
Genetic Inhibition of Solute-Linked Carrier 39 Family Transporter 1 Ameliorates Aβ Pathology in a Model of Alzheimer's Disease
The aggregation or oligomerization of amyloid-β (Aβ) peptide is thought to be the primary causative event in the pathogenesis of Alzheimer's disease (AD). Considerable in vitro evidence indicates that the aggregation/oligomerization of Aβ is promoted in the presence of Zn; however, the functional role of Zn in AD pathogenesis is still not well clarified in vivo. Zn is imported into the brain mainly through the solute-linked carrier (Slc) 39 family transporters. Using a genetically tractable Drosophila model, we found that the expression of dZip1, the orthologue of human Slc39 family transporter hZip1 in Drosophila, was altered in the brains of Aβ42-expressing flies, and Zn homeostasis could be modulated by forcible dZip1 expression changes. An array of phenotypes associated with Aβ expression could be modified by altering dZip1 expression. Importantly, Aβ42 fibril deposits as well as its SDS-soluble form were dramatically reduced upon dZip1 inhibition, resulting in less neurodegeneration, significantly improved cognitive performance, and prolonged lifespan of the Aβ42-transgenic flies. These findings suggest that zinc contributes significantly to the Aβ pathology, and manipulation of zinc transporters in AD brains may provide a novel therapeutic strategy.
Published in the journal:
. PLoS Genet 8(4): e32767. doi:10.1371/journal.pgen.1002683
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002683
Summary
The aggregation or oligomerization of amyloid-β (Aβ) peptide is thought to be the primary causative event in the pathogenesis of Alzheimer's disease (AD). Considerable in vitro evidence indicates that the aggregation/oligomerization of Aβ is promoted in the presence of Zn; however, the functional role of Zn in AD pathogenesis is still not well clarified in vivo. Zn is imported into the brain mainly through the solute-linked carrier (Slc) 39 family transporters. Using a genetically tractable Drosophila model, we found that the expression of dZip1, the orthologue of human Slc39 family transporter hZip1 in Drosophila, was altered in the brains of Aβ42-expressing flies, and Zn homeostasis could be modulated by forcible dZip1 expression changes. An array of phenotypes associated with Aβ expression could be modified by altering dZip1 expression. Importantly, Aβ42 fibril deposits as well as its SDS-soluble form were dramatically reduced upon dZip1 inhibition, resulting in less neurodegeneration, significantly improved cognitive performance, and prolonged lifespan of the Aβ42-transgenic flies. These findings suggest that zinc contributes significantly to the Aβ pathology, and manipulation of zinc transporters in AD brains may provide a novel therapeutic strategy.
Introduction
Alzheimer's disease (AD) is a major neurodegenerative disease affecting the elderly. The accumulation of amyloid-β (Aβ) peptides, which either form the major component of senile plaques (SP) or the oligomer state in patient brains, is hypothesized to be the primary causative event in AD pathogenesis [1]–[3]. However, what drives the Aβ accumulation and how this accumulation links to progression of the disease is not well understood.
Increasing evidence indicates that the disruption of metal homeostasis, particularly in Zn and Cu concentrations, is strongly correlated with the pathophysiological process of AD [4]–[6]. Although copper and, to a lesser extent, iron can induce partial Aβ aggregation, they need a mildly acidic condition (pH 6.6) [7]. Zn2+ is the only metal ion available to aggregate Aβ at pH7.4-the normal physiological pH [8]–[10]. Elevated Zn was found and co-purified with Aβ from AD brain tissues, associated with markedly high Zn level in cerebral spinal fluid (SP) [11]. Measurements from well characterized late stage AD (LAD) also showed a significant increase of Zn in brain sections of hippocampus, multiple neocortical areas and amygdala compared to age-matched normal control subjects [4], [5], [12]–[14]. Although several reports indicate that Zn induces Aβ aggregation at low physiological concentrations [8], [15], [16], later studies showed that higher Zn concentrations are required for significant fibril formation [17], [18]. These pieces of evidence were obtained mostly from in vitro experiments. Therefore how Zn status influences Aβ pathology in vivo throughout life course remains unclear. Dietary intervention of zinc intake with zinc chelators in animals show some encouraging results [19], [20], [21]. However, genetic evidence is still lacking. More importantly, zinc chelators are usually not zinc specific, and may associate with other nonspecific phenotypes [22], precluding accurate mechanism analysis.
Transport of Zn into cells is mediated by a set of zinc transporters called Zrt-Irt like proteins (Zips). Zips are characterized as influx transporters that mediate Zn2+ uptake into cytoplasm from extracellular or vesicular sources [23], [24], and are encoded by the solute-linked carrier (Slc) gene family, Slc39 [23]. At least 14 Zips have been identified in the human genome [23] and 8 in Drosophila [25]. Most Zips are predicted to have eight transmembrane domains (TM) with a histidine-rich loop between TM3 and TM4, and to be located at the plasma membrane [26]. Although the uptake of Zn from the brain's extracellular environment to intracellular compartments in neurons and glia cells is not completely understood, the Zips are thought to be involved in this process [23], [24]. To our knowledge, the relationship between Zips and AD has not been explored to date.
Previously we and others have shown that expression of human Aβ42 in Drosophila brains recapitulates the main symptoms of AD including age-dependent memory loss, formation of amyloid deposits and neurodegeneration [27], [28]. In the current study, we found that the time course expression change of dZip1, an orthologue of human Slc39 family transporter hZIP1 in Drosophila, was reversed in brains of Aβ42 flies as compared with normal control flies. We hypothesize that modulating dZip1 expression level might affect Zn accumulation in the brain and modify the AD pathological process. By creating dZIP overexpression and RNAi transgenic flies, we demonstrated that dZip1 is critically involved in Aβ-induced AD pathological process, and by lowering dZip1 expression Aβ toxicity can be markedly ameliorated.
Results
dZip1 is a putative Zn importer involved in zinc uptake
Using the amino acid sequence of hZip1, BLAST searches revealed 8 putative Zips in Drosophila, among which CG9428-encoded putative protein shared the highest similarity with human Zip1 (29% identity). We designate it as dZip1. Topology analysis of dZip1 revealed the presence of eight putative transmembrane domains, a histidine–rich loop between domains 3 and 4 which was predicted to occur within the cytosol, and extracellular N - and C-terminals (Figure 1A). All these features are typical of Slc39 family members [23], [24].

We measured dZip1 transcript levels in organs of w1118 flies by semi-quantitative RT-PCR (sqRT-PCR). dZip1 is expressed in the gut and other organs including the brain (Figure 1B left). To further explore the physiological role of dZip1 in flies, we created dZip1 over-expression (OE) and RNAi transgenic flies. We confirmed elevated and decreased dZip1 transcript levels accordingly by dZip1 over-expression or RNAi in whole body of transgenic flies driven by actin-Gal4 (Figure 1B Right). We then tested the zinc sensitivity of these flies. Figure 1C shows that dZip1 OE flies were more sensitive to zinc overdose (t-test, p<0.001), while dZip1-RNAi flies were more tolerant to zinc overdose (t-test, p<0.001) in comparison with controls. These results suggest that dZip1 is indeed involved in zinc uptake.
To test whether brain Zn levels could be changed accordingly by specifically modulating brain dZip1 level, the pan-neuronal elav-Gal4 driver was used to drive dZip1 OE and RNAi in fly brains. Figure 1D showed qRT-PCR results of dZip1 transcript level in different transgenic fly brains. Two dZip1-RNAi transgenic lines were used, in which the knockdown effect of dZip1-RNAi #2 transgenic line (∼1/10 of the dZip1 transcript level in control elav-Gal4 flies) was much stronger than dZip1-RNAi #1 transgenic line (∼3/5 of the dZip1 transcript level in control elav-Gal4 flies) (Figure 1D). dZip1 OE transgenic line showed ∼6–7 fold increase of dZip1 transcript level compared with control brains (Figure 1D). Inductively coupled plasma optical emission spectrometry (ICP-OES) result showed that over-expression of dZip1 in the fly brain markedly increased brain Zn accumulation (t-test, P<0.01), while knocking down dZip1 via RNAi decreased brain Zn level compared with the control elav-Gal4 flies (t-test, p<0.05) (Figure 1E). Therefore, specific manipulation of dZip1 expression level in fly brains could affect the brain Zn status.
Aβ42 affects dZip1 expression pattern in the course of aging
Aβ42 expression in fly brains could induce an age-dependent formation of amyloid deposits and neurodegeneration which may correlate with disturbed metal homeostasis, especially for zinc and copper. We therefore checked native dZip1 mRNA levels in brains of elav-Gal4>UAS-Aβ42 (Aβ42) flies and normal elav-Gal4 flies at different ages. We found that the dZip1 mRNA level was developmentally altered in Aβ42 fly brains compared to elav-Gal4 fly brains (Figure 2A). The dZip1 mRNA level in brains of w1118 flies showed similar results as elav-Gal4 flies (data not shown), indicating that this is not due to the effect of the introduced Gal4 gene. These results imply that Aβ expression may indeed lead to a zinc dyshomeostasis in the fly brains. Of note is that the endogenous dZip1 expression was lower in young adult Aβ flies as compared to the control, although the brain zinc level of the Aβ flies at the young stage was not significantly different (Figure 2B), suggesting a complex regulation of zinc metabolism (involving participants such as other zinc importers and exporters besides dZip1) is involved in the brain zinc control.

Using ICP-OES we directly measured the Zn content in brains of 7 - and 30-day old flies. By 7 days of age, differences of Zn content among different groups were still not apparent (Figure 2B). With ageing, the Zn content in all the brains significantly increased; however, in comparison with 7-day old elav-Gal4 flies without Aβ expression, 30-day old Aβ42 flies increased ∼170% of their Zn level, significantly higher than that of 30-day old elav-Gal4 flies (average ∼65% increase of the control Zn). Over-expression of dZip1 further increased Zn accumulation in Aβ42 fly brains, while dZip1 knockdown slowed brain Zn accumulation during aging and significantly reduced Zn accumulation compared to 30-day old Aβ42 flies (t-test, p<0.01). By testing Zn content of these flies at 20-day old (Figure S1), although it's not obvious as that of 30-day, the trend is already apparent and statistically significant. These results indicate an intimate connection among Aβ42 expression, aging and brain zinc accumulation, and the latter can be strongly affected by dZip1 expression interference.
Modulation of dZip1 expression correspondingly alters the course of Aβ42-induced neurodegeneration
Next, we used Hematoxylin and Eosin (H&E) staining to examine whether the extent of brain neurodegeneration in aged Aβ42 flies (visualized as vacuolization in the brain region, arrowhead in Figure 3) could be changed by modulation of dZip1 expression. Compared to the control, Aβ42-expressing brains with dZip1 knockdown were to a large extent normal (Figure 3C), but when dZip1 was overexpressed degenerative changes were significantly more apparent in both the cortex and the neuropil region, where there were more and bigger bubbles (Figure 3B). Counting the number of vacuoles in the cortex and neuropil revealed that over-expression of dZip1 increased brain vacuolization more than 2-fold (Figure 3D, t-test, p<0.001), whereas dZip1 knockdown dramatically decreased brain vacuolization in Aβ42 flies (t-test, p<0.001). Meanwhile, no significant different of neurodegenerative bubbles were found between dZip1 OE alone and age-matched control elav-Gal4 flies in 20-day old fly brains (Figure 3E and 3F). To exclude possible off-target effect of the RNAi action, we confirmed the results with a differently constructed RNAi line, V3986, from the VDRC stock center. V3986 exhibited a similar level of dZip1 reduction as our own dZip1-RNAi line #2 (Figure S2), and could roughly to the same extent rescue Aβ-associated brain vacuolization (Figure S2C and S2D). These results indicated that modulating dZip1 expression could change the brain neurodegenerative process.

Modulation of dZip1 expression level affects the climbing ability and lifespan of Aβ42 flies
It has been shown that Aβ42 flies start to display locomotor dysfunction after three weeks of age and their lifespan is significantly reduced [27], [28]. We therefore tried to examine whether dZip1 expression levels could affect Aβ42 flies' locomotion and lifespan. Two dZip1-RNAi transgenic lines were used, in which the knocking down effect of the dZip1-RNAi #2 transgenic line was much more obvious than the dZip1-RNAi #1 transgenic line (Figure 1D). Assay of climbing ability demonstrated that Aβ42 flies with dZip1 overexpression started to have a locomotor defect at 15-day old age comparable with that of the control elav-Gal4>UAS-Aβ42 flies at between 20–25 days (Figure 4A). In contrast, Aβ42 flies with decreased dZip1 levels through RNAi (#1) had a delayed climbing deficit (Figure 4A). This rescuing effect on climbing ability was even more pronounced when the stronger line dZip1-RNAi #2 was used. As a further control, we tested the climbing ability of the transgenic flies without Aβ42 expression (Figure 4B). Only flies with over-expression of dZip1 manifested mild locomotor defect 30 days after eclosion as compared with the age-matched control elav-Gal4 flies. Again we confirmed the climbing rescuing effect with a different RNAi line V3986 and found it compared similarly with our dZip1-RNAi flies (Figure S2E). We conclude the locomotion defect as a result of Aβ42 toxicity can be modulated through the change of dZip1 expression level.

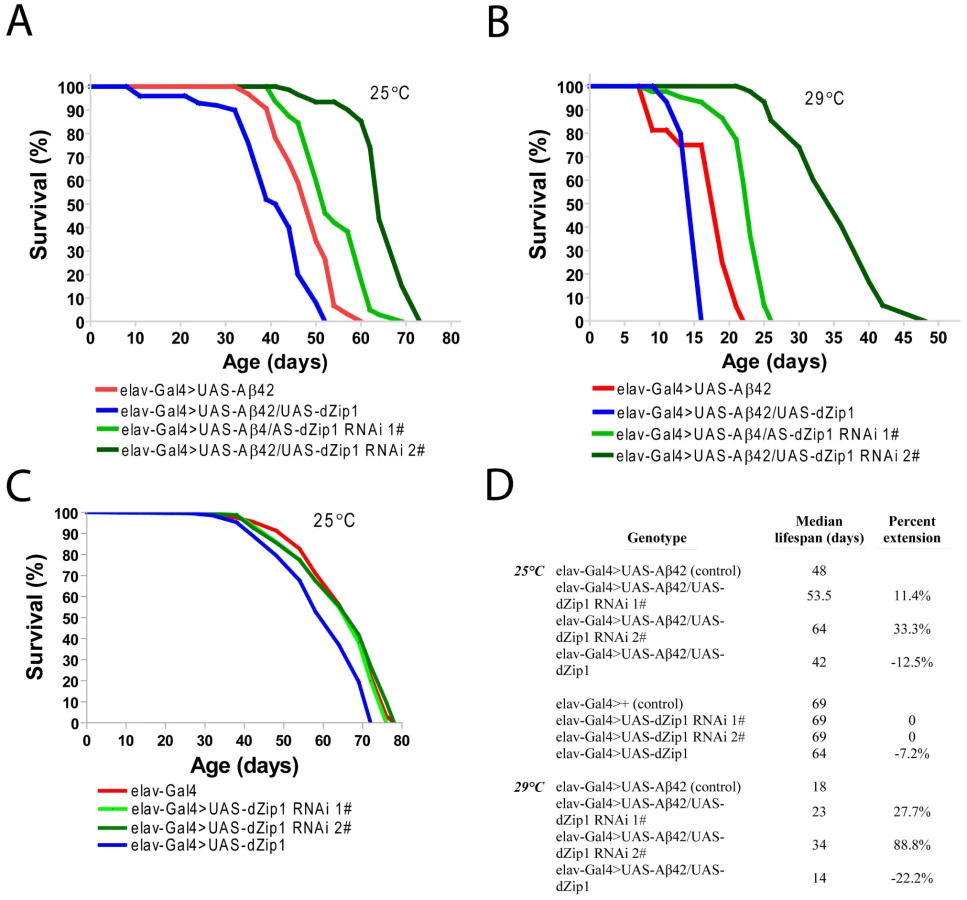
Consistent with the result obtained in the climbing assay, the lifespan of Aβ42 flies was shortened by over-expression of dZip1 and prolonged by RNAi-based knockdown of dZip1 expression (Figure 5A and 5B). The dZip1-RNAi #2 transgenic line exhibited the strongest rescue, with 33.3% and 88.8% increase respectively in the median lifespan of Aβ42 flies reared at 25°C and 29°C (Figure 5D). Similar to that in the climbing assay, the lifespans of flies without Aβ42 expression were largely indistinguishable except for that of the dZip1 OE (elav-Gal4>UAS-dZip1) flies, which displayed a noticeable 7.2% reduction (Figure 5C and 5D). Our results indicate that a reduction of dZip1 expression in Aβ42 flies leads to improved locomotor ability and longer lifespan. Consistently, zinc chelation with clioquinol extended Aβ42 survival (Figure S3). Interestingly, clioquinol appeared rescuing male Aβ42 more effectively than females.
Modulation of dZip1 expression ameliorates Aβ42-induced early memory loss
A cardinal defect in Alzheimer's disease is memory loss. With extensively characterized Pavlovian olfactory aversive conditioning [29], a memory defect in adult Aβ42 flies started to appear as early as 5-day-old. dZip1 knockdown significantly rescued memory loss at this stage (Figure 6A). Paradoxically, overexpression of dZip1 also resulted in obvious memory recovery. As a control, we examined how alteration of dZip1 expression alone (in the absence of Aβ42) might impact memory scores. Overexpressing or knocking down dZip1 did not significantly influence memory of 5-day-old normal flies (Figure 6B and 6C), although overexpression of dZip1 might have a marginal beneficial effect. These results suggest that modulating dZip1 could alleviate the Aβ42 toxicity on memory ability at early stages.

dZip1 overexpression stimulates while dZip1 inhibition decreases Aβ42 deposition
The aforementioned experiments demonstrated dZip1 expression modulation can markedly alter the course of Aβ-associated neurodegeneration. Towards further analysis of the mechanism underlining dZip1 effect on Aβ toxicity, we first tried to determine where Zn was concentrated in these Aβ fly brains. We raised 10-day old Aβ42 flies on normal food supplemented with ZnCl2 and then used Zinquin staining to detect Zn distribution in vivo. The purpose of applying extra Zn is to enhance the fluorescent signal. Without Zinquin treatment, little signal was detected (Figure 7B). Over-expression of dZip1 (Figure 7E and 7F) appeared to produce stronger signals in the neocortex and neuropile region of the fly brain compared with control Aβ42 (Figure 7C and 7D) flies. Quantitative analysis of these signal intensities showed a significant difference (Figure 7K, t-test, p<0.01). Only a faint signal was detected in dZip1-RNAi fly brains (Figure 7G, 7H and 7K, t-test, p<0.05 at 40 h and t-test, p<0.001 at 72 h). Staining of the brains of elav-Gal4 flies without Aβ expression showed faint signals similar to dZip1-RNAi flies (Figure 7I and 7J). These results demonstrate a positive correlation between dZip1 level and Zn accumulation in the neocortex and neuropile region of the fly brain where Aβ deposits were revealed (Figure 8).


To determine if Zinc is causally related to Aβ42 deposition, we overexpressed or knocked down dZip1 under the control of elav-Gal4 in Aβ42 flies and subjected the brains to histochemical analysis. Aβ42 peptides could form diffused amyloid deposits in fly brains [27], [28]. Thioflavin-S (TS) staining of the whole brain was used to specifically visualize the Aβ42 fibril deposits (Figure 8) [23]. Aβ42 deposits were observed in the Kenyon cell body region of Aβ42-expressing brains. The number of TS-positive deposits in Aβ42 flies was significantly increased with aging (Figure 8A and 8A1, 8B and 8B1). Over-expression of dZip1 markedly increased the number of Aβ42 deposits compared with age-matched Aβ42 flies (Figure 8A and A1, ∼212% at 25-day old relative to Aβ42 flies at 25-day old, t-test, p<0.001). Conversely, RNAi-based knockdown of dZip1 in Aβ42 flies significantly decreased the Aβ42 deposits compared with age-matched Aβ42 flies (Figure 8B and B1, ∼48% at 30-day old relative to Aβ42 flies at 25-day old, t-test, p<0.001). Taken together, our results demonstrate that dZip1 over-expression can increase Aβ42 accumulation whereas inhibiting dZip1 can decrease Aβ42 deposition.
Modulation of dZip1 expression also affects the low aggregation form of Aβ
The above TS staining was used to specifically detect the Aβ42 fibril deposits. Recently, an alternative model for the Aβ toxicity is put forth hypothesizing that amyloid oligomers rather than plaques are responsible for the disease [30]. The oligomer form, together with the monomeric form of Aβ, is soluble in SDS whereas the fibril aggregate is not. We next investigated how SDS-soluble Aβ was affected by modulating dZip1 expression. Fly brain lysates were used for a Western blotting analysis. The result showed that the SDS-soluble Aβ42 (low level aggregation forms) was dramatically decreased in accordance to reduced dZip1 level and increased when dZip1 was over-expressed (Figure 8C and 8C1). In a separate experiment, SDS-insoluble but formic acid-soluble Aβ42 (high level aggregation forms) was also examined; much decreased formic acid-soluble Aβ42 level was observed when dZip1 expression was inhibited (Figure S4), consistent with the TS-staining result.
Whole-mount immunohistochemical staining with Aβ42 antibody also revealed a reduction of Aβ42 level in the dZip1-RNAi fly brains (Figure 9). Abundant amyloid deposits were observed in the Kenyon cell body region of 20-day old Aβ42-expressing brains (Figure 9A, arrow). Such deposits were markedly decreased when dZip1 was knocked down (Figure 9C) and greatly increased when dZip1 was over-expressed (Figure 9B). Similar results were found in 30-day old fly brains (Figure 9E and 9F) compared with age-matched Aβ42-expressing control brains (Figure 9D). In the case of co-overexpression of dZip1 and Aβ42, vacuolization became much more pronounced with aging (Figure 9E). As a control for the Aβ42 antibody, we used it against elav-Gal4 fly brains and found no meaningful signal, indicating the antibody reacts specifically with Aβ (Figure S5).

Because dZip1 RNAi led to a general reduction of Aβ42 level, we tried to ask whether this was due to Aβ42 expression inhibition or an increase of Aβ42 degradation. Aβ42 gene was directly under the control of elav-Gal4, and indeed no changes of RNA expression under the various genetic manipulations were observed (Figure 10A and 10B, Figure S2B). We then suspected that zinc might reduce the rate of Aβ42 clearance. Several proteases (NEP1-3, IDE) were proposed to act in the Aβ degradation [31], [32] and we thus explored whether they were affected by dZip1 expression in the Aβ flies. Aβ expression brought some changes to the expression of these genes, but introduction of dZip1-RNAi transgene in the Aβ flies resulted no significant expression increase of these genes (Figure 10C-10F). We did however, observed some decrease of NEP2 expression in dZip1 OE/Aβ flies. Therefore we saw little evidence that the observed rescuing effect of dZip1 RNAi on Aβ42 flies is mediated by an increase of these degrading proteases.

Together, we conclude dZip1 reduction decreases levels of Aβ42, in both the high (fibril aggregates) and low aggregation forms.
Discussion
Previous studies suggest that heavy metals, especially Zn, have a close relationship with the development of Alzheimer's disease [4]–[6], [8]–[10]. However, little genetic evidence exists that demonstrates a functional link between Aβ and proteins involved in zinc assimilation. In this study, we showed that manipulating the Slc39 family protein dZip1 greatly altered the Aβ toxicity. In particular, knocking down dZip1 in brains of Aβ42 flies markedly decreased both Aβ42 deposits and zinc accumulation. dZip1 knockdown ameliorated early memory loss, decreased the number of neurodegenerative vacuoles, significantly enhanced locomotor ability and prolonged life-span in Aβ42-expressing flies. Taken together, our results provide strong evidence to support our hypothesis that knocking down protein dZip1 may mitigate Aβ pathology and Aβ-dependent behavioral defects in a Drosophila model of Alzheimer's disease.
The accumulation and aggregation of Aβ42 peptide in the neocortex has been suggested to be caused by its abnormal interactions with neocortical metal ions especially Zn, which is constitutively found at high levels in the neocortical regions where they play important roles in normal physiology [11], [33], [34]. Our qRT-PCR results showed that the dZip1 transcript level was higher in brains of young control elav-Gal4 and w1118 flies and decreased with age, but lower in brains of young Aβ42 flies and increased with age. Paradoxically, dZip1 transcript level was lower in young adult Aβ42 flies than age matched normal flies. Since at this stage Zn level is not any lower (Figure 2B) in Aβ42 flies, we suspect other Zn homeostasis genes might also be affected. Indeed, besides dZip1 quite a few other Zip or ZnT (Slc30 family transporter) genes are also expressed in the brain (data not shown and the Flyatalas: http://flyatlas.org), likely contributing to Zn uptake or export. Supporting this notion is a recent report showing that the expression level of the Slc30 family protein ZnT3 decreased with age in AD brains [35], [36]. Thus the general Zn status is the result from the combination effect of all these Zn homeostasis genes. It is possible that Aβ42 expression alters the Zn homeostasis starting from an early stage, although we are not totally clear why dZip1 is reduced at early stages but increased at late stages. One thing worthy of consideration is that total zinc level does not reflect well available cellular zinc. In other words, two brains with the same level of total zinc may have very different levels of zinc for use. dZip1 expression regulation may reflect the cell's native response to its own physiological states-to bring more or less zinc into cells. Because Aβ42 can likely bind to zinc, and monomer and oligomers may have different binding characteristics, we speculate that Aβ42 can affect zinc homeostasis even in the absence of noticeable total zinc level alteration. This dyshomeostasis could result or be reflected by native dZip1 expression changes.
Not all pathogenic effects of Aβ in Drosophila correlate directly with its aggregation. An artificial mutation (L17P) with decreased Aβ42 aggregation tendency is associated with lower toxicities, in term of locomotor ability and lifespan, but induces even earlier onset of memory defects than its normal counterpart [37]. Furthermore, although both Aβ40 and Aβ42 affect learning, only Aβ42 causes degeneration [27]; inhibition of PI3K activity ameliorated the Aβ42-induced early memory loss, but did not rescue neurodegeneration [38]. These results lead to the speculation that neuronal dysfunction and neurodegeneration may be mediated by different mechanisms. In our study, while knocking down dZip1 and overexpressing dZip1 were associated with opposite effects on all other aspects of Aβ42-induced toxicity, it is interesting that both knocking down dZip1 or overexpressing dZip1 lessened Aβ42-induced early memory loss. While inhibiting Zn accumulation in fly brains could promote memory in Aβ42-expressing flies by reducing aggregation of Aβ42, the amelioration of memory loss with dZip1-overexpression is likely due to a different mechanism. At the moment, we are not certain how this memory gain with dZip1 over-expression was achieved.
One might ask whether the dramatic effect seen with dZip1 modulation in Aβ42 flies could be reproduced with dietary zinc supplement or chelation. High levels of zinc supplement (such as 2.5 mM) do significantly worsen the viability of Aβ42 flies, however at this level normal flies are also affected (Figure S3C and S3D). Using a zinc chelator clioquiniol, dietary feeding can rescue to some extent Abeta flies, interestingly mostly in male flies (Figure S3A and S3B). Thus genetic intervention is a much more effective method. We interpret this as systemic Zn overloading may cause damages to other tissues before enabling a dramatic Zn increase in the target organ. Likewise, zinc depletion at the organismal level is generally harmful: zinc is an essential nutrient vital to many biological processes. Indeed, high levels of clioquinol greatly impact even the survival of normal flies. Thus, we believe low zinc level can affect fly development and survival so that overall beneficial effect of zinc reduction by dietary measures is significantly less effective than targeted neuronal zinc reduction through genetic interventions.
dZip1 repression results in less Aβ42 level. Although other possibility cannot be excluded, we favor the model that zinc induces oligomerization of Aβ42, as supported by numerous in vitro evidences. More oligomers result more fibril deposits. Perhaps the oligomer and the aggregated Aβ42 are more stable than the monomer, so that the Aβ42 level is dramatically reduced in dZip1 RNAi flies, where a larger fraction of Aβ42 adopt the monomeric form more susceptible to clearance (Figure 11).

In summary, we have demonstrated the modulating effect of dZip1 on Aβ42 toxicity in a Drosophila model of Alzheimer's disease. We observed Aβ42 expression could cause a change of dZip1 expression pattern during ageing. Through genetic manipulation of dZip1 expression, we can modify the pathological process of Aβ42. These results raise the possibility that Zip1, or more broadly Zn transporter genes expressed in the brain, could be a new kind of promising therapeutic target in AD pathology.
Materials and Methods
Drosophila genetics and DNA constructs
Flies were raised and maintained at 25°C or otherwise indicated temperatures. All general stocks were obtained from the Bloomington Drosophila Stock Center, which includes Actin-Gal4, elav-Gal4. UAS-Aβ42 transgenic strain was reported previously [37]. To make UAS-dZip1 transgenic fly, corresponding genomic DNA including a 76 bp intron was cloned into the pUAST vector. The primers used for PCR amplification were: UAS-dZip1-F: 5′-CCGAATTCAAGATGAGCGCTACCGC-3′ and UAS-dZip1-R: 5′ - GGAAGATCTCTA GGAACAGGTTAGGCTG-3′. The UAS-dZip11-RNAi constructs were generated according to Lee and Carthew [39]. The primers used were: WIZ-dZip1-RNAi-F: 5′-GGGTCTAGAATGAGCGCTACCGC-3′ and WIZ-dZip1-RNAi-R: 5′-GGTCTAGACC ACACAGTGCTCACAG-3′. All transgenic flies were generated in w1118 background following standard protocols.
RNA isolation, semi-quantitative and quantitative RT–PCR
Total RNA was extracted from the brain, gut and carcase (whole body minus brain and gut) of 10 adults for each sample using TRIzol Reagent (Invitrogen) according to the manufacturers' instructions and subjected to DNA digestion using DNAse I (Ambion) immediately. The concentration and quality of DNAse-treated total RNA were then tested, and 800 ng total RNA from each sample was used to synthesize cDNA by using Superscript™ II Reverse Transcriptase kit (Invitrogen) with oligo(dT) primers. Semi-quantitative RT-PCR (sqRT-PCR) was performed using primers for rp49 (forward: 5′ - TACAGGCCCAAGATCGTGAA-3′; reverse: 5′ - TCTCCTTGCGCTTCTTGGA-3′) and dZip1 (forward: 5′-ATTATCCTCGCCCTTTCGC-3′; reverse: 5′-TCACCCTCCGCT TCGTCAG-3′). rp49 was used as the loading control. For quantitative RT-PCR (qRT-PCR), 20 fly brains were used for each sample, RNA extraction and cDNA synthesis were the same as described for sqRT-PCR. Primers for amplifying dZip1, NEP1, NEP2, NEP3 and Aβ42 were listed as Table S1. Real-time PCR reactions were monitored on an iCycler (Bio-Rad) by means of SYBR Green (Bio-Rad) dye. mRNA expression levels were determined relative to rp49 expression by relative quantification. Statistical analysis was performed using the Student's t-tests.
Histology and immunostaining
For immunostaining analysis on paraffin sections, antigen retrieval was achieved by boiling the samples in 10 mM sodium citrate (pH 6.6) for 15 min. Immunostaining was performed using an avidin-biotin-peroxidase complex (ABC) kit (Vector Laboratories). For Aβ staining, the primary antibody used was anti-Aβ42 (Promega; 1∶500). Appropriate secondary antibodies were diluted 1∶200, and histochemical detection was done with DAB (Sigma-Aldrich) color development.
Quantification of neurodegeneration
Adult fly heads were fixed in Carnoy solution (ethanol∶chloroform∶acetic acid = 6∶3∶1) overnight at 4 C°, and then embedded in the paraffin and sectioned at 6 m thickness. H&E staining was performed following standard protocols. Neurodegeneration was assessed by quantification of vacuoles with diameter greater than 3 µm in the fly brains. At least five fly brains were analyzed for each genotype.
Fibril Aβ42 deposit detection
Thioflavin-S (TS, Sigma) staining was performed to detect fibril Aβ42 deposits. Fly brains were fixed in 4% paraformaldehyde and permeabilized by 2% triton. Brains were then transferred to 0.25% TS in 50% ethanol overnight. After 1× wash for 10 min in 50% ethanol and 3× wash with PBS, they were mounted with focusclear (Pacgen Biopharmaceuticals Inc.) and covered by cover slips. Slides were inspected with a Zeiss LSM 510 confocal microscope, aided by LSM 510 analysis software. TS-positive deposits located in the mushroom body somatic region were counted for comparison analysis.
Pavlovian olfactory associative memory recording
The training and testing procedures were as previously described [40], [41]. During one training session, a group of 100 flies was sequentially exposed for 60 s to two odors, octanol (OCT) or methylcyclohexanol (MCH), with 45 s of fresh air in between. Flies were subjected to foot-shock (1.5 s pulses with 3.5 s intervals, 60 V) during exposure to the first odor (CS+) but not to the second (CS−). To measure “immediate memory (also referred to as “learning”)”, flies were transferred immediately after training to the choice point of a T-maze and forced to choose between the two odors for 2 min, at which time they were trapped in their respective T-maze arms, anesthetized, and counted. A performance index (PI) was calculated from the distribution of flies in the T-maze. A reciprocal group of flies was trained and tested by using OCT as the CS+ and MCH as the CS+, respectively. PIs from these two groups finally were averaged for an n = 1 and multiplied by 100. A PI of 0 represented a 50∶50 distribution, whereas a PI of 100 represented 100% avoidance of the shock-paired odor.
Western blot analysis
SDS-soluble and SDS-insoluble but formic acid-soluble Aβ42 were prepared as previously reported [27]. Lysates from equal number of fly heads were diluted in SDS sample buffer and separated by 10–20% Tris-Tricine gels (Invitrogen), and transferred to nitrocellulose membranes (Invitrogen). Membranes were boiled in PBS for 3 min. Membranes were blocked with 3% BSA and blotted with primary antibody. Primary antibodies used in this study were mouse anti-Aβ42 (6E10, Covance Research Products) and rabbit anti-Actin (Sigma). After washing in TBST for 3 times, membranes were incubated with secondary antibodies for 1 hr at RT. After 3 times wash in TBST, membranes were incubated with ECL working solution (GE healthcare) and developed with films (Kodak). Data were analyzed with ImageJ sofeware (NIH).
Zinquin staining
10-day old adult flies reared on normal condition were transferred to vials with normal food supplied with 4 mM ZnCl2. Fly brains were then dissected at 40 and 72 h after transferring, respectively. The dissected brains were then incubated with 25 µM Zinquin (Sigma) for 30 min at 37°C, and washed 3 times with 1×PBS buffer for 5 min each time. After that, brains were examined using conventional epifluorescence microscope (Nikon, Diaphot 300) equipped with a Nikon 100×, 1.4 NA Plan Apo oil-immersion objective. Zinquin signals in the neocortex and neuropile region of fly brains were quantitated by using ImagJ software.
Metal stress and content assay
For the metal stress assay, Drosophila was fed on normal medium containing 2.5 mM ZnSO4. Control flies were fed on normal medium in the absence of drug. Mortality was recorded every 24 h or a longer intervals. Each vial contained 20–25 flies, and the experiments were repeated at least three times.
For the metal content analysis, flies were reared on normal food and fly heads were collected at day 7, 20 and 30 after eclosion. Fly heads were dissolved in 1 ml 65% HNO3, boiled in 100°C water bath for 10 min and diluted to 10 ml for metal content analysis with inductively coupled plasma optical emission spectrometry (ICP-OES, IRIS Intrepid II XSP, Thermo Electron Corporation, USA).
Climbing assay
The climbing assay was referenced to Iijima et al. (2004) [27]. Briefly, twenty flies were placed in a plastic vial and gently tapped to the bottom. The number of flies at the top of the vial was counted after 18 s of climbing under red light (Kodak, GBX-2, Safelight Filter). The data shown represent results from a cohort of flies with four repeats tested serially for 5–50 days. The experiment was repeated more than three times.
Longevity assay
Flies of two days after eclosion were used for the experiment. Twenty to 23 flies were placed in a food vial. Each vial was kept at 25 or 29°C, 70% humidity, under a 12-h light–dark cycle. Food vials were changed every 2–3 days, and dead flies were counted at that time. At least 150 flies were prepared for each genotype, and the experiments were carried out more than three times. Percent increases in life span are based on comparing the median survivals. Prism (GraphPad) was used for statistical analysis of lifespan data. Mantel-Cox log-rank statistical analysis was used for testing statistical significance of the differences between the survivorship curves.
Statistical analysis
All data were analyzed by Student's t-test. Statistical results were presented as means ± SEM. Asterisks indicate critical levels of significance (*P<0.05, **P<0.01 and ***P<0.001).
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. HardyJSelkoeDJ 2002 Medicine - The amyloid hypothesis of Alzheimer's disease: Progress and problems on the road to therapeutics. Science 297 353 356
2. HardyJ 2006 A hundred years of Alzheimer's disease research. Neuron 52 3 13
3. WalshDMSelkoeDJ 2007 Abeta Oligomers - a decade of discovery. Journal of Neurochemistry 101 1172 1184
4. DanscherGJensenKBFredericksonCJKempKAndreasenA 1997 Increased amount of zinc in the hippocampus and amygdala of Alzheimer's diseased brains: A proton-induced X-ray emission spectroscopic analysis of cryostat sections from autopsy material. Journal of Neuroscience Methods 76 53 59
5. LovellMARobertsonJDTeesdaleWJCampbellJLMarkesberyWR 1998 Copper, iron and zinc in Alzheimer's disease senile plaques. Journal of the Neurological Sciences 158 47 52
6. FredericksonCJBushAI 2001 Synaptically released zinc: Physiological functions and pathological effects. Biometals 14 353 366
7. AtwoodCSMoirRDHuangXDScarpaRCBacarraNME 1998 Dramatic aggregation of Alzheimer Abeta by Cu(II) is induced by conditions representing physiological acidosis. Journal of Biological Chemistry 273 12817 12826
8. BushAIPettingellWHMulthaupGParadisMDVonsattelJP 1994 Rapid Induction of Alzheimer a-Beta Amyloid Formation by Zinc. Science 265 1464 1467
9. FredericksonCJKohJYBushAI 2005 The neurobiology of zinc in health and disease. Nature Reviews Neuroscience 6 449 462
10. StoltenbergMBushAIBachGSmidtKLarsenA 2007 Amyloid plaques arise from zinc-enriched cortical layers in APP/PS1 transgenic mice and are paradoxically enlarged with dietary zinc deficiency. Neuroscience 150 357 369
11. OpazoCHuangXDChernyRAMoirRDRoherAE 2002 Metalloenzyme-like activity of Alzheimer's disease beta-amyloid. Cu-dependent catalytic conversion of dopamine, cholesterol, and biological reducing agents to neurotoxic H2O2. Journal of Biological Chemistry 277 40302 40308
12. SamudralwarDLDipreteCCNiBFEhmannWDMarkesberyWR 1995 Elemental Imbalances in the Olfactory Pathway in Alzheimers-Disease. Journal of the Neurological Sciences 130 139 145
13. DeibelMAEhmannWDMarkesberyWR 1996 Copper, iron, and zinc imbalances in severely degenerated brain regions in Alzheimer's disease: Possible relation to oxidative stress. Journal of the Neurological Sciences 143 137 142
14. CornettCRMarkesberyWREhmannWD 1998 Imbalances of trace elements related to oxidative damage in Alzheimer's disease brain. Neurotoxicology 19 339 345
15. MantyhPWGhilardiJRRogersSDemasterEAllenCJ 1993 Aluminum, Iron, and Zinc Ions Promote Aggregation of Physiological Concentrations of Beta-Amyloid Peptide. Journal of Neurochemistry 61 1171 1174
16. BushAIMoirRDRosenkranzKMTanziRE 1995 Zinc and Alzheimers-Disease - Response. Science 268 1921 1923
17. ClementsAAllsopDWalshDMWilliamsCH 1996 Aggregation and metal-binding properties of mutant forms of the amyloid Abeta peptide of Alzheimer's disease. Journal of Neurochemistry 66 740 747
18. EslerWPStimsonERJenningsJMGhilardiJRMantyhPW 1996 Zinc-induced aggregation of human and rat beta-amyloid peptides in vitro. Journal of Neurochemistry 66 723 732
19. ChernyRAAtwoodCSXilinasMEGrayDNJonesWD 2001 Treatment with a copper-zinc chelator markedly and rapidly inhibits beta-amyloid accumulation in Alzheimer's disease transgenic mice. Neuron 30 665 676
20. WhiteARDuTLaughtonKMVolitakisISharplesRA 2006 Degradation of the Alzheimer disease amyloid beta-peptide by metal-dependent up-regulation of metalloprotease activity. Journal of Biological Chemistry 281 17670 17680
21. DonnellyPSCaragounisADuTLaughtonKMVolitakisI 2008 Selective intracellular release of copper and zinc ions from bis(thiosemicarbazonato) complexes reduces levels of Alzheimer disease amyloid-beta peptide. Journal of Biological Chemistry 283 4568 4577
22. LiCWangJZhouB 2010 The metal chelating and chaperoning effects of clioquinol: insights from yeast studies. Journal of Alzheimer's Disease 21 1249 1262
23. CousinsRJLiuzziJPLichtenLA 2006 Mammalian zinc transport, trafficking, and signals. Journal of Biological Chemistry 281 24085 24089
24. LichtenLACousinsRJ 2009 Mammalian Zinc Transporters: Nutritional and Physiologic Regulation. Annual Review of Nutrition 29 153 176
25. WangXXZhouB 2010 Dietary Zinc Absorption: A Play of Zips and ZnTs in the Gut. Iubmb Life 62 176 182
26. HuangLPKirschkeCPZhangYFYuYY 2005 The ZIP7 gene (Slc39a7) encodes a zinc transporter involved in zinc homeostasis of the Golgi apparatus. Journal of Biological Chemistry 280 15456 15463
27. IijimaKLiuHPChiangASHearnSAKonsolakiM 2004 Dissecting the pathological effects of human Abeta40 and Abeta42 in Drosophila: A potential model for Alzheimer's disease. Proceedings of the National Academy of Sciences of the United States of America 101 6623 6628
28. FinelliAKelkarASongHJYangHDKonsolakiM 2004 A model for studying Alzheimer's Abeta 42-induced toxicity in Drosophila melanogaster. Molecular and Cellular Neuroscience 26 365 375
29. TullyTQuinnWG 1985 Classical-Conditioning and Retention in Normal and Mutant Drosophila-Melanogaster. Journal of Comparative Physiology A: Sensory Neural and Behavioral Physiology 157 263 277
30. WalshDMSelkoeDJ 2007 Aβ oligomers—a decade of discovery. J Neurochem 101 1172 1184
31. FarrisWMansourianSChangYLindsleyLEckmanEA 2003 Insulin-degrading enzyme regulates the levels of insulin, amyloid b-protein, and the b-amyloid precursor protein intracellular domain in vivo. Proc Natl Acad Sci USA 100 4162 4167
32. IwataNTsubukiSTakakiYWatanabeKSekiguchiM 2003 Identification of the major Abeta1-42-degrading catabolic pathway in brain parenchyma: suppression leads to biochemical and pathological deposition. Nat Med 6 2 143 150
33. FredericksonCJSuhSWSilvaDFredericksonCJThompsonRB 2000 Importance of zinc in the central nervous system: The zinc-containing neuron. Journal of Nutrition 130 1471s 1483s
34. SuhSWJensenKBJensenMSSilvaDSKesslakPJ 2000 Histochemically-reactive zinc in amyloid plaques, angiopathy, and degenerating neurons of Alzheimer's diseased brains. Brain Research 852 274 278
35. BeyerNCoulsonDTRHeggartySRavidRIrvineGB 2009 ZnT3 mRNA levels are reduced in Alzheimer's disease post-mortem brain. Molecular Neurodegeneration 4 53 62
36. AdlardPAParncuttJMFinkelsteinDIBushAI 2010 Cognitive Loss in Zinc Transporter-3 Knock-Out Mice: A Phenocopy for the Synaptic and Memory Deficits of Alzheimer's Disease? Journal of Neuroscience 30 1631 1636
37. IijimaKChiangHCHearnSAHakkerIGattA 2008 Abeta 42 Mutants with Different Aggregation Profiles Induce Distinct Pathologies in Drosophila. PLoS ONE 3 e1703 doi:10.1371/journal.pone.0001703
38. ChiangHCWangLXieZLYauAZhongY 2010 PI3 kinase signaling is involved in Abeta-induced memory loss in Drosophila. Proceedings of the National Academy of Sciences of the United States of America 107 7060 7065
39. LeeYSCarthewRW 2003 Making a better RNAi vector for Drosophila: use of intron spacers. Methods 30 322 329
40. JinPZarnescuDCZhangFPPearsonCELucchesiJC 2003 RNA-mediated neurodegeneration caused by the fragile X premutation rCGG repeats in Drosophila. Neuron 39 739 747
41. TullyTPreatTBoyntonSCDelvecchioM 1994 Genetic Dissection of Consolidated Memory in Drosophila. Cell 79 35 47
42. CoylePZalewskiPDPhilcoxJCForbesIJWardAD 1994 Measurement of zinc in hepatocytes by using a fluorescent probe, Zinquin: relationship to metallothionein and intracellular zinc. Biochemical Journal 303 781 786
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2012 Číslo 4
Nejčtenější v tomto čísle
- A Coordinated Interdependent Protein Circuitry Stabilizes the Kinetochore Ensemble to Protect CENP-A in the Human Pathogenic Yeast
- Coordinate Regulation of Lipid Metabolism by Novel Nuclear Receptor Partnerships
- Defective Membrane Remodeling in Neuromuscular Diseases: Insights from Animal Models
- Formation of Rigid, Non-Flight Forewings (Elytra) of a Beetle Requires Two Major Cuticular Proteins