
Context-Dependent Dual Role of SKI8 Homologs in mRNA Synthesis and Turnover
Eukaryotic mRNA transcription and turnover is controlled by an enzymatic machinery that includes RNA polymerase II and the 3′ to 5′ exosome. The activity of these protein complexes is modulated by additional factors, such as the nuclear RNA polymerase II-associated factor 1 (Paf1c) and the cytoplasmic Superkiller (SKI) complex, respectively. Their components are conserved across uni - as well as multi-cellular organisms, including yeast, Arabidopsis, and humans. Among them, SKI8 displays multiple facets on top of its cytoplasmic role in the SKI complex. For instance, nuclear yeast ScSKI8 has an additional function in meiotic recombination, whereas nuclear human hSKI8 (unlike ScSKI8) associates with Paf1c. The Arabidopsis SKI8 homolog VERNALIZATION INDEPENDENT 3 (VIP3) has been found in Paf1c as well; however, whether it also has a role in the SKI complex remains obscure so far. We found that transgenic VIP3-GFP, which complements a novel vip3 mutant allele, localizes to both nucleus and cytoplasm. Consistently, biochemical analyses suggest that VIP3–GFP associates with the SKI complex. A role of VIP3 in the turnover of nuclear encoded mRNAs is supported by random-primed RNA sequencing of wild-type and vip3 seedlings, which indicates mRNA stabilization in vip3. Another SKI subunit homolog mutant, ski2, displays a dwarf phenotype similar to vip3. However, unlike vip3, it displays neither early flowering nor flower development phenotypes, suggesting that the latter reflect VIP3's role in Paf1c. Surprisingly then, transgenic ScSKI8 rescued all aspects of the vip3 phenotype, suggesting that the dual role of SKI8 depends on species-specific cellular context.
Published in the journal:
. PLoS Genet 8(4): e32767. doi:10.1371/journal.pgen.1002652
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002652
Summary
Eukaryotic mRNA transcription and turnover is controlled by an enzymatic machinery that includes RNA polymerase II and the 3′ to 5′ exosome. The activity of these protein complexes is modulated by additional factors, such as the nuclear RNA polymerase II-associated factor 1 (Paf1c) and the cytoplasmic Superkiller (SKI) complex, respectively. Their components are conserved across uni - as well as multi-cellular organisms, including yeast, Arabidopsis, and humans. Among them, SKI8 displays multiple facets on top of its cytoplasmic role in the SKI complex. For instance, nuclear yeast ScSKI8 has an additional function in meiotic recombination, whereas nuclear human hSKI8 (unlike ScSKI8) associates with Paf1c. The Arabidopsis SKI8 homolog VERNALIZATION INDEPENDENT 3 (VIP3) has been found in Paf1c as well; however, whether it also has a role in the SKI complex remains obscure so far. We found that transgenic VIP3-GFP, which complements a novel vip3 mutant allele, localizes to both nucleus and cytoplasm. Consistently, biochemical analyses suggest that VIP3–GFP associates with the SKI complex. A role of VIP3 in the turnover of nuclear encoded mRNAs is supported by random-primed RNA sequencing of wild-type and vip3 seedlings, which indicates mRNA stabilization in vip3. Another SKI subunit homolog mutant, ski2, displays a dwarf phenotype similar to vip3. However, unlike vip3, it displays neither early flowering nor flower development phenotypes, suggesting that the latter reflect VIP3's role in Paf1c. Surprisingly then, transgenic ScSKI8 rescued all aspects of the vip3 phenotype, suggesting that the dual role of SKI8 depends on species-specific cellular context.
Introduction
Production and turnover of eukaryotic mRNAs are highly conserved processes, which are mainly driven by RNA polymerase II (RNAPolII) and the 3′ to 5′ exosome (exosome), respectively [1], [2]. Regulation of transcription initiation by RNAPolII through promoter sequence-specific transcription factors is a major topic in developmental biology, since it is considered the prime mechanism for differential, cell and organ type-specific gene expression [3]. However, generic accessory factors, which are typically heteromultimeric protein complexes, exist as well. Compared to the RNAPolII machinery, they are less conserved but have been found in all uni - and multicellular eukaryotes investigated so far. In line with their lower conservation, these factors are generally not essential. However, loss of function mutations in their subunits typically result in pleiotropic phenotypes with varying degrees of severity. An example is the Mediator complex, which typically comprises more than 15 subunits and interacts with the C-terminal domain of the largest RNAPolII subunit [4], [5]. In yeast (S. cerevisiae), Mediator is associated with constitutively transcribed genes [6] and yeast Mediator mutants are typically viable but display impaired growth [4]. In multicellular organisms, the composition of Mediator is even more complex and individual subunit loss of function can lead to rather specific phenotypes. For instance, in the model plant Arabidopsis (A. thaliana), in which several additional Mediator subunits have been identified [7], respective mutants display such diverse phenotypes as increased cell proliferation, shifts in embryonic patterning or early flowering [7], [8], [9].
Screens for early flowering mutants also identified Arabidopsis subunit homologs of another conserved multimeric regulator of transcription, the RNAPolII-associated factor 1 complex (Paf1c) [10], [11], [12]. In yeast, Paf1c consists of five subunits [13], whose Arabidopsis homologs are VERNALIZATION INDEPENDENCE (VIP) 4, VIP5, EARLY FLOWERING (ELF) 7, VIP6/ELF8 and PLANT HOMOLOGOUS TO PARAFIBROMIN (PHP) [10], [14], [15], [16]. Among the respective loss of function mutants, php mutants only flower early, whereas vip4, vip5, elf7 and vip6/elf8 mutants all display additional pleiotropic growth defects and aberrant flower development (e.g., variable floral organ number). The early flowering phenotype of vip/elf mutants has been linked to down-regulation of the central flowering time regulator, FLOWERING LOCUS C (FLC), via an epigenetic mechanism, consistent with a role of Paf1c in chromatin modification through changing histone methylation patterns [10], [11], [14]. The latter could also explain the phenotypes in flower development, which can be altered by mutation in epigenetic regulators [17].
Another mutant with dwarf, early flowering and aberrant flower development phenotypes is vip3. VIP3 encodes a WD40 repeat protein, which is the putative Arabidopsis homolog of the yeast Superkiller (Ski) 8 gene [12]. SKI8 is part of the cytosolic SKI complex, which is thought to positively regulate exosome activity [1], [18], [19]. The SKI complex consists of a SKI8 dimer and the SKI2 RNA helicase, which are connected by their mutual interaction with the scaffold protein SKI3 [20]. Interestingly, human hSki8 as well as VIP3 also associate with Paf1c [11], [21], which is not the case for yeast ScSki8 [21], [22]. Rather, ScSki8 has a SKI complex-independent nuclear function in meiotic recombination [23]. This feature is not conserved in VIP3 [24], suggesting that Ski8 activity in plants might be functionally closer to mammals than unicellular eukaryotes.
Compared to its well documented role in Paf1c, the potential role of VIP3 in the SKI complex has not been characterized. Notably, although VIP3 is the top hit in a homology search of the Arabidopsis proteome using ScSki8 as a query, the next best hits are nearly equally significant with better overall coverage and represent structurally similar WD40 repeat proteins. Conversely, if the yeast proteome is queried with VIP3, more than two dozen hits score markedly better than ScSki8. Interestingly, the top hits, like PRP4 or TUP1, have been described as modifiers of pre-mRNA processing or chromatin modifications, respectively [25], [26], which would also be consistent with existing experimental data on VIP3 activity. However, reciprocal BLAST searches with higher eukaryotes clearly identify the respective SKI8 homologs as best hits. Still, experimental evidence for VIP3 involvement in the Arabidopsis SKI complex and the facets of the vip3 phenotype that could be attributed to this role is missing. In this study, we investigate this question by a combination of biochemical, genetic and high throughput techniques.
Results
The Arabidopsis zwg mutant displays pleiotropic growth defects
Analysis of natural genetic variation has become a common tool for isolation of allelic variants in Arabidopsis, facilitated by availability of collections of wild strains, so-called accessions. In the Slavice-0 (Sav-0) accession, we found largely infertile dwarf plants segregating at low frequency when grown in permanent light conditions and low humidity (∼40%) (Figure 1A). Moreover, careful investigation of the segregating population revealed a substantial fraction of non-viable, seedling lethal individuals. At higher humidity (∼60%) and long day conditions, the fraction of seedling lethals decreased considerably, whereas the ratio of dwarfs increased to near Mendelian (typically>20%) proportion, suggesting that the two classes represent the phenotypic spectrum of the same underlying genetic cause. The infertility of the dwarf plants, which rarely produced seeds and if so, very little (Figure 1E–1G), could be overcome to some degree by out-crossing with pollen from wild type looking plants. This allowed us to generate a segregating F2 population derived from a cross to the standard lab accession, Columbia-0 (Col-0). Genetic mapping revealed that the dwarf and infertility phenotypes segregated as a recessive single Mendelian locus on the lower arm of chromosome 4, which we named ZWERGERL (ZWG, Bavarian for “dwarf”).
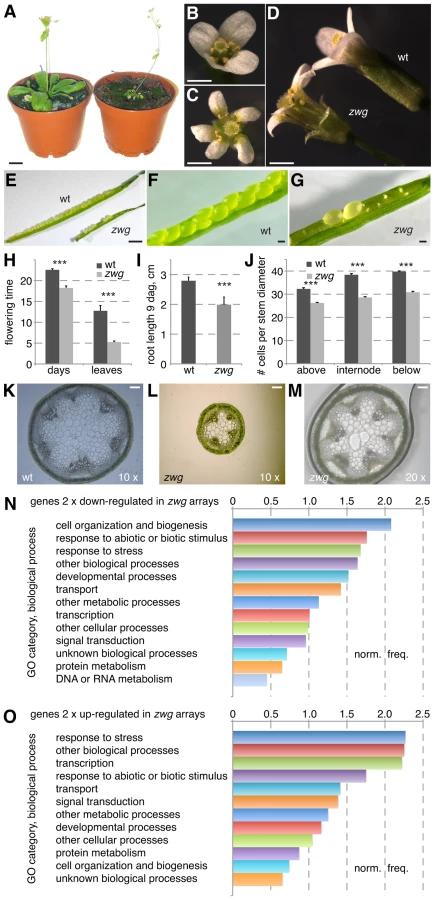
A detailed analysis of the zwg phenotype revealed various floral defects. These included a low penetrance aberrant floral organ number phenotype (Figure 1B–1C) and shorter sepals (Figure 1D). Shorter anthers with few viable pollen accounted for the decreased fertility. The floral phenotypes were accompanied by early flowering, which was evident both in terms of age and rosette leaf number (Figure 1H). By following the development of individual seedlings from germination in tissue culture onwards, we could also detect reduced root elongation in zwg plants (Figure 1I). Radial growth of all organs was affected as well, as exemplified by the dramatically reduced diameter of the main inflorescence stem (Figure 1J–1M). Since the relative decrease in cell number (∼75% of wild type) (Figure 1J) was not as strong as the overall decrease in diameter (∼50% of wild type) (Figure 1K–1M), the reduced organ size in zwg mutants likely represents a combination of impaired cell proliferation and expansion.
Complementing the morphological characterization, we also analyzed the zwg transcriptome by hybridization of CATMA microarrays [27] with cDNA prepared from aerial tissues. Based on four replicate hybridizations, statistically solid expression changes (≥2-fold; p≤0.05) were found for 173 genes that were up-regulated and 425 genes that were down-regulated in zwg as compared to wild type (Table S1). These gene lists did not point to any specific defect in zwg mutants, such as mis-regulation of a particular hormone pathway. Rather, the genes represented an overall balanced sample across functional categories as illustrated by their gene ontology analysis (Figure 1N–1O). The only consistently over-represented category in both the up - and down-regulated sets was response to stress. In summary, our morphological as well as molecular characterization suggests that a general growth defect is responsible for the panoply of zwg mutant phenotypes.
The zwg mutation is a novel null allele of VIP3
To identify the molecular cause of the zwg mutation, we sequenced the genomes of Sav-0 wild type and zwg individuals with short reads [28]. Mapping of the reads onto the Col-0 reference genome revealed an extended region of heterozygosity on the lower arm of chromosome 4 in Sav-0 that encompassed the ZWG locus. The sequence information was exploited to generate polymorphic molecular markers that allowed mapping of the zwg mutation in the zwg x Col-0 population (Figure 2A). Within the zero recombination mapping interval, the sequence reads indicated the presence of a homozygous 7 bp deletion in the coding sequence of At4g29830, previously described as VIP3, in zwg but not in wild type (Figure 2B), which was confirmed by Sanger sequencing of respective PCR fragments. Analysis of a cross between zwg and a vip3 null mutant (SALK_083364) [29] indicated non-complementation, confirming that zwg is indeed a new vip3 allele, which we thus named vip3zwg. The deletion in vip3zwg encompasses nucleotides 861–867 of the open reading frame of the mRNA, which is expressed at similar levels in vip3zwg and wild type. Conceptual translation predicts that the deletion causes a frameshift to produce a 36 kDa instead of a 32 kDa protein with a modified and extended C-terminus, thereby disrupting the last of the five WD40 repeats of VIP3 (Figure 2C). Because of its phenotypic resemblance with the knock out allele, including the down-regulation of FLC expression (Table S1), and its recessive behavior, vip3zwg can be considered a null allele.

VIP3 localizes to both nucleus and cytoplasm and is present in more than one protein complex
To clarify whether VIP3 is indeed the functional Arabidopsis SKI8 homolog, we sought to determine its subcellular localization. To this end, we created a binary construct for expression of a GFP-VIP3 fusion under control of the constitutive 35S promoter. This transgene was introduced into Sav-0 wild type-looking plants that were heterozygous for the zwg mutation as determined by genotyping of the 7 bp deletion on high resolution agarose gels. Western analysis revealed variable expression of GFP-VIP3 fusion protein of the expected size in several independent lines (Figure 2D), within one order of magnitude of the level of endogenous VIP3 as judged from qPCR. In the progeny of these plants, the zwg phenotype segregated in a proportion close to 1/16th rather than 1/4th and was significantly different from the segregation in the parallel grown non-transgenic mother line (Chi-square = 11.54 for df = 1, significant at p<0.001). None of the plants with a zwg phenotype carried the transgene as determined by genotyping. All other plants appeared wild type, suggesting that the fusion protein is functional and rescues all zwg phenotypes. Confocal microscopy showed both cytoplasmic and nuclear (but not nucleolar) localization of GFP-VIP3, in differentiated as well as proliferating cells, in both root and shoot tissues (Figure 2E–2G). Matching the dual subcellular localization, analysis of protein extracts by gel filtration detected the presence of GFP-VIP3 in at least two peaks, one in the ∼690 kDa and another in the 300 kDa range (Figure 2H). Moreover, substantial amounts were observed in smaller (100–200 kDa) fractions. To determine whether any of these fractions could represent the SKI complex or its sub-components, we collected three distinct sets of fractions after gel filtration and performed immunoprecipitations with anti-GFP antibody. Subsequent MALDI-TOF identified peptides of the Arabidopsis SKI3 homolog (At1g76630) in the pool of the smaller fractions (Figure 2I). Notably, protein homology searches unequivocally identify At1g76630 and ScSki3 as unique reciprocal and highly significant hits, suggesting that At1g76630 represents indeed the Arabidopsis SKI3 homolog. No other SKI complex or Paf1c components were identified, which might have resulted from our stringent conditions combined with previous gel filtration. Direct immunoprecipitation from total protein extract using the same conditions indeed not only identified AtSKI3, but also AtSKI2 (At3g46960, see below) and the Paf1c component PHP (Figure 2J). Thus, our analyses suggest that VIP3 is not only part of Paf1c in the nucleus, but also of the cytoplasmic SKI complex and likely represents the true SKI8 homolog.
Random-primed high-throughput RNA sequencing suggests a role of VIP3 in mRNA turnover
To corroborate the consequent notion that VIP3 should have a role in mRNA turnover, we applied a high throughput sequencing strategy to RNA samples isolated from vip3zwg and wild type. Because we aimed to sequence both full length mRNAs and mRNAs undergoing (3′ to 5′) degradation, cDNA from these samples was produced by random-primed rather than poly-T-primed synthesis. Prior to this, mRNA was enriched by removing the bulk of ribosomal RNA with the help of capture columns. The cDNA was then size-fractionated and the 200 bp fraction was used for preparation of the library, which was sequenced to produce single reads of 75 bp (21.3 mio. for wild type; 25.2 mio. for vip3zwg). The reads were mapped onto the Col-0 reference transcriptome, including the 5′ and 3′ UTRs, with relaxed stringency to accommodate nucleotide polymorphisms between Sav-0/vip3zwg and Col-0 [28]. Parallel mapping onto the Col-0 reference genome placed the large majority of reads in exons (80.2% in wild type; 84.5% in vip3zwg), confirming that our sequence data represent RNA molecules and that genomic contamination, if any, is negligible (Table S2). For the follow up analyses, we concentrated on the reads that mapped onto mRNA (27.0% in wild type; 13.8% in vip3zwg), and in particular on the nuclear encoded genes (16.7% of reads in wild type; 6.6% in vip3zwg).
In total, of 26’598 transcripts interrogated, 14’228 were covered by at least one read in both the wild type and vip3zwg sample. In order to obtain a parameter that would allow us to estimate the steady state abundance of full length versus degrading mRNAs, we calculated the ratio between the number of reads mapping onto the 5′-most 20% of a transcript versus those mapping onto the 3′-most 20%. After removal of nonsense values (i.e. 0 or ∞ because of absent coverage of one end) and outliers with extreme values (resulting from excess read abundance combined with obvious mis-mapping, e.g. reads covering the flanking region of a gypsy-like retrotransposon [At4g06477]), the distribution of this 5′ to 3′ coverage index was skewed towards values >1. To some degree this likely represents a technical bias [30], but could also reflect a dominant role of 3′ to 5′ degradation in mRNA turnover. Interestingly, the 5′ to 3′ coverage index was generally higher in the wild type than in the vip3zwg sample (Figure 3A). To verify that this was not a technical artifact, we compared the relative proportion of the accumulated reads in 1% bins along the 10% most highly expressed nuclear encoded transcripts of wild type. The respective profiles for the wild type and vip3zwg sample were similar (Figure 3B), suggesting that the RNA sequencing data from the two samples are comparable.

Prime exosome target transcripts are significantly stabilized in vip3zwg
In order to remove statistically doubtful 5′ to 3′ coverage index values that were due to low transcript abundance, we only considered the 6’500 transcripts for which at least 50% of sequence was covered in both the wild type and vip3zwg samples in follow up analyses. Remaining outliers with index values ≥10 or ≤0.1 were removed as well. From this set, we extracted the group of 5’617 nuclear encoded transcripts that were not strongly affected by depletion of exosome activity [31], as well as 34 chloroplast-encoded transcripts and 68 nuclear-encoded transcripts that were significantly stabilized upon exosome depletion and can be considered prime exosome targets [“the hidden transcriptome”; 31] (Table S3). The 5′ to 3′ coverage index value distribution in the nuclear control group confirmed the earlier picture of higher overall values in wild type as compared to vip3zwg, which is for instance also evident in the ratios between the respective averages or medians (Figure 3C). While no significant difference was found in the chloroplast transcripts, this trend was amplified in the prime exosome targets, which displayed higher average and median index values in wild type and lower ones in vip3zwg as compared to the nuclear control group (Figure 3C).
To evaluate the robustness of the difference between the nuclear control group and the exosome targets, we determined the index value distribution for 1’000 random sets of 68 genes extracted from the nuclear control group. These analyses confirmed the trend towards higher values in wild type, underlined by the finding that the wild type to vip3zwg ratio of averages and medians was nearly always >1 (Figure 3D). Notably, even the maximum ratios observed within the 1’000 sets did not or barely reach the values observed in the exosome target set (1.50 versus 1.53 for the average, 1.40 versus 1.39 for the median). Conversely, within 59 random sets of 10 transcripts extracted from the exosome targets, the trend towards higher values in wild type and lower ones in vip3zwg including the ratios was always evident (Figure 3D). We confirmed this finding by an independent method with independent, triplicate RNA preparations for a set of five randomly chosen genes. For each gene, oligonucleotide pairs for qPCR detection of the respective 5′ and 3′ mRNA ends were designed. The reverse primers for each fragment were used to prime separate cDNA synthesis reactions. Subsequent qPCR allowed quantification of the 5′ and 3′ end abundance for each gene in the replicate samples of the two genotypes. With one exception, the ratio between the 5′ and 3′ end abundance was always higher in wild type than in vip3zwg mutants, as expressed by the ratio between those ratios being greater than 1 (Figure 3E). In summary, these analyses suggest that nuclear encoded mRNAs in general and prime exosome targets in particular are stabilized in vip3zwg mutants.
Atski2 mutants display a dwarf, but no flowering or flower development phenotype
To determine which aspects of the phenotype spectrum of vip3 mutants are due to its involvement in Paf1c or the SKI complex, respectively, we sought to characterize mutants in other SKI subunit homologs of Arabidopsis. Whereas knock out mutants in the SKI3 homolog were not available in reverse genetic collections [29], a line segregating a T-DNA insertion in exon 9 out of 23 of At3g46960 (SALK_118579) was available. Similar to At1g76630 and ScSki3, reciprocal homology searches between Arabidopsis and yeast using ScSki2 or At3g46960 as a query identified each other as the uncontested top hits, suggesting that At3g46960 represents the unique ScSki2 homolog in Arabidopsis (AtSKI2). This notion is also supported by a phylogenetic analysis (Figure 4A; Text S1). Analysis of the SALK_118579 line revealed that it segregates up to ∼25% of dwarf plants (Figure 4B–4C). This phenotype co-segregated perfectly with homozygosity of the T-DNA insert and absence of full length AtSKI2 mRNA. With the caveat that residual RNA production 3′ from the T-DNA insertion site has been reported previously [32], the SALK_118579 line therefore might represent the Atski2 null mutant phenotype. Contrary to the vip3 mutants however, the dwarf phenotype was neither accompanied by a flower development nor an early flowering phenotype (Figure 4D–4E). In line with the latter observation, FLC expression was strongly diminished in vip3zwg, but not in Atski2 mutants (Figure 3F). In summary, these observations suggest that the flower development and early flowering phenotype of vip3 mutants could reflect VIP3's role in Paf1c rather than the SKI complex.

ScSki8 rescues all aspects of the vip3zwg phenotype
Considering that ScSki8 has not been found to associate with Paf1c, we sought to corroborate this notion by testing whether transgenic ScSki8 could rescue the dwarf phenotype of vip3zwg mutants. To this end, the ScSki8 open reading frame was cloned into a binary construct for constitutive expression under control of the 35S promoter. Again, the transgene was introduced into Sav-0 wild type plants that were heterozygous for the vip3zwg mutation. Genotyping of the 7 bp deletion and the transgene in the segregating progeny identified several homozygous vip3zwg mutants carrying the 35S::ScSki8 transgene. These plants developed either as dwarf or as wild type, and this was correlated with transgene expression (Figure 4F–4G). Thus, transgenic expression of ScSki8 could rescue the dwarf phenotype of vip3zwg mutants. Moreover, surprisingly both the early flowering and flower development phenotypes were also rescued (Figure 4H). Therefore, our data suggest that once introduced into Arabidopsis, ScSki8 can fulfill all functions of VIP3, including those not normally encountered in yeast itself.
Discussion
In this study, we present experiments that lead to four main conclusions: First, we show that VIP3 is the bona fide SKI8 homolog of Arabidopsis; second, we demonstrate that next generation sequencing of random-primed RNA samples with short reads can be used to estimate the turnover of mRNA transcripts; third, we show that the phenotypic aspects of VIP3 function in Paf1c and the SKI complex can be separated; and fourth, we provide evidence that the dual role of SKI8 homologs in Paf1c and the SKI complex appears to depend on the species-specific cellular context.
Our interest in VIP3 originates from the discovery of the zwg mutant that segregated in the Arabidopsis Sav-0 accession. It seems unlikely that the 7 bp deletion in vip3zwg represents indeed an allelic variant recovered from a natural environment because of its detrimental phenotypic consequences. A haplo-insufficient beneficial effect of vip3zwg could explain maintenance of the allele by balancing selection, however, we did not observe any obvious phenotypes in the heterozygous plants that would support this idea. Rather, it appears likely that vip3zwg is a spontaneous allele that has arisen during the propagation of the Sav-0 accession in stock centers starting in the 1960s over several decades [33], [34].
At the outset of our study, it was still unclear whether VIP3 is indeed the Arabidopsis SKI8 homolog. While its role in epigenetic regulation of FLC transcription through association with the Paf1c complex had been well documented [11], [14], its potential role in the SKI complex had not been characterized. Because of the evolutionary distance between higher plants, yeast and mammals this could not be considered a given, in particular as VIP3 and SKI8 fall into an abundant class of structurally similar WD40 repeat proteins. This was underlined by the finding that ScSki8 is by far not the closest VIP3 homolog in yeast. For instance, position-specific iterated BLAST identifies more than two dozen yeast proteins that are more homologous to VIP3 than ScSki8 (e.g., a 93.2 score, 69% coverage and 5×10−24 e-value for PRP4 as compared to 48.9 score, 40% coverage and 5×10−8 e-value for ScSki8). It is only our functional analyses that suggest that VIP3 is indeed the bona fide ScSki8 homolog. Consistent with a potential role in both Paf1c and the SKI complex, we found that VIP3 is present in both the nucleus and cytoplasm, and in at least two protein complexes of distinct size. The larger peak fractions around 690 kDA could represent Paf1c, whereas the peak around 300 kDa could represent the SKI complex [21]. A third peak around even smaller (100–200 kDa) size fractions could represent partial components of these complexes or VIP3 dimers, which might accumulate in excess as the GFP-VIP3 transgenes were typically expressed at higher levels than endogenous VIP3. Interestingly, immunoprecipitation of GFP-VIP3 after gel filtration identified association with the Arabidopsis SKI3 homolog, but not the SKI2 homolog. This might mean that the SKI complex dissociates into sub-components during gel filtration and/or that SKI2 is lost during immunoprecipitation washes. Alternatively, it could reflect the fact that SKI8 interaction with SKI3 is direct, while interaction with SKI2 is indirect [20]. However, when directly immunoprecipitated from total protein extract, AtSKI3 as well as AtSKI2 was pulled down in our stringent conditions, underlining that VIP3 is indeed part of the SKI complex.
The notion that VIP3 is a functional subunit of the SKI complex is supported by our genome-wide analysis of mRNA stability in vip3zwg mutants. To estimate mRNA turnover was foremost a technical challenge, because it meant that standard cDNA synthesis using poly-T oligonucleotides directed against the 3′ poly-A tail of mRNAs could not be applied. This also abolished the inherent selection of the mRNA fraction for sequencing from the much larger amount of ribosomal or transfer RNAs. Instead, to also capture mRNAs undergoing 3′ to 5′ degradation, cDNA was synthesized with random-priming, and the mRNA fraction was enriched by removing ribosomal RNAs through capture columns. High throughput sequencing of the cDNA samples and subsequent read mapping onto the reference transcriptome revealed that our method efficiently enriched the mRNA fraction, which generally represents 1–2% in total RNA samples, about 5 to 10-fold. The relative read abundance along transcripts is to some degree determined by technical biases, such as the directionality of cDNA synthesis [30]. However, it should also reflect the steady state equilibrium between mRNA synthesis and breakdown considering that primers were not limiting in cDNA synthesis and that poly-A tails provide priming sites but are not included in the sequence analysis. Generally, the coverage profiles displayed a decrease from 5′ to 3′, suggesting that exosome-mediated 3′ to 5′ degradation is the main driver of mRNA breakdown [18], [35]. To quantify the stability of individual transcripts, we defined a 5′ to 3′ coverage index, which was generally >1, consistent with the overall profile. The comparison of the 5′-most 20% of a transcript versus its 3′-most 20% was designed to avoid skewed values in the case of poorly covered transcripts, and indeed comparatively few outliers were observed. In some cases, these reflected obvious mismappings because of repetitive or redundant sequences (e.g. retrotransposon borders), while in others mismapping might have occurred because of the relaxed stringency that was required to map mRNA sequences from a divergent accession onto the reference transcriptome [28]. Overall, the patterns as well as the quantitative difference between the wild type and vip3zwg samples were robust, even if more selective criteria were applied or if other indexes were considered, such as linear fitting of read coverage. Thus, the index values suggest that in the vip3zwg sample the relative abundance of intact 3′ ends as compared to 5′ ends is higher, pointing to a shifted steady state equilibrium between mRNA transcription and degradation. This finding is consistent with the generic role of the SKI complex in exosome activation [18] and was particularly evident in the group of the most prominent exosome targets, termed the “hidden transcriptome” [31]. In summary, our data support the idea that VIP3 is a SKI complex component that affects mRNA stability and that random-primed RNA-Seq is a valid approach to estimate mRNA turnover.
The implication of VIP3 in the SKI complex suggests that the vip3 phenotype should reflect the combination of VIP3 function in both Paf1c and the SKI complex. The availability of a mutant in the AtSKI2 gene, which can be unequivocally identified by homology searches, enabled us to disentangle the two activities. Interestingly, Atski2 plants displayed dwarfism, but neither early flowering nor aberrant flower development. Thus, the latter aspects of the vip3 phenotype should primarily result from impaired Paf1c function. It is noteworthy however that the Atski2 dwarf phenotype is not as severe as in vip3, and that growth defects have also been observed in mutants of other Paf1c components. It thus appears likely that the SKI complex-related growth defects in vip3 are aggravated by the additionally impaired Paf1c activity.
To clarify more directly which portions of the vip3 phenotype are attributable to impaired Paf1c or SKI complex function, we sought to exploit the fact that ScSki8 does not associate with Paf1c in yeast [21], [22] and presumably also not in Arabidopsis. However, to our surprise ScSki8 was able to fully rescue all aspects of the vip3 phenotype. Thus, it appears that in the cellular context of Arabidopsis, ScSKI8 can fulfill VIP3's role in Paf1c. This could mean that other factors determine whether SKI8 is recruited to Paf1c or not, and that in this sense Arabidopsis is closer to mammals than yeast. Indeed we also tried to complement vip3zwg by constitutive expression of the mouse SKI8 homolog, WDR61. However, for unknown reasons, we never managed to recover transgenic plants in repeated transformation attempts, which could mean that WDR61 expression is poisonous for Arabidopsis. Thus, while cellular context must play an important role, SKI8 function might to some degree also depend on inherent features. Future experiments to determine the interaction patterns of different SKI8 homologs and derivative point mutants of interest are a promising avenue to clarify this issue in detail.
Materials and Methods
Molecular biology and genetics standard procedures, such as plasmid construction, genomic DNA isolation, qPCR, genotyping, or gene mapping were performed as described [36], [37].
Plant materials, growth conditions, and phenotypic analyses
The Sav-0 accession used in this study has been described previously [28]. T-DNA insertion lines were obtained from the Nottingham Arabidopsis Stock Centre and the insertions in lines SALK_083364 [knock out of VIP3 (At4g29830)] and SALK_118579 [knock out of AtSKI2 (At3g46960)] were confirmed by PCR analysis. For propagation and analysis of lines, seeds were germinated on half-strength Murashige & Skoog media in tissue culture and transferred to soil at 10–12 days after germination. Plants were then grown in either permanent light and ∼40% humidity, or 16 hr light–8 hr dark cycles and ∼60% humidity at 22°C. The latter conditions were used for characterization of the phenotypes displayed in the figures. For determination of flowering time, seeds were germinated directly on soil and the number of days or of rosette leaves was scored on the first day when the inflorescence meristem became visible. For root growth measurements, seedlings grown vertically in tissue culture were scored at 9 days after germination using ImageJ software and then transferred onto soil to determine wild type or mutant phenotype in the adult shoot.
Microscopy
For transverse sections, stem segments encompassing the first internode were cut with a razor blade and embedded in 6% agarose. From these samples, 85 µm sections were obtained using a Leica-VT 1000S vibratom and photographed using a Leica Diaplan 3 microscope. Subcellular localization of GPF-VIP3 was determined in shoots and roots of 6 day old seedling using a Zeiss LSM 510 confocal microscope. For the propidium iodide staining the roots were incubated for 2–5 min in a 50 µg/ml solution.
Transgenic constructs
For expression of VIP3 or ScSki8 under control of the 35S promoter, the respective open reading frames amplified from cDNA samples were cloned into vectors pMDC43 or pMD32 [38], respectively. Construct integrity was verified by Sanger sequencing before transfer into Agrobacterium and transformation of Arabidopsis plants by the floral dip method. Constructs were introduced into Sav-0 wild type looking plants that were heterozygous for vip3zwg as determined by genotyping. For genotyping, PCR was performed on genomic DNA using oligonucleotides GAG CTG CGA TTC AGA CAA TGA G and GCC CGG ACA CCG GTT CCA C. The PCR products of 87 bp from the wild type or 80 bp from the vip3zwg allele were resolved on 4% agarose gels. Transformants were selected on hygromycin and homozygous vip3zwg plants among the transformants were selected by genotyping. Transgenic GFP-VIP3 or ScSki8 expression levels were determined by quantitative real time or semi-quantitative RT-PCR, respectively, and normalized compared to the EF1 gene as described [36].
Microarray analyses
For microarray analysis, a mix of equivalent amounts of aerial tissues (rosette leaves, stems, cauline leaves, inflorescences) from 4 week old adult plants was collected and frozen in liquid nitrogen before total RNA was prepped using the QIAGEN RNeasy Plant Mini Kit. cDNA synthesis, labeling, hybridization onto CATMA microarrays [27] and data analysis was then performed as described previously [36].
Random-primed high-throughput RNA sequencing
For RNA-Seq, rosette leaves were harvested from 25 day old phenotypically wild type or vip3zwg plants. Genomic DNA was isolated from one part of each sample to verify genotypes, whereas total RNA was prepped from the remaining tissue using a QIAGEN RNeasy Plant Mini Kit. Ribosomal RNA was subsequently largely removed by treating 10 µg of total RNA with Invitrogen RiboMinus Plant Kits following the manufacturer's instructions. The enriched mRNA samples were then subjected to random-primed cDNA synthesis, amplification, size selection and high throughput sequencing with 75 bp single reads on an Illumina instrument as described [28]. Read mapping onto the Col-0 reference genome or transcriptome (main gene models, TAIR 9.0 release) was performed using the BWA program [39] with a seed length of 50 bp and up to 5 mismatches or gaps allowed.
Protein work
Total protein was extracted from 35S::GFP-VIP3 or 35S::GFP transgenic plants as described before fractionation by gel filtration using an Amersham Superdex 200 10/300 GL FPLC column with a buffer flow rate of 0.5 ml/min [40]. Consecutive 0.5 ml fractions were collected, concentrated and subjected to 10% SDS–PAGE followed by protein immunoblot analysis. Fusion protein was detected using an anti-GFP antibody (dilution 1∶3000) (Living colors, Clontech). For co-immunoprecipitation of GFP-VIP3, three consecutive sets of gel filtration fractions were pooled and incubated for 90 min. at 4°C with 50 µl of μ-magnetic beads conjugated to anti-GFP antibody (μMACS anti-GFP MACS, Miltenyi Biotec). The slurry was passed through a magnetic column, washed 5 times with protein extraction buffer before elution of proteins with hot protein loading buffer. Samples were analyzed by immunoblot (anti-GFP) and silver staining prior to MALDI-TOF analysis. For MALDI-TOF, samples were migrated on a 12% mini polyacrylamide gel for about 2.0 cm, and rapidly stained with Coomassie blue. Entire gel lanes were excised into 5 equal regions from top to bottom and digested with trypsin (Promega) as described [41], [42]. Data-dependent LC-MS/MS analysis of extracted peptide mixtures after digestion with trypsin was carried out on a hybrid linear trap LTQ-Orbitrap XL mass spectrometer (Thermo Fisher Scientific) interfaced to a nanocapillary HPLC equipped with a C18 reversed-phase column (Agilent Technologies). Collections of tandem mass spectra for database searching were generated from raw data with Mascot Distiller 2.3.2, and searched using Mascot 2.3 (Matrix Science) against the 2011_03 release of the UNIPROT database (SWISSPROT+TrEMBL, www.uniprot.org), restricted to Arabidopsis thaliana taxonomy (50’756 sequences after taxonomy filter). Mascot was searched with a fragment ion mass tolerance of 0.50 Da and a parent ion tolerance of 10 ppm. The digestion enzyme trypsin was specified with one missed cleavage. Iodoacetamide derivative of cysteine was specified as a fixed modification. N-terminal acetylation of protein, deamidation of asparagine and glutamine, and oxidation of methionine were specified as variable modifications. The software Scaffold (version Scaffold_3.0.9, Proteome Software Inc.) was used to validate MS/MS based peptide (minimum 90% probability [43]] and protein [min 95% probability [44]) identifications, perform dataset alignment as well as parsimony analysis to discriminate homologous hits.
qPCR analyses
To determine the abundance of 5′ and 3′ ends of selected mRNAs, total RNA was prepared from three independent wild type and vip3zwg replicate samples. Separate cDNA syntheses were performed for each individual gene fragment, followed by qPCRs that were performed as described [36] to detect the respective 5′ and 3′ ends of the transcripts. The following oligonucleotides were used: AT1G01010: GAC AGC TCA ACA CTT TTC CAC TTC and CTT TTA TCC TAA ACA AGA CCC GTA AAG (5′ end); GAA CGA AGC ATG TTT GAT TTA TCA TTG and TTG TTG GTG GTT CAT TGG AGT ACA (3′ end); AT1G24706: GTT CCT CTC CCT TTT CAT CTT ATC G and CAT CTT AAA CCC CTT TCG TGT GTA T (5′ end); GAT TTG CAG ATC CTT TGG TTT GTT C and GCT ATG AAT ATA TCT GAA GTC TGG CAA G (3′ end); AT1G64570: CCA TTT ATC GAT TCT TCA CAG ACA CG and GAT TTC ATG ACT CAA ATT AGG GTT CCA (5′ end); GAT GCT GAG GAT GAG TAA GTT CCT TC and GCT AGT AAT CTG CAT TCA AAC AGC ACT A (3′ end); AT3G02830: CAC TAC CTC TCA CCT CTC TGT TTA CAC and CCA TAG ACG TGA AGA GGA AGA ATG (5′ end); GAA GAA ACA AAG GAA GAA GAA GAA GAG and CCA TAG ACG TGA AGA GGA AGA ATG T (3′ end); AT5G56860: ATT GAT GAG ATA AAC AAA TGA AGA CAC AAA G and CCA TGT GTG TTT GGC TCG TGT C (5′ end); GTT GAT CAG ATC ATC ACA ATA TCC TCA TTA C and GCT ATT AAT TAT CAT ATT AAA CTC TCA CAC ACT CT (3′ end); For detection of FLC expression in relation to the EF1 housekeeping gene, qPCR was performed as described [36] using the same oligonucleotide pairs.
Supporting Information
Zdroje
1. SchaefferDClarkAKlauerAATsanovaBvan HoofA 2011 Functions of the cytoplasmic exosome. Adv Exp Med Biol 702 79 90
2. SelthLASigurdssonSSvejstrupJQ 2010 Transcript Elongation by RNA Polymerase II. Annu Rev Biochem 79 271 293
3. GoodrichJATjianR 2010 Unexpected roles for core promoter recognition factors in cell-type-specific transcription and gene regulation. Nat Rev Genet 11 549 558
4. KimYJBjorklundSLiYSayreMHKornbergRD 1994 A multiprotein mediator of transcriptional activation and its interaction with the C-terminal repeat domain of RNA polymerase II. Cell 77 599 608
5. SoutourinaJWydauSAmbroiseYBoschieroCWernerM 2011 Direct interaction of RNA polymerase II and mediator required for transcription in vivo. Science 331 1451 1454
6. AnsariSAHeQMorseRH 2009 Mediator complex association with constitutively transcribed genes in yeast. Proc Natl Acad Sci U S A 106 16734 16739
7. BackstromSElfvingNNilssonRWingsleGBjorklundS 2007 Purification of a plant mediator from Arabidopsis thaliana identifies PFT1 as the Med25 subunit. Mol Cell 26 717 729
8. GillmorCSParkMYSmithMRPepitoneRKerstetterRA 2010 The MED12-MED13 module of Mediator regulates the timing of embryo patterning in Arabidopsis. Development 137 113 122
9. AutranDJonakCBelcramKBeemsterGTKronenbergerJ 2002 Cell numbers and leaf development in Arabidopsis: a functional analysis of the STRUWWELPETER gene. EMBO J 21 6036 6049
10. HeYDoyleMRAmasinoRM 2004 PAF1-complex-mediated histone methylation of FLOWERING LOCUS C chromatin is required for the vernalization-responsive, winter-annual habit in Arabidopsis. Genes Dev 18 2774 2784
11. OhSZhangHLudwigPvan NockerS 2004 A mechanism related to the yeast transcriptional regulator Paf1c is required for expression of the Arabidopsis FLC/MAF MADS box gene family. Plant Cell 16 2940 2953
12. ZhangHRansomCLudwigPvan NockerS 2003 Genetic analysis of early flowering mutants in Arabidopsis defines a class of pleiotropic developmental regulator required for expression of the flowering-time switch flowering locus C. Genetics 164 347 358
13. JaehningJA 2010 The Paf1 complex: platform or player in RNA polymerase II transcription? Biochim Biophys Acta 1799 379 388
14. OhSParkSvan NockerS 2008 Genic and global functions for Paf1C in chromatin modification and gene expression in Arabidopsis. PLoS Genet 4 e1000077 doi:10.1371/journal.pgen.1000077
15. ParkSOhSEk-RamosJvan NockerS 2010 PLANT HOMOLOGOUS TO PARAFIBROMIN is a component of the PAF1 complex and assists in regulating expression of genes within H3K27ME3-enriched chromatin. Plant Physiol 153 821 831
16. ZhangHvan NockerS 2002 The VERNALIZATION INDEPENDENCE 4 gene encodes a novel regulator of FLOWERING LOCUS C. Plant J 31 663 673
17. GoodrichJPuangsomleePMartinMLongDMeyerowitzEM 1997 A Polycomb-group gene regulates homeotic gene expression in Arabidopsis. Nature 386 44 51
18. HouseleyJLaCavaJTollerveyD 2006 RNA-quality control by the exosome. Nat Rev Mol Cell Biol 7 529 539
19. OrbanTIIzaurraldeE 2005 Decay of mRNAs targeted by RISC requires XRN1, the Ski complex, and the exosome. RNA 11 459 469
20. WangLLewisMSJohnsonAW 2005 Domain interactions within the Ski2/3/8 complex and between the Ski complex and Ski7p. RNA 11 1291 1302
21. ZhuBMandalSSPhamADZhengYErdjument-BromageH 2005 The human PAF complex coordinates transcription with events downstream of RNA synthesis. Genes Dev 19 1668 1673
22. BrownJTBaiXJohnsonAW 2000 The yeast antiviral proteins Ski2p, Ski3p, and Ski8p exist as a complex in vivo. RNA 6 449 457
23. AroraCKeeKMalekiSKeeneyS 2004 Antiviral protein Ski8 is a direct partner of Spo11 in meiotic DNA break formation, independent of its cytoplasmic role in RNA metabolism. Mol Cell 13 549 559
24. JolivetSVezonDFrogerNMercierR 2006 Non conservation of the meiotic function of the Ski8/Rec103 homolog in Arabidopsis. Genes Cells 11 615 622
25. BordonneRBanroquesJAbelsonJGuthrieC 1990 Domains of yeast U4 spliceosomal RNA required for PRP4 protein binding, snRNP-snRNP interactions, and pre-mRNA splicing in vivo. Genes Dev 4 1185 1196
26. SmithRLJohnsonAD 2000 Turning genes off by Ssn6-Tup1: a conserved system of transcriptional repression in eukaryotes. Trends Biochem Sci 25 325 330
27. AllemeerschJDurinckSVanderhaeghenRAlardPMaesR 2005 Benchmarking the CATMA microarray. A novel tool for Arabidopsis transcriptome analysis. Plant Physiol 137 588 601
28. SantuariLPradervandSAmiguet-VercherAMThomasJDorceyE 2010 Substantial deletion overlap among divergent Arabidopsis genomes revealed by intersection of short reads and tiling arrays. Genome Biol 11 R4
29. AlonsoJMStepanovaANLeisseTJKimCJChenH 2003 Genome-wide insertional mutagenesis of Arabidopsis thaliana. Science 301 653 657
30. LevinJZYassourMAdiconisXNusbaumCThompsonDA 2010 Comprehensive comparative analysis of strand-specific RNA sequencing methods. Nat Methods 7 709 715
31. ChekanovaJAGregoryBDReverdattoSVChenHKumarR 2007 Genome-wide high-resolution mapping of exosome substrates reveals hidden features in the Arabidopsis transcriptome. Cell 131 1340 1353
32. XuRRQiSDLuLTChenCTWuCAZhengCC 2011 A DExD/H box RNA helicase is important for K+ deprivation responses and tolerance in Arabidopsis thaliana. FEBS J 278 2296 2306
33. MouchelCFBriggsGCHardtkeCS 2004 Natural genetic variation in Arabidopsis identifies BREVIS RADIX, a novel regulator of cell proliferation and elongation in the root. Genes Dev 18 700 714
34. OssowskiSSchneebergerKLucas-LledoJIWarthmannNClarkRM 2010 The rate and molecular spectrum of spontaneous mutations in Arabidopsis thaliana. Science 327 92 94
35. AndersonJSParkerRP 1998 The 3′ to 5′ degradation of yeast mRNAs is a general mechanism for mRNA turnover that requires the SKI2 DEVH box protein and 3′ to 5′ exonucleases of the exosome complex. EMBO J 17 1497 1506
36. SiboutRPlantegenetSHardtkeCS 2008 Flowering as a condition for xylem expansion in Arabidopsis hypocotyl and root. Curr Biol 18 458 463
37. ScacchiEOsmontKSBeuchatJSalinasPNavarrete-GomezM 2009 Dynamic, auxin-responsive plasma membrane-to-nucleus movement of Arabidopsis BRX. Development 136 2059 2067
38. CurtisMDGrossniklausU 2003 A gateway cloning vector set for high-throughput functional analysis of genes in planta. Plant Physiol 133 462 469
39. LiHDurbinR 2009 Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25 1754 1760
40. HardtkeCSGohdaKOsterlundMTOyamaTOkadaK 2000 HY5 stability and activity in arabidopsis is regulated by phosphorylation in its COP1 binding domain. EMBO J 19 4997 5006
41. ShevchenkoAWilmMVormOMannM 1996 Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal Chem 68 850 858
42. WilmMShevchenkoAHouthaeveTBreitSSchweigererL 1996 Femtomole sequencing of proteins from polyacrylamide gels by nano-electrospray mass spectrometry. Nature 379 466 469
43. KellerANesvizhskiiAIKolkerEAebersoldR 2002 Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal Chem 74 5383 5392
44. NesvizhskiiAIKellerAKolkerEAebersoldR 2003 A statistical model for identifying proteins by tandem mass spectrometry. Anal Chem 75 4646 4658
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2012 Číslo 4
Nejčtenější v tomto čísle
- A Coordinated Interdependent Protein Circuitry Stabilizes the Kinetochore Ensemble to Protect CENP-A in the Human Pathogenic Yeast
- Coordinate Regulation of Lipid Metabolism by Novel Nuclear Receptor Partnerships
- Defective Membrane Remodeling in Neuromuscular Diseases: Insights from Animal Models
- Formation of Rigid, Non-Flight Forewings (Elytra) of a Beetle Requires Two Major Cuticular Proteins