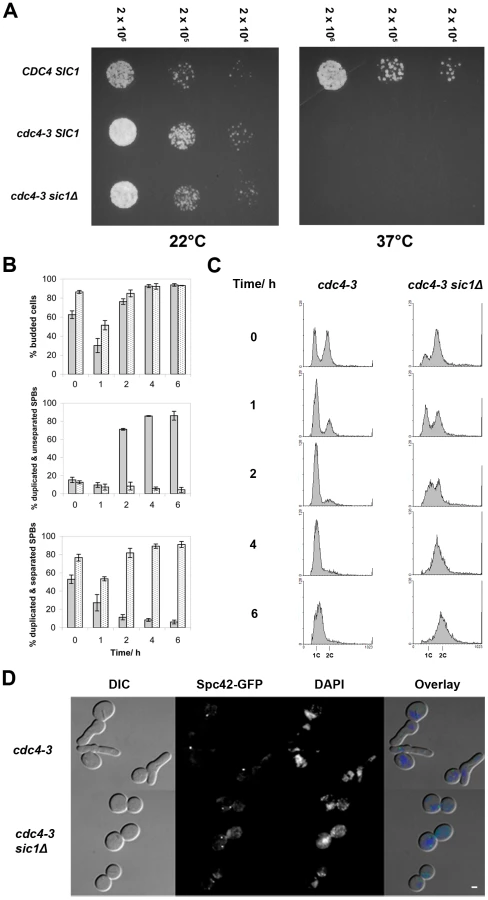
To address the possibility that the SCFCdc4 may still play a role in bridge cleavage, meaning that duplicated SPBs might separate aberrantly in cdc4-3 sic1 cells at the restrictive temperature, we examined the ability of cdc4-3 sic1 cells to resume proliferation at permissive temperatures following prolonged arrest at 37°C. Should aberrant SPB separation occur in cdc4-3 sic1 cells at the restrictive temperature, the separated SPBs may either lack half-bridges or have defective half-bridges, thus leading to subsequent delays in proliferation. We collected cdc4-3 sic1 cells during the temperature shift experiment (described above) at various times after shifting to 37°C for spotting on solid media. Plates were subsequently incubated at permissive temperature (22°C). We found that although viability decreases as cdc4-3 sic1 cells are maintained at 37°C over time, the colonies that do form show a similar range of sizes, regardless of exposure to the restrictive temperature (Figure S1A). There was also no evidence of SPB separation defects when the cdc4-3 sic1 cells taken from these colonies were observed by fluorescence microscopy (Figure S1B).
Kip1 and Cin8 are phosphorylated by Clb2/Cdc28 in vitro
Based on existing genetic evidence in S. cerevisiae, and the finding that cyclin B/Cdk1 regulates centrosome separation in certain metazoan systems by phosphorylating the tail domain of kinesin-5 [35]–[40], we hypothesized that Clb/Cdc28 promotes SPB separation by regulating Kip1 and Cin8 function via direct phosphorylation. There are, however, six consensus CDK phosphorylation sites (S/T-P-X-X) in Kip1 and five in Cin8 (Figure 2A). Although a previous large-scale study by Ubersax et al. identified a large number of yeast proteins that are phosphorylated by Clb2/Cdc28, neither kinesin-5 was identified as a substrate [54]. The BimC box motif found in most kinesin-5 motors is phosphorylated in several metazoan systems but the motif is noticeably absent in both Kip1 and Cin8.

To determine if Kip1 and Cin8 can be phosphorylated by Clb/Cdc28 complexes, we carried out an in vitro phosphorylation assay. Kip1 and Cin8 tagged with 12 copies of the c-Myc epitope were expressed from the GAL1 promoter in yeast cells expressing a hyperstabilized SIC1 allele (SIC1Δ3P) [49] to inhibit phosphorylation by Clb/Cdc28. We also expressed the mutants Kip16A and Cin85A, in which the serine or threonine of every consensus CDK site is mutated to non-phosphorylatable alanine. Wild-type and mutant kinesins were immunoprecipitated separately with anti-c-Myc IgG-agarose beads and then mixed with soluble Clb2/Cdc28 kinase and 32P-γ-ATP.
Both Kip1-myc12 and Cin8-myc12 were phosphorylated in a reproducible manner by Clb2/Cdc28 in vitro (Figure 2B). Furthermore, Kip16A-myc12 was, on average, almost 2–3 fold less phosphorylated compared with wild-type Kip1, whereas Cin85A-myc12 was not phosphorylated above background levels. This observation indicates that phosphorylation of Kip1 by Clb/Cdc28 can occur at sites other than the six consensus CDK sites. We also determined that the observed phosphorylation is likely to be specific to mitotic CDK complexes as a similarly prepared S-phase CDK complex, Clb5/Cdc28, did not phosphorylate either Kip1-myc12 or Cin8-myc12 to any significant extent under similar reaction conditions (Figure S2).
Clb/Cdc28 kinase activity does not regulate Kip1 and Cin8 protein abundance
Both Kip1 and Cin8 are thought to be targeted to the proteasome by APC (anaphase promoting complex)-mediated ubiquitination [46], [47]. It has been proposed that Clb/Cdc28 controls SPB separation indirectly by regulating the in vivo stability of the Kip1 and Cin8 proteins [42], [55]. These findings were based on strains carrying either of two mutant CDC28 alleles. The first allele bears a mutation of tyrosine 19 to glutamate that mimics an inhibitory phosphorylation (cdc28Y19E) [10], [11] and the second is a conditional mutant (cdc28-as1) which is inhibited by the ATP analog, 1-NM-PP1 [48]. However, both cdc28Y19E strains, and cdc28-as1 strains (at the concentration of analog used in these studies) are still capable of DNA replication [12], [41], [48], indicating that these alleles still retain some CDK activity.
Hence, we determined the levels of Kip1 and Cin8 protein in strains deleted for all the B-type cyclin genes, as well as in a separate set of strains that overexpress the hyperstabilized SIC1 allele, SIC1Δ3P [49]. Phenotypic data indicate that these strains lack all Clb/Cdc28 activity as the cells are unable to either initiate DNA replication or enter mitosis [4], [20], [49], [56]. Cells were arrested in G1 with α-factor, and subsequently released into the appropriate medium to eliminate Clb/Cdc28 kinase activity. We observed that Kip1 (Figure 3A) and Cin8 (Figure 3B) protein levels were stable over time in the absence of Clb/Cdc28 kinase activity. In order to verify that our findings are not strain-specific, we repeated our GAL-SIC1Δ3P experiment in a W303a strain background. Just as we had observed in our own strain background, Kip1 and Cin8 protein levels did not change significantly in W303a cells lacking Clb/Cdc28 kinase activity (Figure 3). We verified that BF264-15DU and W303a GAL-SIC1Δ3P strains both arrested with unreplicated DNA and unseparated SPBs (Figure S3). Furthermore, persistent levels of Kip1 and Cin8 do not reflect de-regulated transcription, as KIP1 and CIN8 mRNAs accumulate periodically in Δclb1,2,3,4,5,6 mutant cells [56] (Figure 3C).

Clb/Cdc28 kinase activity does not regulate the localization of Kip1 and Cin8 to the spindle
In certain metazoan systems, the phosphorylation of a consensus CDK site located on the tail domain is required for the localization of kinesin-5 motors to the spindle [35]–[40]. Although this site is absent from the tails of both S. cerevisiae kinesins-5, regions of the Kip1 and Cin8 tail domains have been found to be important to their localization to the nucleus [46], [47], [57]. Moreover, there are two consensus CDK sites (Ser 1037 and Thr 1040) found within the smallest defined nuclear localization sequence (NLS) on the Kip1 tail [46], and one (Ser 972) just N-terminal to the reported Cin8 NLS [47]. Thus, it is possible that Kip1 and Cin8 localization is regulated by Clb/Cdc28-mediated phosphorylation of these residues.
To address this possibility, C-terminal mCherry [58] tags were fused to Kip1 and Cin8 expressed from their respective native promoters, and their localization was determined by fluorescence microscopy (Figure 4). We determined that Kip1-mCherry and Cin8-mCherry are both functional as they both supported growth in a kip1Δ cin8Δ background (Figure 5). We observed that wild-type Kip1 and Cin8, as well as the non-phosphorylatable mutants, Kip16A and Cin85A, localized to the spindle poles and spindle microtubules (Fig 4A). These findings suggest that phosphorylation of Kip1 and Cin8 at their consensus CDK sites is not required for spindle localization.


Clb/Cdc28 has been shown, however, to phosphorylate serine and threonine residues that do not match the consensus Ser/Thr-Pro motif [49], [59], [60]. In order to determine if Clb/Cdc28 kinase activity is necessary for the spindle localization of Kip1 and Cin8, we examined localization both in Δclb1,2,3,4,5,6 and in separate GAL1-SIC1Δ3P strains. Cells were arrested in G1 with α-factor, and then released into medium containing either dextrose (Δclb1,2,3,4,5,6 strains) or galactose (PGAL1-SIC1Δ3P strains) in order to eliminate Clb/Cdc28 kinase activity. The KIP1 and CIN8 genes were fused at their native loci to mCherry and localization of the tagged proteins was monitored by fluorescence microscopy (Figure 4B and 4C). Both Kip1-mCherry and Cin8-mCherry still localized to the SPBs, even in the absence of active Clb/Cdc28 kinase. Taken together, these findings suggest that Clb/Cdc28 complexes do not control SPB separation by regulating the localization of Kip1 and Cin8 to the spindle.
Mutation of consensus CDK sites in Kip1 and Cin8 impairs cell proliferation
To determine if Clb/Cdc28 phosphorylation of Kip1 and Cin8 may regulate other motor functions essential to cell division, we examined the proliferation of cells bearing the non-phosphorylatable kip16A and cin85A alleles using a spot assay (Figure 5). As Kip1 and Cin8 are partially redundant [24], [25], we examined the motor mutants in a strain background where the other motor was deleted. Proliferation was assayed at both ambient temperature (∼22°C) and at 37°C as strains deleted for the CIN8 gene have been reported to exhibit temperature-sensitive growth [24], [25]. We observed that although kip16A-mCherry cin8Δ cells proliferated at a rate similar to that of KIP1-mCherry cin8Δ cells at ambient temperature, they failed to form colonies at 37°C (Figure 5A). In contrast, although cin85A-mCherry kip1Δ cells also proliferated more slowly compared to CIN8-mCherry kip1Δ cells, this defect was not a temperature-sensitive one (Figure 5B). We verified that the observed defects were not caused by the mCherry tag by examining strains expressing untagged Kip16A and Cin85A as their sole kinesin-5 (Figure S4).
To determine which of the CDK consensus site mutations contributed to the proliferation defect, we assayed the proliferation of cells containing individual mutations in each of the six consensus CDK phosphorylation sites in Kip1. The substitution of Ser 388 with alanine alone was sufficient to cause temperature-sensitive lethality in the absence of Cin8 (Figure 5A); no other single CDK site when similarly mutated caused temperature-sensitive lethality (data not shown).
Mutating the homologous serine residue in Cin8, Ser 455, to alanine was sufficient to cause the proliferation defect associated with Cin85A in the absence of Kip1 (Figure 5B). Similarly to Kip1, mutating any other single CDK site on Cin8 to alanine did not cause the Cin85A growth defect. As a control, we also verified that the mCherry tag was not responsible for causing the proliferation defect associated with either Kip1S388A or Cin8S455A (Figure S4).
Although we were unable to test all the effects of mutating all possible combinations of the individual consensus CDK sites on Kip1 and Cin8, we did test tandem mutations of CDK sites located in close proximity to each other in the primary structure of the two proteins. In doing so, we found that, kip1S1037A, T1040A cin8Δ cells proliferated at a significantly slower rate than cells bearing a wild-type copy of KIP1 (Figure 5A and S4A). Unlike kip1S388A cin8Δ cells, however, kip1S1037A, T1040A cin8Δ cells were not fully arrested at 37°C.
The role of kinesin-5 phosphorylation in SPB separation
To determine if the proliferation defects associated with the kip1S388A and kip1S1037A, T1040A alleles are related to a defect in SPB separation, we arrested cells in G1 with α-factor at 25°C. Cells were subsequently released at either 25°C or 37°C (restrictive temperature) and SPB separation was monitored over time by fluorescence microscopy. While kip1S388A cin8Δ cells budded at a rate similar to KIP1 cin8Δ cells at both temperatures tested, most kip1S388A cin8Δ cells were unable to separate SPBs at 37°C (Figure 6A). Even more than two hours after being released from α-factor arrest, less than 10% of kip1S388A cin8Δ cells had separated their SPBs at 37°C, compared to more than 40% of KIP1 cin8Δ cells. A clear SPB separation defect was also observed for kip1S1037A, T1040A cin8Δ cells at 37°C (Figure 6B). At 25°C, cin8Δ cells expressing Kip1S388A appear to be impaired to a greater extent in SPB separation than cells expressing Kip1S1037A, T1040A compared with cin8Δ cells expressing wild-type Kip1, which is consistent with the more severe proliferation defect observed for kip1S388A cin8Δ cells (Figure 5A and Figure S4A).

Although unlikely [61], the single S388A and tandem S1037A, T1040A mutations may cause the loss of Kip1 function by perturbing the protein's in vivo stability. To address this possibility, we compared the levels of Kip1S388A-mCherry and Kip1S1037A, T1040A-mCherry in separate time courses with that of Kip1-mCherry using cells synchronized in α-factor and released at 37°C by western blotting (Figure S5A). In doing so, we verified that both mutant and wild-type protein levels were similar at the restrictive temperature. In addition, as expected from the observed localization of Kip16A, both Kip1S388A and Kip1S1037A, T0140A continued to localize to the SPBs and spindle microtubules even at 37°C (Figure S5B). Taken together, our findings suggest that the phosphorylation of Ser 388 by Clb/Cdc28 is critical for Kip1-mediated separation of SPBs, while the phosphorylation of Ser 1037 and Thr 1040 also contributes to the function of Kip1.
Similar analyses of non-phosphorylatable cin8 alleles were performed, although we were unable to determine conclusively if there is a SPB separation defect in cin85A kip1Δ and cin8S455A kip1Δ cells. Although cin8S455A kip1Δ cells showed a delay in SPB separation (Figure 6C) after release from α-factor, they also appeared to have an uncharacterized defect in progression through G1 as the initiation of both bud emergence (Figure 6C) and DNA replication (data not shown) were both significantly delayed after release from α-factor. Hence, we cannot confirm that Cin8 phosphorylation at Ser 455 plays a role in controlling SPB separation due to the potential confounding effects of the apparent G1/S phase delay.
As expected, flow cytometric analyses of DNA content demonstrated that at 37°C, asynchronous populations of cin8Δ cells expressing either Kip16A or Kip1S388A were enriched for cells with replicated DNA (Figure S6A), supporting the observation that these cells have a defect in mitotic spindle assembly. However, asynchronous log phase populations of kip1Δ cells expressing either Cin85A or Cin8S455A were clearly enriched for cells with unreplicated DNA compared to kip1Δ cells expressing wild-type Cin8 at 37°C (Figure S6B). These analyses confirm that putative CDK phosphorylation site mutations in Cin8 give rise to a defect in the G1/S phase transition. The nature of this defect is unknown.
Kip1 and Cin8 are phosphorylated in vivo in a Clb/Cdc28-dependent manner
Given the genetic and in vitro biochemical evidence we had garnered, we sought to determine if Kip1 and Cin8 are indeed phosphorylated by Clb/Cdc28 complexes in vivo using mass spectrometry. Kip1-myc12 and Cin8-myc12 expressed from the GAL1 promoter in yeast cells for subsequent immunopurification. Additionally, we also expressed the two kinesins-5 in yeast that also expressed either Sic1Δ3P to inhibit Clb/Cdc28 kinases or Clb2-HA3 to promote phosphorylation of Clb/Cdc28 substrates. Immunopurified Kip1-myc12 and Cin8-myc12 were subject to SDS-PAGE followed by protein phosphorylation analysis using microcapillary LC/MS/MS techniques.
Kip1 and Cin8 were determined to both be phosphorylated in vivo at multiple residues in all the samples. A total of eight phosphorylation sites were assigned with high confidence for Kip1 and four were assigned for Cin8 (Table 1; Sequest Xcorr values are available upon request). Although not all of the phosphorylated residues were observed in all the samples, many of the phosphopeptides generated had phosphate(s) assigned to the same residue. Of particular interest to us were the phosphorylations assigned to consensus CDK sites located in the tail domains of the two kinesin-5 motors, namely residues Ser 1037 and Thr 1040 in Kip1, and Ser 972 in Cin8. For Kip1, these sites are particularly relevant since we observed proliferation and SPB separation defects when both Ser 1037 and Thr 1040 were mutated in tandem to Ala (Figure 5 and 6).

In addition, the LC/MS/MS analysis yielded peak intensities (Table S1) which enabled us to compare the relative abundance of phosphopeptides common to all three samples submitted for each protein with that of their unphosphorylated forms. In doing so, we determined that the extent of phosphorylation of TCIPNLSTNENFPLSQFSPK (containing Ser 1037, underlined) from Kip1 and LSNINSNSVQSVISPK (containing Ser 972, underlined) from Cin8 were both greatly reduced in the presence of overexpressed Sic1Δ3P but noticeably increased in the presence of overexpressed Clb2 (Figure 7A), lending support to our hypothesis that Kip1 and Cin8 are both phosphorylated by Clb/Cdc28. As a control, we examined the relative phosphorylation of a non-CDK substrate, the c-Myc epitope tag of both Kip1-myc12 and Cin8-myc12 which was determined to be phosphorylated on the serine residue of each LISEED motif. We verified that the phosphorylation of the c-Myc tag was not CDK-dependent as the c-Myc epitope was the least phosphorylated in cells overexpressing Clb2 (Figure 7B).

Although we were unable to identify phosphopeptides containing Ser 388 from Kip1 or Ser 455 from Cin8, it is often difficult to capture the full extent of protein phosphorylation during mass spectrometry analysis due to both technical and biological limitations [62], [63]. In addition to the phosphopeptides which contain consensus CDK sites, other phosphopeptides were generated from Kip1 and Cin8 that did not include consensus CDK phosphorylation motifs (Table 1; kinases predicted to phosphorylate these non-CDK sites are listed in Table S1 and details of the prediction method are in Text S1). Thus, the results of our mass spectrometry analysis do not rule out the possibility that Kip1 and Cin8 are also phosphorylated at other sites by Clb/Cdc28 complexes.
Homology modeling of Kip1 and Cin8 motor domains
Ser 388 in Kip1 and Ser 455 in Cin8 are found in the N-terminal motor domain of the respective kinesins-5. Hence, in order to understand how phosphorylation at these residues on Kip1 and Cin8 might regulate their functions, we constructed homology models of their motor domains (Figure 8A). Homology modeling was performed using SWISS-MODEL and Swiss PDB Viewer [64]–[66] with X-ray crystal structures of the motor domains of human kinesin-5 HsEg5 [67] and the budding yeast kinesin-14 Kar3 [68] serving as templates. Consistent with the idea that phosphorylation of Ser 388 could regulate motor function, our model of the Kip1 motor domain showed that Ser 388 is solvent-accessible and located at the C-terminal end of strand β8. Here, Ser 388 appears to form part of the core which enables the motor to distinguish between ATP and ADP bound to the nucleotide-binding pocket [69]. The residue itself, however, is not predicted to form essential hydrogen bonds, and does not itself form part of the nucleotide binding pocket. Additional modeling (see Materials & Methods) showed that replacing Ser 388 with an alanine residue has no predicted effects on the backbone structure.

The Cin8 motor domain was modeled on the same template structures as for Kip1. Ser 455 in Cin8 is found in a similar environment to Ser 388 in Kip1, as expected. Closer scrutiny of its neighboring amino acids, however, revealed critical differences (Figure 8B). These differences include the residue at the second position of the P-loop, which is glutamine in Kip1 and Eg5 but methionine in Cin8, and also the two residues on helix α6 (C-terminal to strand β8) closest to Ser 455 (Val 459, Thr 460) and Ser 388 (Ile 392, Ser 393).
Discussion
Previous genetic studies indicate that kinesin-5 motors, Clb/Cdc28 complexes, and the SCF complex are all required for SPB separation and assembly of a mitotic spindle. We have shown that the only important target for SCFCdc4 in SPB separation is the Clb-specific CDK inhibitor, Sic1 (Figure 1). This finding indicates that SCF-mediated destruction of other regulatory proteins or SPB components is not required for SPB separation, and also supports the idea that Clb/Cdc28 complexes promote SPB separation and spindle assembly via regulation of kinesin-5 motors. We have, in fact, determined that kinesin-5 motors are phosphorylated directly by Clb/Cdc28 complexes (Figure 2 and Table 1) and that this phosphorylation plays a role in promoting SPB separation and spindle assembly (Figure 6).
Although certain metazoan kinesin-5 orthologues have been shown to be phosphorylated on their tail domains by cyclin B/Cdk1 [35], [37], [39], the threonine residue at which this phosphorylation occurs is absent from both Kip1 and Cin8. We have shown, however, that Kip1 and Cin8 are both phosphorylated by Clb2/Cdc28 in vitro, and that mutation of their consensus CDK sites significantly reduces the extent of phosphorylation (Figure 2). By genetic mapping, we have identified a solvent-accessible consensus CDK site (Ser 388) in the motor domain of Kip1 that is crucial to its role in SPB separation. Additionally, using a combination of mass spectrometry and genetic analysis, we have found that the phosphorylation of two consensus CDK sites in the tail domain of Kip1 is also important for timely SPB separation. Cells dependent on Kip1S388A or Kip1S1037A, T1040A as their only source of kinesin-5 are severely impaired in their ability to separate duplicated SPBs (Figure 6). Both Kip1 mutants have similar in vivo protein levels to wild-type Kip1 at 37°C and still localize to the SPBs in arrested cells (Figure S5). Hence, we propose that Clb/Cdc28-mediated phosphorylation of Ser 388, Ser 1037, and Thr 1040, regulates some aspect of Kip1 motor activity.
Cells with the homologous serine in Cin8 (Ser 455) substituted with alanine exhibit a severe proliferation defect in the absence of Kip1 which appears to reflect a delay in the transition from G1 into S phase (Figure 6 and S6). Although we do observe an SPB separation defect in cin8S455A kip1Δ mutants, the observed G1/S delay confounds our ability to determine if the separation defect is directly related to the loss of Cin8 motor function. The mechanism underlying this delay has yet to be determined, and the basis for the phenotypic differences between the homologous mutations in Kip1 and Cin8 remains unclear; however, homology modeling suggests that there are important structural differences between the two motors in the vicinity of this consensus phosphorylation site that might contribute to the different phenotypes observed.
Ser 388/Ser 455 forms part of the core of the motor domain, which includes the nucleotide-sensing elements switch I and switch II, and the γ-phosphate-sensing P-loop [69]. Due to the fact that Ser 388/Ser 455 lies near the junction of strand β8, helix α6, and the neck linker (Figure 8), this residue appears to be in a position to influence the transmission of structural changes in the motor core to the neck should it be phosphorylated. We have also noticed that although switch I and switch II are identical in Kip1 and Cin8, Cin8 has a methionine residue (Met 129) in the 2nd position of its P-loop whereas Kip1, like almost all other known kinesins-5, has a glutamine (Gln 142) at this position. The residues on α6 closest to Ser 455 in Cin8 (Val 459, Thr 460) also differ from those closest to Ser 388 in Kip1 (Ile 392, Ser 393). All of these differences are potentially important because these residues are close enough in space to interact; additionally, communication between the respective secondary structural elements to which they belong is essential to the generation of motility.
The Cin8 motor domain has a number of other notable structural differences compared with Kip1 and other kinesins-5. Loop L2 and, in particular, loop L8, are substantially longer in Cin8, while other loops such as L5 and L10 are shorter; the functional significance of these differences in length are still unclear. We do know, however, that L2 and L8 form part of the microtubule-binding surface of kinesins [70], [71] while the structure of L5 is thought to be important in determining ADP-release kinetics during the ATP hydrolysis cycle of Eg5 [72]. In addition to these differences in the structure of their motor domains, CIN8, but not KIP1, has been reported to be involved in a myriad of genetic and physical interactions [73]–[78], suggesting that Cin8 has several cellular functions. The dissection of these multiple functions in future studies may reveal a mechanistic role for direct phosphorylation in spindle assembly.
Additionally, although we were able to determine that Cin8 is phosphorylated at a CDK site in its tail domain, we have yet to observe a phenotype for cin8S972A kip1Δ cells. Previous studies have also been unable to determine a phenotypic consequence for the same mutation [47], [57]. The distinct structure of the tails of Kip1 and Cin8 and their different responses to Clb/Cdc28 phosphorylation may also help explain the existence of two kinesins-5 in S. cerevisiae when many other eukaryotes appear to only have one.
It has been proposed that Clb/Cdc28 kinase activity regulates SPB separation indirectly by inhibiting the activity of the ubiquitin ligase, APCCdh1, thus preventing the ubiquitination of Kip1 and Cin8 and their subsequent degradation [41], [42]. The proposal for Clb/Cdc28 regulation of Kip1 and Cin8 stability was based, however, on observations made with strains bearing the cdc28Y19E allele, and with strains carrying the analog-sensitive cdc28-as1 allele treated with 500 nM 1-NM-PP1. In both cases, a sufficient level of Clb/Cdc28 activity remains to drive DNA replication [12], [41], [48]. By using more stringent means of inhibiting Clb/Cdc28 complexes, we have not observed evidence suggesting that the loss of Clb/Cdc28 activity leads to a decreased abundance of Kip1 and Cin8. Inhibiting Clb/Cdc28 activity by overexpressing a stabilized allele of Sic1 [49] in a W303 strain background similar to that used by Crasta et al. [41], [42] did not affect the abundance of either Kip1 or Cin8. Our observations suggest that the alterations in motor stability observed by Crasta et al. may reflect partially deregulated CDK activity rather than a loss of Clb/Cdc28 activity.
A new study by Robbins & Cross [79] found that most cells whose endogenous CDH1 gene has been replaced with a non-Cdk1-phosphorylatable allele, CDH1-m11 arrest with monopolar spindles, and that Cin8 levels are reduced about fourfold compared with CDH1 cells. However, expressing non-degradable Cin8 at endogenous levels in CDH1-m11 cells failed to drive SPB separation and bipolar spindle assembly. Instead, by expressing non-degradable Clb2, which is also an APCCdh1 target, the authors observed the restoration of SPB separation in CDH1-m11 cells. This observation led Robbins & Cross to conclude that the mitotic cyclins alone are the APCCdh1 targets important for SPB separation.
Regardless of whether Clb/Cdc28 is capable of controlling some aspect of Kip1 and Cin8 stability by regulating APCCdh1 activity, our findings clearly indicate that wild-type Kip1 and Cin8 levels are not sufficient for SPB separation in the absence of Clb/Cdc28 kinase activity (Figure 3 and S2) and are in agreement with those of Robbins & Cross. Thus, we have identified an additional layer of control whereby the direct phosphorylation of kinesin-5 motors is essential for the efficient separation of SPBs and assembly of a short spindle.
It is not fully understood how direct phosphorylation of kinesin-5 motors affects their function in spindle assembly. Although the localization of kinesin-5 motors to the spindle is regulated by cyclin B/Cdk1 phosphorylation in certain metazoan systems [35], [37], [39], [40], we have determined that the localization of Kip1 and Cin8 to the spindle is not dependent on Clb/Cdc28 in budding yeast. Instead, our observations suggest that Clb/Cdc28 phosphorylation regulates some aspect of kinesin-5 motor activity.
Ser 388, the critical consensus CDK site (S/T-P-X-X) we identified in Kip1 is conserved in most known kinesins-5, except for Schizosaccharomyces pombe Cut7 in which the orthologous serine is followed by a serine. This site is also the only one conserved between metazoan and fungal kinesins-5. Moreover, sequence comparison and homology modeling both indicate that the structures of the HsEg5 and XlEg5 motor domains bear a greater resemblance to that of Kip1 than that of Cin8. Thus, it is possible that CDK-phosphorylation at the serine orthologous to Ser 388 in Kip1 may be a common regulatory mechanism for spindle assembly in other eukaryotic organisms.
We have also determined that the phosphorylation of Kip1 at two other CDK sites in its C-terminal globular tail domain is also important to its function in promoting SPB separation. It has been reported that the phosphorylation of the BimC box in Xenopus Eg5 by cyclin B/Cdk1 enhances binding to microtubules both in vitro and in Xenopus egg extract [80]. The tail domains of the kinesins-5 are, however, quite divergent, as they are in other kinesin subfamilies [26]. Hence, it remains to be determined if the phosphorylation of the Kip1 tail domain will have the same effect.
Kinesin-5 motors exhibit a variety of functions that could be regulated, including microtubule binding, microtubule crosslinking, ATP binding and hydrolysis, microtubule-based motility or influencing microtubule dynamics [80]–[84]. Furthermore, there have been several reported examples of different aspects of kinesin motor function being regulated through phosphorylation of either their motor or tail domains, including cases where cyclin B/Cdk1 is the kinase involved [80], [85]–[88]. Detailed biochemical studies will be required to dissect the specific kinesin-5 functions controlled by Clb/Cdc28-mediated phosphorylation. Such studies may also reveal a mechanism linking phosphorylation of the kinesin-5 motor domain to that of its tail domain.
Materials and Methods
Plasmids and DNA manipulation
Standard methods of DNA manipulation were employed in plasmid construction and PCR. Whenever PCR was involved in gene manipulation, the product was sequenced in full to determine the occurrence of PCR errors. More information on plasmid construction can be found in Text S1.
Yeast strains and media
All strains are derivatives of BF264-15DU unless otherwise indicated (Table S2). Strains were constructed by standard yeast methods (detailed in Text S1). Yeast cultures were grown in standard YEP medium (1% yeast extract, 2% peptone, 0.012% adenine, 0.006% uracil supplemented with 2% sugar) unless indicated. For synchrony experiments, bar1 strains were arrested with 25 ng/ml α mating pheromone, also known as α-factor (BioVectra). For experiments involving fluorescence microscopy that used liquid cultures, YEP medium was supplemented with an additional 0.003% adenine (0.015% final).
Asynchronous temperature shift time course experiments
For experiments involving cdc4-3 strains, cells were grown in YEP-dextrose (YEPD) medium overnight at 24°C (permissive temperature). Cells were subsequently diluted to a density of 1×107 cells/ml, and then incubated at 24°C for 90 min. Cultures were subsequently shifted to 37°C (restrictive temperature) to inactivate the temperature-sensitive Cdc4-3. Details of the subsequent return to permissive temperature experiment can be found in Text S1.
Synchronized sugar shift time course experiments
For experiments involving Δclb1,2,3,4,5,6 strains, cells were grown in YEP-galactose (YEPG) medium overnight, and then diluted before being allowed to reach a density of 1×107 cells/ml. They were subsequently arrested with α-factor before adding either 20% dextrose to a final concentration of 2% dextrose in order to inhibit the expression of Clb1 in these strains or an equal volume of water to the control. Fifteen minutes after the addition of dextrose/water, cells were released into pre-warmed (30°C) YEPG.
For experiments involving strains with PGAL1-SIC1Δ3P derived from BF264-15DU, cells were grown in non-inducing YEP-sucrose (YEPS) medium overnight and then diluted before being allowed to reach a density of 1×107 cells/ml. They were subsequently arrested with α-factor, then released into pre-warmed (30°C) YEPG. Unlike BF264-15DU strains [89], W303a-derived strains express invertase and can thus hydrolyze sucrose to yield glucose and fructose. Therefore, we determined that YEPS is not a suitable non-inducing medium for W303a-derived strains since they can break sucrose down to form glucose which represses the GAL1-10 promoter. Instead, these strains had to first be inoculated into filter-sterilized YEPS and grown for a few hours before being shifted to YEP-raffinose (YEPR) for subsequent overnight growth. The cells were arrested with α-factor in YEPR before induction with YEPG.
Synchronized temperature shift time courses
Cells were grown in YEPD medium overnight at 25°C (permissive temperature) and then diluted and allowed to reach a density of 7.5×106 cells/ml. They were subsequently arrested with α-factor. Arrested cultures were divided in two halves and one half was moved to 37°C for 15 min (restrictive temperature) while the other half remained at 25°C. Cells were then released into YEPD pre-warmed to the respective temperatures.
Immunoblotting
Cell samples collected during time courses were spun down, washed with ice cold PBS, then frozen in liquid nitrogen. Lysates were prepared by vortexing cells with acid-washed glass beads (Sigma-Aldrich) in modified RIPA buffer (50 mM Tris-HCl pH 7.5, 20 mM Na4P2O7, 250 mM NaCl, 50 mM NaF, 1% NP-40, 2 mM EDTA, 1 mM Na3VO4, 1 mM DTT, 1.25 mM benzamidine hydrochloride, 0.1 mg/ml PMSF, 1 µg/ml each leupeptin, aprotinin, and pepstatin A). Lysates were cleared by centrifugation at 4°C, and the protein content of the cleared lysates was determined by measuring A280 with a Biophotometer (Eppendorf).
Proteins were separated by SDS-PAGE on 8.5% Tris-HCl gels using the Laemmli method and then transferred to Immobilon-P (Millipore) PVDF membranes for antibody probing. Proteins tagged with c-Myc were detected with mouse anti-c-Myc clone 9E10 (Santa Cruz Biotechnology), while mCherry fusions were detected with rabbit anti-DsRed/RFP (MBL). Cdc28 and Pho85 were detected with mouse anti-PSTAIR (Abcam) for use as loading controls. The secondary antibodies used were horseradish peroxidase-conjugated goat anti-mouse (Pierce Thermo-Scientific) and goat anti-rabbit (Abcam). Blots were visualized with Supersignal West Pico chemiluminescent substrate (Pierce Thermo-Scientific).
Kinase purification for in vitro phosphorylation assay
Soluble Clb2/Cdc28 kinase was prepared, using an abbreviated form of the TAP protocol described by Puig and colleagues which excludes the CaM-binding and elution steps [90]. Clb2-TAP was overexpressed from an episomal plasmid, pGAL-CLB2-TAP [54], together with Cdc28 in a swe1Δ yeast strain (SBY684, a gift from Daniel Lew) to ensure that the purified kinase is not inhibited by Swe1 phosphorylation of Tyr 19 [10]. To provide a negative control, a wild-type strain (SBY1286) carrying a URA3-marked episomal plasmid with the GAL1-10 promoter (YEpUGAL) was used. Both strains were first grown to log phase in synthetic complete dropout medium lacking uracil (SC-Ura) with 2% sucrose, then switched to SC-Ura with 2% galactose. Soluble Clb5/Cdc28 kinase was prepared in a similar manner (see Text S1).
After induction, cells were lysed in modified RIPA buffer by vortexing with glass beads. Clb2/Cdc28 was isolated by binding to IgG-Sepharose beads (Amersham BioSciences/GE Healthcare) followed by overnight cleavage at 4°C with AcTEV protease (Invitrogen) to remove the Protein A portion of the TAP tag. The concentration of Cdc28 was estimated by quantitative western blotting with anti-PSTAIR antibody, using purified GST-Cdk1 (Cell Signaling Technology) as a standard. Densitometric analysis was performed with ImageJ (Wayne Rasband, National Institutes of Health).
Substrate purification for in vitro phosphorylation assay
Kip1-myc12, Cin8-myc12, and their respective CDK site mutants were overexpressed from episomal plasmids under the control of the GAL1 promoter in PGAL1-SIC1Δ3P cells. To avoid the formation of tetramers containing the respective endogenous kinesin-5, Kip1-myc12 and Kip16A-myc12 were overexpressed in kip1Δ strains (SBY1274, 1276), while Cin8-myc12 and Cin85A-myc12 were overexpressed in cin8Δ strains (SBY1280, 1282). Control strains were included that carry the empty vector alone (SBY1278, 1284). Strains were grown to log phase in synthetic complete dropout medium lacking leucine (SC-Leu) with 2% sucrose, arrested with α-factor, and then released into YEPG.
Cells were lysed in modified RIPA buffer. Immunoprecipitation was carried out by diluting lysate in IP buffer (50 mM Tris-HCl pH 7.5, 300 mM NaCl, 0.1% NP-40, 1 mM EDTA, 0.1 mM DTT, 2.5 mM benzamidine hydrochloride, 0.2 mg/ml PMSF, 2 µg/ml each leupeptin, aprotinin, and pepstatin A) before adding anti-c-Myc IgG agarose beads (Sigma-Aldrich). Binding to the beads was carried out at 4°C for 2 h. Beads were then washed twice with modified RIPA buffer with 500 mM NaCl (instead of 250 mM NaCl), and 0.1 mM DTT added, and twice more with modified RIPA buffer with 0.1% SDS, 0.25% DOC and 0.1 mM DTT added. Finally, beads were washed with 50 mM Tris-HCl, pH 7.5 and divided into smaller portions for the phosphorylation reaction described below. Substrate yield was estimated to be about 0.4 µg of Kip1-myc12/Cin8-myc12 by performing identical immunoprecipitations for densitometric comparison against BSA (Sigma-Aldrich) standards after SDS-PAGE and staining with Coomassie Brilliant Blue R250.
In vitro phosphorylation assay
Kinesins (∼0.4 µg) immobilized on IgG-agarose beads were washed with kinase reaction buffer (50 mM Tris-HCl pH 7.5, 10 mM MgCl2, 25 mM β-glycerophosphate, 1 mM DTT). Subsequently, the beads were mixed with ∼12 ng Clb2/Cdc28 in kinase reaction buffer containing 0.2 mM ATP and 0.5 µCi/µl 32P-γ-ATP. One microgram of histone H1 (Roche) was used as a control substrate in a separate reaction. Reactions were incubated at 30°C for 1 h and then halted by adding SDS-PAGE sample buffer before boiling for 5 min. Proteins were separated by electrophoresis on 10% Express PAGE gels (GenScript) and visualized by staining with Coomassie Brilliant Blue R250 (Bio-Rad). Gels were dried and radiolabelling was subsequently detected by autoradiography using a Phosphor Imager and Storm scanner (Molecular Dynamics, GE Healthcare).
Spot assays for proliferation
Strains were grown overnight at room temperature in YEPD and then diluted to allow resumption of log phase growth at room temperature for at least two hours. A 2×106 cells/ml suspension of each strain was then prepared in YEPD and diluted serially to make 2×105 and 2×104 cells/ml suspensions. Three microliters of each suspension was spotted on YEPD plates and the plates were subsequently incubated at either ambient room temperature (∼22°C) or 37°C, as indicated. Plates were imaged using a Bio-Rad GelDoc and the software QuantityOne (Bio-Rad).
Flow cytometry
Cells were prepared for flow cytometric analysis of DNA content using SYTOX Green (Invitrogen) as previously described [91]. Graphs used in figures were generated using WinMDI 2.9 (Scripps Research Institute).
Fluorescence microscopy
For fluorescence microscopy, cells growing in liquid medium were sonicated, then spun down and fixed in 2.0–2.5% paraformaldehyde at room temperature for 5 min, then washed twice with PBS. Cells growing on fresh plates no more than two days old were simply resuspended in water on glass slides before imaging. All fluorescence microscopy was performed on a Zeiss Axio Imager widefield fluorescence microscope controlled with MetaMorph 7.5 (Molecular Devices, MDS Analytical Technologies). Images were captured with a Hamamatsu Orca ER monochrome cooled-CCD camera and analyzed with both MetaMorph 7.5 and Adobe Photoshop 7.0 (Adobe Labs).
Mass spectrometry analysis of protein phosphorylation
Kip1-myc12 and Cin8-myc12 were overexpressed in yeast in a similar manner to that described above. Additionally, the two kinesins-5 were overexpressed in strains that also overexpress either Sic1Δ3P to inhibit Clb/Cdc28 kinase activity or Clb2-HA3 to increase the in vivo phosphorylation of Clb/Cdc28 substrates. Cells were lysed and the myc-tagged proteins were immunopurified as described above, except using different buffers that contained a high concentration of phosphatase inhibitors to better preserve phosphorylated amino acid residues. The anti-phosphatase lysis buffer used had the following composition: 50 mM Tris-HCl pH 7.5, 500 mM NaCl, 20 mM Na4P2O7, 150 mM NaF, 150 mM β-glycerophosphate, 2mM Na3VO4, 1 mM EDTA, 1 mM EGTA, 1% NP-40, 1 mM DTT, 1.25 mM benzamidine hydrochloride, 1 mM PMSF, 1 µg/ml each leupeptin, aprotinin, and pepstatin A. The anti-phosphatase IP buffer used had the following composition: 50 mM Tris-HCl pH 7.5, 300 mM NaCl, 200 mM NaF, 200 mM β-glycerophosphate, 2mM Na3VO4, 1 mM EDTA, 0.1% NP-40, 0.1 mM DTT, 2 mM PMSF, 2 µg/ml each leupeptin, aprotinin, and pepstatin A. Following immunoprecipitation, the anti-c-Myc IgG agarose beads were boiled in SDS-PAGE sample buffer and the liberated proteins separated by SDS-PAGE. Gels were fixed and then stained with Coomassie Brilliant Blue R250. Bands corresponding to Kip1-myc12 and Cin8-myc12 were excised and sent to the Taplin Mass Spectrometry Facility at Harvard University for analysis.
Excised gel bands were cut into approximately 1 mm3 pieces. The samples were reduced with 1 mM DTT for 30 minutes at 60°C and then alkylated with 5mM iodoacetamide for 15 minutes in the dark at room temperature. Gel pieces were then subjected to a modified in-gel trypsin digestion procedure [92]. Gel pieces were washed and dehydrated with acetonitrile for 10 min followed by the removal of acetonitrile. Pieces were then completely dried in a speed-vac before rehydration with 50 mM ammonium bicarbonate solution containing 12.5 ng/µl modified sequencing-grade trypsin (Promega, Madison, WI) at 4°C. Samples were then placed in a 37°C room overnight. Peptides were later extracted by removing the ammonium bicarbonate solution, followed by one wash with a solution containing 50% acetonitrile and 5% acetic acid. The extracts were then dried in a speed-vac (∼1 h). The samples were then stored at 4°C until analysis.
On the day of analysis, the samples were reconstituted in 5 µl of HPLC solvent A (2.5% acetonitrile, 0.1% formic acid). A nano-scale reverse-phase HPLC capillary column was created by packing 5 µm C18 spherical silica beads into a fused silica capillary (100 µm inner diameter×12 cm length) with a flame-drawn tip [93]. After equilibrating the column, each sample was pressure-loaded off-line onto the column. The column was then reattached to the HPLC system. A gradient was formed and peptides were eluted with increasing concentrations of solvent B (97.5% acetonitrile, 0.1% formic acid).
As each peptide was eluted, it was subjected to electrospray ionization, and the resulting ions entered a LTQ-Orbitrap mass spectrometer (ThermoFinnigan, San Jose, CA). Eluting peptides were detected, isolated, and fragmented to produce a tandem mass spectrum of specific fragment ions for each peptide. Peptide sequences were determined by matching protein or translated nucleotide databases with the acquired fragmentation pattern by the software program, Sequest (ThermoFinnigan, San Jose, CA) [94]. The modification of 79.9663 mass units to serine, threonine, and tyrosine was included in the database searches to determine phosphopeptides. Each phosphopeptide that was determined by the Sequest program was also manually inspected to ensure confidence.
Homology modeling
Homology models were constructed using DeepView Swiss PDB Viewer (v4.0) and the Swiss MODEL web server [64]–[66]. Initial amino acid sequence alignments for the Kip1 and Cin8 motor domains were performed using CLUSTALW through the software BioEdit (Tom Hall, Ibis BioSciences) against the primary sequences of the HsEg5/Kif11 and Kar3 motor domains. Initial model construction was then done in DeepView against a solved crystal structure for the HsEg5 motor domain (ExPDB 1ii6B, the base template). Additional template structures (ExPDB 1ii6A for HsEg5 and 3kar_ for Kar3) were subsequently aligned against the base template and the project submitted to Swiss MODEL. The initial models were examined for structural errors, particularly in loop placement. The errors identified were used to adjust the sequence alignments and repeat the modeling process until stable structures with a negative total energy mostly free of backbone problems were obtained. Model quality was evaluated with What Check [95] and DeepView was used to fix side chain errors identified. Residue swaps were performed using the “Mutate” function and the rotamer library included in Swiss PDB Viewer [96]. Images were rendered with Swiss PDB Viewer and Persistence of Vision Raytracer v3.6 (Persistence of Vision Pty. Ltd.).
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. MorganDO
1997 Cyclin-dependent kinases: Engines, clocks, and microprocessors. Annual Review of Cell and Developmental Biology 13 261 291
2. MurrayAW
2004 Recycling the cell cycle: Cyclins revisited. Cell 116 221 234
3. LaceyKR
JacksonPK
StearnsT
1999 Cyclin-dependent kinase control of centrosome duplication. Proceedings of the National Academy of Science USA 96 2817 2822
4. HaaseSB
WineyM
ReedSI
2001 Multi-step control of spindle pole body duplication by cyclin-dependent kinase. Nature Cell Biology 3 38 42
5. AdamsIR
KilmartinJV
2000 Spindle pole body duplication: a model for centrosome duplication? Trends in Cell Biology 10 329 335
6. Simmons KovacsLA
NelsonCL
HaaseSB
2008 Intrinsic and Cyclin-dependent Kinase-dependent Control of Spindle Pole Body Duplication in Budding Yeast. Molecular Biology of the Cell 19 3243 3253
7. WalczakC
2000 Molecular mechanisms of spindle function. Genome Biology 1 reviews101.101 101.104
8. JaspersenSL
WineyM
2004 The budding yeast spindle pole body: Structure, duplication and function. Annual Review of Cell and Developmental Biology 20 1 28
9. O'TooleMWE
2001 The spindle cycle in budding yeast. Nature Cell Biology 3 E23 E27
10. AmonA
SuranaU
MuroffI
NasmythK
1992 Regulation of p34CDC28 tyrosine phosphorylation is not required for entry into mitosis in S. cerevisiae. Nature 355 368 371
11. KeatonMA
BardesESG
MarquitzAR
FreelCD
ZylaTR
2007 Differential susceptibility of S and M phase cyclin/CDK complexes to inhibitory tyrosine phosphorylation in yeast. Current Biology 17 1181 1189
12. LimHH
GohP-Y
SuranaU
1996 Spindle pole body separation in Saccharomyces cerevisiae requires dephosphorylation of the tyrosine 19 residue of Cdc28. Molecular and Cellular Biology 16 6385 6397
13. FitchI
DahmannC
SuranaU
AmonA
NasmythK
1992 Characterization of four B-type cyclin genes of the budding yeast Saccharomyces cerevisiae. Molecular Biology of the Cell 3 805 818
14. RichardsonH
LewDJ
HenzeM
SugimotoK
ReedSI
1992 Cyclin-B homologs in Saccharomyces cerevisiae function in S phase and in G2. Genes & Development 6 2021 2034
15. BaiC
SenP
HofmannK
MaL
GoeblM
1996 SKP1 connects cell cycle regulators to the ubiquitin proteolysis machinery through a novel motif, the F-box. Cell 86 263 274
16. MathiasN
JohnsonSL
WineyM
AdamsAE
GoetschL
1996 Cdc53p acts in concert with Cdc4p and Cdc34p to control G1-to-S-Phase transition and identifies a conserved family of proteins. Molecular and Cellular Biology 16 6634 6643
17. ByersB
GoetschL
1975 Behavior of spindles and spindle plaques in the cell cycle and conjugation of Saccharomyces cerevisiae. Journal of Bacteriology 124 511 523
18. GoeblMG
YochemJ
JentschS
McGrathJP
VarshavskyA
1988 The yeast cell cycle gene CDC34 encodes a ubiquitin-conjugating enzyme. Science 241 1331 1335
19. ByersB
GoetschL
1974 Duplication of spindle plaques and integration of the yeast cell cycle. Cold Spring Harbor Symposium on Quantitative Biology 38 123 131
20. SchwobE
BohmT
MendenhallMD
NasmythK
1994 The B-type cyclin kinase inhibitor p40SIC1 controls the G1 to S transition in S. cerevisiae. Cell 79 233 244
21. VermaR
FeldmanRR
DeshaiesRJ
1997 SIC1 is ubiquitinated in vitro by a pathway that requires CDC4, CDC34, and cyclin/CDK activities. Molecular Biology of the Cell 8 1427 1437
22. FeldmanRR
CorrellCC
KaplanKB
DeshaiesRJ
1997 A complex of Cdc4p, Skp1p, and Cdc53p/Cullin catalyzes ubiquitination of the phosphorylated CDK inhibitor Sic1p. Cell 91 221 230
23. VodermaierHC
2004 APC/C and SCF: Controlling Each Other and the Cell Cycle. Current Biology 14 R787 R796
24. RoofDM
MeluhPB
RoseMD
1992 Kinesin-related proteins required for assembly of the mitotic spindle. The Journal of Cell Biology 118 95 108
25. HoytMA
HeL
LooKK
SaundersWS
1992 Two Saccharomyces cerevisiae kinesin-related gene products required for mitotic spindle assembly. Journal of Cell Biology 118 109 120
26. MikiH
OkadaY
HirokawaN
2005 Analysis of the kinesin superfamily: insights into structure and function. Trends in Cell Biology 15 467 476
27. EnosAP
MorrisNR
1990 Mutation of a gene that encodes a kinesin-like protein that blocks nuclear division in A. nidulans. Cell 60 1019 1027
28. HeckMS
PereiraA
PesaventoP
YannoniY
SpradlingAC
1993 The kinesin-like protein KLP61F is essential for mitosis in Drosophila. The Journal of Cell Biology 123 665 679
29. HaganIM
YanagidaM
1990 Novel potential mitotic motor protein encoded by the fission yeast cut7+ gene. Nature 347 563 566
30. SawinKE
LeGuellecK
PhilippeM
MitchisonTJ
1992 Mitotic spindle organization by a plus-end-directed microtubule motor. Nature 359 540 543
31. JacobsCW
AdamsAE
SzaniszloPJ
PringleJR
1988 Functions of microtubules in the Saccharomyces cerevisiae cell cycle. Journal of Cell Biology 107 1409 1426
32. ReijoRA
CooperEM
BeagleGJ
HuffakerTC
1994 Systematic mutational analysis of the yeast ß-tubulin gene. Molecular Biology of the Cell 5 29 43
33. AmosL
2008 Spindle assembly: Kinesin-5 is in control. Current Biology 18 R1146 R1149
34. SchwobE
NasmythK
1993 CLB5 and CLB6, a new pair of B cyclins involved in DNA replication in Saccharomyces cerevisiae. Genes & Development 7 1160 1175
35. BlangyA
LaneHA
d'HérinP
HarperM
KressM
1995 Phosphorylation by p34cdc2 regulates spindle association of human Eg5, a kinesin-related motor essential for bipolar spindle formation in vivo. Cell 83 1159 1169
36. BlangyA
ArnaudL
NiggEA
1997 Phosphorylation by p34cdc2 protein kinase regulates binding of the kinesin-related motor HsEg5 to the dynactin subunit p150Glued. Journal of Biological Chemistry 272 19418 19424
37. SawinKE
MitchisonTJ
1995 Mutations in the kinesin-like protein Eg5 disrupt localization to the mitotic spindle. Proceedings of the National Academy of Science USA 92 4289 4293
38. GietR
UzbekovR
CubizollesF
Le GuellecK
PrigentC
1999 The Xenopus laevis Aurora-related protein kinase pEg2 associates with and phosphorylates the kinesin-related protein XlEg5. Journal of Biological Chemistry 274 15005 15013
39. SharpDJ
McDonaldKL
BrownHM
MatthiesHJ
WalczakC
1999 The bipolar kinesin KLP61F, cross-links microtubules within interpolar microtubule bundles of Drosophila embryonic mitotic spindles. Journal of Cell Biology 144 125 138
40. GoshimaG
ValeRD
2005 Cell cycle-dependent dynamics and regulation of mitotic kinesins in Drosophila S2 cells. Molecular Biology of the Cell 16 3896 3907
41. CrastaK
HuangP
MorganG
WineyM
SuranaU
2006 Cdk1 regulates centrosome separation by restraining proteolysis of microtubule-associated proteins. EMBO Journal 1 13
42. CrastaK
LimHH
GiddingsTHJr
WineyM
SuranaU
2008 Inactivation of Cdh1 by synergistic action of Cdk1 and polo kinase is necessary for proper assembly of the mitotic spindle. Nature Cell Biology 10 665 675
43. VisintinR
PrinzS
AmonA
1997 CDC20 and CDH1: A family of substrate-specific activators of APC-dependent proteolysis. Science 278 460 463
44. HuangJN
ParkI
EllingsonE
LittlepageLE
PellmanD
2001 Activity of the APCCdh1 form of the anaphase-promoting complex persists until S phase and prevents the premature expression of Cdc20p. Journal of Cell Biology 154 85 94
45. van LeukenR
CligstersL
WothuisR
2008 To cell cycle, swing the APC/C. Biochimica et Biophysica Acta 1786 49 59
46. GordonDM
RoofDM
2001 Degradation of the kinesin Kip1p at anaphase onset if mediated by the anaphase-promoting complex and Cdc20p. Proceedings of the National Academy of Science USA 98 12515 12520
47. HildebrandtER
HoytMA
2001 Cell cycle-dependent degradation of the Saccharomyces cerevisiae spindle motor Cin8p requires APCCdh1 and a bipartite destruction sequence. Molecular Biology of the Cell 12 3402 3416
48. BishopAC
UbersaxJA
PetschDT
MatheosDP
GrayNS
2000 A chemical switch for inhibitor-sensitive alleles of any protein kinase. Nature 407 395 401
49. VermaR
AnnanRS
HuddlestonMJ
CarrSA
ReynardG
1997 Phosphorylation of Sic1p by G1 Cdk is required for its degradation and entry into S phase. Science 278 455 460
50. HartwellLH
MortimerRK
CulottiJ
CulottiM
1973 Genetic control of the cell division cycle in yeast: V. Genetic analysis of cdc mutants. Genetics 74 267 286
51. HerefordLM
HartwellLH
1974 Sequential gene function in the initiation of Saccharomyces cerevisiae DNA synthesis. Journal of Molecular Biology 84 445 461
52. KnappD
BhoiteL
StillmanDJ
NasmythK
1996 The transcription factor Swi5 regulates expression of the cyclin kinase inhibitor p40SIC1. Molecular and Cellular Biology 16 5701 5707
53. GohP-Y
SuranaU
1999 Cdc4, a protein required for the onset of S phase, serves an essential function during G2/M transition in Saccharomyces cerevisiae. Molecular and Cellular Biology 5512 5522
54. UbersaxJA
WoodburyEA
QuangPN
ParazM
BlethrowJD
2003 Targets of the cyclin-dependent kinase Cdk1. Nature 425 859 864
55. CrastaK
SuranaU
2006 Disjunction of conjoined twins: Cdk1, Cdh1 and separation of centrosomes. Cell Division 1 12 20
56. OrlandoDA
LinCY
BernardA
WangJY
ESJ
2008 Global control of cell-cycle transcription by coupled CDK and network oscillators. Nature 453 944 947
57. HildebrandtER
GheberL
HoytMA
2006 Homotetrameric form of Cin8p, a Saccharomyces cerevisiae kinesin-5 motor, is essential for its in vivo function. Journal of Biological Chemistry 281 26004 26013
58. ShanerNC
CampbellRE
SteinbachPA
NG.GB
PalmerAE
2004 Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nature Biotechnology 22 1567 1572
59. HarveySL
CharletA
HaasW
GygiSP
KelloggDR
2005 Cdk1-dependent regulation of the mitotic inhibitor Wee1. Cell 122 407 420
60. NashP
TangX
OrlickyS
ChenQ
GertierFB
2001 Multisite phosphorylation of a CDK inhibitor sets a threshold for the onset of DNA replication. Nature 414 514 521
61. EndowSA
2000 Molecular motors - a paradigm for mutant analysis. Journal of Cell Science 113 1311 1318
62. BlackburnK
GosheMB
2009 Challenges and strategies for targeted phosphorylation site identification and quantification using mass spectrometry analysis. Briefings in Functional Genomics and Proteomics 8 90 103
63. GarciaBA
ShabanowitzJ
HuntDF
2005 Analysis of protein phosphorylation by mass spectrometry. Methods 35 256 264
64. GuexN
PeitschMC
1997 SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis 18 2714 2723
65. SchwedeT
KoppJ
GuexN
PeitschMC
2003 SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Research 31 3381 3385
66. ArnoldK
BordoliL
KoppJ
SchwedeT
2006 The SWISS-MODEL Workspace: A web-based environment for protein structure homology modelling. Bioinformatics 22 195 201
67. TurnerJ
AndersonR
GuoJ
BeraudC
FletterickR
2001 Crystal structure of the mitotic spindle kinesin Eg5 reveals a novel conformation of the neck linker. Journal of Biological Chemistry 276 25496 25502
68. GulickAM
SongH
EndowSA
RaymentI
1998 X-ray Crystal Structure of the Yeast Kar3 Motor Domain Complexed with Mg·ADP to 2.3 Å Resolution. Biochemistry 37 1769 1776
69. ValeRD
FletterickRJ
1997 The Design Plan of Kinesin Motors. Annual Review of Cell and Developmental Biology 13 745 777
70. WoehlkeG
RubyAK
HartCL
LyB
Hom-BooherN
1997 Microtubule Interaction Site of the Kinesin Motor. Cell 90 207 216
71. AlonsoM
van DammeJ
VandekerchkhoveJ
CrossRA
1998 Proteolytic mapping of kinesin/ncd-microtubule interface: nucleotide-dependent conformational changes in the loops L8 and L12. Embo J 17 949 951
72. CochranJC
GilbertSP
2005 ATPase Mechanism of Eg5 in the Absence of Microtubules: Insight into Microtubule Activation and Allosteric Inhibition by Monastrol. Biochemistry 44 16633 16648
73. KorolyevE
Steinberg-NeifachO
EshelD
2005 Mutations in the yeast kinesin-like Cin8p are alleviated by osmotic support. FEMS Microbiology Letters 244 379 383
74. GeiserJR
SchottEJ
KingsburyTJ
ColeNB
TotisLJ
1997 Saccharomyces cerevisiae genes required in the absence of the CIN8-encoded spindle motor act in functionally diverse mitotic pathways. Molecular Biology of the Cell 8 1035 1050
75. BasmajiF
Martin-YkenH
DurandF
DagkessamanskaiaA
PichereauxC
2006 The ‘interactome’ of the Knr4/Smi1, a protein implicated in coordinating cell wall synthesis with bud emergence in Saccharomyces cerevisiae. Molecular Genetics and Genomics 275 217 230
76. CollinsSR
MillerKM
MaasNL
RoguevA
FillinghamJ
2007 Functional dissection of protein complexes involved in yeast chromosome biology using a genetic interaction map. Nature 446 806 810
77. TongAH
LesageG
BaderGD
DingH
XuH
2004 Global mapping of the yeast genetic interaction network. Science 303 808 813
78. PanX
YuanDS
XiangD
WangX
Sookhai-MahadeoS
2004 A robust toolkit for functional profiling of the yeast genome. Mol Cell 16 487 496
79. RobbinsJA
CrossFR
2010 Requirements and reasons for effective inhibition of the Anaphase Promoting Complex activator Cdh1. Molecular Biology of the Cell In Press: published online ahead of print
80. CahuJ
OlichonA
HentrichC
SchekH
DrinjakovicJ
2008 Phosphorylation by Cdk1 increases the binding of Eg5 to microtubules in vitro and in Xenopus egg extract spindles. PLoS ONE 3 e3936 doi:10.1371/journal.pone.0003936
81. van den WildenbergSM
TaoL
KapiteinLC
SchmidtCF
ScholeyJM
2008 The homotetrameric kinesin-5 KLP61F preferentially crosslinks microtubules in antiparallel orientations. Current Biology 18 1860 1864
82. GheberL
KuoSC
HoytMA
1999 Motile properties of the kinesin-related Cin8p spindle motor extracted from Saccharomyces cerevisiae cells. The Journal of Biological Chemistry 274 9564 9572
83. KapiteinLC
PetermanEJG
KwokBH
KimJH
KapoorTM
2005 The bipolar mitotic kinesin Eg5 moves on both microtubules that it crosslinks. Nature 435 114 118
84. GardnerMK
BouckDC
PaliulisLV
MeehlJB
O'TooleET
2008 Chromosome congression by Kinesin-5 motor-mediated disassembly of longer kinetochore microtubules. Cell 135 894 906
85. MennellaV
TanD-Y
BusterDW
AsenjoAB
RathU
2009 Motor domain phosphorylation and regulation of the Drosophila kinesin 13, KLP10A. Journal of Cell Biology 186 481 490
86. MishimaM
PavicicV
GrünebergU
NiggEA
GlotzerM
2004 Cell cycle regulation of central spindle assembly. Nature 430 908 913
87. EspeutJ
GaussenA
BielingP
MorinV
PrietoS
2008 Phosphorylation Relieves Autoinhibition of the Kinetochore Motor Cenp-E. Molecular Cell 29 637 643
88. JangC-Y
CoppingerJA
SekiA
YatesJRI
FangG
2009 Plk1 and Aurora A regulate the depolymerase activity and the cellular localization of Kif2a. Journal of Cell Science 122 1334 1341
89. MondésertG
ClarkeDJ
ReedSI
1997 Identification of genes controlling growth polarity in the budding yeast Saccharomyces cerevisiae: a possible role of N-glycosylation and involvement of the exocyst complex. Genetics 147 421 434
90. PuigO
CasparyF
RigautG
RutzB
BouveretE
2001 The tandem affinity purification (TAP) method: a general procedure of protein complex purification. Methods 24 218 229
91. HaaseSB
ReedSI
2002 Improved flow cytometric analysis of the budding yeast cell cycle. Cell Cycle 1 132 136
92. ShevchenkoA
WilmM
VormO
MannM
1996 Mass Spectrometric Sequencing of Proteins from Silver-Stained Polyacrylamide Gels. Analytical Chemistry 68 850 858
93. PengJ
GygiSP
2001 Proteomics: the move to mixtures. Journal of Mass Spectrometry 36 1083 1091
94. EngJK
McCormackAL
YatesJRI
1994 An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. Journal of the American Society for Mass Spectrometry 5 976 989
95. HooftRW
VriendG
SanderC
AbolaEE
1996 Errors in protein structure. Nature 381 272
96. LovellSC
WordJM
RichardsonJS
RichardsonDC
2000 The penultimate rotamer library. Proteins: Structure, Function and Genetics 40 389 408
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2010 Číslo 5
Nejčtenější v tomto čísle
- Common Genetic Variants near the Brittle Cornea Syndrome Locus Influence the Blinding Disease Risk Factor Central Corneal Thickness
- All About Mitochondrial Eve: An Interview with Rebecca Cann
- Aging and Chronic Sun Exposure Cause Distinct Epigenetic Changes in Human Skin
- The Relationship among Gene Expression, the Evolution of Gene Dosage, and the Rate of Protein Evolution