
DNA Adenine Methylation Is Required to Replicate Both Chromosomes Once per Cell Cycle
DNA adenine methylation is widely used to control many DNA transactions, including replication. In Escherichia coli, methylation serves to silence newly synthesized (hemimethylated) sister origins. SeqA, a protein that binds to hemimethylated DNA, mediates the silencing, and this is necessary to restrict replication to once per cell cycle. The methylation, however, is not essential for replication initiation per se but appeared so when the origins (oriI and oriII) of the two Vibrio cholerae chromosomes were used to drive plasmid replication in E. coli. Here we show that, as in the case of E. coli, methylation is not essential for oriI when it drives chromosomal replication and is needed for once-per-cell-cycle replication in a SeqA-dependent fashion. We found that oriII also needs SeqA for once-per-cell-cycle replication and, additionally, full methylation for efficient initiator binding. The requirement for initiator binding might suffice to make methylation an essential function in V. cholerae. The structure of oriII suggests that it originated from a plasmid, but unlike plasmids, oriII makes use of methylation for once-per-cell-cycle replication, the norm for chromosomal but not plasmid replication.
Published in the journal:
. PLoS Genet 6(5): e32767. doi:10.1371/journal.pgen.1000939
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1000939
Summary
DNA adenine methylation is widely used to control many DNA transactions, including replication. In Escherichia coli, methylation serves to silence newly synthesized (hemimethylated) sister origins. SeqA, a protein that binds to hemimethylated DNA, mediates the silencing, and this is necessary to restrict replication to once per cell cycle. The methylation, however, is not essential for replication initiation per se but appeared so when the origins (oriI and oriII) of the two Vibrio cholerae chromosomes were used to drive plasmid replication in E. coli. Here we show that, as in the case of E. coli, methylation is not essential for oriI when it drives chromosomal replication and is needed for once-per-cell-cycle replication in a SeqA-dependent fashion. We found that oriII also needs SeqA for once-per-cell-cycle replication and, additionally, full methylation for efficient initiator binding. The requirement for initiator binding might suffice to make methylation an essential function in V. cholerae. The structure of oriII suggests that it originated from a plasmid, but unlike plasmids, oriII makes use of methylation for once-per-cell-cycle replication, the norm for chromosomal but not plasmid replication.
Introduction
The regulatory potential of canonical DNA sequences can be greatly expanded by epigenetic modifications. Methylation is the most common modification of DNA and is widely used to control many cellular processes [1]. In bacteria, DNA methylation is restricted to adenine and cytosine residues [2], and can facilitate or interfere with DNA-protein interactions, thereby modulating various DNA transactions [3]. Such transactions include gene expression, DNA restriction, DNA mismatch repair, and chromosome replication and segregation [4], [5].
Most of our knowledge regarding the role of methylation in chromosome replication comes from studies in Caulobacter crescentus and Escherichia coli. In C. crescentus, initiation of DNA replication requires the adenines of the GANTC sequences in the origin of replication to be methylated on both the top and bottom strands by the methylase CcrM. How the methylation helps the origin function is not known, although methylation lowers DNA stability [6], [7] and thereby could facilitate origin-opening, an essential step in the replication initiation process. It is also possible that the methylation changes DNA structure to facilitate protein-DNA interactions at the origin [8]. Irrespective of the mechanism, methylation not only controls the timing of initiation but also restricts initiation to once per cell cycle [9]. Following initiation, the hemimethylated sister origins cannot be reused in the same cell cycle, as the CcrM methylase is not synthesized until the end of the replication cycle.
In E. coli, the methylase is called Dam and acts on the adenines of GATC sequences, which are particularly frequent in the origin of replication, oriC. In this bacterium also the methylation most likely helps in origin-opening [8], [10] but plays a more definite role in restricting the initiation to once per cell cycle [11]. In E. coli, immediate reinitiation is prevented, not by delaying the synthesis of the methylase, but by preventing its action through sequestration of hemimethylated sister origins by a hemimethylation-specific DNA binding protein, SeqA [12]. Sequestration renders DNA unavailable to the methylase. The sequestration also allows initiation synchrony whereby the multiple origins that E. coli maintains during rapid growth fire nearly simultaneously. It is believed that the sequestration process continues at least until all the origins have fired. This happens in a narrow window of time giving rise to the initiation synchrony phenotype [13]. In the absence of Dam, the newly replicated origins, without their hemimethylation marks, remain indistinguishable from the unreplicated ones. The choice of origin for replication being random, once-per-cell-cycle initiation from each origin is no longer guaranteed. As a result, in dam mutants, the initiation becomes asynchronous and cells can have origins that do not fire at all or fire more than once in the same cell cycle. The consequences are the same in seqA mutants, because without sequestration, replicated origins also remain competent for reinitiation.
The lack of discrimination between replicated and unreplicated origins can lead to origin incompatibility [14]. If extra copies of oriC are introduced as plasmids into wild type (WT) E. coli, the plasmid copies do not compete with the chromosomal oriC because of sequestration of newly replicated origins. Without sequestration, in dam or seqA mutants, the plasmid copies remain available for reinitiation, and under selection they can block the growth of cells in which the chromosomal origins did not get a chance to fire. Sequestration-deficient strains are therefore not easily transformed with oriC plasmids [15]. Thus, although not normally required, Dam or SeqA can be essential in a competitive situation.
Vibrio cholerae has two chromosomes (chrI and chrII). The origin of chrI (oriI) shares 58% identity with the E. coli oriC, and both have similarly high densities of GATC sites. The origin of chrII (oriII) also has a high density of GATC sites but has a second feature of a major class of plasmids: repeated initiator-binding sites (iterons) [16]. The dam gene is also essential for V. cholerae, although the reason has remained unknown [17]. Our interest in the role of methylation in V. cholerae chromosomal replication stems from the fact that although the bacterium is a close relative of E. coli, plasmids with either oriI or oriII could transform WT E. coli, but not when it lacked Dam [18]. It remained unclear whether the failure to recover transformants in the case of oriI is because the origin could not function or because of competition (incompatibility) with the closely related chromosomal oriC [14], [18]. Incompatibility is unlikely the case of oriII, since it has little similarity to oriC. Moreover, while oriI and oriC are regulated by the DnaA initiator protein, oriII is regulated by its own specific initiator, RctB [19]. The reason for the Dam requirement of oriII could thus be for the functioning of the origin itself.
Here we show that oriC can be replaced by oriI in the E. coli chromosome, and in this chromosomal context oriI functions without requiring Dam or SeqA. Incompatibility with the chromosomal oriC thus remains a satisfactory explanation of the earlier finding of a Dam requirement for oriI plasmids [18]. For oriII, Dam but not SeqA appears to be required as only fully methylated oriII DNA, but not hemi - or un-methylated DNA, could bind efficiently to the oriII-specific initiator RctB in vitro. Since the binding of RctB is a prerequisite for oriII function, this provides an explanation for why Dam is essential for V. cholerae, chrII being indispensable. Finally, we show that SeqA is necessary to restrict initiation to once per cell cycle for both oriI and oriII, as is the norm for chromosomal origins. Although chrII is believed to have originated from a plasmid, our findings of the methylation requirement for its initiation and cell-cycle specific regulation are unprecedented in studies of plasmids [20], [21]. It appears that a plasmid origin acquired methylation to function as a chromosomal origin, thus providing a novel example of origin evolution in bacteria.
Results
Dam and SeqA are not essential for replication initiation at oriI in E. coli
The E. coli origin of replication, oriC, does not require dam and seqA to initiate replication. In contrast, plasmids driven by oriC are highly deficient in transformation of dam mutants [11]. This is believed to be due to irreversible sequestration of hemimethylated plasmid origins by the SeqA protein after the first round of replication [22]. Indeed, seqA and dam seqA strains can be transformed by oriC plasmids, although the efficiency is lower compared to WT due to incompatibility with the chromosomal copy of the origin [15]. The requirement of dam thus is not intrinsic to oriC function and appears so only in the plasmid context. The dam requirement of V. cholerae oriI has so far been studied only in the plasmid context. However, in contrast to oriC plasmids, oriI plasmids not only failed to transform an E. coli dam mutant but also a seqA or a dam seqA mutant, raising the possibility that the genes could be essential for oriI [11], [18]. We confirmed the plasmid results using E. coli MG1655 (BR1703) and its dam (CVC1415), seqA (BR1704) and dam seqA (CVC1424) mutant derivatives. As before, the dam, seqA and dam seqA mutants could not be transformed with an oriI plasmid, and only the dam mutant could not be transformed with the oriC plasmid (Figure 1A). We suggest below that the oriI plasmid possibly replicated in the absence of dam or seqA, which competed out replication from the chromosomal oriC and led to inviability of the transformants.

To avoid plasmid-mediated competition (incompatibility), we studied oriI by placing it in the E. coli chromosome. Using the Red recombineering system, we replaced the minimal oriC region with the corresponding oriI region (Materials and Methods). The resultant strain, MG1655ΔoriC::oriI-zeo (CVC1400, Table 1; hereafter called MG1655ΔoriC::oriI), could be made dam minus by P1 transduction, using dam-16::aph (CVC1383) as the source of the mutant dam allele [23]. We could also replace the oriC region of MG1655ΔseqA10 with ΔoriC::oriI by P1 transduction. The viability of dam, seqA or dam seqA mutant derivatives of MG1655ΔoriC::oriI (CVC1401, CVC1416 and CVC1425, respectively) indicates that oriI does not require Dam and SeqA for functioning in E. coli.

To understand why oriI and oriC behave similarly in the chromosomal context but differently in the plasmid context, we repeated the transformation experiments using MG1655ΔoriC::oriI cells as the host. The oriI plasmid could now transform the seqA and the dam seqA derivatives of MG1655ΔoriC::oriI efficiently but not the dam derivative (Figure 1A and 1B). The failure to transform the dam derivative can be attributed to permanent sequestration. In contrast to oriI, oriC not only failed to transform the dam derivative but also the damseqA derivative of MG1655ΔoriC::oriI. The results can be understood assuming initiation from oriI to be more efficient than from oriC. Most likely, the weaker oriC failed to compete with oriI in the chromosome (incompatibility) that led to inviability of the transformants. It is known in E. coli that incompatibility problems can be aggravated when the incoming and recipient origins have unequal efficiencies [24].
Dam and SeqA make initiation synchronous and once-per-cell-cycle for oriI in E. coli
oriI and oriC were further analyzed using flow cytometry [25]. Replication initiation and cell division were blocked by antibiotics rifampicin and cephalexin, respectively, but sufficient time was allowed after drug addition to complete replication elongation (replication run-out). This method provides a measure of the fraction of the population that already initiated replication at the time of drug addition. In LB, after the replication run-out, MG1655 cells were distributed mostly into two populations, one with four and the other with eight full chromosomes (Figure 2A). This indicates that cells were born with four origins and they all fired synchronously once, giving rise to the eight chromosome peak. In the dam and seqA mutants, cells had a widely varying number of chromosomes indicating asynchronous initiation (Figure 2C and 2E) [22], [26]. There were also cells with more than eight chromosomes indicating that initiation was no longer restricted to once per cell cycle. In the engineered strain, MG1655ΔoriC::oriI, replication initiation was synchronous (Figure 2B) but not in its dam or seqA derivatives (Figure 2D and 2F). The requirements of dam and seqA for synchronous and once-per-cell-cycle initiation are thus maintained when oriI replaces oriC.

Compared to the WT, replication initiation was less frequent in dam mutants but more frequent in seqA mutants in the case of both the origins. As is oriC, Dam seems to be playing a positive role and SeqA a negative role in replication initiation from oriI.
Dam is required for initiator binding to oriII
It was reported earlier, and we confirmed, that oriII plasmids can not transform an E. coli dam mutant but can transform a seqA mutant [18]. The oriII plasmids also failed to transform the dam seqA mutant, indicating that irreversible sequestration cannot account for the dam requirement. The oriII function could not be tested in the chromosomal context, as was done for oriI, because attempts to replace oriC with oriII failed. In any event, incompatibility between oriC and oriII appears to be an unlikely explanation for the dam requirement, as the structure and control elements of the two origins are different [19]. We show below that the reason for the dam requirement could be for binding of oriII to its specific initiator RctB.
A distinguishing feature of oriII is that its putative RctB binding sites, called 11 - and 12-mers, all contain a GATC site. This prompted us to test whether methylation of the sites might be important for RctB binding (Figure 3A). We first tested binding to the six tandem 12-mers within the minimal oriII by an electrophoretic mobility shift assay. Purified RctB bound efficiently to the 12-mer fragment, when it was fully methylated (Figure 3B). The binding was nearly saturated because most of the DNA molecules were maximally retarded. Binding to hemimethylated DNA, where either the top or the bottom strand carried the methylation marks, and to unmethylated DNA was significantly less. In these cases, most of the bound species appeared as a smear, indicative of weaker binding. The binding improved when the DNA samples were remethylated using Dam in vitro (Figure 3B). The binding of RctB to the three 11-mers or to a pair of 12 - and 11-mers in the negative-control region of oriII was also efficient when the sites were fully methylated (Figure S1A and S1B). Mutating GATC sites to GATG in the 11 - or the 12-mer abolished the binding (Figure S1C). These results indicate that full methylation can significantly improve the affinity of RctB to the 11 - and 12-mers.

To confirm these results in vivo, RctB binding to a plasmid with the six 12-mers was studied in MG1655 or its dam derivative by chromatin immunoprecipitation (ChIP), and the immunoprecipitated DNA analyzed by quantitative PCR. Compared to the vector, the plasmid with the 12-mers was preferentially enriched by immunoprecipitation when the DNA samples were from WT cells (Figure 3D). No significant enrichment was obtained when the DNA samples were from the dam mutant. These results show the importance of methylation for efficient RctB binding in vivo, and therefore, for replication of chrII.
Dam depletion in V. cholerae inhibits oriII preferentially
To test how well the results obtained in vitro and in E. coli reproduce in the native host, the dam gene of V. cholerae was deleted in the presence of a complementing plasmid, pTS-PBADdam (pGD93, Table 2). The replication of this plasmid is temperature sensitive and the cloned V. cholerae dam gene is under the control of an arabinose-inducible and glucose-repressible promoter, PBAD. On LB plates, under the permissive condition (30°C and in the presence of arabinose), the Δdam/pTS-PBADdam strain grew as well as the WT but under the restrictive condition (42°C and in the presence of glucose), single colonies were barely visible (Figure 4A). In LB broth, under the restrictive condition, the mutant grew slower than the WT (with generation times of 27 min and 22 min, respectively), and the growth plateaued to an OD of 0.53 only (Figure 4B). Moreover, the number of viable cells in the mutant culture was only 0.02% of the number of viable WT cells, when initially similar cultures of both were grown for seven and a half hours under restrictive conditions (Figure 4C). The viable cells in the mutant all retained the dam complementing plasmid without selection for it. The results thus appear consistent with an earlier report that Dam is essential for V. cholerae [17].


Under the condition of dam depletion, we expected that initiation at oriII would decrease more than initiation at oriI. This was tested by determining the relative replication efficiencies of the two chromosomes in exponentially growing cells by qPCR. We quantified the amount of DNA at the two origins and the two termini to obtain the ratios oriI/oriII, oriI/terI, oriII/terII and terI/terII. Under the restrictive condition, there was a significant increase (4-fold) in the value of oriI/oriII and of terI/terII, while the values of oriI/terI and oriII/terII remained unchanged (Figure 4D). These results are consistent with our expectation that compared to chrI, replication of chrII is more dependent on Dam.
Hemimethylation period is prolonged at oriI and oriII
The hemimethylation period, the time to remethylate a GATC site after passage of the replication fork, is particularly prolonged at oriC because of the presence of high density of GATC sites within the origin [12]. The prevalence of high density of GATC sites in both oriI and oriII (Figure 5A) prompted us to examine their hemimethylation period, as was done using asynchronous exponential cultures [27], [28].

We examined the hemimethylation period of a GATC site within the origin and, for comparison, another site external to the origin (about 300 kb away) for each of the chromosomes. In oriI, the GATC site chosen is between DnaA boxes R3 and R4, and in oriII, it is between the fourth and the fifth 12-mers (arrows, Figure 5A). Total genomic DNA was extracted and digested with restriction enzymes whose recognition sequences overlap a GATC site and whose cleavage is inhibited when the site is fully methylated but not in one of the two hemimethylated sister sites, generated by passage of the replication fork (Figure 5B). The fraction of hemimethylated (cut) DNA at each of the origin sites was significantly higher than at the external sites (Figure 5C). The values were 11±3% and 56±8% for oriI and oriII, respectively, while at the external markers they were 4±0.8% and 8±3%, respectively (Figure 5D). The results indicate that as in E. coli, the hemimethylation period is prolonged at the two V. cholerae origins but the duration of the period can be significantly different for the two.
From the E. coli paradigm, we expected that SeqA would be required to prolong the hemimethylation periods at both the origins [22]. To test for the requirement, a partial in-frame deletion of seqA was made where the deleted region was substituted with a zeocin drug-resistance cassette, maintaining the seqA reading frame (Figure S2A). The resulting gene was called ΔseqAP and the strain CVC1410. Replication run-out experiments indicated that initiation of one or both the chromosomes has become asynchronous (Figure S2B), and in this respect, V. cholerae appears to be similar to E. coli (Figure 2A and 2E) [22].
For the GATC site tested in oriI, the fraction of hemimethylated DNA increased from 11% in WT to 68% in ΔseqAP (Figure 6A and 6C). Providing Dam or SeqA from a plasmid in the ΔseqAP background decreased the fraction of hemimethylated DNA. The decrease by providing excess of Dam was expected because it converts hemimethylated DNA to fully methylated DNA. The increase in the absence of SeqA and decrease in its presence were unexpected, if SeqA were responsible for prolonging the period. The seqA plasmid did not change the period significantly in the WT background (Figure S3). The results indicate that it is the absence of SeqA that causes the increase of hemimethylated oriI DNA, a result opposite to that found for oriC [29]. The behavior of oriII was similar to that of oriC: The fraction of hemimethylated DNA decreased from 75% in WT to 17% in ΔseqAP (Figure 6B and 6C). Thus seqA effects can be opposite in different origins at specific GATC sites. It remains to be seen whether the results are site-specific or true for the entire origins.

The opposite response of the GATC sites tested in oriI and oriII was also seen in a V. cholerae mutant where seqA was completely deleted (ΔseqAT, CVC2003; Figure S4). oriI also responded opposite to oriC in E. coli (Figure 7). While the percent of hemimethylated DNA at oriC dropped from 13% in MG1655 to 9% in MG1655ΔseqA10, the values at oriI increased from 9% in MG1655ΔoriC::oriI to 25% in its ΔseqA10 derivative. These results suggest that the opposite behavior of oriI and oriC upon seqA deletion is intrinsic to the sequence context of the GATC sites tested in the two origins rather than the sequestration machinery of the two bacteria. Thus depending upon the context, SeqA can both shorten and prolong the hemimethylation period of a GATC site.
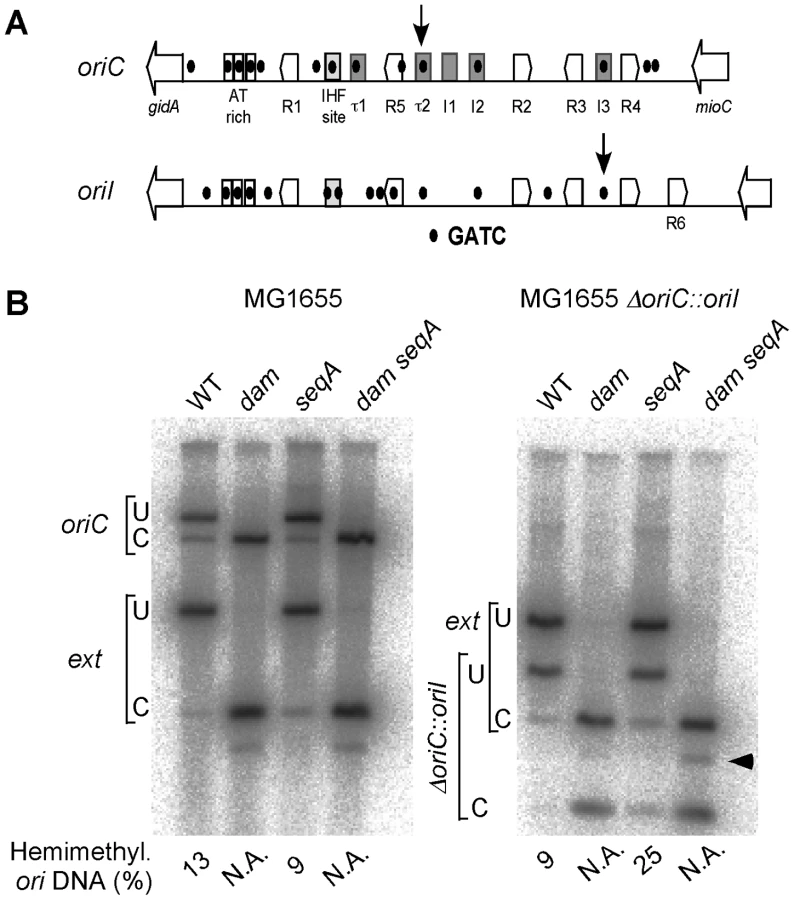
SeqA is required for once-per-cell-cycle initiation from both oriI and oriII
Although a role of SeqA in restraining replication initiation in V. cholerae was suggested by the flow cytometry results (Figure S2B), they did not allow us to distinguish whether one or both the chromosomes were affected. We used fluorescence microscopy to follow replication initiation of the two chromosomes individually. The numbers and positions of oriI and oriII were determined in WT and ΔseqAP strains of V. cholerae by the GFP-P1ParB/parS system [30], [31]. For oriI in WT, 94% of the cells had two to four foci and the rest one or three foci, indicating synchronous and once-per-cell-cycle initiation (Figure 8A and 8E). In contrast, only 45% of ΔseqAP cells showed this pattern (Figure 8B and 8E). The remaining cells had five to nine foci. The significant increase in the number of cells with odd numbers of foci and more than four foci indicates that initiation is no longer synchronous and no longer limited to once per cell cycle in the absence of SeqA.

The regulation of chrII initiation was also affected. While 100% of the cells in the presence of SeqA showed one to two foci (Figure 8C and 8E), this was true for 83% of the ΔseqAP cells (Figure 8D and 8E). The remaining cells showed three to six foci. SeqA thus contributes to synchronous and once-per-cell-cycle initiation of both the chromosomes.
Discussion
Here we have addressed the role of DNA adenine methylation in replication of the two V. cholerae chromosomes. In bacterial replication, adenine methylation can contribute by regulating gene expression, by helping origin opening, and by regulating initiation so that it occurs only once per cell cycle. From the regulatory point of view, the major contribution of methylation is the marking of promoters/origins so that unreplicated DNA can be distinguished from the replicated ones. Newly replicated DNA is uniquely marked with hemimethylated sites that lend themselves to regulation in various ways. In E. coli, the newly replicated initiator (dnaA) promoter and the origin (oriC) are silenced (sequestered) by the SeqA protein, which prevents their reuse for a significant period of the cell cycle. In C. crecsentus, the hemimethylated origin and the initiator promoter are also less active but the mechanisms remain unclear. In V. cholerae, we show that full methylation of oriII promotes initiator binding, providing a new role of the marks in replication initiation, and that SeqA is required for once-per-cell-cycle replication from both the origins (oriI and oriII), as in the case of oriC. By contributing to both initiation and its regulation, methylation thus serves two fundamental requirements for genome maintenance in V. cholerae. A comparison of oriII to plasmid origins also allowed us to address how a plasmid origin could have evolved to drive a chromosome in a cell-cycle specific fashion. We elaborate on these issues below.
Methylation and oriI
Our work started by questioning the essentiality of Dam and SeqA for functioning of oriI since a similar origin, oriC, can do without them [18]. We find that the requirements are not real for oriI but were imposed due to the use of plasmids to check the origin function. When we replaced oriC in the E. coli chromosome with oriI, making it the only origin in the cell, both the dam and seqA genes could be deleted (Figure 1). Thus for the functioning of oriI and oriC, methylation is not essential but it improves chromosomal replication initiation and its control (Figure 2A–2D), including the ability to tolerate extra copies of the origin in trans (Figure 1). In bacteria such as Bacillus subtilis that are naturally devoid of the methylation system, ori plasmids can exert an inhibitory effect (incompatibility) on chromosomal replication [32]. Methylation thus can help bacterial survival in a competitive situation.
Methylation and oriII
Dam plays a previously unrecognized role for oriII. It significantly promotes binding of the chrII-specific initiator, RctB, to the origin, thus possibly serving an essential function (Figure 3). Origin methylation is known to be essential for replication of C. crescentus chromosome, and of plasmids P1 and ColV-K30 in E. coli [33], [34], [35]. The reason is not clear in these cases, but unlikely to be for initiator binding. The initiator binding sites in these systems lack the sequences required for methylation. In contrast, RctB binding sites have an internal Dam recognition site, and methylation of the sites is required for initiator binding (Figure 3 and Figure S1). Thus, for oriII, the mechanism whereby methylation could be essential for its function and, therefore, for the bacterial survival is clear.
SeqA and once-per-cell-cycle replication of oriI and oriII
We show that seqA is not an essential gene in V. cholerae by obtaining viable seqA deletion mutants of V. cholerae. Although earlier studies suggested the gene to be essential, the finding that both oriI and oriII could function without SeqA in E. coli encouraged us to attempt isolation of the deletion mutants [18], [28]. In a deletion mutant, the number of both oriI and oriII per cell was found to be greater than in the WT (Figure 8). The overreplication indicates a breakdown of once-per-cell-cycle replication and reveals that SeqA is a negative regulator of replication. The latter was also concluded when the role of SeqA was studied by SeqA overproduction [28]. There was also an increase in the number of cells with odd number of origins for both the chromosomes, indicating loss of initiation synchrony. Thus, SeqA appears to contribute to both once-per-cell-cycle replication and initiation synchrony.
Hemimethylation periods of oriC, oriI, and oriII
An unexpected finding of this study is that the hemimethylation period of oriI and oriC changed in opposite ways upon seqA deletion: for oriC it decreased whereas for oriI it increased (Figure 6 and Figure 7). The decrease in the case of oriC is expected since SeqA is believed to be the key factor that prolongs the period [22]. A significant increase of the period without requiring SeqA shows that there are other ways to prolong the period, and that SeqA can play an opposite role of shortening the period. The opposite roles of SeqA were seen in isogenic strains of both V. cholerae and E. coli, suggesting that the reason cannot be due to species-specific factors (Figure 6 and Figure 7). The period also changed in opposite ways for oriI and oriII in the same seqA mutants of V. cholerae. SeqA thus has the capacity to both increase and decrease the duration of the period.
SeqA binding to DNA is favored in GATC-dense areas [36], [37]. The density of GATC sites around the diagnostic GATC site happens to be quite different in the three origins. In particular, the diagnostic site in oriI is present in a relatively isolated position (Figure 7A). It is possible that the results therein might be site-specific and not representative of the entire origin.
Proteins other than SeqA that interact with origins can also explain the differences in the hemimethylated periods of the origins. DnaA is known to compete with SeqA for binding to some of the sites in oriC [38], and can significantly prolong the period even without SeqA [37]. Thus, DnaA is a likely candidate for prolonging the period for oriI in the absence of SeqA.
Upon seqA deletion, although the hemimethylation period changed oppositely for oriI and oriII, both the chromosomes over-replicated (Figure 8). The prolongation of the period thus may not always be diagnostic of the role of SeqA in the negative regulation of replication. As stated above, competition with DnaA for oriI binding could be another way for SeqA to exert its negative regulatory role [38]. The correlation of the prolongation of the period and the strength of negative regulation was also poor in the case of oriII. Although, the period reduced drastically in a seqA mutant, the corresponding relaxation of replication was modest (Figure 8). In oriII, the negative control is mediated primarily by limiting RctB, which apparently makes the contribution of sequestration to regulation less significant [19].
Plasmid versus chromosome replication
ChrII has many plasmid-like features including the organization of its origin. Plasmids generally initiate their replication randomly in the cell cycle and control it independently of the chromosome [16], [20], [31], [39]. Plasmid copy number can vary among individual cells due to replication error and unequal segregation. To maintain the mean copy number, plasmids adjust for fluctuations in copy number by replicating more in cells that receive fewer copies than the mean, and replicating less in cells with more copies than the mean. Thus, once-per-cell-cycle replication is not suited for the maintenance of plasmid copy number. We show here that unlike plasmids, chrII replicates once per cell cycle, like other bacterial chromosomes. The high density of GATC sites of oriII is not typical for plasmid origins but is a conserved feature of all sequenced strains of the family Vibrionaceae [18]. It appears that the involvement of methylation has rendered functioning of a plasmid-like origin similar to that of a chromosomal origin.
Why does initiation need to be cell-cycle specific for the chromosome? Completion of cell division demands that the septum forming area be cleared of DNA [40]. Plasmids are generally small and have correspondingly short replication elongation periods. Incompletely replicated plasmids are unlikely to cause steric hindrance to cell division for a significant period, unlike incompletely replicated chromosomes [41]. If chrII were to initiate replication randomly in the cell cycle like the plasmids, late-initiating chrII would likely delay cell division and create heterogeneity in cell generation times. V. cholerae ΔseqA cells did form elongated cells, indicative of a cell division defect (our unpublished results). One reason for this could be steric hindrance to cell division from late-initiating chrII. We suggest that a chromosome replicating from an origin with a plasmid provenance is subject to selection pressure to make the initiation cell-cycle specific, and the acquisition of methylation sites could allow that.
Methylation and bacteria with multiple chromosomes
Understanding the role of methylation can also be important for another reason. It has been suggested that one of the common conspicuous features of the two origins being the high density of GATC sites, their methylation could be a mechanism to coordinate the replication between the two chromosomes [18]. Methylation is essential for the viability of bacteria with multiple chromosomes such as Rhizobium meliloti [42], Brucella abortus [43] and Agrobacterium tumefasciens [44] in addition to V. cholerae [17]. Although there is no evidence yet for direct communication among the chromosomes for replication initiation in any system, it is possible that in these bacteria methylation could be coordinating the replication to the cell cycle, as is does for V. cholerae and possibly other members of the family of Vibrionaceae.
Materials and Methods
Bacterial strains, plasmids, and media
Bacterial strains and plasmids used in this study are listed in Table 1 and Table 2, respectively. Primers are listed in Text S1. E. coli and V. cholerae were grown in LB (10 g tryptone +5 g yeast extract +5 g NaCl per liter, pH adjusted with NaOH to ∼7) or M63 medium (KH2PO4 3 g + K2HPO4 7 g + (NH4)2SO4 2 g + FeSO4 0.5 mg + MgSO4.7H2O 0.25 g, pH adjusted with KOH to ∼7) supplemented with 2 mM MgSO4, 0.1 mM CaCl2, 0.01% thiamine and 0.2% glucose, and additionally 0.1% casamino acids when desired. Antibiotics were used at the following concentrations: ampicillin, 100 µg/ml; chloramphenicol, 25 µg/ml for E. coli, 5 µg/ml for V. cholerae; erythromycin, 20 µg/ml; kanamycin, 25 µg/ml; spectinomycin, 50µg/ml; tetracycline, 15 µg/ml; and zeocin, 25 µg/ml. Diaminopimelic acid (DAP) was used at 0.8 mM, L-arabinose at 2 or 0.2 mg/ml, IPTG at 100 µM and thymidine at 0.3 mM.
Recombineering in E. coli
To replace oriC (coordinates 3923756–3924022) with oriI (coordinates 2961130–364), the latter was amplified from DNA of CVC209 by PCR using primers GD113 and GD114. The PCR product was digested with EcoRI and BamHI, and ligated to similarly digested pEM7-Zeo. The resulting plasmid, pGD83, was digested with SacI and BamHI, and the fragment containing the oriI-zeo region was ligated to a similarly digested vector, pSW23, generating pGD79. The oriI-zeo region of pGD79 was amplified with primers GD124 and GD125, and the product used to replace oriC of CVC1394 by the mini-λ Red recombineering method [45]. The mini-λ prophage was eliminated from the strain by a 30°C to 42°C temperature shift. The resultant strain was called MG1655ΔoriC::oriI-zeo (CVC1400), and the replacement was confirmed by sequencing of the origin region. The genomic DNA of the dam mutant derivative (CVC1401) was confirmed for the absence of adenine methylation by its resistance to DpnI but not to MboI and BfuCI restriction enzymes (data not shown).
Flow cytometry
Cultures of E. coli were grown in LB to OD600≈0.2 and processed for flow cytometry after replication run-out in the presence of rifampicin (150 µg/ml) and cephalexin (10 µg/ml) for three hours as described [46]. The peak fluorescence intensity of an overnight grown E. coli culture in M63 + 0.2% glucose medium (without casamino acids) was taken to represent one genome equivalent.
Electrophoretic mobility shift assay
A fragment with six 12-mers was obtained from pGD61 by digestion with XhoI and NotI.
Fragments with three 11-mers and a pair of 12 - and 11-mers were obtained from pTVC86 and pTVC88, respectively, by digestion with XhoI and BamHI. For methylated and unmethylated fragments, the plasmids were from a dam+ (BR2699) and a dam− (CVC1060) strain, respectively. The fragments were gel-purified, dephosphorylated with Shrimp Alkaline Phosphatase (USB Corporation), and end-labeled with 50 µCi [γ-32P]ATP (PerkinElmer) by using 30 units of T4 polynucleotide kinase (New England Biolabs) and purified through ProbeQuant G-50 micro columns (GE Healthcare). To obtain hemimethylated DNA, oligonucleotide primers, TVC64 and TVC138 (Sigma-Genosys), were end-labeled and purified as above. The labeled primers were then used for PCR one at a time with methylated DNA as template for one cycle to obtain two populations of hemimethylated DNA, one with methylation on the top strand and the other on the bottom strand. The binding reactions were essentially as described [47].
seqA deletion
A partial deletion of seqA was made by deleting codons 51 to 140 and substituting the deleted region with a zeocin cassette maintaining the seqA reading frame as follows. The seqA gene was amplified from CVC209 by PCR with primers GD87 and GD88. The product was digested with EcoRI and cloned in similarly digested vector, pSW4426T. The resultant plasmid was used as template for PCR with primers GD91 and GD92 to amplify the 5′ end of seqA, the plasmid backbone and the 3′end of seqA. After digestion with MfeI, a site of which was present within GD91 and GD92 primers, the PCR product was ligated to the zeocin cassette. The cassette was obtained from pEM7-Zeo by PCR, using primers GD89 and GD90 and digested with EcoRI before ligating to the MfeI fragment. The resulting plasmid, pGD70, containing the ΔseqAP::zeo allele was used to replace seqA of CVC209 by the allele-exchange method [48]. The resulting ΔseqAP::zeo mutant (CVC1410) grew slower than the WT. In LB at 37°C, the doubling times of the mutant was 32±2 min as opposed to 19±2 min for the WT. The ΔseqAP::zeo allele is called hereafter ΔseqAP.
The entire seqA ORF was also deleted and substituted with the zeocin cassette as follows. First, a kilobase region located downstream the stop codon of seqA was amplified by PCR with primers GD228 and GD229, the product digested with EcoRI and BamHI and cloned in a derivate of pEM7-Zeo (pGD111), previously digested with the same enzymes, generating pGD113. pGD111 is essentially same as pEM7-Zeo except that the multi-cloning site upstream of zeo is modified to include KpnI and NdeI restriction sites. Next, a kilobase region located upstream of the start codon of seqA was amplified by PCR with primers GD230 and GD231, the product digested with KpnI and NdeI and cloned in pGD113, previously digested with the same enzymes, generating pGD114. The flanking regions of seqA, now flanking the zeocin cassette, was amplified by PCR with primers GD257 and GD258 and the linear product was introduced by natural transformation in a hapR+ Δdns derivative of N16961 (CVC1121) essentially as described [49], [50]. The transformants were selected for zeocin resistance and checked for the replacement of the seqA gene by the zeocin cassette by PCR and DNA sequencing. The resulting ΔseqAT::zeo mutant (CVC2003) grew as slow as the ΔseqAP::zeo mutant with a doubling time of 32±2 min. The ΔseqAT::zeo allele is called hereafter ΔseqAT.
dam depletion
A complete deletion of the dam ORF and its substitution with a zeocin cassette was obtained by the allele-exchange method in the presence of a complementing plasmid, pGD93. The replication of the plasmid was thermo-sensitive and it carried the V. cholerae dam under the PBAD promoter. pGD93 was made as follows: the dam gene was amplified by PCR with primers GD72 and GD73, and the product after digestion with EcoRI and KpnI was cloned in pBAD24, previously digested with the same enzymes, generating pGD55. Next, the NdeI-HindIII fragment from pGD55 containing the dam gene was cloned in pKOBEGA, previously digested by NdeI and HindIII, generating the pGD93. For allele-exchange, a kilobase region located downstream of the stop codon of dam was amplified by PCR with primers GD261 and GD262, and the product after digestion with EcoRI and BamHI was cloned in a derivative of pEM7-zeo, previously digested with the same enzymes, generating pGD117. A 700 bp region located upstream of the start codon of dam was amplified by PCR with primers GD263 and GD264, and the product after digestion with KpnI and NdeI was cloned in pGD117, previously digested with the same enzymes, generating pGD118. The zeocin cassette with the flanking regions of dam was amplified by PCR with primers GD268 and GD269, and the product cloned as a blunt end fragment in pSW23, previously digested with SmaI, generating the pGD120. The plasmid was digested with SacI and SalI, and the fragment with the zeocin cassette was cloned into pDS132, previously digested also with the same enzymes. The resulting plasmid, pGD121, was used to replace dam of CVC209/pGD93. The resulting strain, CVC2023, was confirmed for the replacement of dam by the zeocin cassette by PCR and by DNA sequencing. To deplete Dam, single colonies grown in the presence of ampicillin (to select pGD93) and arabinose (to express dam) were used to inoculate LB without any drug but containing glucose (to repress dam expression) and the cultures were grown at 42°C (to stop plasmid replication).
Southern blotting
Genomic DNA was isolated from cells of log phase cultures (OD600≈0.3), using the Genelute Bacterial Genomic DNA kit (Sigma). For analyzing chrI and E. coli DNA, 1 µg of DNA was digested 2 hours with 7.5 or 15 units of HphI (New England Biolabs) at 37°C, and the products resolved in a 1.5% agarose gel. For chrII, the conditions were similar except that TaqαI was used at 65°C. The origin probes were prepared by PCR using primers GD36 and GD37 for oriI, GD40 and GD41 for oriII, GD67 and GD68 for oriC, and GD150 and GD151 for ΔoriC::oriI. The primers for external markers on the three chromosomes were GD38 and GD39, GD42 and GD43, and GD128 and GD129, respectively. The probes for ori and the external markers were made radioactive using the RediPrimeII random primer labeling kit (GE Healthcare) and [α-32P] dCTP (PerkingElmer) and mixed separately for the two chromosomes. The band intensities were recorded and quantified as described earlier [46].
Marker frequency determination
Marker frequency was determined by qPCR using a PTC-200 Peltier Thermal Cycler (MJ Research) and a LightCycler 480 SYBR Green I Master (Roche). Genomic DNA was prepared from log phase cultures in LB with Genelute Bacterial Genomic DNA kit (Sigma), and 313 pg was used in each reaction as template. The primers were used at 0.3 µM each. They were proximal to either oriI (GD136 and GD137) or oriII (GD156 and GD157) or terI (GD142 and GD143) or terII (GD140 and GD141) region of the two chromosomes, and were identical to those described [39]. The primer pairs were such that they produced ∼100 to 130 bp fragments in all cases. Cp (crossing point) values were determined and used for calculating the oriI/oriII, oriI/terI, oriII/terII and terI/terII ratios. The ratios were normalized to those of a culture grown to stationary phase in supplemented M63 medium (without casamino acids). Mean ratios were obtained from DNA prepared from three cultures, each grown from independent colonies, and each DNA was analyzed in triplicate.
Chromatin immunoprecipitation
The method was modified from the one described by Lin and Grossman [51]. Briefly, cultures at OD600nm = 0.3 were treated with 1% formaldehyde at room temperature for 30 min. After cell lysis and sonication, RctB complexes were precipitated with antibody against RctB (IP DNA) and Dynabeads-Protein G magnetic beads (Invitrogen), followed by stringent washings (see Text S1 for the detailed ChIP protocol). After reversal of the cross-links by incubation at 65°C overnight, the samples were treated by protease K (Sigma) and then purified with a PCR purification Kit (Qiagen). To quantify the enrichment of RctB binding sites in the IP DNA, 5 µl of 1∶100 dilution of the IP DNA was used to perform locus-specific real-time qPCR with primers GD218 and GD219, specific to the vector backbone of the plasmid carrying the RctB binding sites, and primers GD191 and GD192, specific to a gene in the E. coli genome that served as a reference, as described [52].
Supporting Information
Zdroje
1. SuzukiMM
BirdA
2008 DNA methylation landscapes: provocative insights from epigenomics. Nat Rev Genet 9 465 476
2. MarinusMG
1987 DNA methylation in Escherichia coli. Annu Rev Genet 21 113 131
3. LowDA
CasadesusJ
2008 Clocks and switches: bacterial gene regulation by DNA adenine methylation. Curr Opin Microbiol 11 106 112
4. Lφbner-OlesenA
SkovgaardO
MarinusMG
2005 Dam methylation: coordinating cellular processes. Curr Opin Microbiol 8 154 160
5. CasadesusJ
LowD
2006 Epigenetic gene regulation in the bacterial world. Microbiol Mol Biol Rev 70 830 856
6. EngelJD
von HippelPH
1978 Effects of methylation on the stability of nucleic acid conformations. Studies at the polymer level. J Biol Chem 253 927 934
7. CollinsM
MyersRM
1987 Alterations in DNA helix stability due to base modifications can be evaluated using denaturing gradient gel electrophoresis. J Mol Biol 198 737 744
8. PolaczekP
KwanK
CampbellJL
1998 GATC motifs may alter the conformation of DNA depending on sequence context and N6-adenine methylation status: possible implications for DNA-protein recognition. Mol Gen Genet 258 488 493
9. CollierJ
McAdamsHH
ShapiroL
2007 A DNA methylation ratchet governs progression through a bacterial cell cycle. Proc Natl Acad Sci U S A 104 17111 17116
10. AbelesA
BrendlerT
AustinS
1993 Evidence of two levels of control of P1 oriR and host oriC replication origins by DNA adenine methylation. J Bacteriol 175 7801 7807
11. NielsenO
Lφbner-OlesenA
2008 Once in a lifetime: strategies for preventing re-replication in prokaryotic and eukaryotic cells. EMBO Rep 9 151 156
12. WaldminghausT
SkarstadK
2009 The Escherichia coli SeqA protein. Plasmid 61 141 150
13. Lφbner-OlesenA
HansenFG
RasmussenKV
MartinB
KuempelPL
1994 The initiation cascade for chromosome replication in wild-type and Dam methyltransferase deficient Escherichia coli cells. EMBO J 13 1856 1862
14. Lφbner-OlesenA
von FreieslebenU
1996 Chromosomal replication incompatibility in Dam methyltransferase deficient Escherichia coli cells. EMBO J 15 5999 6008
15. SkarstadK
Lφbner-OlesenA
2003 Stable co-existence of separate replicons in Escherichia coli is dependent on once-per-cell-cycle initiation. EMBO J 22 140 150
16. PaulssonJ
ChattorajDK
2006 Origin inactivation in bacterial DNA replication control. Mol Microbiol 61 9 15
17. JulioSM
HeithoffDM
ProvenzanoD
KloseKE
SinsheimerRL
2001 DNA adenine methylase is essential for viability and plays a role in the pathogenesis of Yersinia pseudotuberculosis and Vibrio cholerae. Infect Immun 69 7610 7615
18. EganES
WaldorMK
2003 Distinct replication requirements for the two Vibrio cholerae chromosomes. Cell 114 521 530
19. DuigouS
KnudsenKG
SkovgaardO
EganES
Lφbner-OlesenA
2006 Independent Control of Replication Initiation of the Two Vibrio cholerae Chromosomes by DnaA and RctB. J Bacteriol 188 6419 6424
20. LeonardAC
HelmstetterCE
1988 Replication patterns of multiple plasmids coexisting in Escherichia coli. J Bacteriol 170 1380 1383
21. HeidelbergJF
EisenJA
NelsonWC
ClaytonRA
GwinnML
2000 DNA sequence of both chromosomes of the cholera pathogen Vibrio cholerae. Nature 406 477 483
22. LuM
CampbellJL
BoyeE
KlecknerN
1994 SeqA: a negative modulator of replication initiation in E. coli. Cell 77 413 426
23. ParkerB
MarinusMG
1988 A simple and rapid method to obtain substitution mutations in Escherichia coli: isolation of a dam deletion/insertion mutation. Gene 73 531 535
24. BatesDB
AsaiT
CaoY
ChambersMW
CadwellGW
1995 The DnaA box R4 in the minimal oriC is dispensable for initiation of Escherichia coli chromosome replication. Nucleic Acids Res 23 3119 3125
25. SkarstadK
BernanderR
BoyeE
1995 Analysis of DNA replication in vivo by flow cytometry. Methods Enzymol 262 604 613
26. BoyeE
Lφbner-OlesenA
1990 The role of dam methyltransferase in the control of DNA replication in E. coli. Cell 62 981 989
27. CampbellJL
KlecknerN
1990 E. coli oriC and the dnaA gene promoter are sequestered from dam methyltransferase following the passage of the chromosomal replication fork. Cell 62 967 979
28. Saint-DicD
KehrlJ
FrushourB
KahngLS
2008 Excess SeqA leads to replication arrest and a cell division defect in Vibrio cholerae. J Bacteriol 190 5870 5878
29. SlaterS
WoldS
LuM
BoyeE
SkarstadK
1995 E. coli SeqA protein binds oriC in two different methyl-modulated reactions appropriate to its roles in DNA replication initiation and origin sequestration. Cell 82 927 936
30. NielsenHJ
LiY
YoungrenB
HansenFG
AustinS
2006 Progressive segregation of the Escherichia coli chromosome. Mol Microbiol 61 383 393
31. SrivastavaP
ChattorajDK
2007 Selective chromosome amplification in Vibrio cholerae. Mol Microbiol 66 1016 1028
32. MoriyaS
AtlungT
HansenFG
YoshikawaH
OgasawaraN
1992 Cloning of an autonomously replicating sequence (ars) from the Bacillus subtilis chromosome. Mol Microbiol 6 309 315
33. ReisenauerA
KahngLS
McCollumS
ShapiroL
1999 Bacterial DNA methylation: a cell cycle regulator? J Bacteriol 181 5135 5139
34. AbelesAL
AustinSJ
1987 P1 plasmid replication requires methylated DNA. EMBO J 6 3185 3189
35. GammieAE
CrosaJH
1991 Roles of DNA adenine methylation in controlling replication of the REPI replicon of plasmid pColV-K30. Mol Microbiol 5 495 503
36. BrendlerT
AustinS
1999 Binding of SeqA protein to DNA requires interaction between two or more complexes bound to separate hemimethylated GATC sequences. EMBO J 18 2304 2310
37. BachT
Morigen
SkarstadK
2008 The initiator protein DnaA contributes to keeping new origins inactivated by promoting the presence of hemimethylated DNA. J Mol Biol 384 1076 1085
38. NieveraC
TorgueJJ
GrimwadeJE
LeonardAC
2006 SeqA blocking of DnaA-oriC interactions ensures staged assembly of the E. coli pre-RC. Mol Cell 24 581 592
39. RasmussenT
JensenRB
SkovgaardO
2007 The two chromosomes of Vibrio cholerae are initiated at different time points in the cell cycle. EMBO J 26 3124 3131
40. MargolinW
2005 FtsZ and the division of prokaryotic cells and organelles. Nat Rev Mol Cell Biol 6 862 871
41. BotelloE
NordströmK
1998 Effects of chromosome underreplication on cell division in Escherichia coli. J Bacteriol 180 6364 6374
42. WrightR
StephensC
ShapiroL
1997 The CcrM DNA methyltransferase is widespread in the alpha subdivision of proteobacteria, and its essential functions are conserved in Rhizobium meliloti and Caulobacter crescentus. J Bacteriol 179 5869 5877
43. RobertsonGT
ReisenauerA
WrightR
JensenRB
JensenA
2000 The Brucella abortus CcrM DNA methyltransferase is essential for viability, and its overexpression attenuates intracellular replication in murine macrophages. J Bacteriol 182 3482 3489
44. KahngLS
ShapiroL
2001 The CcrM DNA methyltransferase of Agrobacterium tumefaciens is essential, and its activity is cell cycle regulated. J Bacteriol 183 3065 3075
45. CourtDL
SwaminathanS
YuD
WilsonH
BakerT
2003 Mini-lambda: a tractable system for chromosome and BAC engineering. Gene 315 63 69
46. SrivastavaP
FeketeRA
ChattorajDK
2006 Segregation of the replication terminus of the two Vibrio cholerae chromosomes. J Bacteriol 188 1060 1070
47. Venkova-CanovaT
SrivastavaP
ChattorajDK
2006 Transcriptional inactivation of a regulatory site for replication of Vibrio cholerae chromosome II. Proc Natl Acad Sci U S A 103 12051 12056
48. Le RouxF
BinesseJ
SaulnierD
MazelD
2007 Construction of a Vibrio splendidus mutant lacking the metalloprotease gene vsm by use of a novel counterselectable suicide vector. Appl Environ Microbiol 73 777 784
49. BlokeschM
SchoolnikGK
2008 The extracellular nuclease Dns and its role in natural transformation of Vibrio cholerae. J Bacteriol 190 7232 7240
50. BlokeschM
SchoolnikGK
2007 Serogroup conversion of Vibrio cholerae in aquatic reservoirs. PLoS Pathog 3 e81 doi:10.1371/journal.ppat.0030081
51. LinDC
GrossmanAD
1998 Identification and characterization of a bacterial chromosome partitioning site. Cell 92 675 685
52. LeeC
KimJ
ShinSG
HwangS
2006 Absolute and relative QPCR quantification of plasmid copy number in Escherichia coli. J Biotechnol 123 273 280
53. GrantSG
JesseeJ
BloomFR
HanahanD
1990 Differential plasmid rescue from transgenic mouse DNAs into Escherichia coli methylation-restriction mutants. Proc Natl Acad Sci U S A 87 4645 4649
54. DemarreG
GueroutAM
Matsumoto-MashimoC
Rowe-MagnusDA
MarliereP
2005 A new family of mobilizable suicide plasmids based on broad host range R388 plasmid (IncW) and RP4 plasmid (IncPalpha) conjugative machineries and their cognate Escherichia coli host strains. Res Microbiol 156 245 255
55. GuzmanLM
BelinD
CarsonMJ
BeckwithJ
1995 Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol 177 4121 4130
56. PhilippeN
AlcarazJP
CoursangeE
GeiselmannJ
SchneiderD
2004 Improvement of pCVD442, a suicide plasmid for gene allele exchange in bacteria. Plasmid 51 246 255
57. EganES
Lφbner-OlesenA
WaldorMK
2004 Synchronous replication initiation of the two Vibrio cholerae chromosomes. Curr Biol 14 R501 502
58. ChaverocheMK
GhigoJM
d'EnfertC
2000 A rapid method for efficient gene replacement in the filamentous fungus Aspergillus nidulans. Nucleic Acids Res 28 E97
59. PalD
Venkova-CanovaT
SrivastavaP
ChattorajDK
2005 Multipartite regulation of rctB, the replication initiator gene of Vibrio cholerae chromosome II. J Bacteriol 187 7167 7175
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2010 Číslo 5
Nejčtenější v tomto čísle
- Common Genetic Variants near the Brittle Cornea Syndrome Locus Influence the Blinding Disease Risk Factor Central Corneal Thickness
- All About Mitochondrial Eve: An Interview with Rebecca Cann
- Aging and Chronic Sun Exposure Cause Distinct Epigenetic Changes in Human Skin
- The Relationship among Gene Expression, the Evolution of Gene Dosage, and the Rate of Protein Evolution