
Exome Sequencing in an Admixed Isolated Population Indicates Variants Confer a Risk for Specific Language Impairment
Children affected by Specific Language Impairment (SLI) have unexpected problems learning to talk and understand language, despite developing normally in all other areas. This disorder runs in families but we do not understand how the genetic contributions work, or which genetic mechanisms might be important. In this paper, we study a Chilean population who are affected by a high incidence of SLI. Such populations may provide increased power to discover contributory genetic factors, under appropriate conditions. We identify a genetic change in the population that causes a change to a protein called NFXL1. This change is usually very rare but is found at a higher frequency than expected in our population, particularly in those people affected by SLI. We then looked at this gene in over 100 individuals from the UK affected by SLI and found four more changes that probably affect the protein. This is a higher number than we would expect by chance. We therefore propose that the NFXL1 gene and the protein it encodes might be important in risk of SLI.
Published in the journal:
. PLoS Genet 11(3): e32767. doi:10.1371/journal.pgen.1004925
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1004925
Summary
Children affected by Specific Language Impairment (SLI) have unexpected problems learning to talk and understand language, despite developing normally in all other areas. This disorder runs in families but we do not understand how the genetic contributions work, or which genetic mechanisms might be important. In this paper, we study a Chilean population who are affected by a high incidence of SLI. Such populations may provide increased power to discover contributory genetic factors, under appropriate conditions. We identify a genetic change in the population that causes a change to a protein called NFXL1. This change is usually very rare but is found at a higher frequency than expected in our population, particularly in those people affected by SLI. We then looked at this gene in over 100 individuals from the UK affected by SLI and found four more changes that probably affect the protein. This is a higher number than we would expect by chance. We therefore propose that the NFXL1 gene and the protein it encodes might be important in risk of SLI.
Introduction
Language deficits form a central feature of many developmental disorders and account for a high number of pediatric referrals and statements of special educational need [1]. These language impairments often represent a secondary clinical feature of a more pertinent developmental disability such as Down syndrome, Autistic Spectrum Disorder or intellectual disability. However, in a proportion of cases, the primary clinical concern is the language difficulties, which occur in the absence of any other developmental deficit or neurological impairment and in the presence of normal non-verbal IQ. In such cases, the diagnosis is Specific Language Impairment (SLI) [2].
SLI affects between 5 and 7% of children in the UK [3] and significantly more boys than girls [4]. The disorder is highly heritable [5] but genetic contributions are expected to be complex in nature with significant heterogeneity between individuals [6]. Common risk variants within ATP2C2 (OMIM#613082), CMIP (OMIM#610112) [7], ABCC13 (OMIM#608835) [8], FLNC (OMIM#102565), RBFOX2 (OMIM#612149) [9] and ROBO2 (OMIM#602431) [10] have been associated with quantitative measures of language skills. Genome-wide association studies of language-impaired probands have also highlighted potential risk variants in NDST4 (OMIM#615039), ZNF385D, COL4A2 (OMIM#120090) [11] and NOP9 [12]. Other studies implicate rare genetic events which may have higher penetrance [13,14]. However, it is clear that the contributions of these various genetic effects are complex. Some may be specific to individuals with certain forms of language deficits, others may contribute across the range of ability [7,8,11,15,16]. The functional impact of these candidate genes has yet to be elucidated and further candidates need to be identified before we can properly understand the molecular pathways underlying SLI.
Clearer links have been made between the presence of language deficits and disruption of the FOXP2 gene (OMIM#605317), a forkhead/winged-helix transcription factor [17,18]. Reduced functional dosage of FOXP2, caused by mutation or chromosomal rearrangements, leads to characteristic deficits in coordinating sequences of orofacial movements, impairing speech, producing a disorder known as developmental verbal dyspraxia (DVD) or childhood apraxia of speech (CAS) [18–22]. Typically the DVD/CAS features of FOXP2 mutation cases are accompanied by wide-ranging problems with spoken and written language [23]. Whilst FOXP2 disruptions are rare and account for only a small proportion of DVD/CAS cases, the investigation of this gene, its expression patterns and interactions, have led to the elucidation of genetic networks that are important to language development and contribute to more common forms of language impairment [23–25]. One of the transcriptional targets of FOXP2 is CNTNAP2 (OMIM#604569), a member of the neurexin family which mediates interactions between neurons and glia during nervous system development [26]. Genetic variation across CNTNAP2 has been associated both with language deficits [15,27–29] and language ability in the general population [30–32]. Variations in, and disruptions of, this gene have also been implicated across a range of neurodevelopmental disorders such as autism, epilepsy and schizophrenia [26], indicating that it is likely to be crucial for brain development. These investigations demonstrate how the identification of genetic mutations underlying a distinct severe form of disorder provide entry points into mechanisms that are relevant to the wider processes underlying the initial deficit.
In 2008, Villanueva et al described a population who are affected by an unusually high prevalence of language impairment [33]. This admixed population inhabits the Robinson Crusoe Island which forms part of the Juan Fernandez Archipelago in the South Pacific Ocean, approximately 400 miles off the coast of Chile. The Island was last colonized in 1876 by 64 individuals of European and South American descent. In the 2002 census, the Island population was 633, the majority of whom were descendants of the founder families. More than 70% of the current population has a surname from the colonizing families and 14% of marriages involve consanguineous unions [34]. In their 2008 study, Villanueva et al completed psychometric profiling of 66 island children aged between 3 and 9 years of age, of whom 40 were descendants of the founder party. They found that 35% of the founder-related children (14 of 40) were affected by specific language impairment. No evidence for a male bias was observed in this group. A further 27.5% of the founder-related child population (11 of 40) had language abilities below that expected for their age but presented with additional developmental concerns or low non-verbal IQ, precluding a diagnosis of SLI. The remaining 37.5% of founder-related children (15 of 40) had typical language development. In contrast, only one of 26 children whose parents are not related to the founder families (3.8%) had evidence of language impairment, a frequency of language impairment that coincided with that seen in mainland Chile (3%) [33]. Furthermore, when the genealogical records of the islanders were recompiled, 90% of the individuals affected by SLI were direct descendants of a single pair of founder brothers who formed part of the founder party [33,35]. Given the clear phenotypic differences between founder-related and non-founder-related children on the Island, we postulated that the founder brothers may have carried a rare causative genetic mutation or, alternatively, combinations of common genetic variations that together confer a high risk of language impairment. A previous genome-wide linkage study of 34 families from the Robinson Crusoe Island identified significant linkage to several chromosome regions, the most consistent of which included a large section (48Mb) of chromosome 7q (SLI4 – OMIM#612514) that included many genes which represent good candidates for language impairment, including FOXP2 and CNTNAP2 [35]. However, in depth genomic profiling has yet to be performed within this population.
In this study, we make use of this admixed isolated population and assess the possibility of a founder mutation, by completing exome sequencing of five individuals from the Robinson Crusoe population affected by SLI. We substantiate the findings of the exome screen by performing association analyses of selected putative functional variants in the wider Robinson Crusoe population. The contribution of identified risk variants is subsequently validated by performing targeted sequencing of candidate genes in a UK-based cohort of individuals affected by SLI.
Results
We selected five related individuals with SLI from the Robinson Crusoe cohort for exome sequencing (Fig. 1). From the exome sequence data, we selected all novel variants (i.e. not reported in publically available or in-house databases) that caused nonsynonymous changes or changes to canonical splice sites and were shared by at least three of the five individuals sequenced. A flow diagram of our methodology can be found in S1 Fig.. All such variants were subsequently genotyped in 111 founder-related cases and controls from the Robinson Crusoe Island (Robinson Crusoe validation cohort) and tested for association to language impairment using a method that takes into account familial relationships. To substantiate the findings of the exome screen and association analyses, we then went on to sequence the coding regions of candidate genes implicated from these investigations in an independent cohort of 117 British children affected by SLI (SLIC cohort).

Exome sequencing
On average, 47,276 (median = 49,543, range = 43,075–50,112) genic variants were identified in each of the five exomes. This included an average of 17,405 (median = 17,326, range = 15,200–19,837) exonic variants, 8,379 (median = 8,089, range = 7,258–9,629) missense variants and 106 (median = 90, range = 72–157) nonsense (including indels) variants per individual. Across all five samples, 90.0% of targeted exome sequencing had coverage of at least 10-fold. The average coverage of targeted sequence was 56.5-fold and 21% of the reads reached this level. Sequencing metrics can be found in S1 Table. To test the hypothesis that the founder brothers carried a rare causative genetic mutation, we focused upon novel variants that caused nonsynonymous protein substitutions or altered canonical splice sites for our downstream analyses. Comparisons between individuals found that no such variants were shared by all five individuals. However, allowing for potential genetic heterogeneity between affected individuals, we identified nine novel nonsynonymous or splice-site variants that were shared by at least 3 of the 5 children sequenced (Table 1). Eight novel nonsynonymous or splice-site variants were validated in the five exome samples by Sanger sequencing. None of these variants overlapped with the regions of suggestive linkage (P<7.3×10−4, chromosomes 2, 6, 7, 8, 9, 12, 13 and 17, as listed in S2 Table) previously identified in this population [35]. S3 Table provides a full list of all shared, high-quality variants that fell within the previously identified regions of linkage. All of these had previously been reported in dbSNP (138) and many were non-genic, intronic or synonymous (see notes column in S3 Table).
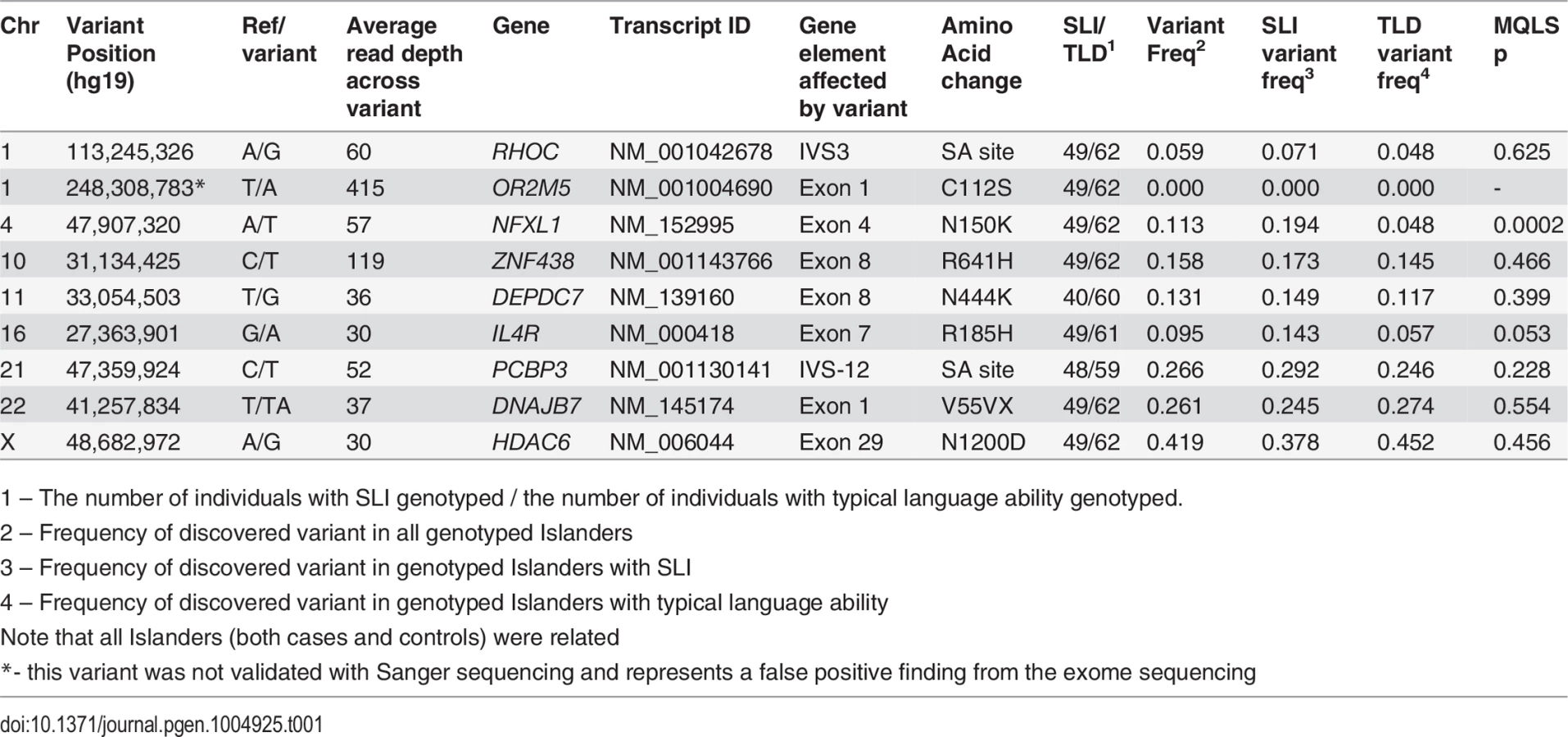
Association analyses of key variants in Robinson Crusoe validation cohort
All shared novel nonsynonymous or splice-site variants identified in the exome screen were subsequently genotyped in 111 members of the Robinson Crusoe population (49 individuals with language-impairment and 62 individuals with typical language ability). This validation cohort was ascertained via 35 children living on the Robinson Crusoe Island who had been diagnosed with SLI or who showed typical language development (as described in methods) and included the five children used in the exome sequencing. All children were descendants of the founder families of the Robinson Crusoe Island and, as such, the cases and controls used in these association analyses were inter-related (Fig. 1). We therefore employed an association algorithm that allowed for relatedness between cases (MQLS, [36]), and that took into account the shared ancestry of the Robinson Crusoe validation cohort (288 individuals over 5 generations). These analyses highlighted one particular coding variant (chr4:g.47,907,320A>T, hg19) that was present at a significantly higher frequency in Islanders with language impairment than in Islanders with typical language ability (Table 1). Thirty nine percent of Islanders with language impairment were found to carry this variant compared to ten percent of Islanders with typical language skills (p = 2.04 × 10−4) (Table 1). Across the Robinson Crusoe validation cohort, the minor allele frequency was 11.3% (25 of 222 chromosomes sampled) (Table 1).
Predicted functional effects of chr4:g.47,907,320A>T
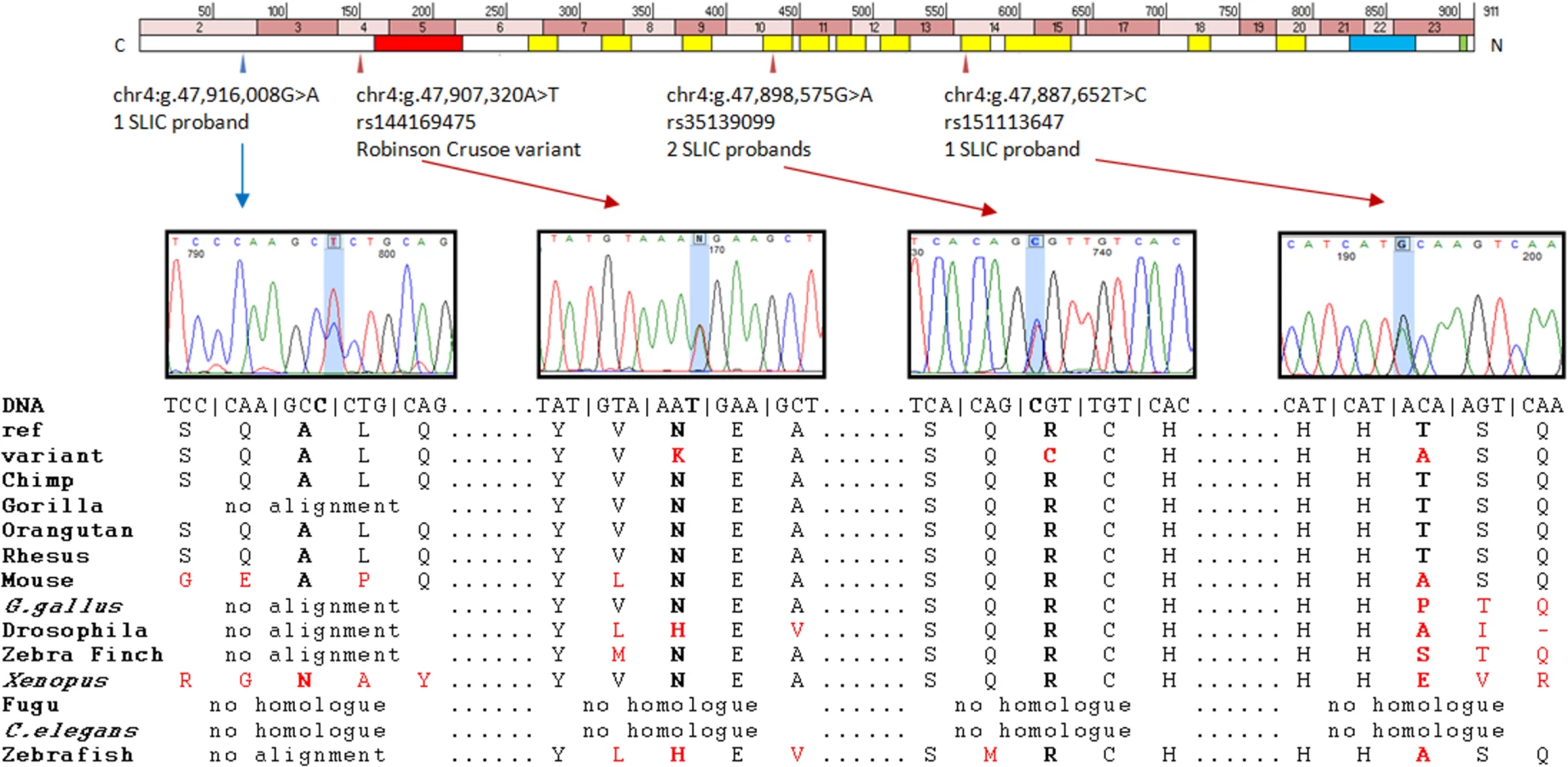
Chr4:g.47,907,320A>T (hg19) falls in exon 4 of the Homo sapiens nuclear transcription factor, X-box binding-like 1 (NFXL1) gene (Fig. 2). The variant causes a nonsynonymous change yielding an asparagine to lysine substitution in the encoded protein (p.N150K, uncharged amino acid to positively charged amino acid). This change is predicted to be “disease-causing” by MutationTaster with a confidence probability of 0.98 (SIFT = 0.67, PolyPhen-2 = 0.178). The position is conserved at both the amino acid and nucleotide level (PhyloP = 0.66, PhastCons = 1); the amino acid N150 is invariant across 36 of the 38 vertebrate species in which an alignment could be made and the thymine nucleotide at this position is conserved across all six ENSEMBL primate species investigated (Human, chimp, gorilla, orangutan, macque and marmoset) (Fig. 2).
Chr4:g.47,907,320A>T, hg19 in control populations
The variant at chr4 : 47,907,320 was not observed in 127 independent European population controls that were genotyped (Table 2). We therefore went on to genotype an additional 320 independent individuals from a Colombian population cohort and 121 independent individuals from a Chilean control population cohort. In these cohorts, the variant was present with a minor allele frequency of 4.2% (27 of 640 chromosome sampled) and 7.4% (18 of 242 chromosome sampled) respectively (Table 2). Subsequent data released by the 1000 genomes project confirmed that this variant is specific to admixed American populations (AMR) with an average minor allele frequency of 4.1%. In the sub-populations of the AMR grouping, the minor allele frequency is reported as 0.9% in Puerto Ricans (PUR – 1 in 110 chromosomes sampled), 3.3% in Colombians in Medellin (CLM – 4 in 120 chromosomes sampled) and 7.6% in individuals of Mexican ancestry in Los Angeles (MXL – 10 of 132 chromosomes sampled) (Table 2). The variant has recently been designated as rs144169475 accordingly.

Linkage analyses of chromosome 4 (46–49Mb)
Parametric and nonparametric linkage analyses were performed for 55 SNPs across the NFXL1 region of chromosome 4 (46–49Mb, hg19) within seven extended pedigrees from the Robinson Crusoe validation cohort (S2 Fig.). In these analyses, we did not observe evidence of linkage (maximum LOD score = 0.62, S3 Fig.).
Sequencing of NFXL1 in a language-impaired cohort (SLIC)
We sequenced the entire coding region of the NFXL1 gene in 117 unrelated probands affected by SLI (from the UK SLI Consortium (SLIC) cohort [7,37–39]), to assess whether we could replicate a role for NFXL1 in SLI etiology. In total, we identified 166 high-quality sequence variants across the NFXL1 gene. 155 of the variants detected were intronic, 4 were in the 3’UTR and 7 affected the coding region. Of the coding variants, three were nonsynonymous and four were synonymous substitutions (Table 3).

Nonsynonymous variants and those with estimated allele frequencies of <5% were verified across all the pools of DNA in which they were observed using Sanger sequencing. This allowed the derivation of accurate allele frequencies within the SLIC cohort.
One of the synonymous variants (chr4:g.47,916,008G>A, hg19) was found in a heterozygous state in one SLIC proband (allele frequency of 0.43%) but had not been documented in any European individuals in the 1000 genomes project [40] or the NHLBI GO ESP Exome Variant Server (EVS), which together consist of data from 4679 control individuals and therefore have the ability to detect rare variants with a population frequency of 0.0001. A comparison of allele frequencies between SLIC probands (1 of 234 chromosomes tested) and controls (0 of 9358 chromosomes tested) yielded a significant P-value of 0.0244. Intriguingly, although it is synonymous, this variant was predicted to be “disease-causing” by MutationTaster with a confidence probability of 0.98 (SIFT = 1.0). This variant falls in the most 5’ coding exon of NFXL1 and is part of a CpG island, indicating that it may be important for the regulation of gene expression. Furthermore, ENCODE data shows that it is part of a H3K4Me3 mark (which is often associated with promoters) and binds multiple transcription factors, particularly POLR2A c-MYC and PHF8 (www.genome.ucsc.edu, accessed April 2014).
The remaining three synonymous variants (rs2053404, rs6818556 and rs35139099) found in SLIC probands were also found at similar frequencies in control databases. All had allele frequencies of >5% and are therefore thought to represent common polymorphisms (Table 3).
One nonsynonymous substitution (chr4:g.47,887,652T>C, hg19 – rs151113647) was found in a heterozygous state in a single SLIC proband (allele frequency of 0.43%) and again, was not observed in 4679 independent European individuals in the control public databases (Table 3), yielding a significant P-value of 0.024 (1 of 234 SLIC chromosomes tested vs 0 of 9358 control chromosomes tested). Further investigations found that this variant had been observed in a heterozygous state in a single African American individual from the EVS. Principal components analysis of genome-wide SNP data in the SLIC proband against the hapmap-3 populations did not detect any African ancestry. The rarity of the rs151113647 variant and its position within a zinc-finger motif (Fig. 2) indicates that it may confer negative effects upon protein function. Nonetheless, because the nucleotide is not highly conserved across species (phyloP = −0.418, phastCons = 0.925), the change was predicted to be a polymorphism by MutationTaster with a confidence probability of 0.99 (SIFT = 0.68, polyphen-2 = 0.00) (Fig. 2).
A second nonsynonymous substitution (chr4 : 47,898,575G>A, hg19 - rs35139099) was observed in a heterozygous state in two independent SLIC probands (allele frequency of 0.85%). This variant was also found in 44 of 4679 independent European control individuals from public databases (allele frequency of 0.47%, Table 3) yielding a P value of 0.3097. Although, it was not observed to occur at a significantly increased frequency in SLIC probands, the rs35139099 variant occurs at a conserved residue (phyloP = 1.466, phastCons = 1) within a zinc-finger motif (Fig. 2) and is therefore predicted to be damaging by MutationTaster with a confidence probability of 0.99 (SIFT = 0.00, Polyphen-2 = 1.00) (Fig. 2).
The remaining nonsynonymous variant (chr4:g.47,901,476G>A, hg19 - rs12651301) was observed to occur across all the sequence pools with an estimated allele frequency of 32% (Table 3). This common variant was also observed in independent European controls from public databases with a frequency of 31% (Table 3) and falls outside of any protein motifs and is thus likely to represent a polymorphism.
The three rare variants identified (rs151113647, rs35139099 and chr4:g.47,916,008G>A, hg19) were sequenced in all available family members of the SLIC proband in whom they were observed (Fig. 3). The chr4:g.47,916,008 variant was inherited from an affected father by two affected children and one child with typical language development (Fig. 3). The rs151113647 variant was inherited from a father, who reports a history of language and literacy problems, by the proband, who attends a special language unit, and his sibling, who also has SLI. The middle child in this family, who also showed evidence of expressive and receptive language deficits, did not inherit the variant (Fig. 3). Two SLIC families carried the rs35139099 variant; in the first, the variant is present in the father, who self-reports a history of dyspraxia, and passed onto both the proband and her elder sib, each of whom has expressive and receptive language problems. The youngest daughter in this family, who was observed to have a similar pattern of language deficits, did not inherit the variant (Fig. 3). In the second family carrying the rs35139099 variant, the change was present in both the proband and his younger sib, who had expressive and receptive language scores ∼1SD below that expected for his age, indicating that it is inherited (Fig. 3). The variant was not present in the mother and we did not have a DNA sample, or phenotypic data, from the father. Nonetheless, haplotype analyses of genome-wide SNP data indicated that the two children shared the same paternal chromosome in this region indicating that the rs35139099 variant was likely inherited from the father.

Discussion
In this paper, we report results from the whole exome sequencing of five individuals from an isolated Chilean island population affected by a high incidence of SLI. We identify a heterozygous nonsynonymous coding variant in the NFXL1 gene that is shared by three of the five individuals sequenced. Association analyses within a larger Robinson Crusoe validation cohort, demonstrated that this variant occurred at a significantly increased frequency in Islanders with language impairment than those with typical language development (P = 0.0002) and is predicted to be “disease-causing”. Subsequent sequencing of NFXL1 in a cohort consisting of 117 independent UK probands (SLIC) with SLI identified four individuals with putative high-risk variants in the heterozygous state; three SLIC individuals carried rare nonsynonymous changes and one SLIC individual carried a novel variant that falls within a regulatory motif. Given the above evidence, we postulate that variants within NFXL1 may contribute to genetic risk of language impairment. We propose that such changes are likely to function as—risk variants with a complex model of inheritance.
We used the Robinson Crusoe ancestry to trace back the relationships between individuals carrying the associated rs144169475 variant. The only common ancestors to the carriers were two founder brothers who had previously been reported to head the SLI lineage on the Island (Fig. 1). These brothers were related to all carriers of the rs144169475 variant (Fig. 1). However no single brother was related to all Islanders carrying the variant allele (Fig. 1). We therefore concluded that both founder brothers are likely to have carried the variant. These data therefore support the founder model of language impairment proposed at the outset of this study. We performed allele dropping simulations within the descendants of these founder brothers and found that a variant with an allele frequency of 3–9% in the founder population would be expected to have a frequency of 8–14% in the current population (S1 Text, S4 Table). This prediction fits well with the observed frequency of 12.5% in the founder-related Islanders and is elevated above that observed in Chilean population controls (7.4%), indicating the presence of a founder effect at this locus. Moreover, we found that the increased frequency of the rs144169475 variant is driven by Islanders with SLI (19.4% in 49 individuals with SLI vs 4.8% in 62 individuals with typical language ability) (Table 2).
Our data further suggest that the effects of rare mutations in NFXL1 may extend to the etiology of SLI in other populations. In a screen of the NFXL1 coding regions in 117 independent UK probands affected by SLI, we observed four individuals who carried rare coding changes generating a combined high risk allele frequency of 1.71%. By contrast, the combined allele frequency of these three variants in 4679 independent European controls (from the 1000 genomes and EVS public databases) is 0.47%, a difference that yields a marginally significant P-value of 0.029 (4 of 234 SLIC chromosomes vs 44 of 9358 control chromosomes). Extending our investigations to include all private coding mutations (i.e. only found in one individual) across the entire NFXL1 transcript, as opposed to the consideration of the three specific mutations considered above, we again observed a marginally increased frequency in the SLIC cohort (2 of 234 chromosomes tested, 0.85%) above that expected given the data reported in public European databases (EVS European American and 1000 Genomes EUR super-population – 28 of 9358 chromosome sequenced, 0.3%, P = 0.0359). Broadening our investigation to include all rare coding changes (<1%) across the entire NFXL1 transcript, revealed a similar trend (1.71% (4 of 234 chromosomes sequenced) in the SLIC cohort, compared to 1.36% (127 of 9358 chromosome sequenced) in public European databases) but this did not reach significance (P = 0.3821).
Given our consistent findings across cohorts, and in line with the data arising from other neurodevelopmental disorders, we suggest that rare variants in NFXL1 may represent genetic risk factors with incomplete penetrance. Given our data, it is likely that these putative risk factors are modulated by other genetic variations and/or environmental factors [41–43]. We could not identify a distinct or specific phenotypic feature that distinguished rs144169475 language-impaired carriers from language-impaired non-carriers. Nor did we observe complete co-segregation between NFXL1 variants and the presence of SLI in either the Robinson Crusoe validation population or the UK SLIC cohort. Thirty nine percent of the Robinson Crusoe validation cohort affected by language impairment carried the rs144169475 variant, as did ten percent of the Robinson Crusoe validation cohort with typical language ability. Similarly, one of the variants observed in the SLIC probands was inherited by a child with typical language development and two children affected by language impairment did not inherit the observed variant. In addition, we observed a high phenocopy rate in the Robinson Crusoe cohort; only 39% of individuals affected by language impairment carried the rs144169475 variant. Incomplete segregation is commonly described in neurodevelopmental disorders such as autism [42,43] and intellectual disability [44,45] and represents a major challenge in the interpretation of high-throughput sequencing data [46].
The NFXL1 gene encodes a NFX-1-type nuclear zinc-finger transcriptional repressor that is expressed at the cytoplasm [47]. Little is known regarding the function of the NFXL1 protein; no disorders have been identified that arise from the mutation of this gene and no animal knock-outs have been described. The protein has zinc-finger domains which mediate DNA binding and carries a RING domain that has E3 ubiquitin ligase function (Fig. 2) [48]. This transcription factor has been shown to be highly expressed in embryonic stem cells prior to differentiation into myelinated oligodendrocytes [49] and shows a high level of expression in the early mouse embryonic development (E11.5) and in human cerebellar structures (www.brainmap.org). NFXL1 is so-called because it is a paralogue of the NF-X1 transcription factor which binds the X-box sequence of class II MHC genes [50]. This feature may be relevant in light of a recent study that found association between HLA loci and SLI [51]. Similarly, an NF-X1 isoform functions in the regulation of the NFĸB pathway [52], as does CMIP, a gene implicated in the etiology of SLI in UK populations [7,53].
Limitations of our study
A natural limitation of all studies of founder or isolated populations is the restricted size of the cohort. Although our study represents a comprehensive profiling of the Robinson Crusoe child population, the total sample consisted of only 111 individuals, 100 of whom were founder-related and 49 of whom had language impairment. Although it should be noted that the power of this particular sample lies in the close relationships between individuals rather than the absolute number of samples, the issue of sample sizes is especially pertinent when one is considering rare variations. Thus it is of particular importance that we observed independent evidence implicating NFXL1 rare variants in another cohort. However, in the absence of a large South American cohort of language-impaired individuals, we were unable to include the rs144169475 variant in our replication investigations (since this SNP is particular to South American populations). Thus, further studies of larger sample sizes that include language-selected controls and South American individuals will be required to fully evaluate the role of rs144169475 and rare NFXL1 coding variants in SLI susceptibility.
Of note, none of the shared variants identified through exome sequencing co-incided with regions of suggestive linkage reported in a previous genomewide linkage study of the Robinson Crusoe population (S2 and S3 Tables) [54]. Nor did we find evidence for linkage to the NFXL1 region of chromosome 4 (S4 Fig.). We must therefore acknowledge that the increased frequency of rs144169475 in language-impaired individuals of the Robinson Crusoe validation cohort does not directly indicate pathogenicity. The result may represent a chance finding or, alternatively, rs144169475 may be a proxy for the causal variant. Since the exome sequencing performed did not capture 100% of the exome, it is possible that the causal variant was not detected here. Full genome sequencing would be required to fully investigate this possibility. However, it is also important to note that a lack of linkage does not preclude the presence of a causal variant and may instead reflect the complexities of analyzing a pedigree of this size and complexity [55]. The pedigree, which explained the known relationships between the founder brothers and the Robinson Crusoe validation cohort, included 288 individuals (321 bits, where a bit is defined as twice the number of non-founders—the number of founders) and so had to be broken into smaller sets for linkage analyses. This segmentation process discards information and can reduce the power to detect linkage if individuals sharing the linked chromosome segment are split between sub-pedigrees [56]. Lastly, since we hypothesize that SLI in this population has a complex genetic basis and involves incomplete and a high phenocopy frequency, it is possible that the power to detect linkage is insufficient. We observed reduced penetrance at the NFXL1 locus (of 25 variant carriers, 19 were diagnosed with SLI, penetrance of 76%) in combination with evidence of a high phenocopy rate in our cohort (of 49 individuals with language impairment, 19 carried the variant, phenocopy rate of 61%). In combination, these factors break down the correspondence between genotype and phenotype, compromising the ability to detect linkage [57].
In summary, the Robinson Crusoe admixed founder population represents a rare resource which may assist in the identification of genetic variants that contribute to SLI susceptibility. Exome sequencing of five individuals from this population identified eight shared coding variants. One of these variants (rs144169475) was found to be significantly associated (P = 0.0002) with language impairment in the wider Robinson Crusoe population. rs144169475 confers a nonsynonymous change (N150K) in the NFXL1 gene at a highly conserved residue. Subsequent sequencing of the NFXL1 coding regions in 117 independent UK SLI cases identified four individuals with rare heterozygous variants predicted to be of functional consequence. We conclude that coding variants within NFXL1 confer an increased risk of SLI within a complex genetic model.
Materials and Methods
Ethics
The work on the Robinson Crusoe Island was approved by the ethics department of the University of Chile. Ethical permission for each SLIC collection was granted by local ethics committees. Guys Hospital Research Ethics Committee approved the collection of families from the Newcomen Centre to identify families from the South East of England with specific language disorder. Ref No. 96/7/11. Cambridge Local Research Ethics Committee approved the CLASP project “Genome Search for susceptibility loci to language disorders” Ref No. LREC96/212. Ethical approval for the Manchester Language Study was given by the University of Manchester Committee on the Ethics of Research on Human Beings. Ref No. 03061 The Lothian Research Ethics Committee approved the project “Genetics of specific language impairment in children in Scotland” for the use of the Edinburgh samples. Ref. No. LREC/1999/6/20. The ethics department of the University of Chile approved the project “Genetic analysis of language-impaired individuals from the Robinson Crusoe Island”. Project Number 001-2010. Informed consent was given by all participants and/or, where applicable, their parents.
Ascertainment of the Robinson Crusoe population
The Robinson Crusoe cohort was ascertained on the basis of phenotypic data from 61 children, between the ages of 3 years and 8 years, 11 months (i.e. the child cohort, described below) all of whom were descendants of the founder families and represents an extended cohort (including children who have turned 3 years of age since 2008) of that described in [33]. First-degree relatives of founder-related children found to meet criteria for SLI or typical language development were then also assessed for language performance (i.e. the family cohort, described below). Age constraints of available standardized tests meant that different language batteries were employed within the child and family cohorts.
Phenotyping and selection of the Robinson Crusoe child cohort
The language ability of 61 children, all of whom were related to a founder individual, was assessed by tests of expressive and receptive language (Toronto Spanish Grammar Exploratory test, TEGE [58]) and phonology (Phonological simplification test (Test para Evaluar Procesos de Simplificación Fonológica—TEPROSIF [59]). Nonverbal IQ was tested using the Colombia Mental Maturity Scale [60]. In addition, all children were subjected to an auditory screen and oral motor exam [61]. All tests were validated and normalized in Chilean populations. On the basis of these tests, all children were classified into one of the three following categories:
“Specific Language Impairment)” (N = 16, 7 male, 9 female, 26.2%) defined as (i) performance >2SD below expected on TEPROSIF (for children aged 6 years or less) or performance >2 years below expected for chronological age on TEPROSIF (for children aged over 6 years) and/or performance below the 10th percentile on either the receptive or expressive scales of the TEGE, (ii) nonverbal IQ not below the 10th percentile, (iii) normal hearing, oral motor skills and neurological development.
“Typical language development” (N = 23, 8 male, 15 female, 37.7%) defined as (i) performance not >2SD below expected on TEPROSIF or performance >2 years below expected for chronological age on TEPROSIF (for children aged over 6 years) and performance above the 10th percentile on both the receptive and expressive scales of the TEGE.
“Nonspecific language impairment” (N = 22, 13 male, 9 female, 36.1%) defined as (i) performance >2SD below expected on TEPROSIF or performance >2 years below expected for chronological age on TEPROSIF (for children aged over 6 years) and/or performance below the 10th percentile on either the receptive or expressive scales of the TEGE, and (ii) nonverbal IQ >1SD below age-expected, and/or (iii) evidence of hearing loss or oral motor disability (e.g cleft lip) or abnormal neurological development.
The observed language deficits in the individuals diagnosed with SLI were typical of those described in other SLI cohorts and involved varied deficits across grammatical, morphosyntactical and receptive aspects of language, but not dialectic variations in intonation, vocabulary or phonology.
Phenotyping and selection of the Robinson Crusoe family cohort
Since we were particularly interested in genetic contributions to SLI, our family cohort consisted of the first-degree relatives of the 39 founder-related children presenting with SLI or typical language development. All available first-degree family members (92 parents and siblings, 47 male, 45 female) were assessed for language difficulties using tests of verbal fluency (Barcelona test [62]) and verbal comprehension (Token test [63]). These family members included 11 parents who were not related to a founder member of the Island (referred to as non-founder-related parents). In addition to these formal language assessments, all individuals (or their parents or spouses) completed a family history interview (provided by P Tallal) [64], which specifically asks questions regarding language difficulties. On the basis of these data individuals were classified as either:
“Language-impaired” (N = 34, 15 male, 19 female, 37.0%, including 4 non-founder-related parents) if they scored below the 10th percentile on either the Barcelona test or the token test or they self-reported a need for writing or reading support at school or a history of language support in the family history questionnaire.
“Typical language ability” (N = 58, 32 male, 26 female, 63.0%, including 7 non-founder-related parents) if they scored above the 10th percentile on both the Barcelona test and the token test and they indicated no requirement for writing, reading or language support in the family history questionnaire.
Exome sequencing of selected Robinson Crusoe children
Five Islanders (3 male, 2 female) from the child cohort who had been diagnosed with SLI were selected for exome sequencing. The selection of individuals for sequencing was based upon the amount and quality of DNA available, the severity of observed language impairment and their known relationships with other affected individuals. The five children were selected to cover the different branches of the founder pedigree and were descendants of the founder families (Fig. 1).
Exome capture was performed using 10μg of genomic DNA with a first generation (v1) Agilent SureSelect human exome kit (Agilent, Santa Clara, CA, USA), which provide an average target coverage of 80% of the exome at 56-fold across all samples. Sequencing of the generated fragments was performed on the SOLiD 4 sequencer (Life Technologies, Carlsbad, CA, USA). Color space reads were mapped to the human reference genome (hg18) in the SOLiD bioscope software (v1.2), which applies an iterative mapping approach. Variants were called using a diBayes algorithm [65] using high stringency settings, requiring calls on each strand. Small insertions and deletions were detected using the SOLiD Small Indel Tool. We assumed a binomial distribution with a probability of 0.5 of sequencing the variant allele at a heterozygous position. Given such a distribution, a minimum of ten reads would be required to provide a 99% probability that two or more reads contain an allele variant call. We filtered variant calls to have at least four unique (i.e. different start sites) variant reads with the variant being present in at least 15% of all reads.
To test the hypothesis that the founder brothers carried a rare causative genetic mutation, for our downstream analyses, we focused upon novel variants that were potentially deleterious. Each exome file was individually filtered to exclude nongenic, intronic (other than canonical splice sites) and synonymous variants. The remaining nonsynonymous and splice-site mutations were further filtered to exclude known sites of variation (as described in dbSNP, (build 130), publically available genome sequences and an in-house sequencing database). The remaining variants were then compared across exome samples to allow the selection of variants that occurred in 3 or more of the 5 children sequenced. A flow diagram of the methodology can be found in S1 Fig.. Shared novel, potentially deleterious variants discovered in the exome data were verified by Sanger sequencing. Primers for Sanger sequencing were designed in primer3 [66]. Primer sequences are available on request.
Association analyses of selected variants in the Robinson Crusoe population
All novel nonsynonymous or canonical splice-site variants found to occur in 3 or more of the 5 exome samples were also genotyped in the wider child and family cohorts from the Robinson Crusoe population. We were able to obtain DNA samples for 35 founder-related children (from the SLI and typical language development child groups described above) and their family members (from the family cohort described above). Forty nine of these individuals (16 children, 22 parents (4 of whom were non-founder-related), 7 siblings and 4 half-siblings) were language impaired and 62 (19 children, 32 parents (7 of whom were non-founder-related), 9 siblings and 2 half-siblings) had language ability in the normal range. These families included the five children used in the exome sequencing. DNA was extracted from EDTA whole blood samples using a standard chloroform extraction protocol. All novel nonsynonymous or canonical splice-site variants identified from the exome screen were sequenced using a standard Sanger protocol in these 111 individuals.
The resultant genotype data were used to perform a family-based test of association within the MQLS-XM package [36,67]. This algorithm calculates a quasi-likelihood score which corrects the Chi-square statistic for relationships between individuals, providing accurate type I error rates [68]. The MQLS-XM extension allows for the accurate application of this statistic to X-linked markers [67]. The MQLS algorithm distinguishes between unaffected controls and controls of unknown phenotype, can incorporate phenotypic data from individuals who have not been genotyped [36] and is robust to the mis-specification of prevalence. It allows for the presence of both linkage and association effects in the test statistic and is computationally straightforward making it particularly suitable for large complex pedigrees in which cases and controls may be inter-related, as is the case in this study [36].
A full pedigree structure was generated that accounted for all known relationships between 111 individuals from the child and family cohorts and the two identified, shared, founder brothers. This pedigree included 288 individuals (141 males, 144 females and 85 founders (i.e. individuals with no parental information available—both original founders and incoming), 203 non-founders) over 5 generations. As described above, 111 individuals (including 11 non-founder-related parents) had full genotype and phenotype data, 11 individuals were also included who had phenotype data but no genotype data and the remaining 166 individuals had no phenotype or genotype data but defined relationships between the 111 genotyped individuals and the founder brothers. In the MQLS-XM analyses, the expected prevalence of SLI was set at 0.25 for males and 0.27 for females. These figures were derived from the child cohort described above.
Any variant that was significantly associated with language impairment in the population cohort was genotyped in 127 independent European population controls (ECACC, HRC-1 DNA Panel),441 independent South American controls; 320 individuals of Colombian descent and 121 individuals of Chilean origin. The Colombian controls were collected as part of a genetic demography study in the Colombian population, where all participants had to have four grandparents of local origin (provided by Luis Carvajal-Carmona and Maria Magdalena Echeverry). The Chilean controls were ascertained from the Santiago area and consisted of DNA from 30 male Chilean students (provided by P Villanueva) and from 91 female adult controls from a breast cancer study (provided by L Jara, University of Chile). Genome-wide SNP data indicated that these samples were of Amerindian and European ancestry. Note that both the European and South American control populations were unselected and, as such, were not screened for language ability.
Linkage analysis of chromosome 4
Genome-wide linkage data for the Robinson Crusoe validation cohort have previously been reported [35]. These previous analyses included 6,090 SNPs and reported suggestive linkage (P<7.3×10−4) between SLI and chromosomes 2, 6, 7, 8, 9, 12, 13 and 17. In the current study, we had access to a new set of denser genotypes from the Robinson Crusoe population, generated with the Affymetrix Axiom GW-LAT 1 array (Affymetrix Inc, Santa Clara, CA, www.affymetrix.com), supplemented with a custom array designed to cover South American-specific variants which together included 1,141,741 SNPs.
929 SNPs across chromosome region chr4 : 46,000,000–49,000,000 (hg19) were selected to cover the chromosome region surrounding the NFXL1 gene (reported transcript—chr4 : 47,849,258–47,916,680, hg19). SNP data were filtered within PLINK [69] to remove markers in close linkage disequilibrium (r2>0.5) resulting in a linkage dataset of 54 independent SNPs that were appended with rs144169475 genotype data and analysed for linkage in MERLIN [70]. Linkage disequilibrium between these SNPs and rs144169475 are provided in S3 Fig.
Since linkage packages were unable to analyse genome-wide data for the 321-bit Robinson Crusoe validation pedigree as a whole, it was broken into sub-pedigrees manually selected on the basis of closest shared ancestor. We employed linkage sub-pedigrees and linkage methods analogous to those described in the previous linage study [35]; Seven extended families of 20–24 bits (where a bit is defined as twice the number of non-founders—the number of founders) were analysed for linkage under parametric and nonparametric models with MERLIN (S2 Fig.) Parametric linkage analyses were performed under a model which reflected the observed nature of rs144169475 (assuming a disease frequency of 26.2% (as observed in the Robinson Crusoe children) and penetrance of 0.76 (as observed in the Robinson Crusoe validation cohort). Nonparametric linkage results are reported as P-values derived from the Kong and Cox exponential model, which can be more powerful in large pedigrees [71]. Expected allele frequencies were derived from the 1000 Genomes AMR super-population (integrated phase 1, accessed March 2014) which includes 181 independent South American individuals (60 Colombians from Medellin, Colombia (CLM), 66 individuals with Mexican ancestry in Los Angeles (MXL) and 55 Puerto Ricans from Puerto Rico (PUR)) [40].
Functional effects of identified variants
Putative functional effects of associated variants were evaluated using MutationTaster [72]. MutationTaster uses a Bayes classifier which integrates information from various biomedical databases and analysis tools to evaluate the possible pathogenicity of coding variants. MutationTaster considers evolutionary conservation at both the nucleotide and amino acid level, splice-site changes, loss of protein motifs or features and changes that might affect the level of mRNA expression and stability within a single tool to classify variants as a “disease mutation” or a “polymorphism”. A p-value is given to indicate “the security” of the prediction [72]. The MutationTaster algorithm was trained using more than 390,000 known disease mutations from HGMD and more than 6,800,000 SNPs and Indel polymorphisms from the 1000 Genomes Project.
For each of the variants highlighted, we also present the SIFT and polyphen-2 scores. In contrast to MutationTaster, the SIFT and PolyPhen algorithms primarily consider protein sequences, motifs and structures to assign pathogenicity and therefore can only be applied to coding changes. SIFT performs a multiple alignment of closely related protein sequences to identify conserved motifs and assign a probability that a given amino acid substitution is pathogenic [73]. PolyPhen-2 uses a Bayes classifier to consider the property of the reference and variant amino acids, the amino acid conservation, protein motifs and 3D protein structure to derive a probability that a mutation is damaging [74]. SIFT scores vary between 0 and 1. Amino acid substitutions are classified as “deleterious” for scores ≤0.05 and “tolerated” for scores >0.05. In Polyphen-2, two training models are available—HumDiv, which is more appropriate for the identification of fully penetrant Mendelian mutations and HumVar, which is more appropriate for the classification of rare alleles at loci potentially involved in complex phenotypes. PolyPhen scores from both of these models vary from 0 to 1, where 0 represents a variant with no functional effect. Functional effects are classified as “benign”, “possibly damaging”, or “probably damaging”, depending on whether the posterior probability falls above or below the appropriate false positive thresholds.
Sequencing of candidate genes in SLIC cohort
In order to further investigate the role of NFXL1 variants in SLI, the coding regions of the NFXL1 gene were subsequently sequenced in 117 unrelated British children affected by SLI. These children formed part of the SLI Consortium (SLIC) collection, which has previously been described in detail [7,37,39]. In short, the probands were collected from four sites across the UK (The Newcomen Centre at Guy’s Hospital, London, the Cambridge Language and Speech Project (CLASP) [75], the Child Life and Health Department at the University of Edinburgh [76] and the Manchester Language Study [77]). All probands were selected to have receptive and/or expressive language skills (as assessed by the Clinical Evaluation of Language Fundamentals (CELF-IV-R) [78]) more than 1.5SD below the normative mean for his or her age and non-verbal IQ (as measured by the Wechsler Intelligence Scale for Children [79]) in the “normal” range (>80).
The concentration of genomic DNA samples from 117 independent SLIC probands was quantified by picogreen and each sample normalized to 10ng/μl. Individual DNAs were pooled prior to PCR amplification. Following PCR, the amplicons were fragmented, end-repaired and adapter-ligated. The prepared and tagged libraries were then multiplexed before paired-end sequencing in a single lane of flow-cell on an Illumina HiSeq 2000 (Illumina Inc, SanDiego, CA, www.illumina.com). Sequences were aligned against human reference sequence (37d5) using STAMPY [80] and variants called by the Syzygy (1.2.6) algorithm to create a VCF file. Syzygy implements a Bayes likelihood calculation to allow a base calling strategy that is particularly suited to the calling of variants in pooled samples, in which the frequency of reads containing a rare variant will be lower than expected [81]. Identified sequence variants were annotated within the SNPeff package allowing the identification of coding variants [82]. Individual DNAs from all pools that contained a nonsynonymous coding variant with an expected frequency of <5% were resequenced by Sanger sequencing using primers designed with the primer3 software [66]. This allowed the verification of the variants, the derivation of true variant frequencies across pools and the identification of the individuals who carried the variant.
The allele frequencies of coding variants discovered in SLIC probands were compared to those observed in 4679 individuals of European ancestry across publically available control databases; the 1000 genomes project (the European (EUR) super-population from integrated phase 1, accessed March 2014) [40] which includes 379 independent European individuals (89 British in England and Scotland, 93 Finnish in Finland, 14 Iberian populations in Spain, 98 Toscani in Italy and 85 Utah residents with Northern and Western European ancestry) and the European American (EA) cohort from the exome variant server (ESP6500 SI-V2, accessed March 2014) (http://evs.gs.washington.edu/EVS/) which includes data from 4300 independent individuals of European American ancestry. The 1000 genomes samples are unselected controls while the EVS samples are selected to include controls, extremes of specific traits (LDL and blood pressure) and specific diseases (early onset myocardial infarction and early onset stroke). Allele frequencies were compared between SLIC probands and controls using a two-tailed Fisher’s exact test with 1 degree of freedom. Calculations were performed in the graphpad online calculator (http://www.graphpad.com/). Where given variants were observed in alternative populations, these data are reported but were not included in the statistical analyses since population admixture and stratification can lead to false positives, especially when investigating rare variants [83].
Supporting Information
Zdroje
1. Harel S, Greenstein Y, Kramer U, Yifat R, Samuel E, et al. (1996) Clinical characteristics of children referred to a child development center for evaluation of speech, language, and communication disorders. Pediatr Neurol 15 : 305–311. 8972529
2. American-Psychiatric-Association (2000) Diagnostic and Statistical Manual of Mental Disorders, 4th Edition, Text Revision (DSM-IVTR). Washington, DC: American Psychiatric Publishing.
3. Law J, Boyle J, Harris F, Harkness A, Nye C (2000) Prevalence and natural history of primary speech and language delay: findings from a review of the literature. Int J Lang Commun Disord 35 : 165–188. 10912250
4. Whitehouse AJ (2010) Is there a sex ratio difference in the familial aggregation of specific language impairment? A meta-analysis. J Speech Lang Hear Res 53 : 1015–1025. doi: 10.1044/1092-4388(2009/09-0078) 20605945
5. Bishop DV, Laws G, Adams C, Norbury CF (2006) High heritability of speech and language impairments in 6-year-old twins demonstrated using parent and teacher report. Behav Genet 36 : 173–184. 16485179
6. Bishop DV (2001) Genetic and environmental risks for specific language impairment in children. Philos Trans R Soc Lond B Biol Sci 356 : 369–380. 11316485
7. Newbury DF, Winchester L, Addis L, Paracchini S, Buckingham LL, et al. (2009) CMIP and ATP2C2 modulate phonological short-term memory in language impairment. Am J Hum Genet 85 : 264–272. doi: 10.1016/j.ajhg.2009.07.004 19646677
8. Luciano M, Evans DM, Hansell NK, Medland SE, Montgomery GW, et al. (2013) A genome-wide association study for reading and language abilities in two population cohorts. Genes Brain Behav 12 : 645–652. doi: 10.1111/gbb.12053 23738518
9. Gialluisi A, Newbury DF, Wilcutt EG, Olson RK, DeFries JC, et al. (2014) Genome-wide screening for DNA variants associated with reading and language traits. Genes Brain Behav 13 : 686–701. doi: 10.1111/gbb.12158 25065397
10. St Pourcain B, Cents RA, Whitehouse AJ, Haworth CM, Davis OS, et al. (2014) Common variation near ROBO2 is associated with expressive vocabulary in infancy. Nat Commun 5 : 4831. doi: 10.1038/ncomms5831 25226531
11. Eicher JD, Powers NR, Miller LL, Akshoomoff N, Amaral DG, et al. (2013) Genome-wide association study of shared components of reading disability and language impairment. Genes Brain Behav 12 : 792–801. doi: 10.1111/gbb.12085 24024963
12. Nudel R, Simpson NH, Baird G, O’Hare A, Conti-Ramsden G, et al. (2014) Genome-wide association analyses of child genotype effects and parent-of-origin effects in specific language impairment (SLI). Genes, Brain, Behavior 13 : 418–429. doi: 10.1111/gbb.12127 24571439
13. Ceroni F, Simpson NH, Francks C, Baird G, Conti-Ramsden G, et al. (2014) Homozygous microdeletion of exon 5 in ZNF277 in a girl with specific language impairment. Eur J Hum Genet 22 : 1165–1171. doi: 10.1038/ejhg.2014.4 24518835
14. Simpson NH, Addis L, Brandler WM, Slonims V, Clark A, et al. (2013) Increased prevalence of sex chromosome aneuploidies in specific language impairment and dyslexia. Dev Med Child Neurol 56 : 346–353. doi: 10.1111/dmcn.12294 24117048
15. Newbury DF, Paracchini S, Scerri TS, Winchester L, Addis L, et al. (2011) Investigation of dyslexia and SLI risk variants in reading—and language-impaired subjects. Behav Genet 41 : 90–104. doi: 10.1007/s10519-010-9424-3 21165691
16. Scerri TS, Morris AP, Buckingham LL, Newbury DF, Miller LL, et al. (2011) DCDC2, KIAA0319 and CMIP are associated with reading-related traits. Biological Psychiatry 70 : 237–245. doi: 10.1016/j.biopsych.2011.02.005 21457949
17. Shu W, Yang H, Zhang L, Lu MM, Morrisey EE (2001) Characterization of a new subfamily of winged-helix/forkhead (Fox) genes that are expressed in the lung and act as transcriptional repressors. J Biol Chem 276 : 27488–27497. 11358962
18. Lai CS, Fisher SE, Hurst JA, Vargha-Khadem F, Monaco AP (2001) A forkhead-domain gene is mutated in a severe speech and language disorder. Nature 413 : 519–523. 11586359
19. Feuk L, Kalervo A, Lipsanen-Nyman M, Skaug J, Nakabayashi K, et al. (2006) Absence of a paternally inherited FOXP2 gene in developmental verbal dyspraxia. Am J Hum Genet 79 : 965–972. 17033973
20. MacDermot KD, Bonora E, Sykes N, Coupe AM, Lai CS, et al. (2005) Identification of FOXP2 truncation as a novel cause of developmental speech and language deficits. Am J Hum Genet 76 : 1074–1080. 15877281
21. Shriberg LD, Ballard KJ, Tomblin JB, Duffy JR, Odell KH, et al. (2006) Speech, prosody, and voice characteristics of a mother and daughter with a 7;13 translocation affecting FOXP2. J Speech Lang Hear Res 49 : 500–525. 16787893
22. Zeesman S, Nowaczyk MJ, Teshima I, Roberts W, Cardy JO, et al. (2006) Speech and language impairment and oromotor dyspraxia due to deletion of 7q31 that involves FOXP2. Am J Med Genet A 140 : 509–514. 16470794
23. Fisher SE (2006) Tangled webs: tracing the connections between genes and cognition. Cognition 101 : 270–297. 16764847
24. Spiteri E, Konopka G, Coppola G, Bomar J, Oldham M, et al. (2007) Identification of the transcriptional targets of FOXP2, a gene linked to speech and language, in developing human brain. Am J Hum Genet 81 : 1144–1157. 17999357
25. Vernes SC, Spiteri E, Nicod J, Groszer M, Taylor JM, et al. (2007) High-throughput analysis of promoter occupancy reveals direct neural targets of FOXP2, a gene mutated in speech & language disorders. Am J Hum Genet 81 : 1232–1250. 17999362
26. Rodenas-Cuadrado P, Ho J, Vernes SC (2014) Shining a light on CNTNAP2: complex functions to complex disorders. Eur J Hum Genet 22 : 171–178. doi: 10.1038/ejhg.2013.100 23714751
27. Stein MB, Yang BZ, Chavira DA, Hitchcock CA, Sung SC, et al. (2011) A common genetic variant in the neurexin superfamily member CNTNAP2 is associated with increased risk for selective mutism and social anxiety-related traits. Biol Psychiatry 69 : 825–831. doi: 10.1016/j.biopsych.2010.11.008 21193173
28. Vernes SC, Newbury DF, Abrahams BS, Winchester L, Nicod J, et al. (2008) A functional genetic link between distinct developmental language disorders. N Engl J Med 359 : 2337–2345. doi: 10.1056/NEJMoa0802828 18987363
29. Peter B, Raskind WH, Matsushita M, Lisowski M, Vu T, et al. (2011) Replication of CNTNAP2 association with nonword repetition and support for FOXP2 association with timed reading and motor activities in a dyslexia family sample. J Neurodev Disord 3 : 39–49. doi: 10.1007/s11689-010-9065-0 21484596
30. Whitehouse AJ, Bishop DV, Ang QW, Pennell CE, Fisher SE (2011) CNTNAP2 variants affect early language development in the general population. Genes Brain Behav 10 : 451–456. doi: 10.1111/j.1601-183X.2011.00684.x 21310003
31. Whalley HC, O’Connell G, Sussmann JE, Peel A, Stanfield AC, et al. (2011) Genetic variation in CNTNAP2 alters brain function during linguistic processing in healthy individuals. Am J Med Genet B Neuropsychiatr Genet 156B: 941–948. doi: 10.1002/ajmg.b.31241 21987501
32. Kos M, van den Brink D, Snijders TM, Rijpkema M, Franke B, et al. (2012) CNTNAP2 and language processing in healthy individuals as measured with ERPs. PLoS One 7: e46995. doi: 10.1371/journal.pone.0046995 23115634
33. Villanueva P, de Barbieri Z, Palomino HM, Palomino H (2008) [High prevalence of specific language impairment in Robinson Crusoe Island. A possible founder effect]. Rev Med Chil 136 : 186–192. 18483672
34. Villanueva P, Fernandez MA, Z DEB, Palomino H (2013) Consanguinity on Robinson Crusoe Island, an Isolated Chilean Population. J Biosoc Sci: 1–10. 23931260
35. Villanueva P, Newbury DF, Jara L, De Barbieri Z, Mirza G, et al. (2011) Genome-wide analysis of genetic susceptibility to language impairment in an isolated Chilean population. Eur J Hum Genet 19 : 687–695. doi: 10.1038/ejhg.2010.251 21248734
36. Thornton T, McPeek MS (2007) Case-control association testing with related individuals: a more powerful quasi-likelihood score test. Am J Hum Genet 81 : 321–337. 17668381
37. SLIC (2002) A genomewide scan identifies two novel loci involved in Specific Language Impairment. Am J Hum Genet 70 : 384–398. 11791209
38. SLIC (2004) Highly significant linkage to the SLI1 locus in an expanded sample of individuals affected by specific language impairment. Am J Hum Genet 74 : 1225–1238. 15133743
39. Falcaro M, Pickles A, Newbury DF, Addis L, Banfield E, et al. (2008) Genetic and phenotypic effects of phonological short-term memory and grammatical morphology in specific language impairment. Genes Brain Behav 7 : 393–402. 18005161
40. Durbin RM, Abecasis GR, Altshuler DL, Auton A, Brooks LD, et al. (2010) A map of human genome variation from population-scale sequencing. Nature 467 : 1061–1073.
41. O’Roak BJ, Deriziotis P, Lee C, Vives L, Schwartz JJ, et al. (2011) Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat Genet 43 : 585–589. doi: 10.1038/ng.835 21572417
42. Leblond CS, Jutta H, Delorme R, Proepper C, Betancur C, et al. (2012) Genetic and Functional Analyses of SHANK2 Mutations Suggest a Multiple Hit Model of Autism Spectrum Disorders. Plos Genetics 8: e1002521. doi: 10.1371/journal.pgen.1002521 22346768
43. Bakkaloglu B, O’Roak BJ, Louvi A, Gupta AR, Abelson JF, et al. (2008) Molecular cytogenetic analysis and resequencing of contactin associated protein-like 2 in autism spectrum disorders. Am J Hum Genet 82 : 165–173. doi: 10.1016/j.ajhg.2007.09.017 18179895
44. Horn D, Kapeller J, Rivera-Brugues N, Moog U, Lorenz-Depiereux B, et al. (2010) Identification of FOXP1 deletions in three unrelated patients with mental retardation and significant speech and language deficits. Hum Mutat 31: E1851–1860. doi: 10.1002/humu.21362 20848658
45. Girirajan S, Brkanac Z, Coe BP, Baker C, Vives L, et al. (2011) Relative Burden of Large CNVs on a Range of Neurodevelopmental Phenotypes. PLoS Genet 7: e1002334. doi: 10.1371/journal.pgen.1002334 22102821
46. Gilissen C, Hoischen A, Brunner HG, Veltman JA (2012) Disease gene identification strategies for exome sequencing. European Journal of Human Genetics 20 : 490–497. 22258526
47. Tang W, Yuan J, Chen X, Shan Y, Luo K, et al. (2005) Cloning and characterization of the CDZFP gene which encodes a putative zinc finger protein. DNA Seq 16 : 391–396. 16323267
48. Mussig C, Schroder F, Usadel B, Lisso J (2010) Structure and putative function of NFX1-like proteins in plants. Plant Biol (Stuttg) 12 : 381–394. doi: 10.1111/j.1438-8677.2009.00303.x 20522174
49. Chaerkady R, Letzen B, Renuse S, Sahasrabuddhe NA, Kumar P, et al. (2011) Quantitative temporal proteomic analysis of human embryonic stem cell differentiation into oligodendrocyte progenitor cells. Proteomics 11 : 4007–4020. doi: 10.1002/pmic.201100107 21770034
50. Song Z, Krishna S, Thanos D, Strominger JL, Ono SJ (1994) A novel cysteine-rich sequence-specific DNA-binding protein interacts with the conserved X-box motif of the human major histocompatibility complex class II genes via a repeated Cys-His domain and functions as a transcriptional repressor. J Exp Med 180 : 1763–1774. 7964459
51. Nudel R, Simpson NH, Baird G, O’Hare A, Conti-Ramsden G, et al. (2014) Associations of HLA alleles with specific language impairment. J Neurodev Disord 6 : 1. doi: 10.1186/1866-1955-6-1 24433325
52. Xu M, Katzenellenbogen RA, Grandori C, Galloway DA (2010) NFX1 plays a role in human papillomavirus type 16 E6 activation of NFkappaB activity. J Virol 84 : 11461–11469. doi: 10.1128/JVI.00538-10 20739528
53. Kamal M, Valanciute A, Dahan K, Ory V, Pawlak A, et al. (2009) C-mip interacts physically with RelA and inhibits nuclear factor kappa B activity. Mol Immunol 46 : 991–998. doi: 10.1016/j.molimm.2008.09.034 19019440
54. Zweier C (2012) Severe Intellectual Disability Associated with Recessive Defects in CNTNAP2 and NRXN1. Mol Syndromol 2 : 181–185. 22670139
55. Cummings AC, Lee SL, McCauley JL, Jiang L, Crunk A, et al. (2011) A genome-wide linkage screen in the Amish with Parkinson disease points to chromosome 6. Ann Hum Genet 75 : 351–358. doi: 10.1111/j.1469-1809.2011.00643.x 21488853
56. Cummings AC, Torstenson E, Davis MF, D’Aoust LN, Scott WK, et al. (2013) Evaluating power and type 1 error in large pedigree analyses of binary traits. PLoS One 8: e62615. doi: 10.1371/journal.pone.0062615 23658753
57. Nyholt D (2008) Principles of linkage analysis. In: Neale B, Ferreira M, Medland SE, Posthuma D, editors. Statistical genetics: gene mapping through linkage and association. New York; Abingdon: Taylor & Francis. pp. 113–134.
58. Pavez M (2003) Test exploratorio de Gramática española de A. Toronto. Aplicación en Chile. Santiago: Ediciones Universidad católica de Chile.
59. Pavez MM, Maggiolo M (2000) Test para evaluar los procesos fonologicos de simplificacion TEPROSIF; De Barbieri Z, editor. Santiago, Chile: Ediciones Escuela de Fonoaudiologia. v. p.
60. Burgemeister B, Blue L, Lorge I (1998) Escala de madurez mental. Columbia: Ed. TEA, Madrid.
61. Villanueva P (2000) Pauta de Examen en Habla y motricidad Orofacial. Santiago: Escuela de Fonoaudiología, Facultad de Medicina, Universidad de Chile.
62. Peña-Casanova J (1991) Programa integrado de exploración Neuropsicológica. Test de Barcelona. Masson, Barcelona.
63. De Renzi E, Vignolo L (1962) The Token Test: a sensitive tests to detect receptive disturbances in aphasics. Brain 85 : 665–678. 14026018
64. Tallal P, Hirsch LS, Realpe-Bonilla T, Miller S, Brzustowicz LM, et al. (2001) Familial aggregation in specific language impairment. J Speech Lang Hear Res 44 : 1172–1182. 11708534
65. Marth GT, Korf I, Yandell MD, Yeh RT, Gu Z, et al. (1999) A general approach to single-nucleotide polymorphism discovery. Nat Genet 23 : 452–456. 10581034
66. Rozen S, Skaletsky H (2000) Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol 132 : 365–386. 10547847
67. Thornton T, Zhang Q, Cai X, Ober C, McPeek MS (2012) XM: association testing on the X-chromosome in case-control samples with related individuals. Genet Epidemiol 36 : 438–450. doi: 10.1002/gepi.21638 22552845
68. Bourgain C, Hoffjan S, Nicolae R, Newman D, Steiner L, et al. (2003) Novel case-control test in a founder population identifies P-selectin as an atopy-susceptibility locus. Am J Hum Genet 73 : 612–626. 12929084
69. Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, et al. (2007) PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81 : 559–575. 17701901
70. Abecasis GR, Cherny SS, Cookson WO, Cardon LR (2002) Merlin—rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet 30 : 97–101. 11731797
71. Kong A, Cox NJ (1997) Allele-sharing models: LOD scores and accurate linkage tests. Am J Hum Genet 61 : 1179–1188. 9345087
72. Schwarz JM, Rodelsperger C, Schuelke M, Seelow D (2010) MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods 7 : 575–576. doi: 10.1038/nmeth0810-575 20676075
73. Dollaghan CA (2011) Taxometric analyses of specific language impairment in 6-year-old children. J Speech Lang Hear Res 54 : 1361–1371. doi: 10.1044/1092-4388(2011/10-0187) 21646422
74. Bedore LM, Leonard LB (2001) Grammatical morphology deficits in Spanish-speaking children with specific language impairment. J Speech Lang Hear Res 44 : 905–924. 11521782
75. Burden V, Stott CM, Forge J, Goodyer I (1996) The Cambridge Language and Speech Project (CLASP). I. Detection of language difficulties at 36 to 39 months. Dev Med Child Neurol 38 : 613–631. 8674912
76. Clark A, O’Hare A, Watson J, Cohen W, Cowie H, et al. (2007) Severe receptive language disorder in childhood—familial aspects and long-term outcomes: results from a Scottish study. Arch Dis Child 92 : 614–619. 17405857
77. Conti-Ramsden G, Botting N (1999) Characteristics of children attending language units in England: a national study of 7-year-olds. Int J Lang Commun Disord 34 : 359–366. 10884906
78. Semel EM, Wiig EH, Secord W (1992) Clinical Evaluation of Language Fundamentals—Revised. San Antonio: Phychological Corporation.
79. Wechsler D (1992) Wechsler Intelligence Scale for Children - Third UK Edition. London: Psychological Corporation.
80. Lunter G, Goodson M (2011) Stampy: a statistical algorithm for sensitive and fast mapping of Illumina sequence reads. Genome Research 21 : 936–939. doi: 10.1101/gr.111120.110 20980556
81. Rivas MA, Beaudoin M, Gardet A, Stevens C, Sharma Y, et al. (2011) Deep resequencing of GWAS loci identifies independent rare variants associated with inflammatory bowel disease. Nat Genet 43 : 1066–1073. 21983784
82. Cingolani P, Platts A, Wang le L, Coon M, Nguyen T, et al. (2012) A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 6 : 80–92. doi: 10.4161/fly.19695 22728672
83. Mathieson I, McVean G (2012) Differential confounding of rare and common variants in spatially structured populations. Nat Genet 44 : 243–246. doi: 10.1038/ng.1074 22306651
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2015 Číslo 3
Nejčtenější v tomto čísle
- Clonality and Evolutionary History of Rhabdomyosarcoma
- Morphological Mutations: Lessons from the Cockscomb
- Maternal Filaggrin Mutations Increase the Risk of Atopic Dermatitis in Children: An Effect Independent of Mutation Inheritance
- Transcriptomic Profiling of Reveals Reprogramming of the Crp Regulon by Temperature and Uncovers Crp as a Master Regulator of Small RNAs