
Transposed Genes in Arabidopsis Are Often Associated with Flanking Repeats
Much of the eukaryotic genome is known to be mobile, largely due to the movement of transposons and other parasitic elements. Recent work in plants and Drosophila suggests that mobility is also a feature of many nontransposon genes and gene families. Indeed, analysis of the Arabidopsis genome suggested that as many as half of all genes had moved to unlinked positions since Arabidopsis diverged from papaya roughly 72 million years ago, and that these mobile genes tend to fall into distinct gene families. However, the mechanism by which single gene transposition occurred was not deduced. By comparing two closely related species, Arabidopsis thaliana and Arabidopsis lyrata, we sought to determine the nature of gene transposition in Arabidopsis. We found that certain categories of genes are much more likely to have transposed than others, and that many of these transposed genes are flanked by direct repeat sequence that was homologous to sequence within the orthologous target site in A. lyrata and which was predominantly genic in identity. We suggest that intrachromosomal recombination between tandemly duplicated sequences, and subsequent insertion of the circular product, is the predominant mechanism of gene transposition.
Published in the journal:
. PLoS Genet 6(5): e32767. doi:10.1371/journal.pgen.1000949
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1000949
Summary
Much of the eukaryotic genome is known to be mobile, largely due to the movement of transposons and other parasitic elements. Recent work in plants and Drosophila suggests that mobility is also a feature of many nontransposon genes and gene families. Indeed, analysis of the Arabidopsis genome suggested that as many as half of all genes had moved to unlinked positions since Arabidopsis diverged from papaya roughly 72 million years ago, and that these mobile genes tend to fall into distinct gene families. However, the mechanism by which single gene transposition occurred was not deduced. By comparing two closely related species, Arabidopsis thaliana and Arabidopsis lyrata, we sought to determine the nature of gene transposition in Arabidopsis. We found that certain categories of genes are much more likely to have transposed than others, and that many of these transposed genes are flanked by direct repeat sequence that was homologous to sequence within the orthologous target site in A. lyrata and which was predominantly genic in identity. We suggest that intrachromosomal recombination between tandemly duplicated sequences, and subsequent insertion of the circular product, is the predominant mechanism of gene transposition.
Introduction
Much of the eukaryotic genome is known to be mobile. This is a characteristic feature of transposable elements, and a large proportion of many eukaryotic genomes are composed of these parasitic elements. However, recent work in plants [1], [2] and Drosophila [3] demonstrates that mobility is also a feature of many non-transposon genes and gene families. An analysis of the Arabidopsis genome suggests that as many as half of all genes had moved to unlinked positions since Arabidopsis diverged from papaya roughly 72 million years ago [4], and that these mobile genes tended to fall into distinct gene families [2]. With the exceptions of unannotated transposons and retroposed genes, the exact mechanism by which single-gene transposition occurred was not deduced, though potential mechanisms include transposon-mediated transduplication or “highjacking” [5], recombination between repeated sequences [6], or nonhomologous end-joining of double-stranded breaks [7]. Unfortunately, ancient gene transposition events, such as those detected in Arabidopsis to date, are an unlikely source of clues because random mutation would be expected to erode all evidence of the mechanism of transposition. In order to detect such evidence, we examined more recent gene transposition events by comparing two relatively closely related (∼5MYA, [8]) Arabidopsis species, A. thaliana and A. lyrata. In this way we were able to identify a large number of recently transposed A. thaliana genes. We found that flanking direct repeats were associated with nearly half of these transposed genes, indicating that these repeats have a role in the process of gene transposition.
Results
Transposed genes within Arabidopsis fall into distinct categories
In order to detect recently transposed nontransposon genes in A. thaliana, we used a semi-automated procedure (Methods). Briefly, we automated a procedure we call the flanking gene method, which compares the location of two sequential genes in a region orthologous between two species such that, given genes A and C, if gene B is present between the two genes in one species but not the other, gene B is denoted as a possible transposed gene (as previously described in [2]). This method can identify genes that are present at a given position in A. thaliana, but absent at the orthologous position in A. lyrata. In order to distinguish between a gain in A. thaliana and a loss in A. lyrata, each region was compared to the orthologous regions in Carica papaya (papaya) and Vitis vinefera (grape), two more distantly related species in the rosid clade (Figure 1) (Methods). The absence of a given gene at the syntenous position in both of the outgroups and in A. lyrata was interpreted as evidence for insertion in A. thaliana; if it was not possible to substantiate the status of the candidate in the outgroup—for example because its expected position in the outgroup was in an unsequenced region—that particular candidate was excluded. We found a total of 420 genes that were present at a given position in A. thaliana and absent at the expected position in A. lyrata. Analysis using the two outgroup species suggested that that 226 of these genes were new insertions in A. thaliana (Table S1). This figure is most conservative, as our methods purposefully discarded questionable data.

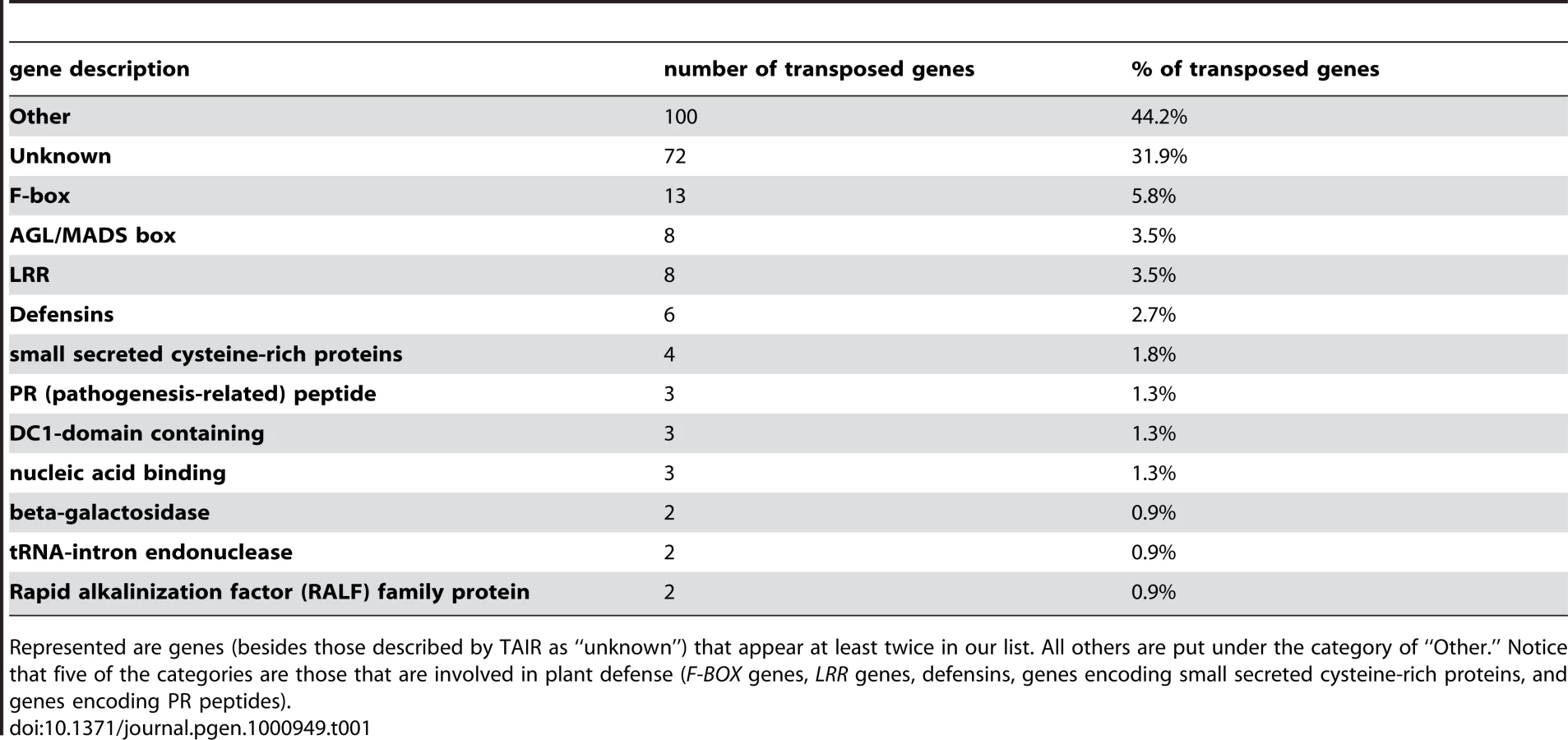
As had been observed previously [2], we found that certain gene families are much more likely to have transposed than others (Table 1). Other than genes that encode unknown proteins, F-box genes were the most common class of transposed genes (6.2%), followed by MADS/AGL genes and LRR-type disease resistance genes (3.5% each), then defensins (2.7%). Defensins, due to their small size and rapidly changing sequences, were mostly undetected in C. papaya. Similarly, the same transcription factor gene families that, according to [2], tend not to transpose in the period following the divergence of A. thaliana and C. papaya (e.g. TF-GRAS genes, WRKY genes, WD40 genes, and GERMINS), are not found at all within our list of 226 genes that had transposed following the divergence of the A. thaliana and A. lyrata lineages. Our data suggests that, correcting for divergence time, the rate (in duplication-transpositions/MY) of gene transposition detected when comparing A. thaliana and A. lyrata (5 MYA) is roughly the same as the rate observed when comparing A. thaliana and C. papaya (72 MYA) [2]. We can calculate that, since the divergence of A. thaliana and A. lyrata, gene transposition has occurred approximately once every 22,000 years (226 transposed genes/5 MYA). Altogether, these data suggest that the more recent gene transposition events we detect in A. thaliana are representative of transpositions that have been occurring over the past 72 million years in the Arabidopsis lineage.
Many transposed genes appear to have recently transposed from a parent site
To examine the nature of these gene transpositions in closer detail, we looked for evidence of recent transposition events from a parental or source position. We focused on transposition events that were not the result of retroposition, but rather DNA-level recombination. These are distinguished as transposed sequences that contain intron and/or non-coding flanking sequences that exist in their parental copy. Assuming that gene transposition happens continuously over time, we expect that a recently transposed gene would retain noncoding sequence similarity to its parent if the parent still existed in the genome. We would also expect that only half of all transposed genes would have remaining donor sites within a given genome if the donor site and the transposed gene were unlinked, as, once transposition occurs, the donor site is then heterozygous for gene in question, and the donor site may be lost via segregation. However, the ratio of identifiable donor sites may be even less than half if the transposition event in question had been relatively ancient, and the noncoding sequences for the donor site and transposed gene had significantly diverged. We felt that the most recent transposition events would also retain evidence of their mechanism of transposition, and we focused on those. To do this, we looked for an unlinked paralog that had sequence similarity with the transposed genes higher than 75% identity across at least 50 base pairs of noncoding sequence. These criteria were used in order to restrict the level of detection to genes that had only recently transposed. Of the 126 transposed genes we examined manually (excluding the “unknown” genes), 106 of our transposed genes did not have a best hit with noncoding sequence similarity greater than or equal to 75%/50 bp. However, 25 (19.8%) had a best hit whose noncoding sequence fit the above criteria, consistent with being relatively recent transposition events, and making them candidate source genes (Table 2, Table S2). In 60% (15/25) of such cases, the parental gene was in a position syntenous in A. lyrata, suggesting that the parental gene itself had not transposed from another position within the last 5 MY. As a control, we also examined 102 genes that had not transposed since the divergence of A. thaliana and A. lyrata (Methods). None of these genes had a best hit whose noncoding sequence similarity was above 75% identity over 50 bp (Table 2, Table S3).

Flanking direct repeats are associated with transposed genes
Next, we examined our transposed genes for signatures of the transposition mechanism. In particular, we searched for the presence of direct repeats flanking the transposed sequence because such repeats have already been shown to be associated with indels in Arabidopsis (though the absence of an outgroup prevented the distinction between insertion and deletions) [9]. In addition, whole-gene transposition in Drosophila [3] has also been associated with direct repeats of highly repetitive DNA. To look for flanking direct repeats around our transposed genes, we used the genome visualization platform GEvo to visually compare the 5′ region ∼500 bp upstream of our target sequence to the sequence ∼500 bp downstream of its 3′ region (Methods). We limited our search to BLAST hits that were greater than or equal to the e-value of a 15/15 bp exact match, and excluded simple sequences. Using these criteria, 17% (22/126) of our total transposed genes had flanking repeats greater than 15 bp (Table 3). However, when we enriched for transposed genes that had an identifiable parental site, 44% (11/25) had flanking direct repeats equal to or greater than 15 bp in length. In contrast, only 5.9% (6/102) of the control, nontransposed genes had flanking repeats greater than 15 bp (Table 3) (p-value 0.00025). The difference was even more dramatic for longer repeats: 36% (9/25) of parental-site transposed genes had flanking repeat sequence over 30 bp in length, versus only 2% (2/102) of the control genes (p-value 0.000051).

Upon closer examination of the nine transposed genes whose flanking repeats were greater than 30 bp, we found that in six cases, the flanking repeat sequence was detected at the parent site at least once (Table 4, Figure 2, Figure S1). Repeat carryover from a parent sequence had been associated with transposed genes in Drosophila [3], suggesting a flanking-repeat excision model. Two of our nine long-direct-repeat genes had inverted repeats associated with them as well as direct repeats (AT5G10330 and AT1G49715, Table 4). Inverted repeats are a hallmark of DNA transposons that are known to “highjack” foreign DNA sequence [5]. In fact, one of our transposed genes in this study, AT1G49715 was a PACK MULE, with characteristically long terminal inverted repeats flanked by 10-bp target-site duplications. Of the unknown genes excluded from this study, we found at least five genes that had clearly been captured by a transposon-like mechanism, based on the fact that were not transcribed, only a portion of them transposed from their putative donor site, and they were flanked by either terminal inverted repeats (TIRs) or long terminal repeats (LTRs) (data not shown). In short, we were able to detect transposons and transposons with genic insertions, so these did not confuse our study of the transposition of more typical genes.


Unexpectedly, in five of the transposed genes with long flanking repeats, the sequence of the repeat could be found at the orthologous site in A. lyrata (AT2G34290, AT2G26010, AT3G10845, AT4G01640, and AT2G16930, Table 4). This A. lyrata sequence could well correspond to the recombination site for the insertion in A. thaliana. In addition, of these five genes, three (AT2G34290, AT2G26010, and AT5G01080) were flanked by repeat sequence that could also be found at the parental site at least once, as though this repeat sequence were a source for intrachromosomal recombination out of the site of origin as well as recombination into the target site orthologous to the A. lyrata homologous sequence (as illustrated in Figure 3). Notably, the repeats surrounding two of these genes (AT2G26010 and AT5G01080) were chimeric between the original flanking sequence and the A. lyrata homeologous target sequence, as demonstrated in Figure 3B. Additionally, among the transposed genes in our study that did not have an identifiable parent source or donor site, nine of these unpaired, transposed genes also had flanking repeat sequence greater than 30 bp in length within 500 bp of the coding region. In eight of these cases, the flanking repeat sequence was found at the orthologous target region in A. lyrata (Table 5). Therefore, among the eighteen total transposed genes that had flanking repeats over 30 bp in length, thirteen (72%) of them had repeat sequence that corresponded to the orthologous site in A. lyrata.


The gene classes that tend to transpose are associated with flanking direct repeats whose sequence is genic, not transposon
Previous work in Drosophila found that the sequence identity of repeats surrounding transposed genes usually comprised transposon sequence [3], and an argument was made suggesting that transposon sequence distributed randomly within the genome could facilitate gene transposition by ectopic recombination. However, when we examined the sequences of the repeats flanking our transposed genes, we generally found these repeats to be made up of host non-transposon sequence of varying kinds, including what appear to be the remnants of fractionated genes (Table 4, Table 5).
We decided to examine all genes in the A. thaliana genome to determine whether the gene classes that tended to transpose were more likely to be flanked by direct repeats, and if so, what the identity of the sequence of those direct repeats were. We made a prediction that the gene classes that tended to transpose would also tend to be flanked by direct repeats; such flanking repeats would endow these gene classes with a propensity for excision and insertion via ectopic recombination.
Using an automated blast search with the parameters described in Methods, we retrieved 1088 genes, including pseudogenes, that were identified as having flanking direct repeats according to the algorithm. These included genes that had not transposed in the 5 MY since the A. thaliana/A. lyrata split. When we examined the classes of genes that tended to have these flanking repeats, we found that, aside from unknown genes, F-box genes were the highest represented as being flanked by direct repeats (4.5%,) closely followed by pseudogenes, then defensins and LRR genes (2.8% and 2.2%, respectively) (Table 6). These percentages are proportional to the percentages that these gene families make up in transposed genes (Table 1). Most notably, these three gene classes are also those that tend to either form, or insert into (interrupt), tandem duplications. Indeed, in 80% of the cases examined, the sequence identity of the flanking repeats is genic, and in the cases of F-box genes and LRR genes, the sequence is similar to the gene itself 40% of the time, as though the flanking repeats are remnants of a tandem duplication.

Discussion
Repeat sequences in general are unstable regions of the genome due to their potential for unequal crossing over, intrachromosomal crossing over, and ectopic recombination [10], [11]. Intrachromosomal recombination between flanking repeats—the sort of event that generates a circular fragment—has been associated in plants and Drosophila with small deletions within transposons. Flanking repeats have also been associated with indels in Arabidopsis, but insertions were not distinguished from deletions [9]. Yang et al. have shown that whole-gene transpositions in Drosophila are primarily associated with 44 - to 433-bp flanking repeats of transposable element sequences [3]. Our results in Arabidopsis are similar to those in Drosophila, but our flanking repeats generally derive from host non-transposon sequence.
Forty-four percent of our transposed genes that had an identifiable donor site had (detectable by our Methods) flanking repeats at their insertion site, and thirty-six percent had flanking repeats greater than 30 bp in length. Most transposed genes (with or without an identifiable donor site) with repeats greater than 30 bp were flanked by direct repeat sequence that was homologous to sequence within the orthologous target site in A. lyrata. One explanation for this pattern might be that the target site was duplicated following the insertion in A. thaliana; such long target-site duplications have been observed in H. pylori genomes after gene transposition [12], and in humans due to retroviral insertion [13]. However, there are simpler explanations, as follows.
In the majority of cases, the repeat sequence flanking the transposed gene consisted of non-transposon host sequence, particularly sequence that corresponds to genic sequence, suggesting that the insertion site had once been part of a tandem repeat, or that the transposed gene had originally resided in a tandem repeat; indeed, the parental gene for three of the nine donor-site transposed genes was part of a tandem duplication (Table 4). Previous work in our lab has demonstrated that mobile genes—transposon and nontransposon alike—in Arabidopsis are often found within tandem repeats from unrelated families [2]; these were called “interruptor” genes. In addition, genes that tend to have duplicated in tandem—such as F-box genes, DEFENSINS, and NB-LRR-type disease-resistance genes—also tend to be those that transpose [2], [14]. Findings by Zhou and coworkers suggested that, in Drosophila, tandem repeats may precede DNA transposition [15]. Other researchers in Drosophila had found the presence of chimeric genes within tandem arrays, and postulated that the mechanism may be via large-loop mismatch repair, where a portion of the DNA sequence between duplicated genes is excised [16]. Further, Cohen and coworkers have demonstrated that tandem duplications are associated with the formation of circles in plants [17] and Drosophila [18], where it is thought that a gene that is part of a tandem duplication can excise by intrachromosomal recombination via the repetitive sequence surrounding it, then potentially integrate into a new region containing sequence homologous to the flanking repeats. In fact, in three of our transposed genes (AT2G34290, AT5G15220, and AT2G26010), a portion of the flanking sequence was found in both the A. lyrata orthologous target sequence and the parental sequence, suggesting the possibility that this same repeat sequence facilitated intrachromosomal recombination and excision from the parental site, then insertion into the homologous target site. Alternatively, ectopic recombination between unlinked sites (say, between one tandem array of a given gene type and another tandem array of the same or similar gene type) may play a role in gene movement; indeed, one hypothesis for the formation of chimeric disease-resistance genes in plants is via ectopic recombination [19].
In over half the cases where a transposed gene had an identifiable parent, no evidence of repeats were found flanking the transposed gene. There are several explanations for this. For instance, the parameters of our experimental design (15/15 exact match) would rule out repeats that were smaller than 15 base-pairs, or repeats that had degenerated beyond what our algorithm could detect. Indeed, in some cases, when we more closely examined the sequence flanking some of the transposed genes which, according to our parameters, were deemed devoid of repeats, we did observe the traces of direct repeats whose sequence similarity would not have been detected by our criteria (data not shown). This is also observed at the parent sites for our transposed genes. In other cases, the donor site contained only one copy of the flanking repeat found at the transposed site. Again, degeneration of the second repeat might have occurred, or the repeat might have been removed entirely via a deletion event [9], [10]. Alternately, recombination with the homologous sequence at the new site might have created chimeric flanking sequences (as illustrated in Figure 2 and Figure 3B). For instance, evidence of flanking direct repeats that are chimeric between the target sequence and the donor repeat sequence is observed in AT2G26010, and AT5G01080, as previously discussed. Particularly after sequence divergence, the original flanking sequence in some instances may not manifest as identifiable direct repeats surrounding the transposed sequence.
Tandemly repeated genes and genes flanked by tandem repeats are a special case because they are regions that are particularly prone to intrachromosomal recombination and the associated circular fragment. As diagrammed in Figure 3, if a sequence in a circular fragment is homologous to a sequence elsewhere in the genome, insertions resulting in flanking repeats are possible. However, reinsertion is likely to be an exceptional event. In most cases, we would expect that the excised circle would simply be lost and thus, these recombination events would result in a net reduction of genomic DNA content. We suggest that this process is the way plants [10] and Drosophila [20] have countered transposon buildup. The gene loss mechanism in plants and other clades able to carry high mutational load is likely to involve the generation of circular fragments of chromosome, and, thus, the potential for transposition.
We calculate the rate of gene transposition as being once every 22,000 years, and hypothesize that the rate of transposition is steady and not punctuated, as the degree of sequence similarity in noncoding sequence between donor and transposed sites varies from 100% identity over all noncoding sequence to 70% identity/50 bp at the very most (Table S2), suggesting that transposition has occurred at different points throughout evolutionary time. This is also consistent with our observation that gene transposition following the divergence of A. lyrata from A. thaliana has occurred at roughly the same rate as it had occurred since following the divergence of A. thaliana from papaya.
Repeats, from a few base-pairs in length to hundreds of base-pairs, are ubiquitous throughout the genome. Via ectopic recombination, these repeats permit the deletion or insertion of a fragment of DNA as small as a few base-pairs, or as large as an entire gene. It seems likely that the genome's potential to re-shape itself in novel ways is built in, not via any special mechanism or adaptation, but as a passive by-product (a spandrel) of the genome's architecture. Obviously the presence of thousands of transposable elements within any genome is a source of recombination; but tandem duplications clearly can play a role as well, particularly at the genic level. Indeed, the fact that many of the repeats surrounding transposed genes in Arabidopsis were associated with what appear to be duplicated gene fragments, and the fact that transposed genes tend to not only form tandem repeats themselves, but insert into tandem repeats, suggests that tandem duplications may particularly facilitate gene excision as well as provide targets for gene insertion.
Methods
The list of 420 transposed genes in A. thaliana
Automation: Genomes of A. thaliana (TAIR masked repeats 50x, v7) and A. lyrata (JGI unmasked, v1, sequenced by the Weigel lab http://www.phytozome.net/alyrata) are co-annotated to each other, so that features (files of the annotations used are available here http://syntelog.com/t/gray_paper_methods/) that are annotated in one but not the other become new features in their respective genomes (this co-annotation step is performed by software available from our source-code repository: http://bpbio.googlecode.com/svn/trunk/co-anno). New fasta files are created from the original genomic features and the new ones discovered via co-annotation. The fasta file from A. thaliana is blasted against the fasta file from A. lyrata using blastn/e-value of 0.1/word size 7. The blast is then “dissolved” by combining small hits to the same gene pairs into single hits, then keeping only those merged hits with a sum length greater than or equal to 96.
The search for transposed genes is performed using the aforementioned dissolved pairs, such that for each gene in a pair, the algorithm is extended out three genes in either direction, creating a list of (gene +3+3)*2 = 14 genes; next, for that group of genes, the flanking gene method is used to find transposed genes in both query and subject by converting gene names to integer positions to get a list of query-subject pairs; for instance: [(1, 123), (2, 125)]. From this, simple addition and matching is used to find any genes that are “missing,” e.g. the gene at position 124 above is unaccounted for, and flanked by consecutive genes 1, 2 from the ortholog. The integer positions are then converted back to gene names, and the transposed gene and the orthologs of its flankers are reported.
Each putative transposed gene is then checked to see whether it lies within a region in a manually generated list of orthologous regions, since a genes in non-orthologous regions cannot be verified as transposed they are discarded. Each putative transposed gene is then blasted against the non-coding sequence in the orthologous flanking region to determine whether it actually appears in the ancestral position even though it is not annotated as such. No BLAST to the transposed gene over 15 bp can be observed between the flankers in A. lyrata, otherwise it might just be gene loss in A. lyrata (hits_within_0<15). Each putatively transposed gene must be flanked by two genes that are not identical to each other in A. thaliana such that A. lyrata flanker_1≠ flanker_2. If it fails this (e.g. A. lyrata flanker_1 = flanker_2) then it is designated an “interrupter” or “I” gene. Any pseudogene, or any gene that has been identified as repetitive or transposable element sequence of any kind by the Arabidopsis genome database TAIR (http://www.arabidopsis.org/), is removed from the list.
Proofing: Once the list of putative transposed genes is at hand, each gene is visualized in our gene visualization platform GEvo (http://synteny.cnr.berkeley.edu/CoGe/GEvo.pl) via a link provided in the output containing the CoGe identifier of the putative transposed gene as well as the identifier for one of the A. lyrata flankers.
Using the orthologous groups C. papaya V. vitifera to confirm gene transposition in A. thaliana
Using blastn parameters of word-size 7 and filtered to exclude simple sequences, each transposed gene was blasted to C. papaya (University of Hawaii v0.4, masked repeats 50x) and V. vitifera (French National Sequence Center v1, masked repeats 50x) to verify the presence of the gene within the genome, masking all non-cd sequence. We then masked the transposed gene itself and blasted 15 Kb on either side of the gene to C. papaya and V. vitifera to find the syntenous regions.
If the gene itself was not found in either papaya or grape, it was labeled “No gene hit” and was discarded from our list. If there was a gene hit to papaya and grape, but the surrounding region of A. thaliana does not hit in the same region in papaya and grape, the gene was labeled “No hits with gene and buffer in same region” and was considered transposed. If there was a hit to the same region of papaya and grape in both the buffer and the gene itself, it was considered to be in the ancestral position and was discarded as not being a true transposition. Each gene had to have a hit in both papaya and grape, and each gene had to be in a nonancestral position in both papaya and grape, otherwise it was discarded from our list. This procedure left us with the 226 confirmed transposed genes that were the basis of this research (Table S1).
Finding duplicates of the transposed genes
The genomic sequence (including non-coding sequence) of all genes considered true transpositions in our list were then blasted to the A. thaliana genome to find the best hit outside of itself. We then examined the best hit to see whether it had sequence similarity with the transposed gene higher than 75% identity across at least 50 base pairs of noncoding sequence. If the best hit fit these criteria, it was deemed to be a putative parental site from which the transposed gene had duplicated (Tables S2, S3).
Finding flanking repeats surrounding the duplicated/transposed genes
We used our genome visualization platform GEvo to visually compare the 5′ region ∼500 bp upstream of our target gene to the sequence ∼500 bp downstream of its 3′ region. We limited our search to BLAST hits that were greater than or equal to the e-value of a 15/15 bp exact match, and excluded simple sequences (Tables S2, S3). The 102 non-transposed genes that were selected for the control were genes chosen from specific gene families that had been confirmed by both [2] and this particular study as being underrepresented for gene transposition. Since this portion of the study was performed manually we restricted our analysis to only 102 genes, similar in number to the total number of transposed genes examined for parental sites and flanking repeats (126).
Automation of flanking repeat discovery around genes in A. thaliana
We wrote a program using blastn, word-size 7, with BLAST hits that were greater than or equal to the e-value of a 15/15 exact match, that would look for repeats between 30–400 bp shared between the 2 kb region up and downstream of coding sequence of each gene (Table S4).
Supporting Information
{kind=link}
Zdroje
1. MeyersBC
2003 Genome-wide analysis of NBS-LRR-encoding genes in Arabidopsis. The Plant cell 15 809 834
2. FreelingM
LyonsE
PedersenB
AlamM
MingR
2008 Many or most genes in Arabidopsis transposed after the origin of the order Brassicales. Genome Res 18 1924 1937
3. YangSA
ArguelloJR
LiX
DingY
ZhouQ
2008 Repetitive element-mediated recombination as a mechanism for new gene origination in Drosophila. Plos Genet 4 e3 doi:10.1371/journal.pgen.0040003
4. MingR
HouS
FengY
YuQ
Dionne-LaporteA
2008 The draft genome of the transgenic tropical fruit tree papaya (Carica papaya Linnaeus). Nature 452 991 996
5. JiangN
BaoZ
ZhangX
EddySR
WesslerSR
2004 Pack-MULE transposable elements mediate gene evolution in plants. Nature 431 569 573
6. MontgomeryEA
HuangSM
LangleyCH
JuddBH
1991 Chromosome Rearrangement by Ectopic Recombination in Drosophila-Melanogaster - Genome Structure and Evolution. Genetics 129 1085 1098
7. MooreJK
HaberJE
1996 Cell cycle and genetic requirements of two pathyways of nonhomologous end-joining repair of double-strand breaks in Saccharomyces cerevisiae. Mo Cell Biol 16 2164 2173
8. KotchM
HauboldB
Mitchell-OldsT
2000 Comparative evolutionary analysis of chalcone synthase and alcohol dehydrogenase loci in Arabidopsis, Arabis and related genera. Mol Biol Evol 17 1483 1498
9. ZiolkowskiPA
KoczykG
GalganskiL
SadowskiJ
2009 Genome sequence comparison of Col and Ler lines reveals the dynamic nature of Arabidopsis chromosomes. Nucleic Acids Research 37 3189 3201
10. DevosKM
BrownJK
BennetzenJL
2002 Genome size reduction through illegitimate recombination counteracts genome expansion in Arabidopsis. Genome Res 12 1075 1079
11. LeisterD
2004 Tandem and segmental gene duplication and recombination in the evolution of plant disease resistance gene. Trends Genet 20 116 122
12. NobusatoA
UchiyamaI
OhashiS
KobayashiI
2000 Insertion with long target duplication: a mechanism for gene mobility suggested from comparison of two related bacterial genomes. Gene 259 99 108
13. MamedovIZ
LebedevYB
SverdlovED
2004 Unusually long target site duplications flanking some of the long terminal repeats of human endogenous retrovirus K in the human genome. Journal of General Virology 85 1485 1488
14. RizzonC
PongerL
GautBS
2006 Striking Similarities in the Genomic Distribution of Tandemly Arrayed Genes in Arabidopsis and Rice. PLoS Comput Biol 2 e115 doi:10.1371/journal.pcbi.0020115
15. ZhouQ
ZhangGJ
ZhangY
XuSY
ZhaoRP
2008 On the origin of new genes in Drosophila. Genome Research 18 1446 1455
16. RogersRL
BedfordT
HartlDL
2009 Formation and Longevity of Chimeric and Duplicate Genes in Drosophila melanogaster. Genetics 181 313 322
17. CohenS
HoubenA
SegalD
2008 Extrachromosomal circular DNA derived from tandemly repeated genomic sequences in plants. Plant Journal 53 1027 1034
18. CohenS
YacobiK
SegalD
2003 Extrachromosomal circular DNA of tandemly repeated genomic sequences in Drosophila. Genome Research 13 1133 1145
19. ParniskeM
JonesJD
1999 Recombination between diverged clusters of the tomato Cf-9 plant disease resistance gene family. Proc Natl Acad Sci 96 5850 5855
20. PetrovDA
ChaoYC
StephensonEC
HartlDL
1998 Pseudogene evolution in Drosophila suggests a high rate of DNA loss. Molecular Biology and Evolution 15 1562 1567
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2010 Číslo 5
Nejčtenější v tomto čísle
- Common Genetic Variants near the Brittle Cornea Syndrome Locus Influence the Blinding Disease Risk Factor Central Corneal Thickness
- All About Mitochondrial Eve: An Interview with Rebecca Cann
- Aging and Chronic Sun Exposure Cause Distinct Epigenetic Changes in Human Skin
- The Relationship among Gene Expression, the Evolution of Gene Dosage, and the Rate of Protein Evolution