
Involvement of Global Genome Repair, Transcription Coupled Repair, and Chromatin Remodeling in UV DNA Damage Response Changes during Development
Nucleotide Excision Repair (NER), which removes a variety of helix-distorting lesions from DNA, is initiated by two distinct DNA damage-sensing mechanisms. Transcription Coupled Repair (TCR) removes damage from the active strand of transcribed genes and depends on the SWI/SNF family protein CSB. Global Genome Repair (GGR) removes damage present elsewhere in the genome and depends on damage recognition by the XPC/RAD23/Centrin2 complex. Currently, it is not well understood to what extent both pathways contribute to genome maintenance and cell survival in a developing organism exposed to UV light. Here, we show that eukaryotic NER, initiated by two distinct subpathways, is well conserved in the nematode Caenorhabditis elegans. In C. elegans, involvement of TCR and GGR in the UV-induced DNA damage response changes during development. In germ cells and early embryos, we find that GGR is the major pathway contributing to normal development and survival after UV irradiation, whereas in later developmental stages TCR is predominantly engaged. Furthermore, we identify four ISWI/Cohesin and four SWI/SNF family chromatin remodeling factors that are implicated in the UV damage response in a developmental stage dependent manner. These in vivo studies strongly suggest that involvement of different repair pathways and chromatin remodeling proteins in UV-induced DNA repair depends on developmental stage of cells.
Published in the journal:
. PLoS Genet 6(5): e32767. doi:10.1371/journal.pgen.1000941
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1000941
Summary
Nucleotide Excision Repair (NER), which removes a variety of helix-distorting lesions from DNA, is initiated by two distinct DNA damage-sensing mechanisms. Transcription Coupled Repair (TCR) removes damage from the active strand of transcribed genes and depends on the SWI/SNF family protein CSB. Global Genome Repair (GGR) removes damage present elsewhere in the genome and depends on damage recognition by the XPC/RAD23/Centrin2 complex. Currently, it is not well understood to what extent both pathways contribute to genome maintenance and cell survival in a developing organism exposed to UV light. Here, we show that eukaryotic NER, initiated by two distinct subpathways, is well conserved in the nematode Caenorhabditis elegans. In C. elegans, involvement of TCR and GGR in the UV-induced DNA damage response changes during development. In germ cells and early embryos, we find that GGR is the major pathway contributing to normal development and survival after UV irradiation, whereas in later developmental stages TCR is predominantly engaged. Furthermore, we identify four ISWI/Cohesin and four SWI/SNF family chromatin remodeling factors that are implicated in the UV damage response in a developmental stage dependent manner. These in vivo studies strongly suggest that involvement of different repair pathways and chromatin remodeling proteins in UV-induced DNA repair depends on developmental stage of cells.
Introduction
A network of DNA damage response (DDR) mechanisms protects organisms against the continuous genotoxic stress induced by reactive metabolites and other genotoxic agents, such as environmental contaminants and ultraviolet (UV) radiation from the sun [1]. The DDR network consists of several DNA repair mechanisms, cell cycle checkpoints and cellular senescence and apoptotic signaling cascades. Nucleotide Excision Repair (NER) is a DNA repair mechanism that is able to remove a wide variety of helix-destabilizing DNA lesions including those induced by UV light.
Eukaryotic NER is a highly conserved multi-step process, involving more than 25 proteins, of which the principal molecular mechanism has been dissected in detail [1], [2]. NER is initiated by two distinct DNA damage recognition mechanisms which use the same machinery to repair the damage. Damage in the transcribed strand of active genes is repaired by Transcription Coupled Repair (TCR), which depends on recruitment of the ATP-dependent chromatin remodeling protein Cockayne Syndrome protein B (CSB) and the WD40 domain containing protein Cockayne Syndrome protein A (CSA) to the site of damage [3]–[5]. TCR is thought to be activated by stalling of elongating RNA polymerase II during transcription [3], [6]. Damage in other, non-transcribed sequences of the genome is repaired by Global Genome Repair (GGR), which requires detection of the lesions by the UV-damaged DNA-binding protein (UV-DDB) complex and a complex containing Xeroderma Pigmentosum group C protein (XPC), human homolog of RAD23 (hHR23) and Centrin-2 [7]–[9]. The XPC protein is essential for the initiation of GGR and subsequent recruitment of other NER factors [10], [11]. The majority of XPC is found in complex with the hHR23B protein, while only a fraction copurifies with the redundant hHR23A protein. Both hHR23 proteins are thought to stabilize XPC and stimulate its function [12]–[14]. Although HR23B is not essential for in vitro NER, in vivo damage is poorly repaired in cells lacking hHR23B [12], indicating that hHR23B is essential for proper NER function. Following detection of a lesion, either via GGR or TCR, the transcription factor IIH (TFIIH) is recruited to open the DNA helix around the damage in an ATP-dependent manner using its Xeroderma Pigmentosum group B and D (XPB and XPD) helicase subunits [1], [2]. Next, Xeroderma Pigmentosum group A (XPA) and Replication Protein A (RPA) are recruited to stabilize the repair complex and properly orient the structure-specific endonucleases Xeroderma Pigmentosum group F (XPF)/Excision Repair Cross-Complementing protein 1 (ERCC1) and Xeroderma Pigmentosum group G (XPG) to excise the damaged strand. The resulting ∼30 nt single strand DNA gap is filled by DNA synthesis and ligation.
In mammals, congenital defects in GGR and TCR lead to an increased sensitivity towards DNA damaging agents such as UV irradiation. Inherited mutations in GGR genes cause Xeroderma Pigmentosum, which is characterized by extreme UV-sensitivity and skin cancer predisposition [15]. Hereditary TCR deficiency causes Cockayne syndrome, which leads to entirely different features such as severe but variable neurodevelopmental symptoms and premature aging. In contrast to mammals, specific TCR defects in yeast have only a marginal effect on DNA damage resistance, despite a relatively larger proportion of the genome that is transcriptionally active [16].
Current knowledge of NER does not provide an explanation for the pleiotropic phenotypic expression of NER-deficiencies. Despite detailed insight in the molecular mechanism of NER, many aspects of the in vivo UV-induced DNA damage response (UV-DDR) are still unclear. It is for instance not well understood how NER functions in nucleosomal DNA and in different tissues of developing organisms. Therefore, a full understanding of the complete UV-DDR and its interplay with NER in living organisms is imperative. The nematode C. elegans seems well suited to analyze the complete UV-DDR in vivo in more detail, because of its short lifetime, well-characterized biology and its amenable use to identify new genes involved in the UV-DDR. Several studies have specifically addressed the role of NER proteins in the UV-DDR in C. elegans. Knockdown of the C. elegans orthologs of mammalian CSB, XPA and XPF increases sensitivity to UV irradiation [17]–[21]. Furthermore, it was shown that the XPA and XPC orthologs function in the C. elegans germ line to induce cell cycle arrest and apoptosis in response to UV irradiation [22]. Together, these studies suggest that NER function is highly conserved in C. elegans. However, a thorough analysis of the function of NER and, more specifically, the role of the GGR and TCR subpathways in response to UV irradiation in different tissues during development has not been performed.
In this study, we make use of mutations in the C. elegans RAD23, XPC and CSB orthologs to show that during early development, in germ cells and embryos, GGR is the major pathway involved in the response to UV irradiation. Defective GGR leads to inefficient lesion removal in germ cells, specific defects in germ cell development and embryonic death after UV irradiation. Intriguingly, in juvenile and adult animals TCR is the major NER pathway involved in the UV response. Analysis of the UV response of embryos shows that, during development, TCR gradually becomes more important than GGR. Finally, we exploit C. elegans to identify novel genes involved in the UV-DDR, specifically in the TCR-related UV response. Our results reveal four genes implicated in SWI/SNF and four genes implicated in ISWI ATP-dependent chromatin remodeling whose involvement in the UV-DDR changes during development.
Results
General NER–deficient and GGR – and TCR–specific mutants
To study the UV-DDR in the context of a whole organism, we tested UV-B sensitivity of mutant C. elegans at different developmental stages. Our initial experiments showed that UV-B irradiation produced better reproducible phenotypes than UV-C irradiation (data not shown), most likely due to the fact that UV-B penetrates deeper through the multiple cell layers of C. elegans. First, we tested UV sensitivity of animals carrying mutations in the general NER genes xpa-1, xpg-1, xpf-1 and ercc-1. Alleles of xpa-1 and xpf-1, but not xpg-1 and ercc-1, have been previously described. xpa-1(ok698) encodes a putative null allele of the C. elegans ortholog of mammalian XPA and was shown to cause severe sensitivity to UV irradiation [20], [22]. him-9(e1487) is an allele of xpf-1, encoding the C. elegans ortholog of mammalian endonuclease XPF [23]. tm1682 and tm1670 are two alleles of xpg-1, the ortholog of the mammalian endonuclease XPG and have not been described before. tm1682 represents a deletion of the first two exons of xpg-1, probably creating a knock-out allele, but also of part of the last exon of the adjacent glycosyl hydrolase gene tre-1 (Figure 1A). Thus, to rule out an effect of tre-1, in our analysis we also included tm1670, which represents a deletion that is predicted to remove exon 2 and a large part of exon 3, encoding for a truncated 679 amino acids in stead of 829 amino acids protein (Figure 1A). Since most of the N-terminal nuclease domain is deleted, the resulting protein is expected to be non-functional. tm2073 represents a deletion in the conserved Rad10 domain of ercc-1, the C. elegans ortholog of mammalian ERCC1 which is in complex with XPF, and is predicted to encode a loss-of-function allele (Figure 1B).

To address the specific contribution of the TCR and GGR pathways in the UV-induced DDR in vivo, we also analyzed C. elegans strains carrying mutations expected to affect either pathway specifically. The genome of C. elegans encodes an ortholog of the GGR-specific mammalian HR23A and HR23B genes, called rad-23. This gene is predicted to encode for a 372 amino acids protein having similar domain organization as mammalian HR23A and HR23B proteins (Figure 1C; [24]). The rad-23(tm2595) allele represents a deletion of the major parts of exon 2 and exon 3 and an insertion of 28 basepairs. Since tm2595 deletes both UBA domains and the XPC binding domain and is predicted to encode a truncated protein of 96 aa, this allele is likely a functional null allele.
The TCR-specific mammalian CSB gene is represented in C. elegans by csb-1 [17]. This gene encodes a 957 amino acids protein containing a SNF2-like ATPase domain, similar to human CSB (Figure 1D). The csb-1(ok2335) allele consists of a 1620 bp deletion which removes exon 5 and 6 and the largest parts of exon 4 and 7. This allele is predicted to encode a truncated protein of 513 amino acids, which is likely a functional null since most of the SNF2 domain is deleted.
GGR but not TCR is essential for survival of UV-irradiated germ cells
To test UV sensitivity of germ cells, adult animals were irradiated and allowed to recover for 24 hours, after which they were put on fresh plates to lay eggs for 3–4 hours (Figure S1A). ‘Germ cell and embryo survival’ was measured by determining the percentage of eggs that hatched over the total amount of eggs laid. As expected, we found that the core NER factors xpa-1, xpg-1, xpf-1 and ercc-1 were necessary for germ cells and embryos to survive even relatively low doses of UV irradiation (Figure 2A and 2B).

Next, we tested UV sensitivity of rad-23(tm2595) and csb-1(ok2335) mutants. Functional rad-23 appeared to contribute only partially to UV resistance (compared to xpa-1), whereas, surprisingly, csb-1 did not seem to contribute at all (Figure 2C). Similar results were obtained using eggs laid immediately after irradiation or after different recovery periods up to 51 hrs after irradiation (data not shown). This suggests that a similar UV response, involving general NER factors and rad-23, but not csb-1, acts in all developing germ cells, oocytes and early embryos.
The specific contribution of rad-23 but not csb-1 suggests that germ and early embryonic cells depend mainly on the GGR pathway of NER to overcome the effects of UV irradiation. Alternatively, it could be that csb-1 is not involved in TCR in C. elegans or that TCR defects are not associated with UV sensitivity. To test whether GGR and TCR act redundantly in the germ line, or whether csb-1 is not involved in UV-damage repair or survival, we generated animals carrying mutations in both rad-23 and csb-1. Irradiation of rad-23; csb-1 double mutants showed that these animals are more UV sensitive than rad-23 single mutants and as sensitive as animals carrying mutations in general NER genes (Figure 2C). This suggests that both TCR and GGR are active in germ cells.
In mammals, RAD23 functions in GGR as part of a heterotrimeric complex containing also Centrin-2 [7], [25] and XPC [8], [26]. The genome of C. elegans contains an ortholog of XPC, xpc-1, for which only recently, during the course of our experiments, a good loss-of-function allele became available. This allele, tm3886, represents a 24 bp insertion and 474 bp deletion in exon 2, probably causing a truncated protein (Figure 1E). To confirm that the specific UV sensitivity of rad-23 is caused by a defect in GGR, we tested the phenotype of the novel xpc-1 mutation. xpc-1 single mutants showed a similar UV sensitivity in the germ line as rad-23 single and rad-23; xpc-1 double mutants, whereas xpc-1; csb-1 double mutants were as UV sensitive as rad-23; csb-1 double mutants (Figure 2D). These results are in line with our previous findings and with the idea that in C. elegans, similar as in mammals, RAD-23 and XPC-1 function in complex during GGR.
Based on our results (summarized in Table 1), we hypothesize that in the germ line GGR plays an essential role in UV survival, whereas TCR only has a secondary, partially redundant function to GGR (Figure 2E). Furthermore, our results are in agreement with the idea that, similar as in mammals, rad-23/xpc-1 and csb-1 act in parallel pathways, GGR and TCR, that converge on a common pathway to repair DNA damage.

GGR maintains germ cells in response to UV irradiation
Previously, it was found that ionizing and UV irradiation both induce apoptosis of a fraction of the pachytene germ cells of C. elegans [22], [27], which are located near the gonad tube bend (Figure 3B, first image). Functional xpa-1 was shown to be required for induction of apoptosis [22], [27], suggesting that the NER process itself is necessary to activate the apoptotic machinery. To test whether induction of germ cell apoptosis requires functional GGR or TCR, we measured induction of apoptosis in the pachytene germ cells of wild type, xpa-1, rad-23, csb-1 and rad-23;csb-1 mutants in response to UVB irradiation. In contrast to wild type animals, xpa-1 mutants exhibited severely reduced apoptosis induction after UVB, as observed previously (Figure 3A; [22]). Furthermore, we found that in csb-1 mutants apoptosis was induced at wild type levels, whereas in rad-23 mutants apoptosis induction was mildly reduced. No apoptosis induction after UV irradiation was observed in rad-23; csb-1 double mutants, similar as in xpa-1 mutants. These results indicate that both the GGR and the TCR pathway are required to induce germ cell apoptosis in response to UV. Together with the mild decrease in apoptosis induction in rad-23 mutants, this is in line with our previous results showing that GGR, acting partially redundant with TCR, is the main NER pathway in the germ line of C. elegans.

Surprisingly, UV irradiation does not induce, but even seems to inhibit apoptosis in xpa-1 and rad-23; csb-1 mutants, and less efficiently in rad-23 mutants. In unirradiated animals, germ cell apoptosis is thought to be a developmental mechanism to maintain germ line homeostasis [28]. Following UV irradiation, NER-dependent apoptosis of pachytene stage germ cells may serve to eliminate damaged cells. After exiting pachytene stage, undamaged germ line nuclei progress to complete meiosis and are fertilized as oocytes in the proximal part of the gonad (reviewed in [29]; Figure 3B, first image). Next, fertilized oocytes initiate embryogenesis. Thus, it was interesting to follow the fate of UV-damaged pachytene germ cells in NER proficient and deficient animals. In wild type and csb-1 animals, oocytes in the proximal part of the gonad appeared morphologically normal after UV irradiation. In contrast, in xpa-1, rad-23 and rad-23; csb-1 mutants, the morphology of oocytes was drastically altered over time after UV irradiation (Figure 3B, arrowheads). Further analysis using DAPI staining to visualize chromatin condensation associated with specific meiotic developmental stages revealed that in xpa-1, rad-23 and rad-23; csb-1 mutant germ cells failed to progress to the oocyte stage for at least up to 30 hrs after irradiation (Figure 3C, arrowheads, and Figure 3D; data not shown). In contrast, in wild type animals and csb-1 mutants morphologically normal diakinesis stage oocytes were readily recognizable at all time points after UV irradiation (Figure 3C and 3D and data not shown). These results suggest that in UV irradiated animals lacking functional XPA-1 or RAD-23 maturation of meiotic germ nuclei is impaired. Further down the gonad tube, the general morphology of embryos in utero was also severely compromised in xpa-1, rad-23 and rad-23; csb-1 mutants (data not shown), suggesting extensive embryonic cell death. This latter finding is in agreement with the fact that fewer eggs are laid with increasing dosages of UV irradiation (data not shown; [30]) and that eggs which are laid show increased mortality rates. Possibly, the lack of UV-induced apoptosis in these mutants leads to a reduced clearance of UV-damaged cells which results in defects in meiotic maturation, morphological changes and ultimately cell death. Together, these results confirm that in germ cells GGR, but not TCR, is the dominant NER pathway necessary to overcome the genotoxic effects of UV irradiation.
To investigate whether the UV hypersensitivity of germ cells of xpa-1 and rad-23 mutants is accompanied or caused by reduced DNA repair, we measured UV damage removal. To this end we applied immunofluorescence to visualize Cyclobutane Pyrimidine Dimers (CPDs), the most abundant UVB-induced DNA lesions [31]. As shown in Figure 4, 18 hours after irradiation a virtual complete removal of CPDs from gonad nuclei was observed in wild type and csb-1 animals, but not in xpa-1, rad-23 and rad-23; csb-1 mutants. These results further corroborate the notion that GGR is the major NER pathway in germ cells of C. elegans.

TCR, but not GGR, is essential for UV survival of larvae
To investigate whether the observed GGR dependence of the UV response is restricted to germ cells or whether it is a common feature of C. elegans cells, we determined UV sensitivity of later developmental stages. We found that early developmental stages of C. elegans are more sensitive to UV irradiation than later stages, in line with what was previously described (data not shown; [21], [30]). To score UV sensitivity of L1 larvae, we developed an assay in which survival of UVB-irradiated L1 larvae was measured (see materials and methods and Figure S1B). Survival was scored by determining the percentage of animals capable of growing to adulthood over the total amount of animals in response to UV irradiation. We found that xpa-1, xpg-1, xpf-1 and ercc-1 L1 larvae were extremely sensitive to UV and arrested development completely in response to relatively low UV doses (Figure 5A and 5B). This developmental arrest is possibly caused by a damage-induced block in transcription, causing breakdown of RNA polymerase II, as was shown following UVC irradiation of xpa-1 mutants [20]. However, at the UVB doses we used (up to 160 J/m2) we were unable to confirm breakdown of RNA polymerase II (data not shown). To our surprise, we found that csb-1 L1 larvae, but not rad-23 L1 larvae, were more sensitive to UV than wild type animals (Figure 5C), opposite to what was observed in germ cells. Similar to rad-23 mutant germ cells, csb-1 L1 larvae showed an intermediate UV sensitivity in between wild type animals and general NER mutants. Again we found that rad-23; csb-1 double mutants were more sensitive than either rad-23 or csb-1 single mutant alone and were comparable to general NER mutants (Figure 5C). Although rad-23 mutant L1 larvae did not show increased lethality, they did appear to develop slightly slower in response to UV irradiation (data not shown). Next, we also tested the recently available xpc-1 mutant. xpc-1 single and rad-23; xpc-1 double mutants did not show enhanced UV sensitivity compared to wild type animals (Figure 5D). rad-23; xpc-1 double mutants even showed a mild but reproducible increase in UV survival. Other functions of rad-23, besides NER [32], [33], might account for this observation, although at the moment we do not understand how these might stimulate UV survival. Importantly, xpc-1; csb-1 double mutants showed extreme UV sensitivity comparable to that of general NER mutants and the rad-23; csb-1 double mutant (Figure 5D). Similar results were obtained using older larval stages and young adults instead of L1 larvae (data not shown). Together, these results (summarized in Table 1) suggest that in contrast to germ cells, TCR is the major NER pathway acting in juvenile and adult C. elegans tissues to counteract the effects of UV irradiation (Table 1, Figure 5E). The GGR pathway seems to act partially redundantly to the TCR pathway.

The observed difference in UV survival of rad-23/xpc-1 and csb-1 during development suggests that as a germ cell grows to become an L1 larva, a switch occurs that favors the dependence on one pathway over the other. To test at which developmental stage csb-1/TCR becomes the primary UV survival pathway instead of rad-23/GGR, we collected eggs from adult animals by hypochlorite treatment and irradiated these at different time points after collection (Figure S1C). We found that in early eggs, rad-23 function is still essential for optimal UV survival (Figure 5F; 1 hr, 20 J/m2), similar to germ cells and embryos. However, in time rad-23 function became gradually dispensable while csb-1 function was more and more essential for optimal UV survival (Figure 5G; 4 and 8 hr, 40 J/m2). Note that during later time points slightly higher UV doses had to be used due to the fact that early embryos are more UV sensitive than later stage embryos. This phenomenon might be due to growth causing less UV penetrance or higher tolerance of transcription and replication blocking lesions [30]. Irradiation of eggs collected by egg laying gave similar results as eggs collected by hypochlorite treatment (data not shown; [30]). In summary, these results suggest that during embryogenesis, before hatching, GGR gradually becomes less and TCR becomes more important for C. elegans to cope with the toxic effects of UV exposure.
Novel chromatin remodelers in UV damage response
The developmental difference between TCR - and GGR-dependent UV-sensitivity of C. elegans suggests that developmental-stage dependent regulatory genes specifically involved in either pathway could be identified using C. elegans. Recently, we have successfully used C. elegans to show that Heterochromatin Protein 1 (HP1), represented by hpl-1 and hpl-2 in C. elegans, is involved in the UV-DDR [34], suggesting a role for chromatin condensation status in UV survival. This implies that proteins involved in chromatin dynamics, e.g. chromatin remodeling and epigenetics, may be implicated in the UV-DDR. These proteins are expected to play important roles in controlling the efficiency of DNA repair, by regulating the access to DNA as well as checkpoint signaling associated with DNA repair [35]. CSB itself is an ATP-dependent chromatin remodeling factor, which is thought to alter nucleosome structure to enable repair [36], [37]. In yeast, fruit flies and mammals, several different ATP-dependent chromatin remodeling complexes, e.g. the SWI/SNF, the ISWI, the NuRD, the CHD and the INO80 families, have been identified, some of which have been implicated in the DDR [35], [36]. To test whether these remodeling complexes are involved in the developmental stage-dependent UV-DDR in C. elegans, we set up a screen in which we systematically tested L1 larvae UV sensitivity of animals in which subunits of these major remodeling complexes or genes carrying motifs predicted to be involved in ATP-dependent chromatin remodeling were knocked down either by mutation or RNAi (Table S1).
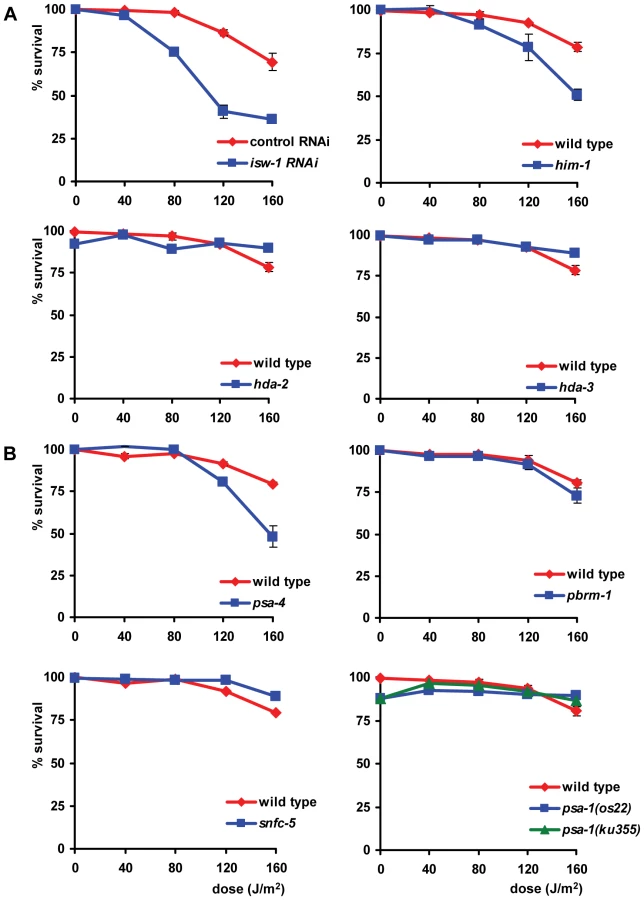
UV survival of L1 larvae in which proteins of the NuRD, the CHD and the INO80 chromatin remodeling family were knocked down closely mimicked that of wild type larvae (data not shown), suggesting no involvement in the UV-DDR. In contrast, knockdown of four proteins of the ISWI family and four proteins of the SWI/SNF family resulted in increased UV-sensitivity (Table 1, Figure 6). We tested two partial loss-of-function alleles of the ISWI/SMARCA5 chromatin remodeling ATPase subunit isw-1 [38]. isw-1(n3297) animals showed reproducible sensitivity to UV irradiation (Figure 6A), but isw-1(n3294) animals did not (Figure S2A). Surprisingly, isw-1(n3297) carries a missense mutation within a non-conserved region of the gene while isw-1(n3294) encodes a missense mutation in a conserved DEXD/H box helicase domain required for chromatin remodeling activity [38]. Since isw-1 null mutants are not viable, we additionally knocked down isw-1 using RNAi and confirmed that isw-1 functions in the UV-DDR (Figure S2B). Furthermore, deletion alleles of hda-2(ok1479) and hda-3(ok1991), which represent orthologs of human class I histone deacetylase [39], and mutation of him-1, the C. elegans ortholog of human cohesin protein SMC1, which are all found in complex with human ISWI/SMARCA5 [40], also increased UV-sensitivity (Figure 6A). To confirm the significance of these findings, we reproduced the observed UV sensitivities in multiple independent experiments. Knockdown of hda-2 and hda-3 by RNAi was also attempted, but was found to produce variable results (data not shown), possibly because efficacy of the RNAi was not always optimal. As the e879 allele used for him-1 was described to be temperature-sensitive [41], we additionally tested him-1 mutants at 25°C and found them to be more UV sensitive than at 20°C (Figure S3A). This increased UV sensitivity at the restricted temperature further confirmed that this gene is indeed implicated in the UV-DDR.

Next, we tested animals carrying a temperature sensitive missense mutation (os13) in the SWI2/SNF2 chromatin remodeling ATPase subunit psa-4 [42], a putative ortholog of human BRM/SMARCA2. Animals tested at a permissive temperature (20°C) were found to be mildly sensitive to UV (Figure 6B), whereas animals tested at a nonpermissive temperature (25°C) showed a strongly enhanced UV sensitivity (Figure S3B). Additionally, mutations in other subunits of SWI/SNF remodeling complexes, e.g. the SMARCC1 ortholog psa-1(os22 and ku355; [42], [43]), the PBRM1 ortholog pbrm-1(tm415) and the SMARCB1 ortholog snfc-5(ok622) also increased UV-sensitivity (Figure 6B, Figure S2C). As both psa-1 alleles were described to be temperature-sensitive, we tested both alleles at 25°C and found them to confer even stronger UV-hypersensitivity than at 20°C (Figure S3B). The UV hypersensitivities of all SWI/SNF mutants were reproduced in multiple, independent experiments, corroborating their significance. Furthermore, knockdown of pbrm-1 and snfc-5 using RNAi also mildly increased UV sensitivity (data not shown). In summary, these results implicate the ISWI and SWI/SNF chromatin remodeling complexes in the UV-DDR of C. elegans. Mutation or RNAi-mediated knockdown of other members of both ATP-dependent chromatin remodeling complexes (Table S1) had no effect, possibly because RNAi was not efficient or because these factors do not play a role in the UV-DDR. Involvement of some factors could not be tested due to lethality.
In addition to the L1 larvae survival experiment, we tested whether the eight identified genes are also involved in the UV-DDR of germ cells and embryos. Since both isw-1 mutants did not lay sufficient eggs on a regular basis, we tested isw-1 involvement using RNAi. Knockdown of the isw-1 and psa-4 ATPase subunits of ISWI and SWI/SNF chromatin remodeling complexes, and of the cohesin member him-1, rendered germ cells sensitive to UV (Table 1, Figure 7). However, mutation of the other ISWI and SWI/SNF subunits had no significant effect on UV survival. These results suggest that ISWI and SWI/SNF chromatin remodeling activity is involved in UV survival of germ cells and embryos, but the response in germ cells seems to involve other subunits than the response in L1 larvae.
Genetic interactions among isw-1, pbrm-1, rad-23, and csb-1
The specific UV sensitivity of L1 larvae but not germ cells caused by knockdown of certain chromatin remodeling genes suggests these genes might be involved in TCR but not GGR. If this is the case, knockdown of these genes in a GGR-deficient background could lead to an even more pronounced UV sensitivity, similar as observed for the rad-23; csb-1 double mutants. Likewise, genes that affect UV sensitivity in L1 larvae as well as germ cells might be generally involved in NER, in both TCR and GGR. Inactivation of these genes in a GGR - or TCR-deficient background should not lead to increased UV sensitivity. To test this, we inactivated isw-1, which affects sensitivity in germ cells and L1 larvae, and pbrm-1, which only affects L1 larvae sensitivity, in rad-23 and csb-1 mutants. RNAi-mediated knockdown of isw-1 in rad-23 and csb-1 animals did not lead to significantly enhanced UV sensitivity compared to the respective controls, in both the L1 as well as the germ cell and embryo survival assay (Figure 8A). Only a mild, but reproducible increase in UV sensitivity was observed in the germ cell and embryo sensitivity of rad-23 mutants in which isw-1 was knocked down. Most of these results, however, are in line with the idea that isw-1 has a general regulatory role in the UV-DDR but not specifically in either TCR or GGR.

Next, we created double mutants for pbrm-1 and rad-23 or csb-1 and compared their UV sensitivity to respective controls (Figure 8B). This showed L1 larvae UV sensitivity of pbrm-1; rad-23 double mutants was comparable to rad-23 single mutants and less severe to that of pbrm-1 single mutants. Unexpectedly, L1 larvae UV sensitivity of pbrm-1; csb-1 double mutants was enhanced compared to csb-1 and pbrm-1 single mutants. These results, which were reproduced in independent experiments, suggest in L1 larvae rad-23 is epistatic to pbrm-1, while pbrm-1 and csb-1 act synergistically to protect against UV exposure. In germ cells and embryos no difference in UV sensitivity between pbrm-1; rad-23 and pbrm-1; csb-1 double mutants and their respective controls was observed. In conclusion, although our results clearly indicate a function for pbrm-1, isw-1 and the other chromatin remodeling genes in the UV-DDR, their precise mode of action is still ambiguous and might not be simply confined to either TCR or GGR.
Discussion
Nucleotide excision repair in C. elegans
The genetic analysis presented in this paper strongly suggests that NER functions mechanistically similarly in the nematode C. elegans as it does in mammals. We and others [17]–[20], [22] find that functional loss of core NER factors renders animals hypersensitive to UV light. Similar as in mammals, NER can be initiated by two distinct pathways, GGR and TCR, which depend on rad-23/xpc-1 and csb-1, respectively. The clear difference between rad-23/xpc-1 and csb-1 UV sensitivities during development and the enhanced UV sensitivity in rad-23/xpc-1; csb-1 double mutants makes it unlikely that the RAD-23 and XPC-1 proteins are involved in both TCR and GGR. Therefore, C. elegans NER seems distinct from NER in budding yeast, where RAD23 and RAD4 (yeast orthologs of hHR23 and XPC, respectively) play a role in TCR as well [44], [45]. Importantly, we observe that the involvement of GGR and TCR in C. elegans is developmentally regulated and differs between germ and somatic cells (Table 1; Figure 9). This developmental regulation was not noticed before in eukaryotes, but might be important for understanding the etiology of different mammalian syndromes associated with NER deficiencies. Following our analysis of the UV-DDR in C. elegans, we identify eight genes involved in ATP-dependent chromatin remodeling that function in the UV-DDR, depending on the developmental stage. Together, our data suggests C. elegans is a powerful model organism to study UV-induced DNA repair and to identify novel genes involved in this process.

Global Genome and Transcription Coupled Repair in C. elegans
We provide evidence that in germ cells, oocytes and early embryo's GGR is the main DNA repair pathway conveying UV resistance. Our analyses of UV-survival, CPD repair, pachytene cell apoptosis and pachytene stage exit all indicate that rad-23 and xpc-1 are necessary and sufficient for germ cells to overcome the effects of UV irradiation. However, it is not exactly clear how UV irradiation of germ cells leads to the embryonic death that is measured in the germ cell and embryo survival assay (Figure 2 and S1A). It is tempting to speculate that the lack of UV-induced apoptosis and defective pachytene stage exit leads to embryonic death. However, animals lacking the C. elegans p53 ortholog also show no UV-induced apoptosis, but have wild type levels of embryonic UV survival [22]. Furthermore, animals carrying a gain-of-function mutation (n1950) in the core cell death pathway gene ced-9, also do not show radiation-induced apoptosis [27] and do not show enhanced UV-induced embryonic lethality (unpublished results). These observations indicate that lack of apoptosis and embryonic death are not necessarily linked.
Our results confirm previous observations that in pachytene cells lacking functional XPA-1 apoptosis is not induced after irradiation [22]. This might imply that active NER is necessary to signal the presence of DNA damage to the apoptotic machinery, via the generation of NER-intermediates such as single stranded DNA [46]. Analysis of the rad-23 and csb-1 single and double mutants suggests that GGR or TCR alone is sufficient to induce apoptosis, although via TCR, e.g. in the rad-23 mutant, it seems to be slightly less efficient. Lack of functional GGR and TCR together inhibits induction of apoptosis. These results contrast the apoptotic response observed in cultured mammalian cells, which undergo increased apoptosis after irradiation when NER, and specifically TCR in differentiated cells, is impaired [47]–[49]. In these cells it is believed that persistence of damage in the transcribed strand of active genes triggers apoptosis. In undifferentiated mouse embryonic stem cells UV-irradiation induces apoptosis in NER-deficient XP-A cells but not in GGR-deficient XP-C cells [47]. Thus, it might be that in undifferentiated cells, similar to C. elegans germ cells, a trigger derived from GG-NER or a repair intermediate is necessary to set off an apoptotic response, contrary to the mainly TCR-driven apoptotic response of differentiated cells. An alternative explanation for the lack of apoptosis in NER deficient C. elegans germ cells might be that UV causes defects in cell cycle progression. Because of this, cells might not reach the late pachytene stage in which they can become apoptotic.
Our results indicate that in C. elegans the involvement of GGR and TCR in survival of UV-induced DNA damage changes during development (Figure 9). A similar developmental change was described for the homologous recombination (HR) and non-homologous end-joining (NHEJ) repair pathways in C. elegans [50]. The error-free HR pathway is mainly active in germ cells and dividing somatic cells, while the error-prone NHEJ pathway becomes predominantly active in non-dividing somatic cells. This difference is probably to ensure that the genome integrity of germ cells and dividing cells is maintained, while genomic damage in non-dividing cells can be tolerated. Similarly, GGR may act in germ cells to ensure that the entire genome is free of lesions. TCR is only necessary to maintain active genes in non-dividing somatic cells. These findings exemplify the advantage of using C. elegans as in vivo tool to study the DNA repair response and are in line with similar observations in mammalian cells. Terminally differentiated human neurons appear to lose the ability to repair DNA lesions throughout the genome whereas they retain the ability to repair active genes [51]. Furthermore, in undifferentiated mouse embryonic stem cells the contribution of GGR to UV survival is larger than that of TCR, whereas in partially differentiated mouse embryonic fibroblasts the contribution of TCR is larger than that of GGR [47]. Although GGR is the major pathway contributing to survival in germ cells, we observed that TCR is also active but not essential for survival in these cells. Vice versa, in later developmental stages TCR is essential for survival, while GGR is also active but not essential for survival. The differences in activity of both repair pathways in later stages correlates to previous observations showing that in adult C. elegans highly transcribed and poorly transcribed genes are both repaired, although highly transcribed genes more efficient [52].
It is still unclear what causes the developmental switch from GGR to TCR. A possible mechanism might be that the switch occurs simultaneously with the onset of transcription in embryos, since TCR depends on transcription. However, transcription takes place in pachytene germ cells as well [29] and is initiated in the embryo already several hours before the csb-1 UV sensitivity becomes visible. A second mechanism might be that the switch is linked to proliferation, as the csb-1 UV sensitivity becomes visible when most cell divisions in the embryo have been completed [53]. However, oocytes, which depend on rad-23, do not divide, while L1 larvae, in which cell division resumes, depend on csb-1. A third mechanism might be the availability of RAD-23 and CSB-1 at the site of damage. Although both rad-23 and csb-1 appear to be expressed in all cells throughout development (data not shown; [17], [52]), there might be a delicate balance between RAD-23 and CSB-1 availability at the site of damage which is for instance influenced by chromatin-dependent accessibility of DNA. This hypothesis, however, does not correlate with the fact that the UV-DDR depends on rad-23 in all different cells of the germ line, while these cells differ significantly with regard to chromatin compaction. Finally, it might simply be that different processes are involved in survival and cell death when comparing germ cells to later stage somatic cells. Part of the UV sensitivity may result from direct interference of photolesions with vital processes such as transcription and replication. However, UV sensitivity may also be partially caused by extensive chromosomal aberrations which are caused by UV irradiation in C. elegans [54]. Germ cells might die from UV irradiation because global genome DNA damage, which is not repaired in a rad-23 genetic background, interferes with meiotic progression and early cell divisions. Later stage somatic cell types probably arrest due to block of transcription, which is persistent in a csb-1 genetic background [20].
ISWI and SWI/SNF chromatin remodeling in the UV damage response
Recent studies have highlighted the role of (ATP-dependent) chromatin remodeling in DNA repair, mainly focusing on the double-strand break response [35], [36]. Using a dedicated genetic screen we identified eight genes implicated in chromatin remodeling whose involvement in the UV-DDR was unknown or at least ambiguous. Several lines of evidence suggest these genes genuinely function in the UV-DDR, instead of indirectly influencing UV survival because of their involvement in other processes such as transcription. First, inactivation of five genes caused UV hypersensitivity specifically in L1 larvae while inactivation of three other genes also caused germ cell hypersensitivity (Table 1). This specific difference between L1 and germ cell UV response would not be expected if UV hypersensitivity resulted indirectly from the impairment of other processes. Second, many other genes whose knockdown probably causes pleiotropic phenotypes (see Table S1) were not found to be involved in the UV response. This also argues for a specific role of the eight identified chromatin remodeling genes in the UV-DDR. Finally, comparisons to literature and other DNA repair mechanisms suggest these genes might facilitate access of proteins to DNA or be involved in DNA damage signaling (see discussion below). The mild UV hypersensitivity of the chromatin remodeling mutants, which contrasts the severe hypersensitivity of NER mutants, is in line with such a regulatory role.
We identified four genes implicated in ISWI-dependent chromatin remodeling, isw-1, hda-2, hda-3 and him-1. Mutation of him-1 was shown before to cause UV sensitivity [21], while isw-1, hda-2 and hda-3 were also identified in previous damage response screens [55], [56]. The human isw-1 ortholog SMARCA5 is part of a chromatin remodeling complex that includes the hda-2/-3 ortholog HDAC1 and the cohesin subunit him-1 ortholog SMC1 [40]. Therefore, our findings suggest that an ISWI/cohesin complex involving these proteins is involved in the UV-DDR. However, since these proteins participate in several different other protein complexes, they might regulate the UV response independently of each other. This is suggested by the fact that isw-1 and him-1 loss-of-function causes sensitivity in germ cells, embryo's and L1 larvae, whereas hda-2 and hda-3 loss-of-function only affects L1 larvae. Alternatively, it could be that different ISWI/Cohesin complexes regulate different aspects of the UV-damage response that differ between germ cells and somatic cells and only involve hda-2/hda-3 in somatic cells (Figure 9). Several previous observations support a role for ISWI/Cohesin in the UV-DDR. For instance, the Drosophila ACF complex, containing the isw-1 ortholog ISWI, was found to facilitate NER in dinucleosomal DNA in vitro [57]. Furthermore, SMC is known to be phosphorylated following ionizing or UV irradiation and is thought to play a role in the S-phase checkpoint response in mammalian cells [58], [59]. The evolutionary conserved function of ISWI/Cohesin activity in different DNA damage responses in different species suggests it is involved in one or more steps which are common among DNA damage pathways and possibly involve slightly different complexes: (i) ISWI and/or cohesin may function to mediate a DNA damage induced checkpoint response and (ii) ISWI and/or cohesin may function to facilitate efficient repair by altering chromatin structure. Follow-up functional studies will be required to explore the exact molecular role of ISWI/cohesin in the UV-DDR.
Our analysis further implicated four genes involved in SWI/SNF mediated chromatin remodeling in the UV-DDR. pbrm-1, psa-1 and snfc-5, orthologs of human PBRM1, SMARCC1 and SMARCB1, respectively, only showed UV sensitivity when irradiated as L1 larvae, similar to hda-2 and hda-3. Since the L1 larvae survival assay seems specific for TCR, this would suggest that these genes are specifically involved in TCR or a TCR-associated process (Figure 9). However, our genetic analysis of pbrm-1; rad-23 and pbrm-1; csb-1 double mutants suggests that pbrm-1 acts in parallel to csb-1 but not rad-23 in L1 larvae. To clarify these seemingly contradicting results, more detailed follow-up experiments to determine the precise function of pbrm-1 are necessary. psa-4, ortholog of human BRM/SMARCA2, showed also UV sensitivity in the germ line, indicating that it might have a more general role in the UV-DDR. Possibly, different ATP-dependent chromatin remodeling complexes play a role during TCR compared to GGR, or throughout development, while they may share some of the same subunits. In mammals, several different SWI/SNF-like complexes have been identified containing either BRM/SMARCA2, the ortholog of psa-4 [42], or BRG1 as ATPase subunit. Furthermore, involvement of other subunits such as SMARCC1 (psa-1), PBRM1 (pbrm-1) and SMARCB1 (snfc-5) also differs between different SWI/SNF complexes. SWI/SNF chromatin remodeling has been implicated in the UV-DDR before, but the exact mechanism by which it functions remain unknown. Mammalian cells lacking SMARCB1 or the BRM-paralog BRG1 are hypersensitive to UV irradiation, possibly because SWI/SNF functions in the checkpoint response [60], [61]. Yeast SWI/SNF chromatin remodeling, on the other hand, was shown to stimulate excision repair in vitro and in cells, possibly because of rearrangement of chromatin at a damaged site to allow repair [62], [63]. Therefore, it remains unclear whether SWI/SNF chromatin remodeling directly participates in the repair of a lesion or whether it modulates the checkpoint response, or whether it functions in both processes but involves complexes of different composition. We expect that the identification of specific SWI/SNF genes involved in the UV-DDR will lead to a better understanding of the role of SWI/SNF in the DNA repair mechanism.
In summary, our analysis showed that C. elegans is especially well suited to genetically dissect genes and pathways involved in the UV-DDR at different stages of development. Based on the observed evolutionary conserved role of UV-DDR in C. elegans, it is expected that further analysis using the nematode will increase our understanding of how this response is organized in living organisms.
Materials and Methods
C. elegans alleles, RNAi
All strains were cultured according to standard methods [64]. Alleles used were csb-1(ok2335), ercc-1(tm2073), hda-2(ok1479), hda-3(ok1991), him-1(e879), him-9(e1487), isw-1(n3294), isw-1(n3297), pbrm-1(tm415), psa-1(ku355), psa-1(os22), psa-4(os13), rad-23(tm2595), snfc-5(ok622), xpa-1(ok698), xpc-1(tm3886) and xpg-1(tm1670). snfc-5, xpa-1, xpc-1, ercc-1, rad-23 and csb-1 mutants were backcrossed four times, pbrm-1 was backcrossed three times. Double mutants were genotyped using PCR (primer sequences available upon request). RNAi bacteria were obtained from the Caenorhabditis elegans RNAi feeding library (Geneservice). Control RNAi was vector pPD129.36 (a gift from A. Fire).
Germ cell and embryo survival assay
To measure UV sensitivity of germ cells and early embryos, staged young adults were washed and transferred to empty agar plates (Figure S1A). Next, animals were irradiated at the indicated dose using two Philips TL-12 (40W) tubes emitting UVB light, after which they were transferred to plates plated with OP50 bacteria. Following a 24 hr recovery period, animals were allowed to lay eggs for 2–3 hrs on fresh 6 cm plates containing food. In each experiment, for each dose 6 plates containing 3–5 adults per plate were used. The number of eggs laid was determined and 24 hours later the number of unhatched eggs, to calculate the survival percentage.
Egg survival and L1 larvae survival assay
To measure UV sensitivity of eggs or L1 larvae, eggs were collected from gravid adult animals by hypochlorite treatment and transferred to fresh plates seeded with HT115(DE3) bacteria (Figure S1B and S1C). HT115(DE3) bacteria were specifically used because these bacteria form a uniform thin lawn on NGM plates, which increases reproducibility of the survival assay, as the thicker lawn formed by OP50 bacteria was found to partially shield C. elegans from UV irradiation. We did not observe any typical effects using HT115(DE3) bacteria related to the UV sensitivity of animals. To measure egg survival (Figure 5D), animals were irradiated at indicated time points following hypochlorite treatment. The number of unhatched and hatched eggs was determined 24 hours later to calculate the survival percentage. To measure L1 larvae survival, animals were irradiated 16 hrs (at 20°C) after hypochlorite treatment. Animals that developed beyond the L2 stage (survivors) and animals that arrested development at the L1/L2 stage were counted to determine survival percentage. For experiments performed at 25°C (Figure S3), animals were cultured at 20°C and transferred to 25°C 45 minutes before irradiation. Hypochlorite treatment had no effect on survival rates (data not shown) and similar results were obtained by regular egg laying. Statistical analysis was performed using a one-way ANOVA test.
Immunofluorescence and DAPI staining
To visualize CPD DNA damage, gonads were extruded by cutting the heads and tails of young adult animals using a fine gauge needle. Gonads were fixed in 3% paraformaldehyde, 0.1% Triton X-100 for 15 minutes, washed and permeabilized 2 times 10 minutes in PBS, 0.1% Triton X-100. Next, gonads were incubated for 5 minutes in PBS, 0.07 M NaOH, to denature DNA. Gonads were then washed in PBS, 0.5% BSA, 0.15% glycin and incubated >2 hrs with CPD antibody (Cosmo Bio Co.) in PBS, 0.5% BSA, 0.15% glycin. Subsequently, animals were washed 2 times 10 minutes in PBS, 0.1% Triton X-100 and incubated >2 hrs with Alexa488 fluorescent secondary antibody (Molecular Probes). Finally, animals were mounted on a glass slide using Vectashield with DAPI (Vector laboratories). For DAPI staining, animals were fixed, permeabilized and mounted on a slide using Vectashield with DAPI.
Microscopy and germ line apoptosis
Images in Figure 3C and Figure 4 were acquired using a Zeiss LSM 510 META confocal microscope. Images in Figure 3B 1 were acquired using a Zeiss Axio Imager.Z1 and Nomarski optics. To determine germ line apoptosis, staged young adult animals were irradiated using 160 J/m2 UVB. Six hours later germ cell apoptosis was scored using Nomarski optics.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. HoeijmakersJH
2001 Genome maintenance mechanisms for preventing cancer. Nature 411 366 374
2. NouspikelT
2009 DNA repair in mammalian cells : Nucleotide excision repair: variations on versatility. Cell Mol Life Sci 66 994 1009
3. FousteriM
VermeulenW
van ZeelandAA
MullendersLH
2006 Cockayne syndrome A and B proteins differentially regulate recruitment of chromatin remodeling and repair factors to stalled RNA polymerase II in vivo. Mol Cell 23 471 482
4. TroelstraC
van GoolA
de WitJ
VermeulenW
BootsmaD
1992 ERCC6, a member of a subfamily of putative helicases, is involved in Cockayne's syndrome and preferential repair of active genes. Cell 71 939 953
5. HenningKA
LiL
IyerN
McDanielLD
ReaganMS
1995 The Cockayne syndrome group A gene encodes a WD repeat protein that interacts with CSB protein and a subunit of RNA polymerase II TFIIH. Cell 82 555 564
6. SvejstrupJQ
2007 Contending with transcriptional arrest during RNAPII transcript elongation. Trends Biochem Sci 32 165 171
7. ArakiM
MasutaniC
TakemuraM
UchidaA
SugasawaK
2001 Centrosome protein centrin 2/caltractin 1 is part of the xeroderma pigmentosum group C complex that initiates global genome nucleotide excision repair. J Biol Chem 276 18665 18672
8. SugasawaK
NgJM
MasutaniC
IwaiS
van der SpekPJ
1998 Xeroderma pigmentosum group C protein complex is the initiator of global genome nucleotide excision repair. Mol Cell 2 223 232
9. WakasugiM
KawashimaA
MoriokaH
LinnS
SancarA
2002 DDB accumulates at DNA damage sites immediately after UV irradiation and directly stimulates nucleotide excision repair. J Biol Chem 277 1637 1640
10. VolkerM
MoneMJ
KarmakarP
van HoffenA
SchulW
2001 Sequential assembly of the nucleotide excision repair factors in vivo. Mol Cell 8 213 224
11. GuzderSN
SungP
PrakashL
PrakashS
1998 Affinity of yeast nucleotide excision repair factor 2, consisting of the Rad4 and Rad23 proteins, for ultraviolet damaged DNA. J Biol Chem 273 31541 31546
12. NgJM
VermeulenW
van der HorstGT
BerginkS
SugasawaK
2003 A novel regulation mechanism of DNA repair by damage-induced and RAD23-dependent stabilization of xeroderma pigmentosum group C protein. Genes Dev 17 1630 1645
13. ReardonJT
MuD
SancarA
1996 Overproduction, purification, and characterization of the XPC subunit of the human DNA repair excision nuclease. J Biol Chem 271 19451 19456
14. SugasawaK
MasutaniC
UchidaA
MaekawaT
van der SpekPJ
1996 HHR23B, a human Rad23 homolog, stimulates XPC protein in nucleotide excision repair in vitro. Mol Cell Biol 16 4852 4861
15. de BoerJ
HoeijmakersJH
2000 Nucleotide excision repair and human syndromes. Carcinogenesis 21 453 460
16. van GoolAJ
VerhageR
SwagemakersSM
van de PutteP
BrouwerJ
1994 RAD26, the functional S. cerevisiae homolog of the Cockayne syndrome B gene ERCC6. Embo J 13 5361 5369
17. LeeMH
AhnB
ChoiIS
KooHS
2002 The gene expression and deficiency phenotypes of Cockayne syndrome B protein in Caenorhabditis elegans. FEBS Lett 522 47 51
18. ParkHK
SuhD
HyunM
KooHS
AhnB
2004 A DNA repair gene of Caenorhabditis elegans: a homolog of human XPF. DNA Repair (Amst) 3 1375 1383
19. ParkHK
YookJS
KooHS
ChoiIS
AhnB
2002 The Caenorhabditis elegans XPA homolog of human XPA. Mol Cells 14 50 55
20. AstinJW
O'NeilNJ
KuwabaraPE
2008 Nucleotide excision repair and the degradation of RNA pol II by the Caenorhabditis elegans XPA and Rsp5 orthologues, RAD-3 and WWP-1. DNA Repair (Amst) 7 267 280
21. HartmanPS
HermanRK
1982 Radiation-sensitive mutants of Caenorhabditis elegans. Genetics 102 159 178
22. StergiouL
DoukoumetzidisK
SendoelA
HengartnerMO
2007 The nucleotide excision repair pathway is required for UV-C-induced apoptosis in Caenorhabditis elegans. Cell Death Differ 14 1129 1138
23. YoudsJL
O'NeilNJ
RoseAM
2006 Homologous recombination is required for genome stability in the absence of DOG-1 in Caenorhabditis elegans. Genetics 173 697 708
24. MasutaniC
ArakiM
SugasawaK
van der SpekPJ
YamadaA
1997 Identification and characterization of XPC-binding domain of hHR23B. Mol Cell Biol 17 6915 6923
25. NishiR
OkudaY
WatanabeE
MoriT
IwaiS
2005 Centrin 2 stimulates nucleotide excision repair by interacting with xeroderma pigmentosum group C protein. Mol Cell Biol 25 5664 5674
26. MasutaniC
SugasawaK
YanagisawaJ
SonoyamaT
UiM
1994 Purification and cloning of a nucleotide excision repair complex involving the xeroderma pigmentosum group C protein and a human homologue of yeast RAD23. Embo J 13 1831 1843
27. GartnerA
MilsteinS
AhmedS
HodgkinJ
HengartnerMO
2000 A conserved checkpoint pathway mediates DNA damage–induced apoptosis and cell cycle arrest in C. elegans. Mol Cell 5 435 443
28. GumiennyTL
LambieE
HartwiegE
HorvitzHR
HengartnerMO
1999 Genetic control of programmed cell death in the Caenorhabditis elegans hermaphrodite germline. Development 126 1011 1022
29. HubbardEJ
GreensteinD
2000 The Caenorhabditis elegans gonad: a test tube for cell and developmental biology. Dev Dyn 218 2 22
30. HartmanPS
1984 UV irradiation of wild type and radiation-sensitive mutants of the nematode Caenorhabditis elegans: fertilities, survival, and parental effects. Photochem Photobiol 39 169 175
31. CadetJ
SageE
DoukiT
2005 Ultraviolet radiation-mediated damage to cellular DNA. Mutat Res 571 3 17
32. WadeSL
PooreyK
BekiranovS
AubleDT
2009 The Snf1 kinase and proteasome-associated Rad23 regulate UV-responsive gene expression. Embo J 28 2919 2931
33. DantumaNP
HeinenC
HoogstratenD
2009 The ubiquitin receptor Rad23: at the crossroads of nucleotide excision repair and proteasomal degradation. DNA Repair (Amst) 8 449 460
34. LuijsterburgMS
DinantC
LansH
StapJ
WiernaszE
2009 Heterochromatin protein 1 is recruited to various types of DNA damage. J Cell Biol 185 577 586
35. DinantC
HoutsmullerAB
VermeulenW
2008 Chromatin structure and DNA damage repair. Epigenetics Chromatin 1 9
36. WangGG
AllisCD
ChiP
2007 Chromatin remodeling and cancer, Part II: ATP-dependent chromatin remodeling. Trends Mol Med 13 373 380
37. CitterioE
Van Den BoomV
SchnitzlerG
KanaarR
BonteE
2000 ATP-dependent chromatin remodeling by the Cockayne syndrome B DNA repair-transcription-coupling factor. Mol Cell Biol 20 7643 7653
38. AndersenEC
LuX
HorvitzHR
2006 C. elegans ISWI and NURF301 antagonize an Rb-like pathway in the determination of multiple cell fates. Development 133 2695 2704
39. ShiY
MelloC
1998 A CBP/p300 homolog specifies multiple differentiation pathways in Caenorhabditis elegans. Genes Dev 12 943 955
40. HakimiMA
BocharDA
SchmiesingJA
DongY
BarakOG
2002 A chromatin remodelling complex that loads cohesin onto human chromosomes. Nature 418 994 998
41. ChanRC
ChanA
JeonM
WuTF
PasqualoneD
2003 Chromosome cohesion is regulated by a clock gene paralogue TIM-1. Nature 423 1002 1009
42. SawaH
KouikeH
OkanoH
2000 Components of the SWI/SNF complex are required for asymmetric cell division in C. elegans. Mol Cell 6 617 624
43. CuiM
FayDS
HanM
2004 lin-35/Rb cooperates with the SWI/SNF complex to control Caenorhabditis elegans larval development. Genetics 167 1177 1185
44. MuellerJP
SmerdonMJ
1996 Rad23 is required for transcription-coupled repair and efficient overrall repair in Saccharomyces cerevisiae. Mol Cell Biol 16 2361 2368
45. VerhageR
ZeemanAM
de GrootN
GleigF
BangDD
1994 The RAD7 and RAD16 genes, which are essential for pyrimidine dimer removal from the silent mating type loci, are also required for repair of the nontranscribed strand of an active gene in Saccharomyces cerevisiae. Mol Cell Biol 14 6135 6142
46. MatsumotoM
YaginumaK
IgarashiA
ImuraM
HasegawaM
2007 Perturbed gap-filling synthesis in nucleotide excision repair causes histone H2AX phosphorylation in human quiescent cells. J Cell Sci 120 1104 1112
47. de WaardH
SonneveldE
de WitJ
Esveldt-van LangeR
HoeijmakersJH
2008 Cell-type-specific consequences of nucleotide excision repair deficiencies: Embryonic stem cells versus fibroblasts. DNA Repair (Amst) 7 1659 1669
48. ConfortiG
NardoT
D'IncalciM
StefaniniM
2000 Proneness to UV-induced apoptosis in human fibroblasts defective in transcription coupled repair is associated with the lack of Mdm2 transactivation. Oncogene 19 2714 2720
49. LjungmanM
ZhangF
1996 Blockage of RNA polymerase as a possible trigger for u.v. light-induced apoptosis. Oncogene 13 823 831
50. ClejanI
BoerckelJ
AhmedS
2006 Developmental modulation of nonhomologous end joining in Caenorhabditis elegans. Genetics 173 1301 1317
51. NouspikelT
HanawaltPC
2000 Terminally differentiated human neurons repair transcribed genes but display attenuated global DNA repair and modulation of repair gene expression. Mol Cell Biol 20 1562 1570
52. MeyerJN
BoydWA
AzzamGA
HaugenAC
FreedmanJH
2007 Decline of nucleotide excision repair capacity in aging Caenorhabditis elegans. Genome Biol 8 R70
53. SulstonJE
SchierenbergE
WhiteJG
ThomsonJN
1983 The embryonic cell lineage of the nematode Caenorhabditis elegans. Dev Biol 100 64 119
54. StewartHI
RosenbluthRE
BaillieDL
1991 Most ultraviolet irradiation induced mutations in the nematode Caenorhabditis elegans are chromosomal rearrangements. Mutat Res 249 37 54
55. van HaaftenG
RomeijnR
PothofJ
KooleW
MullendersLH
2006 Identification of conserved pathways of DNA-damage response and radiation protection by genome-wide RNAi. Curr Biol 16 1344 1350
56. PothofJ
van HaaftenG
ThijssenK
KamathRS
FraserAG
2003 Identification of genes that protect the C. elegans genome against mutations by genome-wide RNAi. Genes Dev 17 443 448
57. UraK
ArakiM
SaekiH
MasutaniC
ItoT
2001 ATP-dependent chromatin remodeling facilitates nucleotide excision repair of UV-induced DNA lesions in synthetic dinucleosomes. Embo J 20 2004 2014
58. KimST
XuB
KastanMB
2002 Involvement of the cohesin protein, Smc1, in Atm-dependent and independent responses to DNA damage. Genes Dev 16 560 570
59. YazdiPT
WangY
ZhaoS
PatelN
LeeEY
2002 SMC1 is a downstream effector in the ATM/NBS1 branch of the human S-phase checkpoint. Genes Dev 16 571 582
60. Klochendler-YeivinA
PicarskyE
YanivM
2006 Increased DNA damage sensitivity and apoptosis in cells lacking the Snf5/Ini1 subunit of the SWI/SNF chromatin remodeling complex. Mol Cell Biol 26 2661 2674
61. GongF
FahyD
LiuH
WangW
SmerdonMJ
2008 Role of the mammalian SWI/SNF chromatin remodeling complex in the cellular response to UV damage. Cell Cycle 7 1067 1074
62. GongF
FahyD
SmerdonMJ
2006 Rad4–Rad23 interaction with SWI/SNF links ATP-dependent chromatin remodeling with nucleotide excision repair. Nat Struct Mol Biol 13 902 907
63. HaraR
SancarA
2002 The SWI/SNF chromatin-remodeling factor stimulates repair by human excision nuclease in the mononucleosome core particle. Mol Cell Biol 22 6779 6787
64. BrennerS
1974 The genetics of Caenorhabditis elegans. Genetics 77 71 94
65. HartmanPS
HeveloneJ
DwarakanathV
MitchellDL
1989 Excision repair of UV radiation-induced DNA damage in Caenorhabditis elegans. Genetics 122 379 385
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2010 Číslo 5
Nejčtenější v tomto čísle
- Common Genetic Variants near the Brittle Cornea Syndrome Locus Influence the Blinding Disease Risk Factor Central Corneal Thickness
- All About Mitochondrial Eve: An Interview with Rebecca Cann
- Aging and Chronic Sun Exposure Cause Distinct Epigenetic Changes in Human Skin
- The Relationship among Gene Expression, the Evolution of Gene Dosage, and the Rate of Protein Evolution