
F-Box Protein Specificity for G1 Cyclins Is Dictated by Subcellular Localization
Levels of G1 cyclins fluctuate in response to environmental cues and couple mitotic signaling to cell cycle entry. The G1 cyclin Cln3 is a key regulator of cell size and cell cycle entry in budding yeast. Cln3 degradation is essential for proper cell cycle control; however, the mechanisms that control Cln3 degradation are largely unknown. Here we show that two SCF ubiquitin ligases, SCFCdc4 and SCFGrr1, redundantly target Cln3 for degradation. While the F-box proteins (FBPs) Cdc4 and Grr1 were previously thought to target non-overlapping sets of substrates, we find that Cdc4 and Grr1 each bind to all 3 G1 cyclins in cell extracts, yet only Cln3 is redundantly targeted in vivo, due in part to its nuclear localization. The related cyclin Cln2 is cytoplasmic and exclusively targeted by Grr1. However, Cdc4 can interact with Cdk-phosphorylated Cln2 and target it for degradation when cytoplasmic Cdc4 localization is forced in vivo. These findings suggest that Cdc4 and Grr1 may share additional redundant targets and, consistent with this possibility, grr1Δ cdc4-1 cells demonstrate a CLN3-independent synergistic growth defect. Our findings demonstrate that structurally distinct FBPs are capable of interacting with some of the same substrates; however, in vivo specificity is achieved in part by subcellular localization. Additionally, the FBPs Cdc4 and Grr1 are partially redundant for proliferation and viability, likely sharing additional redundant substrates whose degradation is important for cell cycle progression.
Published in the journal:
. PLoS Genet 8(7): e32767. doi:10.1371/journal.pgen.1002851
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002851
Summary
Levels of G1 cyclins fluctuate in response to environmental cues and couple mitotic signaling to cell cycle entry. The G1 cyclin Cln3 is a key regulator of cell size and cell cycle entry in budding yeast. Cln3 degradation is essential for proper cell cycle control; however, the mechanisms that control Cln3 degradation are largely unknown. Here we show that two SCF ubiquitin ligases, SCFCdc4 and SCFGrr1, redundantly target Cln3 for degradation. While the F-box proteins (FBPs) Cdc4 and Grr1 were previously thought to target non-overlapping sets of substrates, we find that Cdc4 and Grr1 each bind to all 3 G1 cyclins in cell extracts, yet only Cln3 is redundantly targeted in vivo, due in part to its nuclear localization. The related cyclin Cln2 is cytoplasmic and exclusively targeted by Grr1. However, Cdc4 can interact with Cdk-phosphorylated Cln2 and target it for degradation when cytoplasmic Cdc4 localization is forced in vivo. These findings suggest that Cdc4 and Grr1 may share additional redundant targets and, consistent with this possibility, grr1Δ cdc4-1 cells demonstrate a CLN3-independent synergistic growth defect. Our findings demonstrate that structurally distinct FBPs are capable of interacting with some of the same substrates; however, in vivo specificity is achieved in part by subcellular localization. Additionally, the FBPs Cdc4 and Grr1 are partially redundant for proliferation and viability, likely sharing additional redundant substrates whose degradation is important for cell cycle progression.
Introduction
The ubiquitin-proteasome system plays an essential role in controlling passage through the eukaryotic cell cycle [1]. A significant fraction of cell cycle-regulated ubiquitination is carried out by SCF (Skp1-Cullin-F-box protein) family ubiquitin ligases, which target numerous cell cycle regulators for proteasomal degradation. All SCF ligases consist of three core subunits: a structural cullin subunit (Cdc53 in yeast, Cul1 in mammals), an adaptor protein (Skp1) and a RING finger protein (Rbx1), plus one of a family of modular substrate-specificity subunits called F-box proteins (FBPs) [2]–[7]. There are large numbers of FBPs in all eukaryotes, and each is believed to target the SCF to a specific set of substrates by interacting with distinct epitopes in those proteins. In almost all instances, FBPs recognize proteins that have been post-translationally modified, usually by phosphorylation, which enables ubiquitination to be regulated by substrate modification [8].
In budding yeast, the FBPs Cdc4 and Grr1 have well-established cell cycle-regulatory roles [1]. Both FBPs recognize phosphorylated epitopes in their substrates, however they bind to these epitopes through distinct phosphorecognition domains: a WD40 repeat domain in Cdc4 and a leucine rich repeat domain in Grr1 [8]. Interestingly, although Grr1 and Cdc4 are thought to have entirely non-overlapping sets of substrates, each is capable of interacting with targets that have been phosphorylated by cyclin dependent kinase (Cdk). This group of substrates includes several proteins that regulate entry into S phase including the Grr1 substrates Cln1 and Cln2 [9], as well as the Cdc4 substrates Sic1 [10] and Cdc6 [11]. In addition to this group of defined SCF targets, Cdk phosphorylates hundreds of yeast proteins [12], [13], and many of these are rapidly degraded [14], suggesting that there is a widespread connection between Cdk phosphorylation and protein degradation. However, the majority of these proteins have not been identified in genome-wide screens for Cdc4 or Grr1 targets [15], [16], suggesting that they may be targeted for degradation by alternate ubiquitin ligases.
One such Cdk-phosphorylated protein is the G1 cyclin Cln3. Similar to cyclin D1 in mammals, Cln3 is the furthest upstream cyclin, which senses growth cues and triggers entry into the cell cycle. Cells become committed to progress through the cell cycle upon phosphorylation of the transcriptional repressor protein Whi5 by Cln3/Cdc28, which leads to Whi5 inactivation and increased expression of downstream genes including the related cyclins Cln1 and Cln2 [17], [18]. Consistent with Cln3 having a critical role in cell cycle entry, its levels are very tightly controlled. In addition to being regulated by transcription [19], [20] and subcellular localization [21]–[23], Cln3 is rapidly degraded. This proteolytic degradation is critical to restrain Cln3 activity, since expression of a truncated and stable form of the Cln3 protein drives cells through G1 phase prematurely, resulting in a significant reduction in cell size [24]–[26]. Despite the physiological importance of Cln3 degradation, the ubiquitin ligase that targets Cln3 for degradation has not been identified.
Previous studies have implicated an SCF ligase in Cln3 degradation [26], [27], however no FBP has been identified that recognizes Cln3. Here, we show that Cdc4 and Grr1 redundantly target Cdk-phosphorylated Cln3 for degradation. Mutation of either FBP alone has no detectable effect on Cln3 levels or stability, yet Cln3 is completely stable in double mutant cells. Surprisingly, we find that both Cdc4 and Grr1 interact with all 3 G1 cyclins (Cln1, Cln2 and Cln3) in cell extracts, however only Cln3 is redundantly targeted in vivo, because it is the only G1 cyclin that localizes primarily to the nucleus. Cln2 is cytoplasmic and exclusively targeted by Grr1 [9], [28]. However, we show that Cdc4 can target Cln2 for degradation when cytoplasmic Cdc4 localization is forced in vivo. Finally, we observed a synthetic growth defect in cdc4 grr1 double mutant cells that is not suppressed by deletion of CLN3. In sum, these data demonstrate that the binding specificities of FBPs do not necessarily dictate which proteins are in vivo targets, and suggest that Cdc4 and Grr1 have additional redundant targets whose regulated degradation is necessary for normal cell cycle control.
Results
SCFGrr1 and SCFCdc4 Redundantly Target Cln3 for Degradation
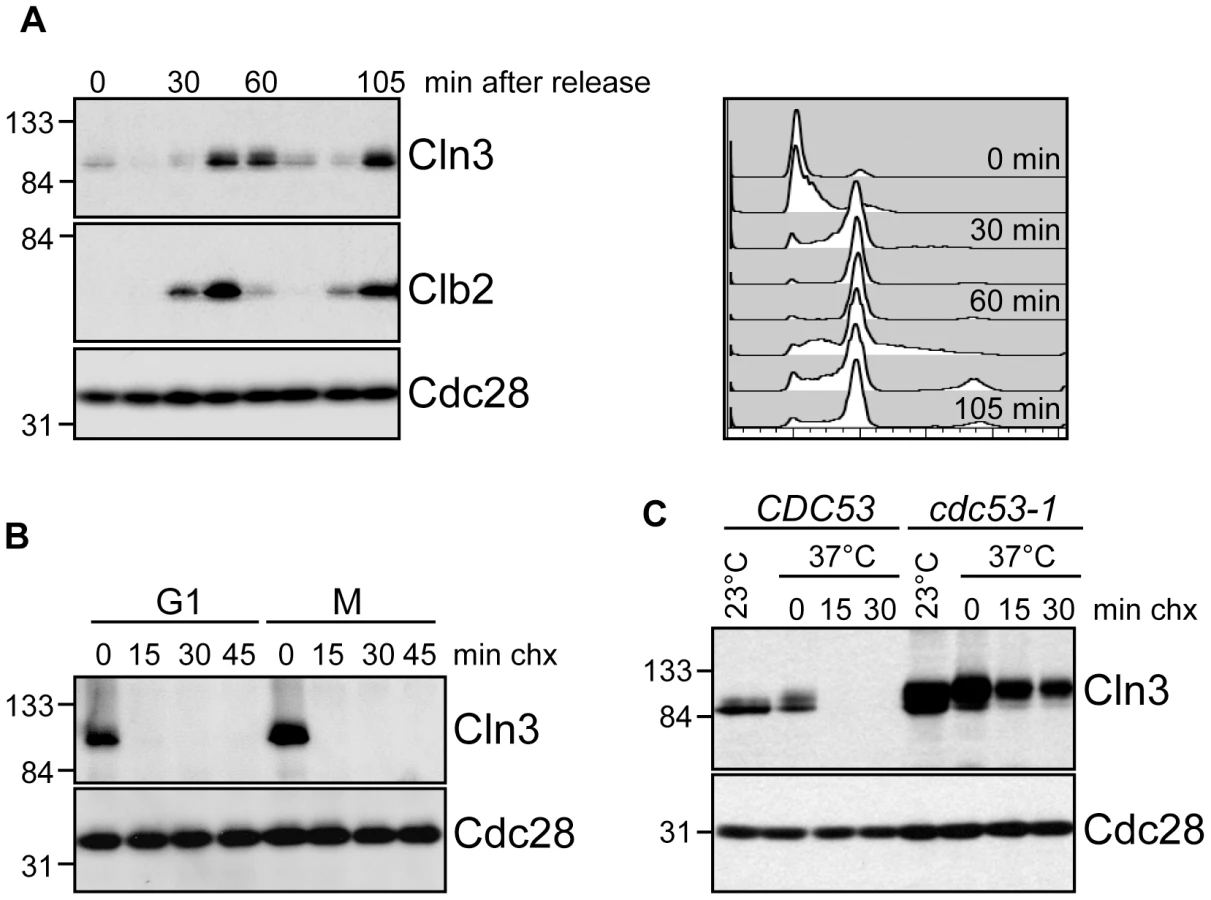
To better understand the regulation of Cln3 degradation, we examined Cln3 protein levels throughout the cell cycle and found that they paralleled the reported transcriptional expression profiles [19], [20], [29], rising in mitosis, shortly after the mitotic cyclin Clb2, and persisting through G1 phase (Figure 1A; Figure S1A). Cln3 was rapidly degraded in cells arrested in either G1 or mitosis, demonstrating that it is degraded throughout the cell cycle (Figure 1B; Figure S1B). Previous data suggested that an SCF ubiquitin ligase is responsible for targeting Cln3 [26], [27]. Consistent with these observations, we found that phosphorylated Cln3 is stable and accumulates in cells expressing a temperature-sensitive allele of CDC53 (cdc53-1) upon shift to the restrictive temperature (Figure 1C; Figure S1C). Similarly, transcriptional shut-off of the genes encoding Cdc53 or Cdc34 (the E2 enzyme that functions with SCF ligases) stabilized Cln3 (Figure S1D).
We next attempted to identify the specific FBP that targets Cln3 for degradation. Cln3 levels were compared among strains carrying single deletions of every non-essential yeast F-box protein (Figure 2A; data not shown), however no single deletion caused a significant increase in Cln3 protein levels. Furthermore, inactivation of the essential FBP Cdc4 did not stabilize Cln3 (Figure 2B; Figure S1D). Together, these data suggested that Cln3 might be redundantly targeted by two or more FBPs. The most likely candidate FBPs to redundantly regulate Cln3 were Grr1 and Cdc4, since each of these proteins is known to target Cdk-phosphorylated substrates [9]–[11], [30], [31]. Indeed, we found that simultaneous inactivation of Grr1 and Cdc4 led to complete stabilization of Cln3, whereas deletion of GRR1 or inactivation of Cdc4 alone had no significant effect on Cln3 levels or stability (Figure 2B). Cln3 protein was almost undetectable in both single mutant strains after only 2 minutes of cycloheximide treatment (Figure 2C), and reintroduction of either Cdc4 or Grr1 into grr1Δ cdc4-1 cells led to an equivalent reduction of Cln3 levels (Figure S2A). Consistent with these observations, overexpression of Cln3 in either grr1Δ cells, or cells with limiting amounts of Cdc4 (cdc4-1 grown at the permissive temperature) was lethal (Figure 2D), indicating that both FBPs are required for the cell to tolerate high levels of Cln3. Together, these data demonstrate that Cln3 is redundantly regulated by Cdc4 and Grr1.

Cdc4 and Grr1 Interact with Cdk-Phosphorylated Cln3
In order to demonstrate that Cdc4 and Grr1 directly target Cln3, we examined the binding of Cln3 to each FBP. Because complexes between ubiquitin ligases and substrates are unstable and often difficult to detect, we assayed binding of Cln3 to GST-tagged Grr1 or Cdc4 proteins that lack the F-box domain and therefore cannot interact with the remainder of the SCF ubiquitin ligase complex (Cdc4ΔF and Grr1ΔF) [15], [32]. In contrast to the full-length proteins, expression of GST-Cdc4ΔF or GST-Grr1ΔF had no effect on levels of Cln3 (Figure S2A), confirming that they were not incorporated into active SCF complexes. Upon pull-down of GST-tagged proteins from cellular lysates, Cln3 associated with both Cdc4ΔF and Grr1ΔF proteins but not with GST (Figure 3A, lane 2), supporting the model Cdc4 and Grr1 directly target Cln3 in vivo.

Previous studies suggested that the C-terminal tail of Cln3 is required for its degradation [26] . To verify this result and narrow down the region required for Cln3 degradation we constructed a series of C-terminal truncation mutants (Figure S2B) and examined their half-lives in vivo. With the exception of the largest deletion of 177 amino acids (equivalent to the previously characterized allele Cln3-1; [26]), all mutants were still turned over at significant rates (Figure 3B). Although each of these mutants was slightly more stable than wild-type Cln3, cells expressing these mutants did not show any obvious changes in cell cycle progression (Figure S2E–S2F), suggesting that partial stabilization does not impact the cell cycle in vivo. However, since each truncated protein was more stable than wild-type Cln3 in either grr1Δ or cdc4-1 cells (Figure 2B), this indicates that these mutations must partially interfere with degradation by both FBPs.
The large size of the C-terminal tail makes it possible that each FBP recognizes a distinct epitope within this domain. Therefore, to further narrow down the requirements for binding to each FBP, we analyzed the binding of the C-terminal Cln3 truncation mutants to Cdc4 and Grr1. Interestingly, we found that all truncations disrupted binding to Cdc4 (Figure 3A, middle panels), including the smallest deletion of just 22 amino acids. In contrast, all truncations except for the largest deletion (Cln3-1) bound well to Grr1, albeit at reduced levels (Figure 3A, bottom panels). These data suggest that while Grr1 and Cdc4 both require the Cln3 C-terminus for binding, they do not bind identical epitopes.
Cdk-phosphorylation of the Cln3 C-terminus is also required for its degradation [27], [33]. Since both Cdc4 and Grr1 bind Cdk-phosphorylated epitopes, this suggested that both FBPs might bind to the Cln3 C-terminus in a phospho-dependent manner. To test this, we first constructed a stable, Cdk-deficient allele of Cln3. The Cln3 C-terminus includes 10 serine/threonine-proline motifs, constituting the minimal Cdk-consensus site. A previous report demonstrated that mutation of the single full Cdk consensus site (S/TPxK) in the C-terminus of Cln3, serine 468, partially stabilized a Cln3 C-terminus-β-Gal fusion protein [27]. Notably, mutation of this site in the context of the full-length protein only had a minor effect on Cln3 stability and did not affect cell cycle position (Figure S2C–S2D). We then changed all of the remaining C-terminal Cdk consensus sites to alanine residues within the endogenous CLN3 locus and assayed the stability of the mutated Cln3 proteins. Mutation of the 9 most C-terminal sites completely stabilized Cln3 (Figure 4A, Cln3-9A), and led to a cell cycle profile consistent with a significantly shortened G1 phase (Figure 4B). Next, we tested whether Cln3-9A could interact with Cdc4 and Grr1. Consistent with our prediction, significantly less Cln3-9A bound to each FBP, in comparison to wild-type Cln3 (Figure 4C, compare lanes 6 & 8, 10 & 12). These data strongly suggest that phosphorylation of the Cln3 C-terminus contributes to its interaction with both Cdc4 and Grr1.

Although Cdk phosphorylation of the C-terminus is required for Cln3 degradation, our binding data suggested that each FBP recognizes a distinct epitope within this domain, so we further analyzed the requirements for these C-terminal phosphosites in degradation by each FBP. Both Cdc4 and Grr1 are thought to target regions of their substrates that include multiple Cdk-phosphorylated residues [9]–[11], [30], [31], so we mutated two clusters of Cdk phosphosites in attempt to specifically interfere with binding to one or the other FBP (Figure S2B). First, we mutated five Cdk sites in the N-terminal half of the C-terminal domain, spanning residues 447–468 (designated Cln3-5A). Interestingly, we found that Cln3-5A was partially stabilized in wild-type cells (Figure 4A), and Cln3-5A-expressing cultures contained a slightly reduced fraction of cells in G1 phase (Figure 4B). Since degradation of wild-type Cln3 is unaffected in either cdc4-1 or grr1Δ cells, the partial stabilization of the Cln3-5A protein suggested that these mutations partially interfered with targeting by both Cdc4 and Grr1. Consistent with this possibility, Cln3-5A was further stabilized in both cdc4-1 and grr1Δ single mutant cells (Figure 4D, top panels).
Next, we mutated three sites at the extreme C-terminus (designated Cln3-3A). Notably, these sites are adjacent to the region required for Cdc4 interaction (Figure 3A), and are separated by two amino acids each, which matches the doubly phosphorylated degron motif that is preferred by Cdc4 [34], [35]. In wild-type cells Cln3-3A was rapidly degraded and its expression had no effect on cell cycle progression (Figure 4A–4B). However, Cln3-3A was degraded differently in cells lacking GRR1 and CDC4: Cln3-3A turnover was unaffected in cdc4-1 cells, but almost completely blocked in grr1Δ cells (Figure 4D, bottom panels). This demonstrates that Cln3-3A cannot be targeted by Cdc4 and is consistent with the prediction that degradation by Cdc4, but not Grr1, requires phosphorylation of these three C-terminal sites.
Together, these data suggest that although the Cdk-phosphorylated C-terminus of Cln3 is required for its targeting by Cdc4 and Grr1, the two FBPs recognize different epitopes within this domain. However, since Cln3 is thought to be constitutively bound to Cdk, it is possible that these sites are all constitutively phosphorylated in cis and that this phosphorylation allows Cln3 to be degraded by both FBPs throughout the cell cycle. A second possibility is that Cln3 is phosphorylated in trans by an alternate cyclin/Cdk complex in order to be targeted for degradation. Phosphorylation in trans could lead to cell cycle-regulated Cln3 degradation. To test whether cis phosphorylation of Cln3 by Cdk is required for its turnover, we constructed an N-terminal truncation mutant of Cln3 that removes the cyclin box and therefore can not bind to Cdk (Cln3ΔN). Interestingly, blocking cis phosphorylation in this manner led to almost complete stabilization of Cln3 protein (Figure 4E). The subtle degradation that was observed was dependent upon Cdk-phosphorylation, since mutation of the 9 C-terminal Cdk sites further stabilized the protein, suggesting that this protein could be phosphorylated by alternative cyclin/Cdk complexes. However, since Cln3ΔN is considerably more stable that full length Cln3, we conclude that the primary mode of Cln3 degradation depends upon phosphorylation in cis.
C-terminal Domains of G1 Cyclins Confer F-box Protein Specificity
Although Cdc4 and Grr1 have both been shown to bind and ubiquitinate Cdk-phosphorylated proteins, they are thought to recognize non-overlapping sets of targets [36]. This raises the question of what is unique about Cln3 that enables it to be recognized by both F-box proteins. To address this issue, we compared Cln3 and the related cyclin Cln2, which is targeted for degradation exclusively by Grr1 [9]. Like Cln3, Cln2 has an N-terminal cyclin box and a C-terminus that includes a PEST domain and Cdk phosphorylation sites (Figure 5A; [37]). Moreover, the 169 C-terminal amino acids of Cln2 constitute a transferrable degron, which can direct Grr1-mediated degradation of a heterologous protein [38]. Together, this suggests that all of the FBP specificity resides within the C-terminal domains of both cyclins.

To test this possibility, chimeric proteins were created by exchanging the C-terminal degron domains of Cln2 and Cln3 (Figure 5A) and the stability of these chimeric proteins was assayed in strains lacking Grr1, Cdc4, or both FBPs. As expected for a Grr1 target, Cln2 was degraded rapidly in wild-type cells, but completely stable in grr1Δ cells (Figure 5B, top panels). Moreover, Cln2 was degraded in cdc4-1 cells, although the protein level was higher overall (which was expected because cdc4-1 cells arrest in G1/S phase when CLN2 transcription peaks). In contrast to Cln2, Cln3 was not affected by loss of either Grr1 or Cdc4, but was completely stabilized in grr1Δ cdc4-1 cells (Figure 2B). However, when the C-terminus of Cln3 was replaced with the C-terminus of Cln2 (Cln3-2C), the Cln3 degradation profile was nearly identical to that of Cln2 (Figure 5B, middle panels). In wild-type cells, the half-life of Cln3-2C was longer than Cln3 (compare Figure 2B to Figure 4B) and, importantly, Cln3-2C was completely stable in grr1Δ cells, demonstrating that it could no longer be targeted by Cdc4. When the C-terminus of Cln2 was replaced with the C-terminus of Cln3 (Cln2-3C), the opposite result was observed (Figure 5B, bottom panels). In wild-type cells, Cln2-3C was considerably less stable than Cln2. In addition, unlike Cln2, Cln2-3C was degraded in grr1Δ cells (although it was slightly more stable than in wild-type cells). However, Cln2-3C was stable in grr1Δ cdc4-1 cells. Together, these data demonstrate the C-termini of Cln2 and Cln3 confer their FBP specificity, and indicate that there is a unique feature in the Cln3 C-terminus that promotes Cdc4-mediated turnover.
Subcellular Localization Regulates G1 Cyclin Degradation
Our analysis of Cln3 truncation mutants demonstrated that the last 22 amino acids of Cln3 are important for interaction with Cdc4 but not Grr1 (Figure 3A). Interestingly, this 22 amino acid sequence in Cln3 includes a bipartite nuclear localization signal (NLS), which is important for Cln3 nuclear localization (Figure S3; [21], [22]). In contrast, Cln2 does not have an NLS motif and it is predominantly cytoplasmic [22], [28]. This raises the possibility that the localization of G1 cyclins may also contribute to FBP specificity in vivo. The localization of the FBPs supports this model: Grr1 is both cytoplasmic and nuclear, whereas Cdc4 is exclusively nuclear [39]. Therefore, since Cln2 is primarily cytoplasmic, it may only be accessible to Grr1. In contrast, since Cln3 is primarily nuclear, this may enable targeting by both Grr1 and Cdc4. Consistent with this model, the Cln3Δ22 mutant, which lacks the NLS sequence and is primarily cytoplasmic (Figure S3; [22]), is almost completely stable in grr1Δ cells (Figure 5C). Notably, we find that Cln3Δ22 is considerably more stable than full-length Cln3 expressed in cells lacking Cdc4 (compare Figure 5C and Figure 2B), despite that fact that it can bind to Grr1 as well as the full-length protein in whole cell extracts (Figure 3A). This suggests that nuclear Cln3 is more susceptible to degradation than cytoplasmic Cln3, perhaps because most cytoplasmic Cln3 is tethered to the ER and not fully active [23].
Another prediction from our data is that Cdc4 may be capable of targeting Cln2 for degradation, but does not do so in vivo only because Cln2 is predominantly localized to the cytoplasm. To test this idea, we first tested whether Cln2 could interact with Cdc4. Importantly, Cln2 associated with both Cdc4ΔF and Grr1ΔF proteins in extracts (Figure 6A, lanes 6 & 10). Moreover, Cln2-4T3S, a stable Cln2 protein that has mutations in 7 Cdk-consensus sites [37], was unable to bind to Grr1 or Cdc4 (Figure 6A, lanes 8 &12), suggesting that Cdc4 and Grr1 bind to similar Cdk-phosphorylated epitopes. In addition, we found that the third G1 cyclin, Cln1, also interacted with both Cdc4 and Grr1 (Figure S4). Thus, like Cln2, Cln1 is a potential target of both Cdc4 and Grr1, but may be regulated exclusively by Grr1 in vivo by virtue of its cytoplasmic localization.

Since Cdc4 interacts with Cln1 and Cln2 in extracts, this suggests that Cdc4 might be capable of targeting all three G1 cyclins in vivo, if it were localized in the same subcellular compartment as all three cyclins. To test this, we utilized a previously characterized Cdc4 protein (NES-Cdc4) that is fused to a nuclear export signal and localizes to the cytoplasm [39]. Expression of NES-Cdc4 in grr1Δ cells led to decreased Cln2 protein levels, whereas expression of wild-type Cdc4 had no effect (Figure 6B). In addition, this downregulation was dependent upon Cdk-phosphorylation of Cln2, since levels of Cln2-4T3S were unaffected by NES-Cdc4 expression (Figure 6B). Together, these data show that localization of yeast G1 cyclins determines which of the FBPs can target each in vivo, and that the binding specificities of the FBPs do not dictate which proteins are in vivo targets.
Genetic Interaction between GRR1 and CDC4
The finding that Cdc4 and Grr1 can bind to some of the same Cdk-phosphorylated proteins, and that they redundantly target Cln3 for degradation in vivo, suggests that these two FBPs may share a number of overlapping targets that are important for cell cycle progression. In support of this possibility, we found that cells carrying a GRR1 deletion and having compromised Cdc4 function (grr1Δ cdc4-1, grown at the permissive temperature for cdc4-1) demonstrate a synergistic growth defect well beyond that predicted for a combination of the individual (minor) growth phenotypes (Figure 7). This type of negative genetic interaction is consistent with Cdc4 and Grr1 having partially redundant roles in one or more common cellular functions [40]. Importantly, deletion of CLN3 did not reverse this growth defect (Figure 7), supporting the idea that additional proteins are redundantly targeted by Cdc4 and Grr1. Alternatively, it is possible that Cdc4 and Grr1 have specific substrates that cause a synergistic growth defect when their levels are elevated. However, the large number of unstable, Cdk-phosphorylated proteins in the cell [13], [14], [41], combined with the ability of Grr1 and Cdc4 to bind some targets in common, raises the intriguing possibility that these two FBPs target a significant fraction of the cell cycle proteome for degradation.

Discussion
Cln3 turnover is essential for accurate entry into the cell cycle and for proper cell size control [24]–[26], however the identity of the ubiquitin ligase that targets Cln3 for destruction has remained a long-standing question. Here, we show that Cln3 is redundantly targeted by two F-box proteins with key cell cycle-regulatory roles, Cdc4 and Grr1. Interestingly, inactivation of either FBP alone has no detectable effect on Cln3 expression or half-life, yet Cln3 is completely stable in double mutant cells (Figure 2B–2C). This result is quite surprising because, although redundant regulation of degradation has been previously found for other proteasome substrates in both yeast and mammals [42]–[44], in most cases elimination of one ubiquitin ligase or the other leads to a partial stabilization of the substrate, which we do not observe with Cln3. This may be because targeting of these other substrates by two ligases is only partially redundant in the sense that the ligases may recognize different epitopes on the substrate, or respond to different physiological cues.
In the case of Cln3, our data suggests that both Cdc4 and Grr1 recognize the Cdk-phosphorylated C-terminus of Cln3 (Figure 4). However, the two FBPs do not recognize identical epitopes. Cdc4 targeting requires three phosphosites at the extreme C-terminus of Cln3, whereas mutation of these sites have no effect on targeting by Grr1 (Cln3-3A, Figure 4D, bottom panels). Interestingly, mutation of a second cluster of five phosphosites partially interferes with targeting by both Cdc4 and Grr1 (Cln3-5A, Figure 4D, top panels). This suggests that the two FBPs may also interact with some overlapping residues. Alternatively, these five phosphosites may be required as priming sites that promote the phosphorylation of the three C-terminal sites that Cdc4 requires, in a mechanism similar to what has recently been demonstrated for the Cdc4-specific target Sic1 [35]. Further work will be required to dissect the mechanism of targeting by each FBP. However, since the Cln3 C-terminus is likely to be constitutively phosphorylated by Cdk in cis (Figure 4E), and we have not been able to identify any condition when only one FBP targets Cln3, this suggests that Cdc4 and Grr1 are truly redundant for Cln3 degradation.
The fact that there is no significant change in Cln3 levels in either single mutant, along with the observed genetic interaction between CDC4 and GRR1 (Figure 7), raises the possibility that there are additional redundant targets of Cdc4 and Grr1 that have not yet been identified. A likely possibility is that other Cdk-phosphorylated, nuclear proteins are dual-regulated targets. Given the large number of Cdk-phosphorylated proteins that are expressed in a cell cycle-dependent pattern [12], [13], [19], [20], it is possible that Cdc4 and Grr1 recognize a much larger fraction of all cell cycle-regulated proteolysis than was previously recognized. However, Cdk-phosphorylation cannot be the only factor that determines whether a protein can be an in vivo substrate of both Grr1 and Cdc4, since established Cdk-phosphorylated Cdc4 targets, such as Sic1 and Far1, cannot be targeted by Grr1 in vivo or in vitro [4], [39]. In addition, we find that deletion of SIC1 does not reverse the synthetic sickness of grr1Δ cdc4-1 strains (data not shown). Whether the synthetic growth defect observed for cdc4 and grr1 is due to upregulation of a single common target protein that is crucial for proliferation, or multiple targets (either shared or specific) that each play a modest role in the cell cycle is a key question requiring further study.
In contrast to Cln3, which is targeted by both Cdc4 and Grr1, the related cyclins Cln1 and Cln2 are targeted exclusively by Grr1 in vivo [9]. Interestingly, we find that Cdc4 can interact with both Cln1 and Cln2 in extracts (Figure S4; Figure 6). Moreover, previous studies with recombinant proteins reported a weak association between Cln2 and Cdc4 [4] and demonstrated that Cdc4 can ubiquitinate Cln2 in vitro [39]. Interestingly, since Cln2 has been shown to phosphorylate Whi5 in the nucleus [45], this suggests that Cdc4 may have an unappreciated role in targeting a small but important nuclear fraction of Cln1/2. Alternatively, the small fraction of nuclear Cln2 may be protected from degradation in some way. In addition, these data indicate that FBPs do not necessarily have the exquisite binding specificity for substrates that has been proposed, and that co-localization of FBPs and substrates also contributes to substrate specificity in vivo. In the future it will be of interest to investigate whether any other Grr1 substrates can interact with Cdc4, or be targeted by Cdc4 upon co-localization.
Importantly, the finding that FBP specificity for G1 cyclins depends upon the localization of cyclins may aid in our understanding of the mechanism of degradation of the mammalian cyclin D1 protein. Several FBPs have been implicated in cyclin D1 degradation [46]–[51], and it is possible that it is regulated by similar redundant mechanisms. It is interesting to note that degradation of the furthest upstream G1 cyclins appears to be quite complex and include redundancy. Since both Cln3 and cyclin D1 are crucial regulators that act as sensors of extracellular growth signals and trigger entry into the cell cycle, cells may have evolved redundant mechanisms to degrade these cyclins in order to buffer cells against dramatic fluctuations in cyclin synthesis and inappropriate cell cycle entry.
Materials and Methods
Yeast Strains and Plasmids
A complete list of strains, including the specific experiments each was used in, is provided in Table S1. Unless otherwise indicated, all strains are in the S288c background and have a 13Myc-HIS3MX C-terminal tagging cassette integrated at the CLN3 or CLN2 genomic locus. Strains carrying point mutations in phosphorylation sites were generated by integrating a PCR product containing the desired mutations, the 13Myc tag, and the HIS3MX marker at the CLN3 or CLN2 genomic locus. The integration of each mutation was then confirmed by sequencing.
Plasmids expressing GST and GST-tagged Grr1 have been described previously [32]. To construct pYES2-GST-CDC4, the CDC4 sequence was amplified from genomic DNA by PCR and cloned into the pYES2-GST vector. pYES2-CDC4ΔF was constructed similarly, except that the sequence corresponding to amino acids 323–475 was amplified and cloned into pYES2-GST. To construct the GST-NES-CDC4 plasmid, a DNA fragment including the NES and the CDC4 N-terminal sequences was subcloned from pBM138 (provided by Matthias Peter, [39]) into pYES2-GST-CDC4.
All strains were grown in YM-1 complete medium with 2% dextrose, with the exception of strains carrying GST plasmids, which were grown in C medium lacking uracil with 2% dextrose or raffinose [15]. To arrest cells in G1, 20 µg/ml alpha-factor (United Biochemical Research, Inc.) was added for 2–3 hours. To arrest cells in mitosis, 10 µg/ml nocodazole (US Biological) was added to cells for 2 hours. Strains carrying temperature-sensitive alleles of CDC53 or CDC4 were grown at 23°C and shifted to 37°C for 2 hours to inactivate the respective proteins. Strains carrying tetracycline-regulated alleles of CDC53, CDC4 and CDC34 were treated with 10 µg/ml doxycycline (EMD Biosciences) for 8 hours to shut off transcription from the tetracycline-regulated promoters (Figure S1D). For GST-pulldown assays to analyze Cln3 binding (Figure 3A; Figure 4C), grr1Δ cdc4-1 strains carrying GST plasmids were grown in 2% raffinose and then induced by the addition of 2% galactose for 20–22 hours. For GST-pulldown assays to analyze Cln1 and Cln2 binding (Figure 6A; Figure S4), grr1Δ strains carrying GST plasmids were grown in 2% raffinose and then induced by the addition of 2% galactose for 8 hours. To examine levels of Cln2 and Cln2-4T3S following expression of GST, GST-CDC4, and GST-NES-CDC4 (Figure 6B), grr1Δ cells carrying GST plasmids were grown in 2% raffinose and expression was induced by the addition of 0.05% galactose for the indicated number of hours.
Western Blotting
Equivalent optical densities of cells were pelleted, lysed in pre-heated SDS sample buffer (50 mM Tris pH 7.5, 5 mM EDTA, 5% SDS, 10% glycerol, 0.5% β-mercaptoethanol, bromophenol blue, 1 µg/ml leupeptin, 1 µg/ml bestatin, 1 mM benzamidine, 1 µg/ml pepstatin A, 17 µg/ml PMSF, 5 mM sodium fluoride, 80 mM β-glycerophosphate and 1 mM sodium orthovanadate) and heated to 95°C for 5 minutes. Glass beads were then added and samples were bead-beat for 3 minutes in a Mini-BeadBeater-96 (Biospec), followed by centrifugation. For cycloheximide assays with two minute time points, cell pellets were lysed in cold TCA buffer (10 mM Tris pH 8.0, 10% trichloroacetic acid, 25 mM ammonium acetate, 1 mM EDTA) and incubated on ice. Samples were then centrifuged and the pellets resuspended in Resuspension Solution (0.1 M Tris pH 11.0, 3% SDS). Samples were heated to 95°C for 5 minutes, allowed to cool to room temperature, and clarified by centrifugation. Supernatants were added to 4× SDS-PAGE Sample Buffer (0.25 M Tris pH 6.8, 8% SDS, 40% glycerol, 20% β-mercaptoethanol) and heated to 95°C for 5 minutes. Extracts were then subjected to SDS–polyacrylamide gel electrophoresis (SDS–PAGE), followed by transfer to nitrocellulose membranes, and Western blotting with antibodies against Myc (Clone 9E10, Covance) Cdc28 (sc-6709, Santa Cruz Biotechnology), Clb2 (sc-9071, Santa Cruz Biotechnology), Cdc53 (sc-6717, Santa Cruz Biotechnology), Flag (Clone M2, Sigma) and GST (Clone 4C10, Covance).
Cycloheximide-Chase Assays
Cells were grown to mid-log phase, or arrested as indicated, then treated with 50 µg/ml cycloheximide. Samples were fixed for flow cytometry, and cell pellets from equivalent optical densities of cells were collected for Western blotting at indicated time points.
Cell Cycle Analysis
Cells were fixed with 70% ethanol and stored at 4°C overnight. Cells were then sonicated, treated with 0.25 mg/ml RNase A for 1 hour at 50°C, followed by digestion with 0.125 mg/ml Proteinase K for 1 hour at 50°C and labeling with 1 µM Sytox Green (Invitrogen). Data was collected using a FACScan (Becton Dickinson) and analyzed with FlowJo (Tree Star, Inc.) software.
GST-Pulldown Assays
Cell pellets containing 20–30 optical densities of cells were lysed in HEPES lysis buffer (25 mM HEPES pH 7.6, 400 mM NaCl, 0.2% Triton X-100, 1 mM EDTA, 10% glycerol, 1 µg/ml leupeptin, 1 µg/ml bestatin, 1 mM benzamidine, 1 µg/ml pepstatin A, 17 µg/ml PMSF, 5 mM sodium fluoride, 80 mM β-glycerophosphate and 1 mM sodium orthovanadate) by bead beating in a cold block for 3 minutes and clarified by centrifugation at 4°C. Extracts were incubated with 20 µl of a 50% slurry of glutathione-sepharose 4B in lysis buffer (GE Healthcare), while rotating at 4°C for 2 hours. Beads were collected by centrifugation and washed seven times with 1 ml lysis buffer. Proteins were eluted by boiling in 2× SDS-PAGE sample buffer and analyzed by Western blotting against the GST and Myc tags. 2% input of each extract is also shown.
Immunofluorescence
Cells expressing Myc-tagged Cln3 proteins were fixed in 3.7% formaldehyde for 1 hour at 23°C followed by two washes in potassium phosphate buffer (83 mM K2HPO4, 17 mM KH2PO4, pH 7.5) and one wash in sorbitol phosphate buffer (1.2 M sorbitol, 83 mM K2HPO4, 17 mM KH2PO4, pH 7.5). Cells were then spheroplasted with zymolyase and adhered to poly-L-lysine coated slides. Cells on slides were permeabilized with methanol and acetone, then blocked in PBS-BSA (10 mg/ml bovine serum albumin, 0.04 M K2HPO4, 0.01 M KH2PO4, 0.15 M NaCl, 0.1%NaN3). Cells were then incubated with rabbit anti-Myc antibody (sc-789, Santa Cruz Biotechnology) overnight, followed by 5 washes in PBS-BSA, and incubation with Alexafluor 488-conjugated goat anti-mouse secondary antibody (Invitrogen). Cells were again washed 5 times with PBS-BSA, then stained with 1 µg/ml DAPI and mounted with ProLong Gold Antifade reagent (Invitrogen). Microscopy was carried out using a Zeiss Axioskop 2 fluorescence microscope with an 100× 1.3NA Plan-NEOFLAUR objective. Images were taken with a RT Monochrome SPOT camera (Diagnostic Instruments, Inc.) and accompanying software. Image analysis was done with Adobe Photoshop software. All images were captured for the same exposure times and adjustments to contrast and brightness were performed equally on all panels.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. ReedSI 2003 Ratchets and clocks: the cell cycle, ubiquitylation and protein turnover. Nat Rev Mol Cell Biol 4 855 864
2. BaiCSenPHofmannKMaLGoeblM 1996 SKP1 connects cell cycle regulators to the ubiquitin proteolysis machinery through a novel motif, the F-box. Cell 86 263 274
3. FeldmanRMCorrellCCKaplanKBDeshaiesRJ 1997 A complex of Cdc4p, Skp1p, and Cdc53p/cullin catalyzes ubiquitination of the phosphorylated CDK inhibitor Sic1p. Cell 91 221 230
4. SkowyraDCraigKLTyersMElledgeSJHarperJW 1997 F-box proteins are receptors that recruit phosphorylated substrates to the SCF ubiquitin-ligase complex. Cell 91 209 219
5. SkowyraDKoeppDMKamuraTConradMNConawayRC 1999 Reconstitution of G1 cyclin ubiquitination with complexes containing SCFGrr1 and Rbx1. Science 284 662 665
6. OhtaTMichelJJSchotteliusAJXiongY 1999 ROC1, a homolog of APC11, represents a family of cullin partners with an associated ubiquitin ligase activity. Mol Cell 3 535 541
7. SeolJHFeldmanRMZachariaeWShevchenkoACorrellCC 1999 Cdc53/cullin and the essential Hrt1 RING-H2 subunit of SCF define a ubiquitin ligase module that activates the E2 enzyme Cdc34. Genes Dev 13 1614 1626
8. WillemsARSchwabMTyersM 2004 A hitchhiker's guide to the cullin ubiquitin ligases: SCF and its kin. Biochim Biophys Acta 1695 133 170
9. BarralYJentschSMannC 1995 G1 cyclin turnover and nutrient uptake are controlled by a common pathway in yeast. Genes Dev 9 399 409
10. VermaRFeldmanRMDeshaiesRJ 1997 SIC1 is ubiquitinated in vitro by a pathway that requires CDC4, CDC34, and cyclin/CDK activities. Mol Biol Cell 8 1427 1437
11. DruryLSPerkinsGDiffleyJF 1997 The Cdc4/34/53 pathway targets Cdc6p for proteolysis in budding yeast. EMBO J 16 5966 5976
12. HoltLJTuchBBVillenJJohnsonADGygiSP 2009 Global analysis of Cdk1 substrate phosphorylation sites provides insights into evolution. Science 325 1682 1686
13. UbersaxJAWoodburyELQuangPNParazMBlethrowJD 2003 Targets of the cyclin-dependent kinase Cdk1. Nature 425 859 864
14. BelleATanayABitinckaLShamirRO'SheaEK 2006 Quantification of protein half-lives in the budding yeast proteome. Proc Natl Acad Sci U S A 103 13004 13009
15. BenantiJACheungSKBradyMCToczyskiDP 2007 A proteomic screen reveals SCFGrr1 targets that regulate the glycolytic-gluconeogenic switch. Nat Cell Biol 9 1184 1191
16. TangXOrlickySLiuQWillemsASicheriF 2005 Genome-wide surveys for phosphorylation-dependent substrates of SCF ubiquitin ligases. Methods Enzymol 399 433 458
17. CostanzoMNishikawaJLTangXMillmanJSSchubO 2004 CDK activity antagonizes Whi5, an inhibitor of G1/S transcription in yeast. Cell 117 899 913
18. de BruinRAMcDonaldWHKalashnikovaTIYatesJ3rdWittenbergC 2004 Cln3 activates G1-specific transcription via phosphorylation of the SBF bound repressor Whi5. Cell 117 887 898
19. PramilaTWuWMilesSNobleWSBreedenLL 2006 The Forkhead transcription factor Hcm1 regulates chromosome segregation genes and fills the S-phase gap in the transcriptional circuitry of the cell cycle. Genes Dev 20 2266 2278
20. SpellmanPTSherlockGZhangMQIyerVRAndersK 1998 Comprehensive identification of cell cycle-regulated genes of the yeast Saccharomyces cerevisiae by microarray hybridization. Mol Biol Cell 9 3273 3297
21. EdgingtonNPFutcherB 2001 Relationship between the function and the location of G1 cyclins in S. cerevisiae. J Cell Sci 114 4599 4611
22. MillerMECrossFR 2001 Mechanisms controlling subcellular localization of the G(1) cyclins Cln2p and Cln3p in budding yeast. Mol Cell Biol 21 6292 6311
23. VergesEColominaNGariEGallegoCAldeaM 2007 Cyclin Cln3 is retained at the ER and released by the J chaperone Ydj1 in late G1 to trigger cell cycle entry. Mol Cell 26 649 662
24. CrossFR 1988 DAF1, a mutant gene affecting size control, pheromone arrest, and cell cycle kinetics of Saccharomyces cerevisiae. Mol Cell Biol 8 4675 4684
25. NashRTokiwaGAnandSEricksonKFutcherAB 1988 The WHI1+ gene of Saccharomyces cerevisiae tethers cell division to cell size and is a cyclin homolog. EMBO J 7 4335 4346
26. TyersMTokiwaGNashRFutcherB 1992 The Cln3-Cdc28 kinase complex of S. cerevisiae is regulated by proteolysis and phosphorylation. EMBO J 11 1773 1784
27. YaglomJLinskensMHSadisSRubinDMFutcherB 1995 p34Cdc28-mediated control of Cln3 cyclin degradation. Mol Cell Biol 15 731 741
28. MillerMECrossFR 2000 Distinct subcellular localization patterns contribute to functional specificity of the Cln2 and Cln3 cyclins of Saccharomyces cerevisiae. Mol Cell Biol 20 542 555
29. ChoRJCampbellMJWinzelerEASteinmetzLConwayA 1998 A genome-wide transcriptional analysis of the mitotic cell cycle. Mol Cell 2 65 73
30. HenchozSChiYCatarinBHerskowitzIDeshaiesRJ 1997 Phosphorylation - and ubiquitin-dependent degradation of the cyclin-dependent kinase inhibitor Far1p in budding yeast. Genes Dev 11 3046 3060
31. LyonsNAMorganDO 2011 Cdk1-dependent destruction of Eco1 prevents cohesion establishment after S phase. Mol Cell 42 378 389
32. KishiTYamaoF 1998 An essential function of Grr1 for the degradation of Cln2 is to act as a binding core that links Cln2 to Skp1. J Cell Sci 111 Pt 24 3655 3661
33. CrossFRBlakeCM 1993 The yeast Cln3 protein is an unstable activator of Cdc28. Mol Cell Biol 13 3266 3271
34. HaoBOehlmannSSowaMEHarperJWPavletichNP 2007 Structure of a Fbw7-Skp1-cyclin E complex: multisite-phosphorylated substrate recognition by SCF ubiquitin ligases. Mol Cell 26 131 143
35. KoivomagiMValkEVentaRIofikALepikuM 2011 Cascades of multisite phosphorylation control Sic1 destruction at the onset of S phase. Nature 480 128 131
36. SkaarJRPaganJKPaganoM 2009 SnapShot: F box proteins I. Cell 137 1160 1160 e1161
37. LankerSValdiviesoMHWittenbergC 1996 Rapid degradation of the G1 cyclin Cln2 induced by CDK-dependent phosphorylation. Science 271 1597 1601
38. BersetCGriacPTempelRLa RueJWittenbergC 2002 Transferable domain in the G(1) cyclin Cln2 sufficient to switch degradation of Sic1 from the E3 ubiquitin ligase SCF(Cdc4) to SCF(Grr1). Mol Cell Biol 22 4463 4476
39. BlondelMGalanJMChiYLafourcadeCLongarettiC 2000 Nuclear-specific degradation of Far1 is controlled by the localization of the F-box protein Cdc4. EMBO J 19 6085 6097
40. DixonSJCostanzoMBaryshnikovaAAndrewsBBooneC 2009 Systematic mapping of genetic interaction networks. Annu Rev Genet 43 601 625
41. HoltLJKrutchinskyANMorganDO 2008 Positive feedback sharpens the anaphase switch. Nature 454 353 357
42. StarostinaNGKipreosET 2012 Multiple degradation pathways regulate versatile CIP/KIP CDK inhibitors. Trends Cell Biol 22 33 41
43. XieYRubensteinEMMattTHochstrasserM 2010 SUMO-independent in vivo activity of a SUMO-targeted ubiquitin ligase toward a short-lived transcription factor. Genes Dev 24 893 903
44. DaiMSJinYGallegosJRLuH 2006 Balance of Yin and Yang: ubiquitylation-mediated regulation of p53 and c-Myc. Neoplasia 8 630 644
45. SkotheimJMDi TaliaSSiggiaEDCrossFR 2008 Positive feedback of G1 cyclins ensures coherent cell cycle entry. Nature 454 291 296
46. LinDIBarbashOKumarKGWeberJDHarperJW 2006 Phosphorylation-dependent ubiquitination of cyclin D1 by the SCF(FBX4-alphaB crystallin) complex. Mol Cell 24 355 366
47. KanieTOnoyamaIMatsumotoAYamadaMNakatsumiH 2011 Genetic reevaluation of the role of F-box proteins in cyclin D1 degradation. Mol Cell Biol
48. OkabeHLeeSHPhuchareonJAlbertsonDGMcCormickF 2006 A critical role for FBXW8 and MAPK in cyclin D1 degradation and cancer cell proliferation. PLoS ONE 1 e128 doi:10.1371/journal.pone.0000128
49. SantraMKWajapeyeeNGreenMR 2009 F-box protein FBXO31 mediates cyclin D1 degradation to induce G1 arrest after DNA damage. Nature 459 722 725
50. WeiSYangHCChuangHCYangJKulpSK 2008 A novel mechanism by which thiazolidinediones facilitate the proteasomal degradation of cyclin D1 in cancer cells. J Biol Chem 283 26759 26770
51. YuZKGervaisJLZhangH 1998 Human CUL-1 associates with the SKP1/SKP2 complex and regulates p21(CIP1/WAF1) and cyclin D proteins. Proc Natl Acad Sci U S A 95 11324 11329
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2012 Číslo 7
Nejčtenější v tomto čísle
- Guidelines for Genome-Wide Association Studies
- The Role of Rice HEI10 in the Formation of Meiotic Crossovers
- Identification of Chromatin-Associated Regulators of MSL Complex Targeting in Dosage Compensation
- GWAS Identifies Novel Susceptibility Loci on 6p21.32 and 21q21.3 for Hepatocellular Carcinoma in Chronic Hepatitis B Virus Carriers