
Comparative Genomics of Plant-Associated spp.: Insights into Diversity and Inheritance of Traits Involved in Multitrophic Interactions
We provide here a comparative genome analysis of ten strains within the Pseudomonas fluorescens group including seven new genomic sequences. These strains exhibit a diverse spectrum of traits involved in biological control and other multitrophic interactions with plants, microbes, and insects. Multilocus sequence analysis placed the strains in three sub-clades, which was reinforced by high levels of synteny, size of core genomes, and relatedness of orthologous genes between strains within a sub-clade. The heterogeneity of the P. fluorescens group was reflected in the large size of its pan-genome, which makes up approximately 54% of the pan-genome of the genus as a whole, and a core genome representing only 45–52% of the genome of any individual strain. We discovered genes for traits that were not known previously in the strains, including genes for the biosynthesis of the siderophores achromobactin and pseudomonine and the antibiotic 2-hexyl-5-propyl-alkylresorcinol; novel bacteriocins; type II, III, and VI secretion systems; and insect toxins. Certain gene clusters, such as those for two type III secretion systems, are present only in specific sub-clades, suggesting vertical inheritance. Almost all of the genes associated with multitrophic interactions map to genomic regions present in only a subset of the strains or unique to a specific strain. To explore the evolutionary origin of these genes, we mapped their distributions relative to the locations of mobile genetic elements and repetitive extragenic palindromic (REP) elements in each genome. The mobile genetic elements and many strain-specific genes fall into regions devoid of REP elements (i.e., REP deserts) and regions displaying atypical tri-nucleotide composition, possibly indicating relatively recent acquisition of these loci. Collectively, the results of this study highlight the enormous heterogeneity of the P. fluorescens group and the importance of the variable genome in tailoring individual strains to their specific lifestyles and functional repertoire.
Published in the journal:
. PLoS Genet 8(7): e32767. doi:10.1371/journal.pgen.1002784
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002784
Summary
We provide here a comparative genome analysis of ten strains within the Pseudomonas fluorescens group including seven new genomic sequences. These strains exhibit a diverse spectrum of traits involved in biological control and other multitrophic interactions with plants, microbes, and insects. Multilocus sequence analysis placed the strains in three sub-clades, which was reinforced by high levels of synteny, size of core genomes, and relatedness of orthologous genes between strains within a sub-clade. The heterogeneity of the P. fluorescens group was reflected in the large size of its pan-genome, which makes up approximately 54% of the pan-genome of the genus as a whole, and a core genome representing only 45–52% of the genome of any individual strain. We discovered genes for traits that were not known previously in the strains, including genes for the biosynthesis of the siderophores achromobactin and pseudomonine and the antibiotic 2-hexyl-5-propyl-alkylresorcinol; novel bacteriocins; type II, III, and VI secretion systems; and insect toxins. Certain gene clusters, such as those for two type III secretion systems, are present only in specific sub-clades, suggesting vertical inheritance. Almost all of the genes associated with multitrophic interactions map to genomic regions present in only a subset of the strains or unique to a specific strain. To explore the evolutionary origin of these genes, we mapped their distributions relative to the locations of mobile genetic elements and repetitive extragenic palindromic (REP) elements in each genome. The mobile genetic elements and many strain-specific genes fall into regions devoid of REP elements (i.e., REP deserts) and regions displaying atypical tri-nucleotide composition, possibly indicating relatively recent acquisition of these loci. Collectively, the results of this study highlight the enormous heterogeneity of the P. fluorescens group and the importance of the variable genome in tailoring individual strains to their specific lifestyles and functional repertoire.
Introduction
Pseudomonas is a large genus within the γ subclass of Proteobacteria known for its ubiquity in the environment, utilization of a striking variety of organic compounds as energy sources [1], [2], and production of an array of secondary metabolites [3]–[5]. Some species include well-known pathogens such as P. syringae, which comprises many pathovars that are important plant pathogens, and P. aeruginosa, an opportunistic human pathogen. Others are not associated with disease and are prevalent in natural habitats, including soil, water, and plant surfaces. Certain strains live in a commensal relationship with plants, protecting them from infection by pathogens that would otherwise cause disease [6]–[8]. As such, Pseudomonas spp. function as key components of ecological processes that suppress plant diseases in agricultural and natural environments [9]–[11], and several strains are used commercially to manage plant diseases in agriculture [12].
The genus Pseudomonas currently comprises more than 100 named species that have been divided into lineages, groups and subgroups based on multilocus sequence analysis [13]–[15]. Many of the plant commensal strains fall into the Pseudomonas fluorescens group, which currently includes more than fifty named species [13]. Given this diversity, it is not surprising that individual plant-associated strains within the P. fluorescens group differ in many respects, including their capacity to suppress plant disease. For example, effective antagonists are typically identified only after screening large collections of isolates for plant disease suppression, indicating that only a subset of strains within the P. fluorescens group provide biological control. Successful biological control strains have certain characteristics in common: the capacity to colonize plant surfaces, specifically the infection court of target pathogens; and the production of antibiotics toxic to target pathogens or the induction of systemic resistance responses in the plant [8], [16]. Antibiotics, which function as major determinants of biological control, fall into diverse classes, including the phenazines [17], [18], polyketides [3], [19], cyclic lipopeptide biosurfactants [20], and many others. Some strains of Pseudomonas spp. also produce phytohormones [21]–[23] or metabolites that alter plant hormone levels [24], [25], directly influencing the growth and development of their plant associates [26]. Other strains induce resistance responses in plants against disease [27], [28]. Plant-commensal strains of Pseudomonas spp. are intricately enmeshed in plant and soil biology through all of these diverse activities, and their functions as biological control agents have distinguished them as microorganisms with significant effects on agricultural productivity.
Given the spectrum of ecological, metabolic, and biochemical characteristics of this genus, it is not surprising that diversity among Pseudomonas spp. extends to the genomic sequence level. The complete genomes of many species have now been sequenced [29], [30], and only 25% to 35% of the genome of each strain is composed of core genes shared by all members of the genus. Comparisons among the genomes of four strains within the P. fluorescens group (Pseudomonas protegens Pf-5 (previously called P. fluorescens Pf-5 [31]) and P. fluorescens strains SBW25, Pf0-1 and WH6 [32]–[34]) highlight the tremendous diversity of these bacteria. Of the 5741–6009 predicted protein-coding genes (referred to herein as the predicted proteome) identified in each genome, only 3115 are present in all four, composing a core genome representing only 52% to 54% of each strain. Furthermore, nearly a third (1488 to 1833 genes) of the predicted proteome for each strain is unique to that strain, again highlighting the heterogeneity of this group of bacteria.
The genomes of Pseudomonas spp., like those of many other bacteria, display a highly mosaic structure, being composed of relatively stable core regions interspersed with regions that vary among the strains [29], [32]–[34]. Regions that are unique to a specific strain are thought to shape that strain's distinctive characteristics, including its interactions with plant pathogens that are targets of biological control. Many of the unique genomic regions bear features of horizontally-acquired DNA (i.e., atypical trinucleotide content, lack of repetitive extragenic palindromic (REP) elements, or the presence of transposons, prophages, or genomic islands). Therefore, these features may be exploited as markers of genomic regions that define the distinctive attributes of an individual strain. For example, novel natural products including the cyclic lipopeptide orfamide A [35] and derivatives of rhizoxin [36], [37], and traits, such as LlpA bacteriocins [38] and the FitD insect toxin [39], have been discovered through genomics-guided approaches focused on strain-specific regions of the genome of P. protegens Pf-5. The combined repertoire of the core and variable regions of a genome reflects the ecological history of the strain and the various environments or selective pressures that it has encountered over evolutionary time.
To date, the sequenced strains represent only a fraction of the diversity within the P. fluorescens group, and much of the group's metabolic, ecological, and genetic diversity remains unexplored. Here, we provide a comparative analysis of strains within the group, and new genomic sequences for seven plant-associated strains. The seven newly-sequenced strains originate from habitats including soil, root and leaf surfaces from two continents, and exhibit biological control activities against bacterial, fungal and oomycete pathogens through varied mechanisms including antibiotic production, induced systemic resistance, and competitive exclusion (Table 1). Several of the strains were obtained from disease-suppressive soils that exhibit natural processes of biological control due to the presence of indigenous microflora antagonistic to soilborne plant pathogenic fungi or nematodes. Our results confirm the strain-to-strain variation observed previously in the P. fluorescens group, with several hundred genes unique to each of the new genomes. Within each genome, we discovered genes for traits that can be explored in the future for their roles in biological control and other heterotrophic interactions. To explore the evolutionary origin of these genes, we mapped their genomic distributions along with the sites of REP elements and mobile genetic elements (MGEs) to determine if the traits fell into the more ancestral or recently-acquired regions of the genomes. Finally, we complemented our genomic analysis with phenotypic screens to link the gene inventories to key phenotypes exhibited by plant-associated strains in the P. fluorescens group.

Results/Discussion
Genomic features
A summary of the features of each of the seven newly-sequenced genomes of biocontrol strains of Pseudomonas spp. is provided in Table 2. The characteristics (size, GC content, predicted number of coding sequences, and number of rRNA operons) are within the range of previously-sequenced genomes of Pseudomonas spp. [29]. Nevertheless, the seven genomes vary in size by approximately one megabase (ranging from 6.02–6.99 Mb) with the number of CDSs ranging from 5333–6224, indicating substantial strain-to-strain variation. The genomes of P. chlororaphis strains 30-84 and O6 and P. protegens Pf-5 are larger and have a higher GC content than those of the other strains. Only strain A506 has a plasmid, which will be described in detail in a separate publication.

Phylogenetic analysis
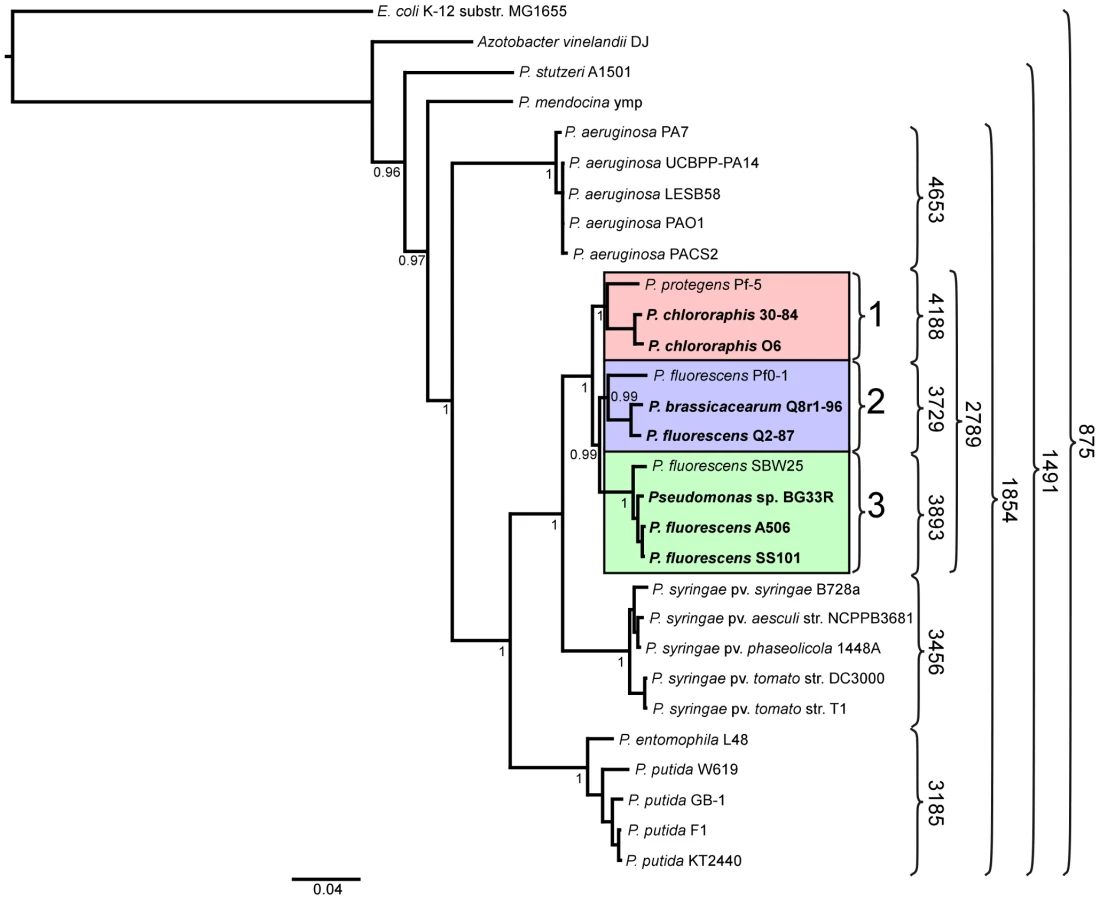
We inferred a phylogenetic tree using a Bayesian approach for representative strains of Pseudomonas spp. having fully sequenced genomes based on multilocus sequence analysis (MLSA) [40] (Figure 1). Along with three of the previously-sequenced strains within the P. fluorescens group (Pf-5, Pf0-1, and SBW25), the seven strains of this study fall into a single large clade composed of three sub-clades. The two strains of P. chlororaphis fall into Sub-clade 1, with strain Pf-5 more distantly associated with the group. Sub-clade 2 is composed of P. fluorescens Q2-87 and P. fluorescens Q8r1-96 (revealed as Pseudomonas brassicacearum Q8r1-96 in this study) and the previously-sequenced strain P. fluorescens Pf0-1, which is not as closely related to strains Q2-87 and Q8r1-96 as those two strains are related to one another. All of the strains in Sub-clades 1 and 2 were isolated from plant roots or soil in the USA (Table 1). In Sub-clade 3, strain A506, which was isolated from a leaf surface in California, USA, and strain SS101, isolated from wheat roots in The Netherlands, are most closely related. Sub-clade 3 also includes the previously-sequenced strain SBW25, isolated from a leaf of sugar beet in England, and Pseudomonas sp. BG33R (also called P. synxantha BG33R), isolated from roots of a peach tree in South Carolina, USA (Table 1). These results are reasonably consistent with a Bayesian phylogeny based on 16S rRNA (Figure S1) and very consistent with a maximum likelihood phylogeny constructed by concatenating 726 protein sequences present in the fully-sequenced strains of Pseudomonas spp. (Figure S2). These phylogenies also are congruent with those from a recent report in which a large number of strains representing many species of Pseudomonas were evaluated by MLSA [13]. In the MLSA study, strains of P. fluorescens and P. chlororaphis also were found to be in a distinct clade clearly distinguished from other Pseudomonas spp. Our MLSA analysis also is consistent with a recent report that assigned strain Pf-5 to the new species P. protegens, which is related to P. chlororaphis but also exhibits distinct properties [31].
Core and pan-genome analysis
A core genome containing 2789 predicted protein coding genes was identified for the P. fluorescens group from a ten-way best-match BLASTp search (Figure 1, Figure 2). This core genome represents only 45% to 52% of the predicted proteome of each strain, further illustrating a large degree of genomic diversity in this group of bacteria. The size of the core genome in the P. fluorescens group is considerably smaller than that of P. aeruginosa, which we have estimated to be 4653 putative protein-coding genes based on comparative BLASTp searches among five sequenced isolates (Figure 1), but is closer to the core genome sizes we estimated for strains of P. syringae and P. putida/entomophila, 3456 and 3185 CDSs, respectively (Figure 1). This estimate is also somewhat smaller than earlier estimates based upon the previously-sequenced genomes of strains within the P. fluorescens group [29], [32], [34], [41], which is to be expected as the number of strains available for comparison increases. Genes conserved among all of the genomes encode proteins contributing mainly to fundamental housekeeping functions, such as protein and nucleic acid synthesis, whereas genes encoding hypothetical proteins and those associated with mobile elements are underrepresented in the core genome (Table S1).

Of the 2789 core genes, only 20 are specific to the P. fluorescens group (Table S2); the other 2769 genes have orthologs in at least one other sequenced genome of Pseudomonas spp. Annotated functions of the 20 core genes include biofilm formation, hypothetical or conserved hypothetical proteins and regulation (Table S2). We attribute the remarkably small number of core genes distinguishing this group from other Pseudomonas spp. to the diversity of strains within the P. fluorescens group and the highly plastic nature of their genomes. This diversity also is reflected in the large size of the pan-genome, which, at 13,872 putative protein-coding genes, is substantially larger than that estimated here for P. aeruginosa (7,824 CDSs). The pan-genome of the P. fluorescens group also exceeds that estimated here for P. syringae (9,386 CDSs) based on the five strains considered in our analysis (Figure 1), but is only slightly larger than the pan-genome of 19 strains of P. syringae (12,829 CDSs) estimated by Baltrus et al. [30]. Of the 13,872 CDSs composing the pan-genome of the P. fluorescens group, 5798 have no orthologs in other genomes of Pseudomonas spp., which probably is due to a high level of differentiation of genes in the group and a high frequency of horizontal gene acquisition from other taxa. It is also likely that the large gene inventory in Pseudomonas spp. is not yet reflected in the relatively small number of genomes sequenced to date.
Pairwise comparisons of predicted proteomes supported the phylogenetic relationships among strains illustrated in the MLSA analysis. For example, strains within a sub-clade (Figure 1) share 69–90% of their predicted proteomes, whereas strains in different sub-clades share only 64–73% of their proteomes (Figure 1, 3). Correspondingly, the core genomes for each sub-clade are substantially larger than the core genome for the group as a whole, ranging from 3729 to 4188 CDSs among the three sub-clades (Figure 1). Pair-wise BLASTp analyses also offered some support for the relatively distant relationship of strain Pf-5 with Sub-clade 1 and of strain Pf0-1 with Sub-clade 2. Indeed, using the level of shared gene content as an indicator of relatedness, strain Pf0-1 is more closely related to strains in Sub-clade 1 than to Q8r1-96 or Q2-87. Of note, the size of core genomes of Sub-clades 1 and 2 increased by 1045 or 912 CDSs, respectively, when only the two more closely-related strains in each of these sub-clades were used for comparison (Figure 1, Table S4).
Whole genome alignments of the strains in the P. fluorescens group were conducted to gauge the level of synteny. There is a relatively high level of synteny around the origin of replication for strains within a single sub-clade (Figure S3, Figure S4, Figure S5), but very little synteny is evident between genomes of strains in different sub-clades. As has been described for a number of bacterial genomes, including P. fluorescens [32], [33], the majority of unique genes and genome rearrangements have occurred around the terminus of replication. This is evident from the distribution of core genes, which are concentrated near the origin of replication of each genome (Figure 3). Nonetheless, the current assemblies suggest that inversion events may have taken place near the origin of replication in strain Q2-87 (Figure S4).

The combination of the phylogenetic analysis and the comparative BLASTp dataset provided an opportunity to identify genes that differentiate each sub-clade. The three genomes in Sub-clade 1 share 73 genes that are not present in any other sequenced Pseudomonas genome (Table S5). These include genes encoding biosynthesis of the antimicrobial pyrrolnitrin and the insect toxin FitD. Within this clade, the two P. chlororaphis strains share 255 genes that are not found in other sequenced strains of Pseudomonas spp. (Table S6). These genes, which may be characteristic of the species, include a cytochrome c oxidase system, bacteriocins, type I secretion system components and several secondary metabolite biosynthesis gene clusters. The three genomes in Sub-clade 2 share 38 genes that are not present in any other sequenced Pseudomonas genome (Table S7). These genes include a lipase and putative type VI secretion system effectors. Strains Q2-87 and Q8r1-96 share 195 genes that are not found in other Pseudomonas genomes, including components of type I and type III secretion systems (Table S8). Strains in Sub-clade 3 share 87 genes that are not found in other strains of Pseudomonas spp., including genes for pili biosynthesis, components of type III secretion systems, and ribose utilization (Table S9).
Each of the ten genomes of the P. fluorescens group includes ca. 300 to 900 genes (6 to 15% of the predicted proteome) that are unique to that strain (Figure 2). This estimate of strain-specific genes is smaller than earlier estimates (ca. 19–29% of the predicted proteome) [32], [34], [41], which is not surprising because the number of unique genes is expected to fall as the number of strains available for comparison increases. Both the large number of strain-specific genes and the large size of the pan-genome indicate a high level of genomic diversity consistent with the observed biological diversity of the P. fluorescens group, including the distinctive biocontrol properties of the strains.
Defining the core and lineage-specific regions of the genomes
We used four criteria to distinguish regions of the Pseudomonas genomes that are more ancestral from those that may have been more recently acquired: i) distribution of the genes unique to each strain as well as the core genes shared among all strains, ii) atypical trinucleotide composition, iii) presence of putative MGEs, and iv) distribution of repetitive extragenic palindromic (REP) elements (Figure 3).
REP elements
REP elements are short nucleotide sequences, typically 20–60 nt long, that are abundant in the intergenic regions of many Pseudomonas spp. [32], [33], [42]–[45]. Functions of REP elements remain in question but they may provide sites for DNA gyrase or DNA polymerase I binding, or for recombination [42]–[44]. REP elements appear to accumulate within the non-coding regions of genomes over time; they are rarely associated with regions of atypical trinucleotide content but display a similarly global distribution to core genes [33]. Therefore, REP elements have been used as markers of older, more stable regions of the genome [33]. Nonetheless, selective pressures are likely to prevent their incorporation within important housekeeping regions where they may disrupt the function of essential cellular processes [32]. For example, there are typically no REP sequences located near the chromosomal replication origin.
The genomes of the P. fluorescens group were examined for the presence of REP elements using a combination of basic repeat searches and Hidden Markov Model (HMM) searches. At least one type of REP element occurring at least 250 times was observed within the non-coding regions of each genome, except that of P. fluorescens Pf0-1 (Table S10, Table S11). In several genomes, two distinct REP elements were identified. To examine the level of conservation of REP elements within the group, HMMs trained on REP sequences from each strain were used to search the genomic sequences of all other strains. This analysis revealed that one primary REP sequence, referred to here as REPa, was conserved, but not identical, among the strains; HMMs trained on REPa sequences from one strain typically identified a large number of these sequences within the genomes of other strains (Table S11). Interestingly, the HMMs trained on REPa sequences from strains in the P. fluorescens group also detected a large number of copies of this element in genomes of P. putida and a small number of copies in genomes of P. syringae (Table S11).
In addition to the primary REPa elements, secondary REP elements were identified in a number of the genome sequences. The first of these, REPb, was identified in both P. chlororaphis strains and at lower abundance in P. protegens Pf-5 (Table S10, Table S11). Given that these strains are phylogenetically related within Sub-clade 1 (Figure 1), this sequence may be sub-clade specific. In contrast, two other secondary REP sequences, REPc and REPd, display unique and scattered distributions among strains in Sub-clades 2 and 3. Another secondary REP element, REPe, was identified only within the genome of P. fluorescens SBW25 (Table S10, Table S11).
REP sequences frequently are organized into pairs or clusters displaying inverted orientations [46]. This organization may be related to their mechanism of dispersal. Recent work has provided evidence for the involvement of a family of IS200/IS605-like REP-associated tyrosine transposases (RAYTs) in REP sequence maintenance and within-genome propagation, where REP pairs (REP doublets forming hairpins; REPINs) are likely to be the minimal mobilizable unit [42], [44]. As recently described in P. fluorescens SBW25 [44], the majority of REP sequences identified within the newly-sequenced genomes were found as oppositely oriented pairs separated by a uniform distance, typically 60–70 bp (Figure S6).
We identified at least one RAYT gene in each genome sequence except that of P. fluorescens Pf0-1. Phylogenetic analysis of the RAYT protein sequences revealed a major clade containing an orthologous RAYT protein in the other nine genomes (Figure 4). In those nine genomes, the RAYT-encoding gene in this major clade is flanked by copies of the REPa element, suggesting that these RAYT orthologs could be involved in the maintenance and propagation of REPa sequences. Interestingly, the sub-clade structure of the major RAYT clade closely resembles that seen in the MLSA tree of Pseudomonas strains (Figure 1, Figure 4), suggesting that these RAYT genes may have been a stable part of the genomes since their divergence. Additional support for this hypothesis comes from the observation that related REPa-associated RAYT genes from Sub-clades 2 and 3 are located within regions of local synteny. Notably, the genome of strain Q8r1-96 harbors two RAYT genes flanked by copies of the REPa sequence (Figure 4). One of these RAYTs (PflQ8_4225) is similar to that encoded by Q2-87 and, as stated above, is encoded in a region of local synteny. The second RAYT in the Q8r1-96 genome is similar to the Sub-clade 1 RAYT proteins and, therefore, may have been acquired laterally from a Sub-clade 1-like strain (Figure 4). Previous studies described a relationship between the number of REP elements within a genome and the presence of a cognate RAYT gene [42]. This trend also is apparent in the strains of this study, most of which carry between 500 and 1500 copies of the REPa sequence element and a single cognate RAYT protein. A larger number of REPa sequences were found in the genome of Q8r1-96, which has two putative cognate RAYT genes. In contrast, very few REP elements are present in the genome of P. fluorescens Pf0-1, which has no RAYT gene. Additionally, RAYT genes associated with REPb, REPd and REPe sequences, which are abundant within their respective genomes, were identified in a number of strains (Figure 4). No RAYT genes were found to be associated with REPc sequences, which are at relatively low abundance in the genomes of several strains (Table S11). Interestingly, the Pf-5 genome has 999 copies of REPa but has a mutation in the RAYT gene, which introduced a stop codon and is likely to inactivate its function. It is possible that REPa sequences were dispersed in the Pf-5 genome prior to the mutation in the RAYT, which may have occurred relatively recently. Overall, however, these observations support the role of RAYT proteins in the propagation and maintenance of their cognate REP sequences.
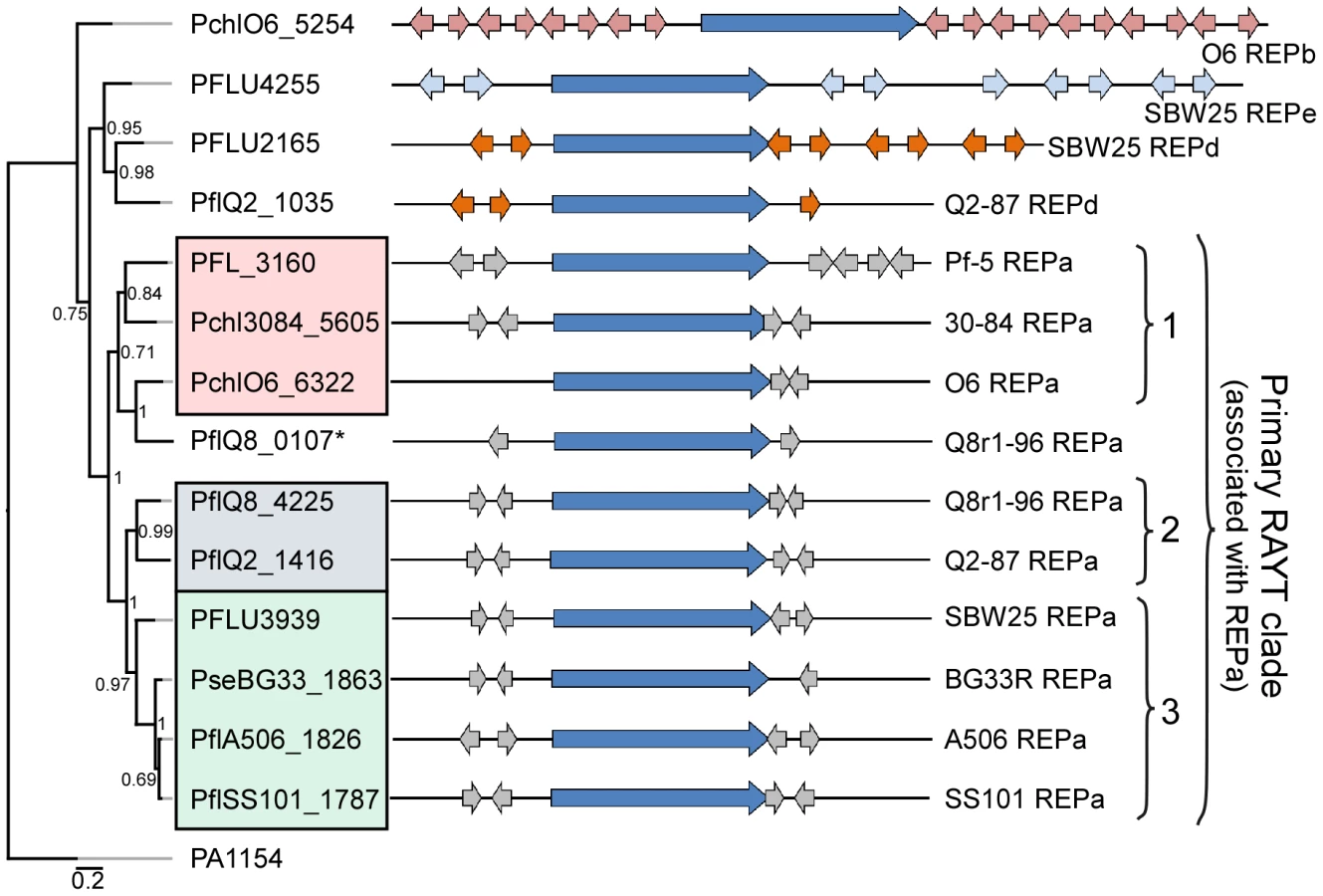
The REP elements are not uniformly distributed in the genomes of the P. fluorescens group, and regions lacking REP sequences are striking in the genomes evaluated here (Figure 3), as described previously for Pf-5 [33] and SBW25 [32]. These regions, termed REP deserts, vary in number among the genomes. For example, using an arbitrary lower limit of 25 kb to define a REP desert, the four genomes in Sub-clade 3 have 31 to 66 REPa deserts, totaling 1.2 to 3.2 Mb in size (20% to 47% of the genome). Defining deserts for the secondary REPs, which are present in fewer copies than REPa in all genomes, is difficult; but in many cases, regions lacking secondary REP elements encompass REPa deserts (Figure 3). The REPa deserts commonly correspond to atypical regions of the genome, defined by atypical nucleotide composition, and some of the REPa deserts contain mobile genetic elements (Figure 3).
Mobile genetic elements (MGEs)
MGEs were defined in this study as genome segments encoding putative functions linked to the intra - and extracellular movement of DNA in bacteria and/or bearing traces of recent horizontal gene transfer events.
Each Pseudomonas genome in this study contains a unique set of transposons (Table S12). The number of transposon copies per genome ranges from six (SS101) to twenty (O6) with about half of the copies likely to be rendered inactive by frameshift mutations and/or deletions. Members of the IS3, IS4, IS5 and IS66 families are most common. Among notable transposon-related features is a 5.2 kb composite transposon from Q8r1-96 with a putative pathway for catabolism of the broadleaf herbicide bromoxynil. The transposon, which is the only composite transposon in these genomes, is comprised of two IS elements of the IS5 family flanking a group of genes that encode a LysR-like transcriptional regulator, a transporter of the sodium solute superfamily, and a bromoxynil-specific nitrilase (Transposon 1). Another interesting transposon-related feature is found in strain 30-84, where two putative insecticidal toxin genes are found adjacent to genes encoding site-specific integrases and a Tn402-like transposase (Island 2). The type of genes present, their overall arrangement, and lack of flanking inverted repeats suggest that this genomic region may represent an integron remnant.
Genomes of all of the strains contain one to four prophages and/or prophage remnants, each ranging in size from 3.4 to 72.3 kb. Collectively, the seven newly-sequenced genomes have 18 prophages, most of which have a set of cargo genes that are distinct from those in prophages of other strains (Figure S7). Notable exceptions are the prophages integrated in the mutS/cinA region (Prophage 1 of each genome), each of which carries a subset of five distinct bacteriophage gene cassettes (Figure 5). These prophages display the mosaic structure that characterizes prophages in other Pseudomonas spp. [47]. The remaining prophages in the seven genomes carry a diverse array of cargo genes that encode putative bacteriocins, UV resistance proteins, adenine - and cytosine-specific DNA methyltransferases, and conserved hypothetical proteins (Table S13). In addition, a prophage remnant in strain SS101 contains two gene clusters encoding components of chaperone-usher machinery (Island 2). Each cluster encodes an usher, a chaperone, and two fimbrial subunits that may be involved in the production of cell surface-associated appendages similar to Cup fimbriae of P. aeruginosa [48]. In P. aeruginosa, these fimbriae are involved in bacterial surface attachment and biofilm formation [49], [50]. Homologous loci are present in P. syringae and P. putida, but their precise roles, as well as the role of the chaperone-usher machinery in SS101, remain to be discovered.

In addition to transposons and prophages, the genomes carry three to seven genomic islands ranging from 2.3 to 154.3 kb in size (Table S13). Collectively, the seven newly-sequenced genomes have 32 genomic islands. Among the cargo genes of these islands are those with predicted functions as components of restriction-modification systems (Island 1 in 30-84; Island 1 in Q8r1-96), assorted transporters (Island 1 in 30-84; Islands 3 and 6 in SS101; Islands 3 and 4 in BG33R), transcriptional regulators (Island 2 in O6; Island 4 in Q8r1-96; Island 2 and 4 in Q2-87; Islands 2, 4, and 6 in SS101; Island 7 in BG33R), two-component signal transduction systems (Island 3 in 30-84; Islands 1, 2, and 3 in O6; Island 6 in SS101), a methyl-accepting chemotaxis protein (Island 4 in SS101), a polyphosphate kinase (Island 3 in 30-84; Island 2 in O6), a TonB-dependent outer-membrane receptor (Island 1 in O6; Island 3 in BG33R), a putative β-lactamase (Island 4 in SS101), a UV irradiation resistance protein (Island 4 in SS101), and a diverse array of conserved hypothetical proteins (all strains). Other notable features include gene clusters for a mevalonate-independent pathway of isoprenoid production and a type VI secretion system (Island 3 of strain O6), a chaperone-usher fimbrial biogenesis pathway (Island 6 of strain SS101), and an indole-3-acetic acid uptake and catabolism pathway (Island 3 of BG33R). Finally, several genomic islands also contain transposons (Island 2 in 30-84; Island 3 in O6; Islands 1 and 2 in Q2-87; Island 6 in SS101) and genes of bacteriophage or plasmid origin (Island 2 in 30-84; Islands 1 and 2 in O6; Island 3 in A506; Islands 1, 2, and 3 in Q8r1-96; Islands 1 and 5 in Q2-87; Islands 4 and 6 in SS101; and Islands 1, 3, and 6 in BG33R) (Table S13).
Plasmid-like elements were identified in strains A506 and BG33R. Strain A506 carries a 57-kb cryptic plasmid, pA506, which has features in common with the pPT23A family of plasmids, members of which are widespread in P. syringae [51]. pA506 and the pPT23A plasmids share genes involved in replication, mating pair formation and conjugative transfer. pA506 also contains a type IV secretion gene cluster interrupted by the insertion of 11 genes that encode components of type IV conjugative pili similar to those of the pathogenicity island PAPI-1 from P. aeruginosa PA14 [52]. Other plasmid-borne genes have putative functions as integrases and components of a lesion-bypass DNA polymerase RulAB that may contribute to tolerance of UV-induced DNA damage in A506.
Strain BG33R harbors a 154-kb genomic island, Island 3, which belongs to a class of mosaic elements known as integrative conjugative elements (ICEs) [53]. ICEs resemble conjugative plasmids carrying bacteriophage-like integrase genes and are capable of site-specific integration into bacterial genomes. In the genome of BG33R, Island 3 is integrated into one of the five tRNA-Gly genes. Genes for site-specific integration, plasmid maintenance and conjugation span almost half of the island and are similar to their counterparts in the PFGI-1 ICE of P. protegens Pf-5 [54]. However, unlike PFGI-1, Island 3 lacks genes encoding conjugative pili and therefore appears to be anchored in the genome of BG33R. The presence of a fragment of pilS suggests that, at some point in time, Island 3 contained a functional conjugative pilus gene cluster that subsequently underwent deletion. Island 3 also contains genes encoding putative pathways for uptake and catabolism of IAA, quinolones, and haloaromatic compounds, a MexCD-like multi-drug resistance efflux pump and other transporters, a pertussis toxin subunit-like protein, several transposases, and regulatory and conserved hypothetical proteins.
CRISPRs (Clustered Regularly Interspaced Short Palindromic Repeats), loci responsible for prokaryotic immunity to phage infection, were not found in any of these strains. Putative CRISPRs identified using the CRISPRFinder program [55] are present within called genes, rather than in intergenic regions as expected, and are not contiguous to genes encoding typical CRISPR-associated proteins that are required for CRISPR functionality.
As expected for elements acquired horizontally, the MGEs map to regions of the genomes having atypical nucleotide composition and devoid of REP elements (Figure 3). The genes carried by MGEs contribute to the heterogeneity of strains in the P. fluorescens group, comprising 2% to 6% of each of the genomes, ranging from 131.8 kb (in Q2-87) to 379.2 kb (in BG33R). Nevertheless, they make up a small proportion of the genetic variation seen within the group, and many of the strain-specific regions of the genomes do not exhibit the distinct hallmarks of MGEs (i.e., transposons, integrases, prophages, or conjugative elements) described above.
Phylogenetic distribution of traits involved in plant-microbe interactions
We surveyed the genomes of each strain for the presence of loci associated with biological control, including secondary metabolite biosynthesis and bacteriocin production. We also identified loci contributing to the interactions of Pseudomonas spp. with plant and animal host cells and the environment, such as secretion systems for export of exoenzymes, proteinaceous effectors, and toxins. The locations of these loci were mapped onto the genomes of each strain, along with the locations of unique and core genes, regions of atypical trinucleotide composition, MGEs, and REP elements, to provide insight into the evolution of traits contributing to the distinctive biology of each strain.
Secondary metabolite biosynthesis
Compounds toxic to phytopathogenic fungi, oomycetes, and bacteria are important contributors to biological control, and collectively, the strains evaluated herein are known to produce phenazines [17], [18], hydrogen cyanide, the chlorinated tryptophan derivative pyrrolnitrin, and the polyketides 2,4-diacetylphloroglucinol, rhizoxin and pyoluteorin [3]. Gene clusters for each of these compounds were identified in the genomic sequences of the producing strains (Figure 6). In addition to these known gene clusters, a locus similar to the characterized 2-hexyl-5-propyl-alkylresorcinol biosynthesis gene cluster of Pseudomonas chlororaphis subsp. aurantiaca BL915 [56] was identified in the two P. chlororaphis genomes. 2-hexyl-5-propyl-alkylresorcinol exhibits moderate antifungal and antibacterial activity and, if produced by P. chlororaphis O6 and 30-84, could contribute to their suppression of fungal and bacterial plant pathogens.

Cyclic lipopeptides (CLPs), composed of a lipid tail linked to a cyclic oligopeptide, are a class of compounds produced by many strains of Pseudomonas spp. that exhibit surfactant, antimicrobial, anti-predation, and cytotoxic properties [20], [35], [57], [58]. The structural diversity of the CLPs is due to differences in the length and composition of the lipid moiety as well as in the type, number and configuration of the amino acids in the peptide chain. These compounds are synthesized via a non-ribosomal mechanism of peptide synthesis and genes encoding non-ribosomal peptide synthetases (NRPSs) are clustered with those having efflux and regulatory functions in the CLP biosynthetic loci of Pseudomonas spp. Genes coding for production of the CLP orfamide A are present in a single gene cluster in the Pf-5 genome [35], whereas orthologs for the CLPs massetolide A and viscosin are present in two distinct locations in the genomes of P. fluorescens SS101 and SBW25, respectively [59], [60] (Figure 7). We identified gene clusters for CLP biosynthesis in the genomes of BG33R and Pf0-1, and found that strain BG33R exhibited phenotypes (swarming motility, hemolytic activity, and surfactant activity) associated with CLP production. Although these phenotypes were not expressed by Pf0-1, they were exhibited by a derivative of Pf0-1 containing the gacA+ gene from strain Pf-5 (Figure 7, Table S14) but not by a derivative of Pf0-1 containing the gacS+ gene from strain Pf-5 (Table S14). Similarly, other phenotypes typically expressed by Pseudomonas spp. under the control of the Gac/Rsm signal transduction pathway [61] were not exhibited by Pf0-1 but were exhibited by the gacA+-complemented derivative of Pf0-1 (Table S14). From these results, we concluded that the previously-sequenced strain Pf0-1 [32] has a mutation in gacA, which encodes a component of the GacA/GacS global regulatory system required for the production of many secondary metabolites and exoenzymes in Pseudomonas spp. [61]. Consequently, throughout this study we relied on the gacA+ derivative of Pf0-1 to explore relationships between gene inventory and phenotypes for this strain. Although the structures of the CLPs produced by BG33R and Pf0-1 are unknown, the amino acid composition of the peptide moiety could be predicted from the sequences of the NRPSs in the CLP gene clusters. The predicted structure of the BR33R CLP includes a 9-amino acid peptide similar to that of massetolide [59] or pseudophomin A and B [62], and the Pf0-1 CLP includes an 11-amino acid peptide that is distinct from other CLPs described to date (Figure 7).

The fluorescent pseudomonads are characterized by their production of fluorescent pigments in the large and diverse pyoverdine class [63], which function as siderophores for iron acquisition by the bacterial cell. Many genes are involved in the biosynthesis, utilization and regulation of the pyoverdine iron-acquisition system [64], and these Pseudomonas spp. have a full complement of pyoverdine genes, which are present in three to seven clusters dispersed in the genomes. Many Pseudomonas spp. produce secondary siderophores that also contribute to iron nutrition [64], such as enantio-pyochelin, which is produced by Pf-5 [65]. Among these secondary siderophores is pseudomonine, which is produced by the same NRPS pathway used for the biosynthesis of two other siderophores, acinetobactin and anguibactin, with the primary substrate dictating the final product from a common biosynthetic mechanism [66]. Gene clusters for the biosynthesis and uptake of a pseudomonine-like compound [67] are present in the genomes of BG33R and A506, and clusters for the biosynthesis and transport of the siderophore achromobactin [68] are present in P. chlororaphis strains O6 and 30-84. The production of these secondary siderophores has not been confirmed. However, we identified a number of putative binding sites for the ferric uptake regulator (FUR) in the intergenic regions of these gene clusters using HMMs trained on sequences identified in the genome of P. protegens Pf-5 [69], suggesting that the genes are iron-regulated, as expected for a siderophore biosynthesis region. In addition, four genomes (Pf-5, BG33R, SBW25 and SS101) have a full complement of genes required for the biosynthesis and efflux of a hemophore (Figure 6), a protein that, when exported from the cell, can chelate heme with high affinity and then be bound and taken up by specific outer membrane receptors [70].
Within the genomes, we identified many orphan gene clusters, defined as loci with characteristic sequences of secondary metabolism genes but without known biosynthetic products. Eight orphan clusters have genes for NRPSs; two have genes for polyketide synthases (PKSs); and one contains a hybrid NRPS-PKS (Figure 6). All strains except Pf-5 have a cluster homologous to pvfABCD, which contains an NRPS-encoding gene and is required for the biosynthesis of a putative signaling molecule in P. entomophila [71]. A homologous gene cluster (mgoBCAD) is required for production of the phytotoxin mangotoxin by strains of P. syringae pv. syringae causing apical necrosis of mango [72], [73], but the recently-described mangotoxin biosynthesis gene cluster (mboABCDEF) [74] is not present in any of the ten genomes of the P. fluorescens group. The structure of mangotoxin is not known, but we attempted to detect its production by strains of the P. fluorescens group using an established plant bioassay. Mangotoxin-associated phytotoxicity was not observed on tomato leaves inoculated with any of the ten strains. These results agree with a recent report that strains Pf-5 and Pf0-1 do not produce mangotoxin [74]. The functions of the pvfABCD homologs in the strains of the P. fluorescens group are unknown, but possibilities include a signaling role as proposed for P. entomophila [71].
Three of the NRPS-containing orphan gene clusters in the newly-sequenced genomes are likely to encode the biosynthesis of secondary siderophores, based upon similarities to siderophore biosynthetic loci in other bacteria and the presence of genes encoding TonB-dependent outer-membrane proteins, which commonly function in siderophore uptake. One of these, a 36.5-kb region in the genome of BG33R, includes genes for the biosynthesis of salicylic acid, an intermediate in the biosynthesis of pyochelin and other siderophores. Of note, putative FUR binding sites also were identified upstream of several genes within this gene cluster, providing further support for a role of the cluster in iron homeostasis. Bioinformatic analysis of the second putative siderophore-biosynthesis cluster, which is present in Q8r1-96 and SBW25, predicts that the NRPS product is a nine amino acid peptide, possibly ornicorrugatin, which is produced by SBW25 [75]. The NRPS-encoding genes in the third cluster, present in the genome of Q2-87, are predicted to synthesize a six amino acid peptide via a biosynthetic pathway similar to that for siderophore biosynthesis by Ralstonia eutropha [76].
Pseudomonas spp. are well known for their prolific production of diverse secondary metabolites, only a fraction of which are synthesized via the NRPS and PKS mechanisms of biosynthesis considered in this analysis. Although the products of orphan gene clusters in the seven genomes of this study could not be predicted from the nucleotide sequence data, the loci provide promising subjects for identification of novel natural products. In keeping with the roles of known secondary metabolites produced by these strains, the metabolites could certainly serve important functions in the ecology of these bacteria, including their interactions with other soil - or plant-associated microorganisms.
Of the many secondary metabolite and siderophore biosynthetic gene clusters present in the genomes, only the clusters for pyoverdine production are present in all strains (Figure 6). Certain other clusters (e.g., HCN biosynthesis) are in a conserved location in the genomes of all strains composing a sub-clade, possibly indicating acquisition during the divergence of the sub-clade from its progenitors. Other clusters (e.g., phenazine, 2-hexyl-5-propyl-alkylresorcinol, 2,4-diacetylphloroglucinol, and achromobactin) are present in conserved locations within the genomes of the most closely-related strains within a sub-clade, and may have been acquired more recently in the evolution of those strains. The majority of secondary metabolite gene clusters have a patchy distribution among the ten genomes, indicating a complex pattern of inheritance including several independent acquisition events and/or loss of the clusters from the genomes of certain strains (Figure 6). Therefore, the distribution of secondary metabolism gene clusters in the genomes of these Pseudomonas spp. cannot be explained by a single type of inheritance, but results from many processes operating throughout the evolution of these strains ([3] and references therein).
Bacteriocins
Among the arsenal of anti-microbials produced by Pseudomonas spp. are the bacteriocins, narrow-spectrum proteinaceous toxins that typically kill bacteria closely related to the producing strain. Bacteriocins toxic to bacterial phytopathogens can contribute to biocontrol [77] and can play an important role in the fitness of a strain by killing or inhibiting bacterial co-inhabitants that compete for limited resources in the environment. Each of the ten genomes of the P. fluorescens group has two to seven predicted bacteriocins (Figure 6). Collectively, the genomes include genes for many of the structurally-diverse bacteriocins known to be produced by Pseudomonas spp., including the S1/2/3/AP41 pyocins [78], [79], S5 pyocins [80], colicin M-like bacteriocins [81], and the lectin-like Llp bacteriocins [38] (Figure 6). Strain A506 has a region related to those encoding microcin B17 production in the Enterobacteria [82]; this bacteriocin has not been described previously in Pseudomonas spp. We also identified putative novel bacteriocins in the predicted proteomes of the P. fluorescens group by the presence of receptor, translocation, and active domains characteristic of these proteinaceous toxins. One group of putative bacteriocins (designated N1 for novel group 1, Figure 6, Figure S8) has members in all strains studied except for Pf-5. The predicted translocation domain (Pfam: PF06958) shared by proteins in the N1 group is similar to those of other bacteriocins produced by Pseudomonas spp., whereas the active and receptor-binding domains are variable. Some members of the N1 group have a DNase domain (Pfam: PF12639) distantly related to those found in pyocins S1/2/AP41, whereas others have a cytotoxic domain (Pfam: PF09000) similar to the active domain found in colicin E3 of E. coli, which has RNase activity directed at the 16S ribosomal subunit [83]. This cytotoxic domain is not present in any known bacteriocin produced by Pseudomonas spp. The second group of putative bacteriocins (designated N2) is found in four strains (Figure 6, Figure S8). All of the proteins in the N2 group have receptor-binding and translocation (Pfam: PF06958) domains similar to, but distinct from, those in carocin S1, a bacteriocin produced by Pectobacterium carotovorum [84]. The active domains are predicted to encode DNase activity; these domains are similar to the active domain of pyocin S3 (∼50% ID) or carocin S1 (∼40% ID) for the N2 proteins of 30-84, O6 and SS101, but similar to those of pyocin S1/S2/AP41 for the N2 proteins in Pf0-1 (Pfam: PF12639). A third predicted type of novel bacteriocin (N3, Figure 6), present in the genome of BG33R, has an active domain similar to the pore-forming domain of colicin N in the C terminus (Pfam: PF01024) but similar to a portion of colicin M at the N terminus [85]. The functions of the diverse bacteriocins present in the genomes of the P. fluorescens group remain largely uncharacterized, although enzymatic activity was demonstrated for the colicin M-like bacteriocin from Q8r1-96 [81] and antibacterial activity for an Llp bacteriocin produced by strain Pf-5 [38]. The widespread presence and diversity of these proteinaceous toxins suggest that bacteriocins may play an important role in the intraspecific interactions and competitiveness of Pseudomonas spp.
In the genomes of the P. fluorescens group, many of the genes coding for bacteriocins are clustered with genes encoding immunity, forming prototypic toxin-antitoxin gene pairs. Others are distal from any known immunity gene, suggesting that immunity may be conferred for multiple related bacteriocins from a single immunity gene or that novel resistance genes may exist in these genomes. There are striking differences among strains in the numbers and types of bacteriocins produced, with no clear correlations to the phylogenetic relationships among the strains. Indeed, many of the bacteriocin genes fall in genomic islands or other atypical regions of the genomes (Figure 6), indicating that these genes may be the result of horizontal mechanisms of inheritance and dispersal.
Metabolism of phytohormones, volatiles, and plant signaling compounds
Plant-associated bacteria can influence plant growth and development directly by producing or degrading plant hormones or other factors that modulate plant regulatory mechanisms [7]. Indole-3-acetic acid (IAA) is the primary auxin in plants, controlling many important physiological processes, and IAA production by plant-associated bacteria can have profound effects on plant growth and development [22]. We screened the genomes of the P. fluorescens group for pathways involved in the production of IAA [22] and detected genes for tryptophan-2-monooxygenase (IaaM) and indole-3-acetamide hydrolase (IaaH), which convert tryptophan to IAA via the two-step indole-3-acetamide pathway, in the genomes of P. chlororaphis strains 30-84 and O6. IAA is known to be produced by strain O6 via the indole-3-acetamide pathway [86] and we detected auxin in cultures of strain O6, as expected; however, we did not detect auxin in cultures of 30-84. Although we detected no obvious mutations in iaaM and iaaH of strain 30-84, the sequences differ slightly from those in strain O6 (e.g., substitution for a conserved proline at site 80 of IaaH) and may be non-functional. Differences in auxin production also could be due to variation in expression of the IAA biosynthesis genes by the two strains under the conditions of our study. An IAA catabolic (iac) gene cluster in the genome of strain BG33R (Figure 6) encodes putative IAA degradation enzymes, a regulatory protein, a dedicated outer membrane porin, and an ABC transporter. The overall genetic organization differs from that of the iac cluster of P. putida 1290, but resembles a putative IAA degradation locus of Marimonas sp. MWYL1 [25]. The cluster resides next to a phage-like integrase gene on genomic Island 3 of BG33R, suggesting that it was acquired via horizontal transfer.
Strains 30-84, O6, and Pf-5 also carry genes for catabolism of the plant hormone and antimicrobial metabolite phenylacetic acid (PAA) [87], [88](Figure 6) and we found that the strains can grow on a medium containing PAA as a sole carbon source. These genes, like the well-characterized paa operon of P. putida U [89], control conversion of PAA to Krebs cycle intermediates via phenylacetyl-CoA (PAA-CoA) and encode a PAA-CoA ligase, a PAA-CoA oxygenase/reductase, and enzymes catalyzing cleavage and further degradation of the aromatic ring [90]. The paa clusters of strains in Sub-clade 1 also include genes encoding components of a PAA-specific transporter.
Aminocyclopropane-1-carboxylic acid (ACC) is the immediate precursor of the plant hormone ethylene. Stressed plants accumulate ethylene, which inhibits root elongation and accelerates abscission, aging and senescence [91]. ACC deaminase-producing rhizobacteria lower plant ethylene levels by converting ACC into ammonia and α-ketobutyrate, thereby stimulating root growth and improving tolerance to environmental or pathogen-induced stress. Among Pf-5 and the seven newly-sequenced strains, only strain Q8r1-96 carries the acdS gene, which encodes ACC deaminase. Q8r1-96 grew on DF salts medium [92] with 3 mM ACC as the sole source of nitrogen and produced measurable amounts of α - ketobutyrate (2062.4±539.1 nmol mg protein−1 hr−1) during deamination of ACC . On the other hand, strains Q2-87 and SS101, which do not have acdS, did not grow on the DF-ACC medium and exhibited no detectable ACC deaminase activity.
Acetoin and 2,3-butanediol are volatiles often produced by bacteria during mixed acid-type fermentation. Both compounds have been implicated as plant growth-promoting metabolites [27], [93]. The synthesis of acetoin and 2,3-butanediol is best understood in the Enterobacteriaceae and Bacillus spp., where it proceeds via the formation of α-acetolactate from pyruvate and further conversion to acetoin and 2,3-butanediol [24]. The transformations are catalyzed by the catabolic α-acetolactate synthase (BudB/AlsS), α-acetolactate decarboxylase (BudA/AlsD) and acetoin reductase (BudC/YdjL) in members of the Enterobacteriaceae and Bacillus spp. [94]–[96]. P. chlororaphis O6 is known to produce 2,3-butanediol [27], and a putative acetoin reductase gene is present in the genome of O6 and other strains in Sub-clade 1. However, we did not detect orthologs of budAB/alsSD, which catalyze the synthesis of α-acetolactate and acetoin from pyruvate in other bacteria, in the genomes of O6 or 30-84. One plausible explanation for this apparent discrepancy is that α-acetolactate is formed by another pathway in strains O6 and 30-84, possibly via the α-acetohydroxyacid synthase encoded by ilvBN [97]. We detected orthologs of ilvBN in all ten genomes of the P. fluorescens group. α-Acetolactate is unstable and spontaneously decomposes in the presence of oxygen into acetoin or diacetyl (also called 2,3-butanedione) [24], which would provide the necessary substrate for the acetoin reductase and formation of 2,3-butanediol by strains in Sub-clade 1. Six strains featured in this study carry aco genes for an acetoin dehydrogenase (AoDH) enzyme complex that converts acetoin to acetaldehyde and acetyl-CoA. A four-gene cluster encoding an AcoR regulatory protein and AcoABC proteins that represent, respectively, the E1α, E1β, and E2 subunits of the AoDH enzyme complex, are present in these genomes. Four strains (Pf-5, 30-84, Q8r1-96,and Q2-87) also have an uncharacterized gene, acoX, and a 2,3-butanediol dehydrogenase gene, bdh, which may allow catabolism of 2,3-butanediol as well as acetoin. Interestingly, the dedicated E3 (dihydrolipoamide dehydrogenase) component of AoDH is missing from all of the genomes, and a common E3 subunit is presumably shared by AoDH and the pyruvate dehydrogenase and 2-oxoglutarate dehydrogenase enzyme complexes [24].
The non-protein amino acid γ-aminobutyric acid (GABA) is secreted in millimolar amounts by plant tissues in response to abiotic and biotic stresses [98]. This metabolite reduces the activity of herbivorous insects and the virulence of bacterial and fungal pathogens [99]. Indeed, gabT mutants of P. syringae pv. tomato DC3000, which lack production of GABA aminotransferase, exhibit reduced expression of type III secretion and effector genes and reduced virulence in Arabidopsis [100]. This observation is consistent with the idea that GABA plays a role in plant-bacterial communication. Genomes of all ten strains included in this study have gabT and gabD, which encode a putative GABA aminotransferase and a succinate semialdehyde dehydrogenase involved in GABA utilization. Interestingly, the genomes of Q8r1-96, Q2-87, 30-84 and O6 carry three gabT paralogs, two of which are linked to gabD-like genes. An almost identical gab gene arrangement is found in the genome of the plant pathogen P. syringae pv. tomato DC3000, but a recent study by Park et al. [100] implicated only one gabTD-like locus in the catabolism of GABA. The function of GABA in the interactions of biocontrol Pseudomonas spp. with their plant hosts remains to be established.
Exoenzymes
Secreted enzymes are an important group of molecules involved in nutrient acquisition and the interactions of bacteria with their microbial co-inhabitants and eukaryotic hosts. Each of the ten genomes has a conserved cluster for the exoprotease AprA and its secretion via a type I mechanism (Figure 6). The strains also tested positive for exoprotease production, whereas aprA deletion mutants of strains Pf-5 and A506 lacked exoprotease production (Table S14), indicating that the conserved aprA gene is responsible for this phenotype. AprA (previously called AprX) production by A506 has a confounding role in the biological control of fire blight disease of pear and apple. The protease degrades pantocin A, a peptide antibiotic produced by the biological control agent Pantoea vagans C9-1 that is toxic to the fire blight pathogen Erwinia amylovora [101]. AprA-mediated proteolysis of pantocin A results in diminished biological control of fire blight when pome fruits are treated with A506 in combination with P. vagans [102]. In contrast, a mixed inoculum composed of the aprA mutant of A506 with P. vagans results in more effective and consistent biological control of fire blight than achieved with either of the biocontrol strains applied individually. This enhanced biological control is attributed to the combined activity of two compatible biocontrol strains that suppress disease by complementary mechanisms [102]. Seven genomes, representing all three sub-clades, contain additional genes with predicted functions as exoproteases (Figure 6), but their roles in the biology of the strains remain unknown at present.
Chitinases produced by certain Pseudomonas spp. can hydrolyze fungal cell walls, thereby contributing to the biological control of fungal diseases of plants [103]. Collectively, the genomes contain two chitinase genes, with one form distributed among strains in all three clades (Figure 6). A second chitinase, which is orthologous to chiC of P. aeruginosa [104], is present in a region with unusual trinucleotide composition in the three strains in Sub-clade 1, suggesting recent acquisition by this lineage. We evaluated all ten strains for chitinase production, and found that strains having at least one of these chitinases exhibited chitinolytic activity in culture (Table S14).
One strain, SBW25, exhibited pectolytic activity on potato, and a gene for pectate lyase [105] is present in the genome of SBW25, whereas neither pectolytic activity nor the pectate lyase gene was present in the other genomes (Figure 6).
Secretion systems
Many extracellular enzymes are transported out of the cell through type II secretion systems (T2SSs) and, collectively, the ten genomes evaluated in this study have four T2SSs. Three of the T2SSs are related to the Xcp and Hxc systems of P. aeruginosa [106], whereas the fourth system, present only in the genomes of Sub-clade 3, is novel. Each genome has one to three T2SSs, and candidate substrates include lipase, esterases, alkaline phosphatases, and, in SBW25, a pectate lyase.
Type III and Type VI secretion systems, which function in the delivery of effector molecules into plant, animal, or bacterial cells, are prevalent in Gram-negative bacteria, including environmental strains of Pseudomonas spp. having no known pathogenic or symbiotic associations with eukaryotic cells [34], [69], [107]–[109]. We identified several types of both secretory systems in the genomes of the plant-associated strains of the P. fluorescens group.
The type III secretion system (T3SS) is used by a variety of Gram-negative bacteria for delivery of effector molecules into a eukaryotic host cell [110]. Six strains examined in this study (i.e. A506, Q8r1-96, Q2-87, SS101, SBW25 and BG33R) carry rsp/rsc (rhizosphere-expressed secretion protein and rsp - conserved) gene clusters that vary in length between 18 and 28 kb and resemble the hrc/hrp T3SS of the plant pathogen P. syringae. The rsp/rsc clusters of these six strains belong to the Hrp1 family (Figure 8), which includes T3SSs from pathogenic and saprophytic plant-associated Pseudomonas spp. The Hrp1 family is phylogenetically diverse and encompasses multiple lineages of T3SSs that are often encoded by genomic islands [108], [111]. The T3SSs of strains in Sub-clades 2 and 3 are integrated into different sites in the genomes and differ in the arrangement of genes within the rsc/rspZ operon. These T3SSs may represent independent acquisitions in the two sub-clades and may relate to sub-clade-specific host or biocontrol properties. No T3SS was detected in the genomes of Sub-clade 1.

Q2-87 is unique amongst the biocontrol strains in possessing a second T3SS gene cluster, in addition to rsp/rsc, that is similar to the inv/mxi/spa cluster of human and animal pathogens such as Salmonella enterica [112]. A type III effector gene in the inv/mxi/spa-like gene cluster of S. enterica also is present in the Q2-87 genome. This is the first example of an inv/mxi/spa-like T3SS in Pseudomonas spp.
We identified putative Type III effectors in genomes of several strains in the P. fluorescens group through bioinformatic analyses (Figure 6). Of the three effectors shown previously to be secreted by Q8r1-96, two are homologs of the P. syringae effectors HopAA1-1 and HopM1; the third, RopB, is a novel effector [108]. Homologs of RopAA-1, RopM and RopB also are present in the genome of Q2-87. A homolog of ExoU, a P. aeruginosa effector with phospholipase activity that causes rapid death in eukaryotic cells [113], is present in all strains with T3SSs except for strain Q8r1-96. The A506, SS101 and BG33R genomes have 14 to 15 additional genes preceded by putative Hrp(Rsp)L-dependent promoters; Q8r1-96 also contains one such gene. These genes encode conserved hypothetical proteins with N-termini typical of T3SS-secreted proteins (i.e., abundance of Ser and polar residues at the N-termini, only one acidic residue in the first 12 positions, and an aliphatic amino acid in position 3 or 4)(Table S15, Figure 6). Clearly, experimental evidence is required to prove/disprove the possibility that these proteins indeed represent novel type III effectors.
Despite the widespread occurrence and high level of conservation of T3SSs in strains of the P. fluorescens group, their functions remain enigmatic. In SBW25, the T3SS has been shown to operate in the sugar beet rhizosphere and its inactivation compromised the ability of this strain to efficiently colonize plant roots [109],[114],[115]. On the other hand, the T3SSs of P. brassicacearum strain Q8r1-96 and P. fluorescens KD are expressed during root colonization, but the corresponding mutants are not altered in their rhizosphere competence [108], [116]. In environmental strains of P. aeruginosa, ExoU and other T3SS effectors are required for colonization and killing of protozoa [117]. Similarly, the T3SSs of P. fluorescens may function in defense of the bacteria against predation and competition in their natural habitats in the soil, rhizosphere, or on aerial plant tissues. In line with this possibility, the T3SS of P. fluorescens strain KD is involved in suppression of Pythium ultimum, a soilborne oomycete pathogenic to many plant species [116].
Type VI secretion systems (T6SSs) are prevalent and conserved among Gram-negative bacteria. During the first years after their discovery, they were thought to be involved primarily in delivery of virulence effectors to eukaryotic hosts. More recently, the prevalence of T6SSs in genomes of environmental bacteria, which are likely to encounter intense competition and predation in natural habitats, has become increasingly evident; and T6SSs are now thought to play a role in interbacterial interactions [118]–[120]. Each of the genomes of the P. fluorescens group includes one to three clusters of genes encoding a T6SS. Collectively, the genomes include four types of T6SSs, three of which are similar to the three well-characterized T6SS loci of P. aeruginosa (termed HSI-I, HSI-II and HSI-III) [121]. A locus similar to the HSI-I T6SS, which was described previously in strain Pf-5 [69], is present in nine of the genomes evaluated herein; only strain Pf0-1 lacks an ortholog of HSI-I (Figure 6). These HSI-I loci lack tagJ1, which encodes an accessory lipoprotein in the P. aeruginosa HSI-I, and four genomes have two additional genes at this location in the gene cluster. Seven genomes (30-84, O6, Q2-87, SS101, Pf0-1, A506, and BG33R) have loci similar to HSI-II, and SBW25 has an incomplete copy of an HSI-II-like T6SS. The HSI-II loci have several gene substitutions relative to the locus in P. aeruginosa. Strains A506, SS101 and BG33R lack clpV, but contain a gene encoding an Hcp family protein, which is absent from the rest of the genomes. In contrast to P. aeruginosa, the HSI-II loci of P. chlororaphis 30-84 and O6 include several genes encoding hypothetical proteins and a PAAR motif protein similar to evpJ, a non-essential gene found in the T6SS of Edwardsiella tarda [122]. A third locus, in the genomes of strains Q2-87, Q8r1-96 and Pf0-1, contains genes encoding all of the necessary components of the HSI-III T6SS of P. aeruginosa but lacks an ortholog of PA2372, which is not essential to the function of the transport system [123]. P. chlororaphis O6 has a fourth T6SS related to a T6SS locus (y3658–y3677) in Yersinia pestis strain Kim [124] and tss-4 from Burkholderia pseudomallei strain K96243 [125]. This T6SS is in a region of the O6 genome with atypical trinucleotide composition that is flanked by transposases, indicating that it may have been recently acquired. Effector proteins delivered by the T6SSs of P. fluorescens or P. chlororaphis are unknown and orthologs of Tse1, Tse2, and Tse3, which are secreted via the H1-T6SS of P. aeruginosa [119], were not found in these genomes.
Insect toxicity
Certain strains in the P. fluorescens group are toxic to insects and, in some cases, this toxicity is associated with gene clusters encoding the Mcf (makes caterpillars floppy) toxin or Tc (toxin complexes) first described in insect pathogens such as Serratia entomophila and bacterial endosymbionts of entomopathogenic nematodes such as Photorhabdus spp. and Xenorhabdus spp. [39], [126]–[128]. fitD (fluorescens insect toxin), which is closely related to mcf, is present in the genome of P. protegens Pf-5 and associated with that strain's lethality against the tobacco hornworm Manduca sexta [39]. The fitABCDEFGH locus, which includes genes for regulation and efflux of the FitD toxin [39], is located within a 90-gene insertion into the genome of Pf-5, portions of which have features (phage integrase and phage remnants, unusual nucleotide composition) indicative of horizontal acquisition. P. chlororaphis strains O6 and 30-84 also have complete fitABCDEFGH loci that are part of 24–28 gene insertions into the same location in both genomes. Genes distantly related to fitD (27–28% identity) are present in the genomes of Q8r1-96, Q2-87, and Pf0-1, but other genes of the fit locus are not present in these strains.
Whereas the fit cluster is present only in members of Sub-clade 1, loci similar to the Tc clusters are present in seven of the ten genomes including representatives of each sub-clade. Collectively, the genomes have six distinct types of Tc clusters distinguished by the number and organization of component genes and the location of the clusters in the genomes. Pseudomonas sp. BG33R contains two Tc clusters, which have distinct compositions and are located distally in the genome. The cluster in P. chlororaphis 30-84 is flanked by phage integrases within a unique 74.1-kb region (Island 2; Table S13) having a transposase at one terminus. These features, along with the unusual GC content and trinucleotide composition, suggest a horizontal mechanism of inheritance. Tc clusters appear to be widely distributed in bacterial genomes, and their functions in the ecology of the producing strains remain largely unknown. To date, a role for these genes in Pseudomonas spp. has been established only for the tccC gene from Pseudomonas taiwanensis, which confers an insect lethality phenotype when expressed in E. coli [128]. Insect toxicity has been reported for Pf-5 [39], [127], but is not known for any of the other strains of this study, and the potential roles of toxins and other phenotypes in the interactions of these bacteria with insects is an intriguing area for future study.
Correlating phylogenies with metabolic profiles and gene inventories
The diversity of bacteria within the P. fluorescens group has been recognized for many decades, and a polyphasic approach including catabolic profiles has been used to classify these bacteria since the 1960s [129]. We conducted catabolic profiling assays using the Biolog Phenotype Microarray (PM) system and found that, despite their genomic diversity, the strains displayed similar core carbon metabolic profiles (Figure S9). In contrast, the strains differed in some of their subsidiary catabolic abilities, particularly in utilization of plant-derived compounds. For instance, sucrose, D-tartaric acid and M-tartaric acid were utilized by several strains, whereas L-tartaric acid was not utilized. Only Pf0-1 utilized citraconic acid; only A506 utilized 2-deoxy-D-ribose; and only SBW25 and Q8r1-96 utilized L-homoserine. From these catabolic profiles, additional phenotypic characterization of the strains, and bioinformatic analyses of the ten genomes, we identified genes correlated with many of the traits used for classification of species, sub-species and biovars within the P. fluorescens group (Table S16).
All strains in Sub-clade 1 (Figure 1) have clusters for utilization of phenylacetic acid, benzoate, and trehalose, which are characteristics of P. chlororaphis and the newly-described species P. protegens, to which Pf-5 has been assigned [31]. Strains 30-84 and O6 each have a levan sucrase gene, clusters for phenazine biosynthesis and an L-arabinose transporter, whereas Pf-5 lacks these loci; lack of phenazine production, levan sucrase activity and L-arabinose assimilation are among the phenotypes differentiating Pf-5 from P. chlororaphis subsp. aureofaciens [31], [130] (Table S16). Strain O6 has a cluster encoding a nitrate reductase and nitrite transporter system, and we found that it could reduce nitrate to nitrite. In contrast, strain 30-84 lacks the gene cluster and the nitrate-reducing activity, a phenotype known to vary among strains of P. chlororaphis subsp. aureofaciens [130]. Additionally, D-serine was utilized by the two P. chlororaphis strains, the only two strains in this analysis that contain a D-serine ammonia lyase gene and an adjacent D-serine deaminase transcriptional activator.
Within Sub-clade 2, the genomes of Q2-87 and Q8r1-96 have a full complement of genes for denitrification, and we detected complete reduction of nitrate by both strains. The strains have genes or gene clusters for levan sucrase and the utilization of ethanol, sorbitol, and mannitol; they also utilized these compounds as sole carbon sources (Figure S6, Table S16). These phenotypes are characteristic of P. brassicacearum, a species of root-associated bacteria that, like Q2-87 and Q8r1-96, produces 2,4-diacetylphloroglucinol [131] and fits into the P. corrugata sub-group defined by Mulet et al. [13]. A very close relationship between P. brassicacearum NFM421 and strain Q8r1-96 also was revealed in the phylogenies based on the full genome sequences of these strains, whereas P. brassicacearum NFM421 appears more distantly related to strain Q2-87 (Figure S2). Consequently, we adopted the species designation of P. brassicacearum for strain Q8r1-96. In contrast to strains Q2-87 and Q8r1-96, Pf0-1 lacks genes for nitrate reduction, levan sucrase, and utilization of myo-inositol, sorbitol, and ethanol, and is likely to fall into a separate sub-group within the P. fluorescens group. Again, these gene inventories were congruent with the phenotypes exhibited by Pf0-1 in our assays (Table S16).
Three of the four strains in Sub-clade 3 exhibited phenotypes typical of P. fluorescens Biovar I [129], testing positive for levan sucrase, utilization of sorbitol, L-arabinose, xylose, L-tryptophan, and adonitol, but lacking the capacity to reduce nitrate. The fourth strain, BG33R, shared all of these phenotypes except for levan sucrase production and therefore falls into Biovar V-6, a group whose commonalities with Biovar I have been noted previously [132]. We made putative links between the use of each of these carbon sources and specific catabolic genes in each of these strains. Therefore, by coupling the genomic analysis with phenotypic tests, we identified a set of traits and gene inventories that are useful in differentiating the strains in a manner congruent with their phylogenies (Table S16, Figure S10).
Conclusions
It appears that Pseudomonas spp. occupy varied niches by virtue of an expanded pan-genome, with the variable genome providing functions that tailor fitness to specialized habitats occupied by a subset of strains. At 13,872 genes, the pan-genome of ten plant-associated strains within the P. fluorescens group makes up a substantial portion of the pan-genome of the genus as a whole, which was estimated at 25,907 in this study. Comparisons of many of the sequenced strains of Pseudomonas spp. (Figure 1) identified a core genome of only 1491 genes, representing less than 35% of any individual genome, further emphasizing the heterogeneity of the genus and the important role of the variable genome in tailoring individual strains to their specific lifestyles. This heterogeneity was highlighted further by the discovery that only 20 genes are shared by all strains within the P. fluorescens group and absent from the genomes of other Pseudomonas spp., suggesting that gene flow among Pseudomonas spp. is a significant factor modulating gene inventory. Although the ten strains in this study exhibit many commonalities with respect to their plant commensal lifestyle, their genetic repertoires are varied and plastic. There are clearly multiple pathways to success with respect to establishing bacterial populations on plant surfaces.
Comparison of ten genomes within the P. fluorescens group provided ample evidence that the tremendous ecological and physiological diversity of these bacteria extends to the genomic level. Genomic diversity commensurate with the biological diversity of the P. fluorescens group also was recognized in earlier studies comparing the genomes of strains sequenced previously, leading Silby et al. [29], [31] to propose that these strains fall into a species complex. Here, we defined three sub-clades within the P. fluorescens group on the basis of phylogenies inferred from MLSA and genomic comparisons between ten strains in the group. Distinctions between the three lineages were supported by a number of criteria such as genomic synteny, sizes of the lineage-specific core genomes, and gene inventories. As genome sequences for additional strains within the P. fluorescens group are incorporated into these phylogenetic analyses in the future, the number of lineages undoubtedly will increase. For example, Pf-5 and Pf0-1 are not as closely related to other members of their sub-clades as those members are to one another, and it is likely that these strains reside in distinct lineages that will become more defined as genomes for sister strains become available in the future. Alternatively, distinctions between the three lineages defined here may become blurred as more genomes are added. Recently, genomes of many other plant-associated strains within the P. fluorescens group have been published [133]–[136], and many more are likely to become available in the near future. Although we retained most species names that have been published previously for the strains included in this study, our genomic analysis highlighted discrepancies in the taxonomy of the P. fluorescens group and we recognize that some species designations are likely to change in the future. The need for taxonomic revision within the P. fluorescens group is well recognized, and the findings of this study illustrate the important role that comparative genomics is likely to play in defining the relationships between strains comprising this heterogeneous group of bacteria.
Like other Pseudomonas spp., strains within the P. fluorescens group have large genomes conferring an extensive functional repertoire. In other bacterial genera, genomes are smaller in pathogenic vs. environmental isolates or strains. In contrast, there is no striking pattern correlating genome size to a known pathogenic vs. saprophytic lifestyle in Pseudomonas spp. Plant pathogenic strains of P. syringae, opportunistic human pathogens of P. aeruginosa, and the insect pathogen P. entomophila have genome sizes ranging from 5.9 Mb to 6.9 Mb. Therefore, rather than exhibiting a reduced genome size reflecting a specialized pathogenic lifestyle, genomes of these known pathogens within the Pseudomonas spp. are similar in size to those of environmental isolates. This similarity is not surprising given that plant pathogenic strains of P. syringae are known to live epiphytically on plant surfaces and P. aeruginosa can be isolated from soil, water and other environmental substrates, indicating that pathogenesis is only one aspect of the lifestyle of these species. The varied functions conferred by a large genome appear to be required by members of the genus to handle the range of environments that these bacteria encounter.
This study included a survey of the genomes for traits associated with biological control and other multitrophic interactions of the P. fluorescens group with plants, microbes, and insects. The distribution of these traits was superimposed on maps defining the ancestral and recently-acquired regions of each genome to develop a view of the evolution of these traits in the P. fluorescens group. Regions containing core CDSs shared among all strains, which comprise 45% to 52% of each predicted proteome, represent the more ancestral components of each genome. Almost all of the traits associated with biological control or other multitrophic interactions map to genomic regions present in only a subset of the strains or unique to a specific strain (Figure 3). This finding is consistent with the established literature, which provides numerous examples of strain specificity related to biological control activity. Certain traits (e.g., HCN production) are associated with specific sub-clades, possibly reflecting an ancestral status within specific lineages (Figure 6). Most of the traits have a patchy distribution among the strains, and loci for many of these traits were probably acquired through horizontal gene transfer. A fraction of the identified traits (e.g., certain bacteriocins and Tc insect toxins) are encoded by genes located in clearly-defined MGEs (Table S13), but genes encoding the vast majority of biocontrol traits map outside of the MGEs to other regions of the variable genome. Some of these loci have characteristics suggesting recent acquisition, such as atypical GC content and trinucleotide composition and the lack of REP elements. Other loci map to regions that are similar to the core genome in these respects, possibly reflecting acquisition from related bacteria with similar GC content and trinucleotide skew, acquisition in the distant past with subsequent alteration in sequences, or vertical inheritance accompanied by subsequent deletion from certain strains. On the other hand, many of the variable genomic regions may have resulted from horizontal transfer of sequences, other than defined MGEs, that were introduced into the cell and became integrated through recombination or other mechanisms independent of transposons or integrons. Indeed, one role proposed for REP elements is as sites for homologous recombination [137]. This mechanism of evolution appears particularly likely in the genomes of the P. fluorescens group described herein, as many of the genes unique to each genome are present as singles, doubles or large groups inserted into core regions lacking any diagnostic features of MGEs. While the specific mechanisms for inheritance of biocontrol traits are obscure, the findings of this study underscore the exclusive occurrence of many traits in specific strains or sub-clades, which is consistent with the strain-specificity of biological control that has been observed for decades.
The variable regions of the P. fluorescens group genomes represent valuable resources for future discovery of new aspects of the biology of these bacteria and their interactions with other organisms. A case in point is provided by strain Pf-5, as the genomic sequence data have facilitated the discovery of four novel traits with potential roles in biological control. These are the cyclic lipopeptide orfamide A [35]; the bacteriocin LlpA [38]; analogs of rhizoxin [36], a macrolide that inhibits microtubule assembly in eukaryotic cells; and the FitD insect toxin [39]. An emphasis of this study was to associate variations in the genomes of strains within the P. fluorescens group to phenotypes key to the metabolism or lifestyle of these bacteria. Coupling comparative genomics with phenotype testing, we confirmed many of the known phenotypes of the biological control strains. More importantly, novel gene clusters were identified in each strain, providing opportunities for future exploration of unknown mechanisms by which these bacteria interact with their co-inhabitants, plant hosts and other organisms in the natural environment.
Materials and Methods
Selection and characterization of strains
Seven strains were selected for genomic sequencing based upon their characterized and distinctive biological control properties and their isolation from different habitats (bulk or rhizosphere soil or aerial plant surfaces) (Table 1). The seven strains and three previously-sequenced strains (Pf-5, Pf0-1, and SBW25) evaluated in this study exhibited the conserved phenotypes of the P. fluorescens group: positive for fluorescence under UV light, arginine dihydrolase activity, and oxidase activity; and negative for growth at 41°C and induction of a hypersensitive response on tobacco, determined through standard methods [138] (Table S16). The ten strains were subjected to a panel of biochemical and biological assays (nitrate reduction, levan sucrase production, potato soft rot, gelatinase activity, and catabolic spectra) [138], [139] to assign each to a biovar of P. fluorescens or to a species of Pseudomonas [129] (Table 1 and Table S16). Strains A506, 30-84, SS101, and BG33R are rifampicin-resistant (100 µg/ml) derivatives of field isolates; previously, spontaneous mutants with resistance to rifampicin were selected to facilitate tracking of these strains in field studies. Strain A506 is known to have a single nucleotide insertion in rpoS, which causes a frameshift resulting in a truncated form of the stationary-phase sigma factor RpoS [140]. During the course of this work, we discovered that strain Pf0-1 has a mutation in gacA, which encodes a component of the GacA/GacS global regulatory system in Pseudomonas spp. [61]. We sequenced gacA and gacS from the strain Pf0-1 in our collection and confirmed that the sequences are identical to those in the published genome of Pf0-1 [32]. It is not possible to know whether the mutations in A506 and Pf0-1 were present in the strains prior to isolation or if they developed in the laboratory during storage, but all strains have been maintained as frozen stocks (−80°C) throughout this study and for many years preceding.
Genome sequencing
The genome sequences were determined using shotgun sequencing with a combination of Sanger sequencing (to 4× coverage of the genome size) and 454 pyrosequencing technologies with paired end reads [141], [142]. A hybrid genome assembly was prepared from these datasets using Newbler 2.3 (Roche) and Celera Assembler 5.42. Each genome was subsequently evaluated for additional assembly improvement with the Celera Assembler 5.42 assembly versions providing the starting points. Multiple gaps were closed by merging overlapping contigs and resolving repetitive gaps. Further physical and sequencing gaps were closed by sequencing of PCR products spanning the gaps. The order of scaffolds in the genomes of strains 30-84 and Q8r1-96 (Figure 3) was confirmed by PCR.
Accession numbers
The whole genome shotgun sequencing projects have been deposited at DDBJ/EMBL/GenBank under the accessions AHHJ00000000 (30-84), AHOT00000000 (O6), AGBM00000000 (Q2-87), AHPO00000000 (Q8r1-96), AHPP00000000 (BG33R) and AHPN00000000 (SS101). Accession numbers for the complete genome sequences are: CP003041 for the chromosome of A506 and CP000076 for the updated genome sequence of Pf-5.
Bioinformatic analysis
Identification of putative protein-encoding genes and annotation of the genomes were performed as previously described [143]. A set of open reading frames predicted to encode proteins was initially identified using GLIMMER [144]. Open reading frames consisting of fewer than 30 codons and those containing overlaps were eliminated. Functional assignment, identification of membrane-spanning domains, determination of paralogous gene families, and identification of regions of unusual trinucleotide composition were performed as previously described [143]. The annotation of each of the genomes has undergone significant manual curation, removing small spurious overlapping ORFs and improving gene function calls. Manual curation of the genomes was performed using the MANATEE program (http://manatee.sourceforge.net/jcvi/index.shtml). The annotation of the previously-published genome of Pf-5 was updated and manually curated as part of this study. Phylogenetic analyses on the pyocin proteins were performed using MEGA 5 [145]; domain analyses were performed using the InterProScan program, found on the InterPro website [146]. Secondary metabolite production clusters were examined using the antiSMASH program [147]. The amino acid composition of products from NRPS sequences were predicted using NRPSpredictor 2 [148]. Transposons were identified using the ISfinder database (http://www-is.biotoul.fr/) [149]; only expectation values of 10−5 and below were considered as significant matches during searches. The Pseudomonas genome database [150] was consulted to obtain information on previously-published genomes for comparative purposes.
The seven genomes were compared to other genomes of Pseudomonas species using a multiway BLASTp analysis, and putative orthologs were identified with an E-value cutoff of 10−5. Synteny analyses were performed using Progressive MAUVE [151]. Phylogenetic relationships among all sequenced Pseudomonas species were investigated by generating phylogenetic trees with MrBayes 3.1.2 [152] using 1) 16S rRNA and 2) concatenated alignments of 10 highly conserved housekeeping genes: acsA, aroE, dnaE, guaA, gyrB, mutL, ppsA, pyrC, recA, and rpoB. We also used Hal, a Markov Clustering algorithm based on e-values from reciprocal all-by-all BLASTP analysis [153], to determine phylogenetic relationships among the sequenced strains of Pseudomonas spp.
REP elements were defined by searching for repeat sequences greater than 30 nt in length that occurred more than 10 times within individual genomes using RepeatScout [154]. Overlapping repeat regions were identified using sequence alignments and assembled to generate consensus repeat motifs. The consensus sequences were used to search the genomes with an identity cut-off of >90%. Sequences identified were aligned using ClustalX and HMMs were generated from alignments using HMMER2.
Phenotypic assays supporting gene function or biotype designations
Ten strains within the P. fluorescens group (Table 1) were tested for phenotypes associated with gene functions. In addition, we tested derivatives of strain Pf0-1 containing cloned gacA or gacS genes from strain Pf-5. pJEL5965 has a 1.6-kb EcoRI/HindIII fragment containing the gacA gene from Pf-5 cloned into pME6000; pJEL5999 has a 6.7-kb KpnI fragment containing the gacS gene from Pf-5 cloned into pME6000 [155]. Mutants of strains Pf-5 and A506 were included as controls in phenotypic tests, including gacS and aprA mutants of A506 [101], and ii) gacA [69], ofaA [69], chiC, hcnB, and aprA mutants of Pf-5. The hcnB mutant was created as described for the ofaA mutant [69], except that the PCR product was digested with HindIII and cloned into the HindIII site of pEX18Tc [156]. The aprA and chiC mutants contain in-frame deletions and were made using previously-described methods for creating an in-frame deletion [157], except that the PCR products were digested with BamHI or XbaI and cloned into the BamHI or XbaI site of pEX18Tc [156]. Primers used for mutant construction are listed in Table S17. The deletions were confirmed by PCR amplification and sequencing of the mutant alleles. All phenotypic tests were done on duplicate cultures grown at 27°C (unless another temperature is specified), experiments were repeated, and representative results are presented (Table S12, Table S16).
Exoenzymes
Extracellular protease was assessed visually as a cleared zone around bacterial colonies on half-strength BBL Litmus milk agar (Becton, Dickinson and Company, Sparks, MD USA) following incubation for 2 or 4 days. aprA mutants of strains A506 [101] and Pf-5 served as negative controls. Gelatinase activity was assessed in 12% gelatin incubated at 20°C and examined at 48 h and 1 week post inoculation [138]. Lipase activity was assessed in LB agar containing 1% w/v Tween 80, added before autoclaving. A positive result was observed as the formation of a white precipitate around a bacterial colony; plates were examined at 48 h and 1 week post inoculation. Chitinase activity was estimated from cultures grown in KB broth for 4 days with shaking using a methylumbelliferone-based chitinase assay kit (Sigma, St. Louis, MO). A chitinase-deficient mutant (chiC) of Pf-5 served as a negative control.
Secondary metabolites
Cyclic lipopeptide (CLP) production was assessed as surfactant activity in the droplet collapse assay [35] and hemolytic activity, detected as a clearing zone surrounding colonies grown for 48 hr at 27°C on BBL™ Blood Agar Base (Becton, Dickinson and Company, Sparks, MD, USA). CLP production also was visualized as clear zones surrounding colonies grown on CAS agar amended with iron, as described by Hartney et al. [158]. Swarming motility was assessed on standard succinate medium (SSM) [159] containing 0.6% agar following 2 days of incubation at room temperature, as described previously [60]. Mutants deficient in cyclic lipopeptide production serving as negative controls were: an orfamide deficient mutant (ofaA) of strain Pf-5 [69], a viscosin-deficient mutant (viscA) of strain SBW25 [60], and a massetolide-deficient mutant (massA) of strain SS101 [59].
Indole production was assayed in supernatants of cultures of strains in KB broth with 0.2 mg/ml L-tryptophan for 48 h. Salkowski's reagent [160] was added to the supernatants in a 2∶1 ratio and OD530 nm was measured after 30 min incubation at room temperature.
We attempted to detect mangotoxin-associated activity using an established bioassay [72] evaluating symptoms following wound-inoculation of tomato leaves (cultivars Oregon Spring and Legacy).
Hydrogen cyanide production was detected as described by Sarniguet et al. [161]. A mutant of Pf-5 (hcnB) deficient in hydrogen cyanide production served as a negative control.
ACC deaminase activity
The amount of α-ketobutyrate generated by the enzymatic hydrolysis of 1-aminocyclopropane-1-carboxylic acid in cell-free extracts was monitored as described by Honma and Shimomura [162].
Biolog phenotyping and carbon source utilization
Strains of Pseudomonas spp. were grown on LB agar at 25°C overnight. Cells were inoculated into 1× IF-0 media (Biolog, Inc., Hayward, CA, USA) and the transmittance of the suspension measured using a Biolog Turbidimeter (Biolog, Inc.). Cells were added until a uniform suspension of 42% transmittance was achieved. The cell suspension was added to 1× IF-0 media containing Dye A (Biolog, Inc.) in a ratio of 1∶5 to produce a cell suspension with a final transmittance of 85%. 100 µl of cell suspension was transferred to each well of Biolog plates PM01 and PM02A (Biolog, Inc.). Plates were incubated using the OmniLog Phenotype MicroArray System (Biolog, Inc.) at 25°C for 48 h, with measurements recorded at 15 min intervals. Data was visualized using OmniLog File Management/Kinetic Analysis software v1.20.02 and analyzed using OmniLog Parametric Analysis software v1.20.02 (Biolog, Inc.). The total area under the curve was used to compare strain phenotypes.
Growth on selected compounds as sole carbon sources was tested on minimal medium 925 [163] amended with the compounds at 0.1% w/v, unless otherwise noted.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. WuXMonchySTaghaviSZhuWRamosJ 2010 Comparative genomics and functional analysis of niche-specific adaptation in Pseudomonas putida. FEMS Microbiol Rev 35 299 323
2. LessieTGPhibbsPVJr 1984 Alternative pathways of carbohydrate utilization in Pseudomonads. Annu Rev Microbiol 38 359 388
3. GrossHLoperJE 2009 Genomics of secondary metabolite production by Pseudomonas spp. Nat Prod Rep 26 1408 1446
4. RaaijmakersJMVlamiMde SouzaJT 2002 Antibiotic production by bacterial biocontrol agents. Antonie Van Leeuwenhoek 81 537 547
5. BenderCLAlarcón-ChaidezFGrossDC 1999 Pseudomonas syringae phytotoxins: mode of action, regulation, and biosynthesis by peptide and polyketide synthases. Microbiol Mol Biol Rev 63 266 292
6. WellerDM 1988 Biological control of soilborne plant pathogens in the rhizospere with bacteria. Annu Rev Phytopathol 26 379 407
7. LugtenbergBKamilovaF 2009 Plant-growth-promoting rhizobacteria. Annu Rev Microbiol 63 541 556
8. HaasDDefagoG 2005 Biological control of soil-borne pathogens by fluorescent pseudomonads. Nat Rev Microbiol 3 307 319
9. WellerDMRaaijmakersJMGardenerBBMThomashowLS 2002 Microbial populations responsible for specific soil suppressiveness to plant pathogens. Annu Rev Phytopathol 40 309 348
10. MendesRKruijtMde BruijnIDekkersEvan der VoortM 2011 Deciphering the rhizosphere microbiome for disease-suppressive bacteria. Science 332 1097 1100
11. MazzolaM 2004 Assessment and management of soil microbial community structure for disease suppression. Annu Rev Phytopathol 42 35 59
12. StockwellVOStackJP 2007 Using Pseudomonas spp. for integrated biological control. Phytopathology 97 244 249
13. MuletMLalucatJGarcía-ValdésE 2010 DNA sequence-based analysis of the Pseudomonas species. Environ Microbiol 12 1513 1530
14. GuttmanDSMorganRLWangPW 2008 The evolution of the Pseudomonads. FatmiMBCollmerAIacobellisNSMansfieldJWMurilloJ Pseudomonas syringae pathovars and related pathogens – identification, epidemiology and genomics Dordrecht Springer 307 319
15. YamamotoSKasaiHArnoldDLJacksonRWVivianA 2000 Phylogeny of the genus Pseudomonas: intrageneric structure reconstructed from the nucleotide sequences of gyrB and rpoD genes. Microbiology (Reading, England) 146 Pt 10 2385 2394
16. HaasDKeelC 2003 Regulation of antibiotic production in root-colonizing Pseudomonas spp. and relevance for biological control of plant disease. Annu Rev Phytopathol 41 117 153
17. MavrodiDVBlakenfeldtWThomashowLS 2006 Phenazine compounds in fluorescent Pseudomonas spp.: biosynthesis and regulation. Annu Rev Phytopathol 44 417 445
18. PiersonLPiersonE 2010 Metabolism and function of phenazines in bacteria: impacts on the behavior of bacteria in the environment and biotechnological processes. Appl Microbiol Biotechnol 86 1659 1670
19. WellerDMLandaBBMavrodiOVSchroederKLDe La FuenteL 2007 Role of 2,4-diacetylphloroglucinol-producing fluorescent Pseudomonas spp. in the defense of plant roots. Plant Biol (Stuttg) 9 4 20
20. RaaijmakersJMDe BruijnINybroeOOngenaM 2010 Natural functions of lipopeptides from Bacillus and Pseudomonas: more than surfactants and antibiotics. FEMS Microbiol Rev 34 1037 1062
21. LoperJESchrothMN 1986 Influence of bacterial sources of indole-3-acetic acid on root elongation of sugar beet. Phytopathology 76 386 389
22. SpaepenSVanderleydenJRemansR 2007 Indole-3-acetic acid in microbial and microorganism-plant signaling. FEMS Microbiol Rev 31 425 448
23. KangBRYangKYChoBHHanTHKimIS 2006 Production of indole-3-acetic acid in the plant-beneficial strain Pseudomonas chlororaphis O6 is negatively regulated by the global sensor kinase GacS. Curr Microbiol 52 473 476
24. XiaoZXuP 2007 Acetoin metabolism in bacteria. Crit Rev Microbiol 33 127 140
25. LeveauJHJGerardsS 2008 Discovery of a bacterial gene cluster for catabolism of the plant hormone indole 3-acetic acid. FEMS Microbiol Ecol 65 238 250
26. ArshadMFrankenbergerWTJ 1998 Plant growth-regulating substances in the rhizosphere: microbial production and functions. Adv Agron 62 45 151
27. HanSHLeeSJMoonJHParkKHYangKY 2006 GacS-dependent production of 2R, 3R-butanediol by Pseudomonas chlororaphis O6 is a major determinant for eliciting systemic resistance against Erwinia carotovora but not against Pseudomonas syringae pv. tabaci in tobacco. Mol Plant-Microbe Interact 19 924 930
28. BakkerPAHMPieterseCMJvan LoonLC 2007 Induced systemic resistance by fluorescent Pseudomonas spp. Phytopathology 97 239 243
29. SilbyMWWinstanleyCGodfreySACLevySBJacksonRW 2011 Pseudomonas genomes: diverse and adaptable. FEMS Microbiol Rev 35 652 680
30. BaltrusDANishimuraMTRomanchukAChangJHMukhtarMS 2011 Dynamic evolution of pathogenicity revealed by sequencing and comparative genomics of 19 Pseudomonas syringae isolates. PLoS Path 7 e1002132 doi:10.1371/journal.ppat.1002132
31. RametteAFrapolliMSauxMF-LGruffazCMeyerJ-M 2011 Pseudomonas protegens sp. nov., widespread plant-protecting bacteria producing the biocontrol compounds 2,4-diacetylphloroglucinol and pyoluteorin. Syst Appl Microbiol 34 180 188
32. SilbyMWCerdeño-TarragaAMVernikosGSGiddensSRJacksonRW 2009 Genomic and genetic analyses of diversity and plant interactions of Pseudomonas fluorescens. Genome Biol 10 R51
33. PaulsenITPressCMRavelJKobayashiDYMyersGSA 2005 Complete genome sequence of the plant commensal Pseudomonas fluorescens Pf-5. Nat Biotechnol 23 873 878
34. KimbrelJGivanSHalgrenACreasonAMillsD 2010 An improved, high-quality draft genome sequence of the germination-arrest factor-producing Pseudomonas fluorescens WH6. BMC Genomics 11 522
35. GrossHStockwellVOHenkelsMDNowak-ThompsonBLoperJE 2007 The genomisotopic approach: a systematic method to isolate products of orphan biosynthetic gene clusters. Chem Biol 14 53 63
36. LoperJEHenkelsMDShafferBTValerioteFAGrossH 2008 Isolation and identification of rhizoxin analogs from Pseudomonas fluorescens Pf-5 by using a genomic mining strategy. Appl Environ Microbiol 74 3085 3093
37. BrendelNPartida-MartinezLPScherlachKHertweckC 2007 A cryptic PKS-NRPS gene locus in the plant commensal Pseudomonas fluorescens Pf-5 codes for the biosynthesis of an antimitotic rhizoxin complex. Org Biomol Chem 5 2211 2213
38. ParretAHATemmermanKDe MotR 2005 Novel lectin-like bacteriocins of biocontrol strain Pseudomonas fluorescens Pf-5. Appl Environ Microbiol 71 5197 5207
39. Pechy-TarrMBruckDMaurhoferMFischerEVogneC 2008 Molecular analysis of a novel gene cluster encoding an insect toxin in plant-associated strains of Pseudomonas fluorescens. Environ Microbiol 10 2368 2386
40. BennasarAMuletMLalucatJGarcia-ValdesE 2010 PseudoMLSA: a database for multigenic sequence analysis of Pseudomonas species. BMC Microbiol 10 118
41. MavrodiDVPaulsenITRenQLoperJE 2007 Genomics of Pseudomonas fluorescens Pf-5. RamosJLFillouxA Pseudomonas, a model system in biology Spinger, The Netherlands Springer 3 30
42. NunvarJHuckovaTLichaI 2010 Identification and characterization of repetitive extragenic palindromes (REP)-associated tyrosine transposases: implications for REP evolution and dynamics in bacterial genomes. BMC Genomics 11 44
43. Aranda-OlmedoITobesRManzaneraMRamosJLMarquésS 2002 Species-specific repetitive extragenic palindromic (REP) sequences in Pseudomonas putida. Nucleic Acids Res 30 1826 1833
44. BertelsFRaineyPB 2011 Within-genome evolution of REPINs: a new family of miniature mobile DNA in bacteria. PLoS Genet 7 e1002132 doi:10.1371/journal.pgen.1002132
45. TobesRParejaE 2005 Repetitive extragenic palindromic sequences in the Pseudomonas syringae pv. tomato DC3000 genome: extragenic signals for genome reannotation. Res Microbiol 156 424 433
46. GilsonESaurinWPerrinDBachellierSHofnungM 1991 Palindromic units are part of a new bacterial interspersed mosaic element (BIME). Nucleic Acids Res 19 1375 1383
47. CeyssensP-JLavigneR 2010 Bacteriophages of Pseudomonas. Future Microbiol 5 1041 1055
48. RuerSStenderSFillouxAde BentzmannS 2007 Assembly of fimbrial structures in Pseudomonas aeruginosa: functionality and specificity of chaperone-usher machineries. J Bacteriol 189 3547 3555
49. KulasekaraHDVentreIKulasekaraBRLazdunskiAFillouxA 2005 A novel two-component system controls the expression of Pseudomonas aeruginosa fimbrial cup genes. Mol Microbiol 55 368 380
50. ValletIOlsonJWLorySLazdunskiAFillouxA 2001 The chaperone/usher pathways of Pseudomonas aeruginosa: Identification of fimbrial gene clusters (cup) and their involvement in biofilm formation. Proc Natl Acad Sci USA 98 6911 6916
51. ZhaoYMaZSundinGW 2005 Comparative genomic analysis of the pPT23A plasmid family of Pseudomonas syringae. J Bacteriol 187 2113 2126
52. CarterMQChenJLoryS 2010 The Pseudomonas aeruginosa pathogenicity island PAPI-1 is transferred via a novel type IV pilus. J Bacteriol 192 3249 3258
53. GuglielminiJQuintaisLGarcillán-BarciaMPde la CruzFRochaEPC 2011 The repertoire of ICE in prokaryotes underscores the unity, diversity, and ubiquity of conjugation. PLoS Genet 7 e1002222 doi:10.1371/journal.pgen.1002222
54. MavrodiDVLoperJEPaulsenITThomashowLS 2009 Mobile genetic elements in the genome of the beneficial rhizobacterium Pseudomonas fluorescens Pf-5. BMC Microbiol 9 8
55. GrissaIVergnaudGPourcelC 2007 CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res 35 W52 W57
56. Nowak-ThompsonBHammerPEHillDSStaffordJTorkewitzN 2003 2,5-dialkylresorcinol biosynthesis in Pseudomonas aurantiaca: novel head-to-head condensation of two fatty acid-derived precursors. J Bacteriol 185 860 869
57. RaaijmakersJMde BruijnIde KockMJD 2006 Cyclic lipopeptide production by plant-associated Pseudomonas spp.: diversity, activity, biosynthesis, and regulation. Mol Plant-Microbe Interact 19 699 710
58. MazzolaMde BruijnICohenMFRaaijmakersJM 2009 Protozoan-induced regulation of cyclic lipopeptide biosynthesis is an effective predation defense mechanism for Pseudomonas fluorescens. Appl Environ Microbiol 75 6804 6811
59. de BruijnIde KockMJde WaardPvan BeekTARaaijmakersJM 2008 Massetolide A biosynthesis in Pseudomonas fluorescens. J Bacteriol 190 2777 2789
60. de BruijnIde KockMJDYangMde WaardPvan BeekTA 2007 Genome-based discovery, structure prediction and functional analysis of cyclic lipopeptide antibiotics in Pseudomonas species. Mol Microbiol 63 417 428
61. LapougeKMario SchubertMAllainFHTDieter HaasD 2008 Gac/Rsm signal transduction pathway of gamma-proteobacteria: from RNA recognition to regulation of social behaviour. Mol Microbiol 67 241 253
62. PedrasMSCIsmailNQuailJWBoyetchkoSM 2003 Structure, chemistry, and biological activity of pseudophomins A and B, new cyclic lipodepsipeptides isolated from the biocontrol bacterium Pseudomonas fluorescens. Phytochemistry 62 1105 1114
63. ViscaPImperiFLamontIL 2007 Pyoverdine siderophores: from biogenesis to biosignificance. Trends Microbiol 15 22 30
64. CornelisPMatthijsSVan OeffelenL 2009 Iron uptake regulation in Pseudomonas aeruginosa. BioMetals 22 15 22
65. YouardZAMislinGLMajcherczykPASchalkIJReimmannC 2007 Pseudomonas fluorescens CHA0 produces enantio-pyochelin, the optical antipode of the Pseudomonas aeruginosa siderophore pyochelin. J Biol Chem 282 35546 35553
66. WuestWMSattelyESWalshCT 2009 Three siderophores from one bacterial enzymatic assembly line. J Am Chem Soc 131 5056 5057
67. Mercado-BlancoJvan der DriftKMOlssonPEThomas-OatesJEvan LoonLC 2001 Analysis of the pmsCEAB gene cluster involved in biosynthesis of salicylic acid and the siderophore pseudomonine in the biocontrol strain Pseudomonas fluorescens WCS374. J Bacteriol 183 1909 1920
68. BertiADThomasMG 2009 Analysis of achromobactin biosynthesis by Pseudomonas syringae pv. syringae B728a. J Bacteriol 191 4594 4604
69. HassanKAJohnsonAShafferBTRenQKidarsaTA 2010 Inactivation of the GacA response regulator in Pseudomonas fluorescens Pf-5 has far-reaching transcriptomic consequences. Environ Microbiol 12 899 915
70. WandersmanCDelepelaireP 2004 Bacterial iron sources: from siderophores to hemophores. Annu Rev Microbiol 58 611 647
71. Vallet-GelyIOpotaOBonifaceANovikovALemaitreB 2010 A secondary metabolite acting as a signalling molecule controls Pseudomonas entomophila virulence. Cell Microbiol 12 1666 1679
72. ArrebolaECazorlaFMRomeroDPerez-GarciaAde VicenteA 2007 A nonribosomal peptide synthetase gene (mgoA) of Pseudomonas syringae pv. syringae is involved in mangotoxin biosynthesis and is required for full virulence. Mol Plant-Microbe Interact 20 500 509
73. ArrebolaECarrionVCazorlaFPerez-GarciaAMurilloJ 2012 Characterisation of the mgo operon in Pseudomonas syringae pv. syringae UMAF0158 that is required for mangotoxin production. BMC Microbiol 12 10
74. CarriónVJArrebolaECazorlaFMMurilloJde VicenteA 2012 The mbo operon is specific and essential for biosynthesis of mangotoxin in Pseudomonas syringae. Plos ONE 7 e36709 doi:10.1371/journal.pone.0036709
75. CornelisP 2010 Iron uptake and metabolism in pseudomonads. Appl Microbiol Biotechnol 86 1637 1645
76. BhattGDennyTP 2004 Ralstonia solanacearum iron scavenging by the siderophore Staphyloferrin B is controlled by PhcA, the global virulence regulator. J Bacteriol 186 7896 7904
77. HertAPMarutaniMMomolMTRobertsPDOlsonSM 2009 Suppression of the bacterial spot pathogen Xanthomonas euvesicatoria on tomato leaves by an attenuated mutant of Xanthomonas perforans. Appl Environ Microbiol 75 3323 3330
78. ParretAHADe MotR 2002 Bacteria killing their own kind: novel bacteriocins of Pseudomonas and other γ-proteobacteria. Trends Microbiol 10 107 112
79. Michel-BriandYBaysseC 2002 The pyocins of Pseudomonas aeruginosa. Biochimie 84 499 510
80. SanoYKobayashiMKageyamaM 1993 Functional domains of S-type pyocins deduced from chimeric molecules. J Bacteriol 175 6179 6185
81. BarreteauHBouhssAFourgeaudMMainardiJ-LTouzeT 2009 Human - and plant-pathogenic Pseudomonas species produce bacteriocins exhibiting colicin M-like hydrolase activity towards peptidoglycan precursors. J Bacteriol 191 3657 3664
82. DuquesneSDestoumieux-GarzonDPeduzziJRebuffatS 2007 Microcins, gene-encoded antibacterial peptides from enterobacteria. Nat Prod Rep 24 708 734
83. CarrSWalkerDJamesRKleanthousCHemmingsAM 2000 Inhibition of a ribosome-inactivating ribonuclease: the crystal structure of the cytotoxic domain of colicin E3 in complex with its immunity protein. Structure 8 949 960
84. ChuangD-yChienY-cWuH-P 2007 Cloning and expression of the Erwinia carotovora subsp. carotovora gene encoding the low-molecular-weight bacteriocin Carocin S1. J Bacteriol 189 620 626
85. KöckJOlschlägerTKampRMBraunV 1987 Primary structure of colicin M, an inhibitor of murein biosynthesis. J Bacteriol 169 3358 3361
86. DimkpaCOZengJMcLeanJEBrittDWZhanJ 2012 Production of indole-3-acetic acid via the indole-3-acetamide pathway in the plant-beneficial bacterium Pseudomonas chlororaphis O6 is inhibited by ZnO nanoparticles but enhanced by CuO nanoparticles. Appl Environ Microbiol 78 1404 1410
87. KimYChoJ-YKukJ-HMoonJ-HChoJ-I 2004 Identification and antimicrobial activity of phenylacetic acid produced by Bacillus licheniformis isolated from fermented soybean, Chungkook-Jang. Curr Microbiol 48 312 317
88. WightmanFLightyDL 1982 Identification of phenylacetic acid as a natural auxin in the shoots of higher plants. Physiol Plantarum 55 17 24
89. OliveraERMinambresBGarciaBMunizCMorenoMA 1998 Molecular characterization of the phenylacetic acid metabolic pathway in Pseudomonas putida U: the phenylacetyl-CoA catabolon. Proc Natl Acad Sci USA 95 6419 6424
90. IsmailWEl-Said MohamedMWannerBLDatsenkoKAEisenreichW 2003 Functional genomics by NMR spectroscopy: Phenylacetate catabolism in Escherichia coli. Eur J Biochem 270 3047 3054
91. GlickBR 1995 The enhancement of plant growth by free-living bacteria. Can J Microbiol 41 109 117
92. DworkinMFosterJW 1958 Experiments with some microorganisms which utilize ethane and hydrogen. J Bacteriol 75 592 603
93. RyuC-MFaragMAHuC-HReddyMSWeiH-X 2003 Bacterial volatiles promote growth in Arabidopsis. Proc Natl Acad Sci USA 100 4927 4932
94. BlomqvistKNikkolaMLehtovaaraPSuihkoMLAiraksinenU 1993 Characterization of the genes of the 2,3-butanediol operons from Klebsiella terrigena and Enterobacter aerogenes. J Bacteriol 175 1392 1404
95. RennaMCNajimudinNWinikLRZahlerSA 1993 Regulation of the Bacillus subtilis alsS, alsD, and alsR genes involved in post-exponential-phase production of acetoin. J Bacteriol 175 3863 3875
96. NicholsonWL 2008 The Bacillus subtilis ydjL (bdhA) gene encodes acetoin teductase/2,3-butanediol dehydrogenase. Appl Environ Microbiol 74 6832 6838
97. NelsonDDuxburyT 2008 The distribution of acetohydroxyacid synthase in soil bacteria. Antonie Van Leeuwenhoek 93 123 132
98. BouchéNFrommH 2004 GABA in plants: just a metabolite? Trends Plant Sci 9 110 115
99. ShelpBJBownAWMcLeanMD 1999 Metabolism and functions of gamma-aminobutyric acid. Trends Plant Sci 4 446 452
100. ParkDHMirabellaRBronsteinPAPrestonGMHaringMA 2010 Mutations in γ-aminobutyric acid (GABA) transaminase genes in plants or Pseudomonas syringae reduce bacterial virulence. The Plant Journal 64 318 330
101. AndersonLMStockwellVOLoperJE 2004 An extracellular protease of Pseudomonas fluorescens inactivates antibiotics of Pantoea agglomerans. Phytopathology 94 1228 1234
102. StockwellVOJohnsonKBSugarDLoperJE 2011 Mechanistically compatible mixtures of bacterial antagonists improve biological control of fire blight of pear. Phytopathology 101 113 123
103. NielsenMNSorensenJFelsJPedersenHC 1998 Secondary metabolite - and endochitinase-dependent antagonism toward plant-pathogenic microfungi of Pseudomonas fluorescens isolates from sugar beet rhizosphere. Appl Environ Microbiol 64 3563 3569
104. FoldersJAlgraJRoelofsMSvan LoonLCTommassenJ 2001 Characterization of Pseudomonas aeruginosa chitinase, a gradually secreted protein. J Bacteriol 183 7044 7052
105. NikaidouNKamioYKzakiK 1993 Molecular cloning and nucleotide sequence of the pectate lyase gene from Pseudomonas marginalis N6301. Bioscience Biotechnology and Biochemistry 57 957 960
106. FillouxA 2011 Protein secretion systems in Pseudomonas aeruginosa: an essay on diversity, evolution and function. Front Microbiol 2 155
107. CusanoAMBurlinsonPDeveauAVionPUrozS 2011 Pseudomonas fluorescens BBc6R8 type III secretion mutants no longer promote ectomycorrhizal symbiosis. Environ Microbiol Rep 3 203 210
108. MavrodiDVJoeAMavrodiOVHassanKAWellerDM 2011 Structural and functional analysis of the type III secretion system from Pseudomonas fluorescens Q8r1-96. J Bacteriol 193 177 189
109. PrestonGMBertrandNRaineyPB 2001 Type III secretion in plant growth-promoting Pseudomonas fluorescens SBW25. Mol Microbiol 41 999 1014
110. CornelisGR 2010 The type III secretion injectisome, a complex nanomachine for intracellular ‘toxin’ delivery. Biol Chem 391 745 751
111. ArakiHTianDGossEMJakobKHalldorsdottirSS 2006 Presence/absence polymorphism for alternative pathogenicity islands in Pseudomonas viridiflava, a pathogen of Arabidopsis. Proc Natl Acad Sci USA 103 5887 5892
112. TroisfontainesPCornelisGR 2005 Type III secretion: More systems than you think. Physiology 20 326 339
113. HauserAR 2009 The type III secretion system of Pseudomonas aeruginosa: infection by injection. Nat Rev Micro 7 654 665
114. RaineyPB 1999 Adaptation of Pseudomonas fluorescens to the plant rhizosphere. Environ Microbiol 1 243 257
115. JacksonRWPrestonGMRaineyPB 2005 Genetic characterization of Pseudomonas fluorescens SBW25 rsp gene expression in the phytosphere and in vitro. J Bacteriol 187 8477 8488
116. RezzonicoFBinderCDefagoGMoenne-LoccozY 2005 The type III secretion system of biocontrol Pseudomonas fluorescens KD targets and phytopathogenic chromista Pythium ultimum and promotes cucumber protection. Mol Plant-Microbe Interact 18 991 1001
117. MatzCMorenoAMAlhedeMManefieldMHauserAR 2008 Pseudomonas aeruginosa uses type III secretion system to kill biofilm-associated amoebae. ISME J 2 843 852
118. SchwarzSHoodRDMougousJD 2010 What is type VI secretion doing in all those bugs? Trends Microbiol 18 531 537
119. RussellABHoodRDBuiNKLeRouxMVollmerW 2011 Type VI secretion delivers bacteriolytic effectors to target cells. Nature 475 343 347
120. RecordsAR 2011 The type VI secretion system: a multipurpose delivery system with a phage-like machinery. Mol Plant-Microbe Interact 24 751 757
121. BlevesSViarreVSalachaRMichelGPFFillouxA 2010 Protein secretion systems in Pseudomonas aeruginosa: a wealth of pathogenic weapons. Int J Med Microbiol 300 534 543
122. ZhengPSunJGeffersRZengA-P 2007 Functional characterization of the gene PA2384 in large-scale gene regulation in response to iron starvation in Pseudomonas aeruginosa. J Biotechnol 132 342 352
123. HsuFSchwarzSMougousJ 2009 TagR promotes PpkA-catalysed type VI secretion activation in Pseudomonas aeruginosa. Mol Microbiol 72 1111 1125
124. PieperRHuangS-TRobinsonJMClarkDJAlamiH 2009 Temperature and growth phase influence the outer-membrane proteome and the expression of a type VI secretion system in Yersinia pestis. Microbiology 155 498 512
125. ShalomGShawJGThomasMS 2007 In vivo expression technology identifies a type VI secretion system locus in Burkholderia pseudomallei that is induced upon invasion of macrophages. Microbiology 153 2689 2699
126. ffrench-ConstantRHDowlingAWaterfieldNR 2007 Insecticidal toxins from Photorhabdus bacteria and their potential use in agriculture. Toxicon 49 436 451
127. OlcottMHHenkelsMDRosenKLWalkerFLSnehB 2010 Lethality and developmental delay in Drosophila melanogaster larvae after ingestion of selected Pseudomonas fluorescens strains. PLoS ONE 5 e12504 doi:10.1371/journal.pone.0012504
128. LiuJ-RLinY-DChangS-TZengY-FWangS-L 2010 Molecular cloning and characterization of an insecticidal toxin from Pseudomonas taiwanensis. J Agric Food Chem 58 12343 12349
129. StanierRYPalleroniNJDoudoroffM 1966 The aerobic pseudomonads: a taxonomic study. J Gen Microbiol 43 159 271
130. PeixAValverdeARivasRIgualJMRamírez-BahenaM-H 2007 Reclassification of Pseudomonas aurantiaca as a synonym of Pseudomonas chlororaphis and proposal of three subspecies, P. chlororaphis subsp. chlororaphis subsp. nov., P. chlororaphis subsp. aureofaciens subsp. nov., comb. nov. and P. chlororaphis subsp. aurantiaca subsp. nov., comb. nov. Int J Syst Evol Microbiol 57 1286 1290
131. AchouakWSutraLHeulinTMeyerJMFrominN 2000 Pseudomonas brassicacearum sp. nov. and Pseudomonas thivervalensis sp. nov., two root-associated bacteria isolated from Brassica napus and Arabidopsis thaliana. Int J Syst Evol Microbiol 50 9 18
132. BarrettELSolanesRETangJSPalleroniNJ 1986 Pseudomonas fluorescens biovar V: its resolution into distinct component groups and the relationship of these groups to other psychotropic Pseudomonas associated with food spoilage. J Gen Microbiol 132 2709 2721
133. ShenXChenMHuHWangWPengH 2012 Genome sequence of Pseudomonas chlororaphis GP72, a root-colonizing biocontrol strain. J Bacteriol 194 1269 1270
134. Redondo-NietoMBarretMMorriseyJPGermaineKMartínez-GraneroF 2012 Genome sequence of the biocontrol strain Pseudomonas fluorescens F113. J Bacteriol 194 1273 1274
135. RongXGurelFBMeuliaTMcSpadden GardenerBB 2012 Draft genome sequences of the Pseudomonas fluorescens biocontrol strains Wayne1R and Wood1R. J Bacteriol 194 724 725
136. OrtetPBarakatMLalaounaDFochesatoSBarbeV 2011 Complete genome sequence of a beneficial plant root-associated bacterium, Pseudomonas brassicacearum. J Bacteriol 193 3146
137. KofoidEBergthorssonUSlechtaESRothJR 2003 Formation of an F′ plasmid by recombination between imperfectly repeated chromosomal Rep sequences: a closer look at an old friend (F′128 pro lac). J Bacteriol 185 660 663
138. GerhardtPMurrayRGEWoodWKriegNR 1994 Methods for general and molecular bacteriology Washington, D.C. American Society for Microbiology 791
139. SchaadNWJonesJBChunW 2001 Laboratory guide for identification of plant pathogenic bacteria St Paul, Minnesota USA APS Press 398
140. HagenMJStockwellVOWhistlerCAJohnsonKBLoperJE 2009 Stress tolerance and environmental fitness of Pseudomonas fluorescens A506, which has a mutation in RpoS. Phytopathology 99 679 688
141. FraserCMCasjensSHuangWMSuttonGGClaytonR 1997 Genomic sequence of a Lyme disease spirochaete, Borrelia burgdorferi. Nature 390 580 586
142. GoldbergSMDJohnsonJBusamDFeldblyumTFerrieraS 2006 A Sanger/pyrosequencing hybrid approach for the generation of high-quality draft assemblies of marine microbial genomes. Proc Natl Acad Sci USA 103 11240 11245
143. PaulsenITSeshadriRNelsonKEEisenJAHeidelbergJF 2002 The Brucella suis genome reveals fundamental similarities between animal and plant pathogens and symbionts. Proc Natl Acad Sci USA 99 13148 13153
144. SalzbergSLDelcherALKasifSWhiteO 1998 Microbial gene identification using interpolated Markov models. Nucleic Acids Res 26 544 548
145. TamuraKPetersonDPetersonNStecherGNeiM 2011 MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28 2731 2739
146. HunterSJonesPMitchellAApweilerRAttwoodTK 2012 InterPro in 2011: new developments in the family and domain prediction database. Nucleic Acids Res 40 D306 D312
147. MedemaMHBlinKCimermancicPde JagerVZakrzewskiP 2011 antiSMASH: rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res 39 W339 W346
148. RöttigMMedemaMHBlinKWeberTRauschC 2011 NRPSpredictor2—a web server for predicting NRPS adenylation domain specificity. Nucleic Acids Res 39 W362 367
149. SiguierPPerochonJLestradeLMahillonJChandlerM 2006 ISfinder: the reference centre for bacterial insertion sequences. Nucleic Acids Res 34 D32 D36
150. WinsorGLLamDKWFlemingLLoRWhitesideMD 2011 Pseudomonas genome database: improved comparative analysis and population genomics capability for Pseudomonas genomes. Nucleic Acids Res 39 D596 D600
151. DarlingAEMauBPernaNT 2010 progressiveMauve: Multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE 5 e11147 doi:10.1371/journal.pone.0011147
152. RonquistFHuelsenbeckJP 2003 MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19 1572 1574
153. RobbertseBYoderRJBoydAReevesJSpataforaJW 2011 Hal: an automated pipeline for phylogenetic analyses of genomic data. PLoS Currents Tree of Life doi:10.1371/currents.RRN1213
154. PriceALJonesNCPevznerPA 2005 De novo identification of repeat families in large genomes. Bioinformatics 21 i351 i358
155. MaurhoferMReimmannCSchmidli-SachererPHeebSHaasD 1998 Salicylic acid biosynthetic genes expressed in Pseudomonas fluorescens strain P3 improve the induction of systemic resistance in tobacco against tobacco necrosis virus. Biol Control 678 684
156. HoangTTKarkhoff-SchweizerRRKutchmaAJSchweizerHP 1998 A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally-located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene 212 77 86
157. KidarsaTAGoebelNCZabriskieTMLoperJE 2011 Phloroglucinol mediates crosstalk between the pyoluteorin and 2,4-diacetylphloroglucinol biosynthetic pathways in Pseudomonas fluorescens Pf-5. Mol Microbiol 81 395 414
158. HartneySLMazurierSKidarsaTAQuecineMCLemanceauP 2011 TonB-dependent outer-membrane proteins and siderophore utilization in Pseudomonas fluorescens Pf-5. BioMetals 24 193 213
159. MeyerJMAbdallahMA 1978 The fluorescent pigment of Pseudomonas fluorescens: biosynthesis, purification, and physicochemical properties. J Gen Microbiol 107 319 328
160. GordonSAWeberRP 1951 Colorimetric estimation of indoleacetic acid. Plant Physiol 26 192 195
161. SarniguetAKrausJHenkelsMDMuehlchenAMLoperJE 1995 The sigma factor sigma(s) affects antibiotic production and biological control activity of Pseudomonas fluorescens Pf-5. Proc Natl Acad Sci USA 92 12255 12259
162. HonmaMShimomuraT 1978 Metabolism of 1-aminocyclopropane-1-carboxylic acid. Agric Biol Chem 42 1825 1831
163. LangleyRAKadoCI 1972 Studies on Agrobacterium tumefaciens. Conditions for mutagenesis by N-methyl-N′-nitrosoguanidine and relationships of A. tumefaciens mutants to crown gall tumor induction. Mutat Res 14 277 286
164. ThomashowLSWellerDMBonsallRFPiersonLS 1990 Production of the antibiotic phenazine-1-carboxylic acid by fluorescent Pseudomonas species in the rhizosphere of wheat. Appl Environ Microbiol 56 908 912
165. TuckerBRadtkeCKwonSIAndersonAJ 1995 Suppression of bioremediation by Phanerochaete chrysosporium by soil factors. J Hazard Mater 41 251 265
166. KimMSKimYCChoBH 2004 Gene expression analysis in cucumber leaves primed by root colonization with Pseudomonas chlororaphis O6 upon challenge inoculation with Corynespora cassiicola. Plant Biol 6 105 108
167. HowellCRStipanovicRD 1979 Control of Rhizoctonia solani in cotton seedlings with Pseudomonas fluorescens and with an antibiotic produced by the bacterium. Phytopathology 69 480 482
168. HowellCRStipanovicRD 1980 Suppression of Pythium ultimum-induced damping-off of cotton seedlings by Pseudomonas fluorescens and its antibiotic, pyoluteorin. Phytopathology 70 712 715
169. RaaijmakersJMWellerDM 1998 Natural plant protection by 2,4-diacetylphloroglucinol-producing Pseudomonas spp. in take-all decline soils. Mol Plant-Microbe Interact 11 144 152
170. VincentMNHarrisonLABrackinJMKovacevichPAMukerjiP 1991 Genetic analysis of the antifungal activity of a soilborne Pseudomonas aureofaciens strain. Appl Environ Microbiol 57 2928 2934
171. WilsonMLindowSE 1993 Interactions between the biological control agent Pseudomonas fluorescens A506 and Erwinia amylovora in pear blossoms. Phytopathology 83 117 123
172. MazzolaMZhaoXCohenMFRaaijmakersJM 2007 Cyclic lipopeptide surfactant production by Pseudomonas fluorescens SS101 is not required for suppression of complex Pythium spp. populations. Phytopathology 97 1348 1355
173. KruijtMHaTRaaijmakersJM 2009 Functional, genetic and chemical characterization of biosurfactants produced by plant growth-promoting Pseudomonas putida 267. J Appl Microbiol 107 546 556
174. de SouzaJTde BoerMde WaardPvan BeekTARaaijmakersJM 2003 Biochemical, genetic, and zoosporicidal properties of cyclic lipopeptide surfactants produced by Pseudomonas fluorescens. Appl Environ Microbiol 69 7161 7172
175. TranHFickeAAsiimweTHofteMRaaijmakersJM 2007 Role of the cyclic lipopeptide massetolide A in biological control of Phytophthora infestans and in colonization of tomato plants by Pseudomonas fluorescens. New Phytol 175 731 742
176. KluepfelDAMcInnisTMZehrEI 1993 Involvement of root-colonizing bacteria in peach orchard soils suppressive of the nematode Criconemella xenoplax. Phytopathology 83 1250 1245
177. JonesDTTaylorWRThorntonJM 1992 The rapid generation of mutation data matrices from protein sequences. Comput Appl Biosci 8 275 282
178. BossisELemanceauPLatourXGardanL 2000 The taxonomy of Pseudomonas fluorescens and Pseudomonas putida: current status and need for revision. Agronomie 20 51 63
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2012 Číslo 7
Nejčtenější v tomto čísle
- Guidelines for Genome-Wide Association Studies
- The Role of Rice HEI10 in the Formation of Meiotic Crossovers
- Identification of Chromatin-Associated Regulators of MSL Complex Targeting in Dosage Compensation
- GWAS Identifies Novel Susceptibility Loci on 6p21.32 and 21q21.3 for Hepatocellular Carcinoma in Chronic Hepatitis B Virus Carriers