
Hyperdiverse Gene Cluster in Snail Host Conveys Resistance to Human Schistosome Parasites
Schistosomes are water-borne blood-flukes that are transmitted by snail vectors. They infect over 200 million people in more than 70 countries and cause severe and chronic disability. Snails naturally vary in resistance to this parasite even within species, so bolstering snail resistance in the wild would block transmission. We artificially selected snails for resistance and observed a rapid evolutionary response, with the greatest change occurring in the same genomic region in two independent trials. We subsequently confirmed that the selected haplotype conveys resistance to infection by schistosomes. The extraordinarily high sequence divergence among haplotypes in this region appears to be elevated due to ongoing natural selection, likely via host-parasite co-evolution. We observed the highest variation in genes encoding putative parasite recognition proteins, suggesting that these control the resistance phenotype in a manner reminiscent of immune gene complexes in other taxa. Thus, this gene cluster presents a potential new target to interfere with parasite transmission at the vector stage.
Published in the journal:
. PLoS Genet 11(3): e32767. doi:10.1371/journal.pgen.1005067
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1005067
Summary
Schistosomes are water-borne blood-flukes that are transmitted by snail vectors. They infect over 200 million people in more than 70 countries and cause severe and chronic disability. Snails naturally vary in resistance to this parasite even within species, so bolstering snail resistance in the wild would block transmission. We artificially selected snails for resistance and observed a rapid evolutionary response, with the greatest change occurring in the same genomic region in two independent trials. We subsequently confirmed that the selected haplotype conveys resistance to infection by schistosomes. The extraordinarily high sequence divergence among haplotypes in this region appears to be elevated due to ongoing natural selection, likely via host-parasite co-evolution. We observed the highest variation in genes encoding putative parasite recognition proteins, suggesting that these control the resistance phenotype in a manner reminiscent of immune gene complexes in other taxa. Thus, this gene cluster presents a potential new target to interfere with parasite transmission at the vector stage.
Introduction
Schistosomiasis is by far the most important helminth parasitic disease of humans. Schistosomes infect over 200 million people worldwide [1,2], causing a chronic, debilitating disease that can lead to lifelong disability [3,4]. The disability-adjusted-life years lost to this disease are estimated at 13–56 million, a value rivaling that of malaria [3]. There are no effective vaccines against schistosomes, and effective treatment still relies on regular dosing with a single drug, praziquantel [5]. Praziquantel resistance in schistosomes can be easily selected for in the lab, suggesting that natural populations which infect humans could also evolve drug resistance [6]. In fact, there is now credible evidence of reduced drug susceptibility in some heavily-treated human populations [7]. This problem may increase substantially as mass treatment with praziquantel escalates under initiatives such as the Schistosomiasis Control Initiative [8] and the Gates Foundation’s SCORE project (http://score.uga.edu). Alternate control strategies are therefore needed, including tactics for blocking transmission via the aquatic snails that serve as intermediate hosts.
Understanding the molecular mechanisms by which snails and schistosomes interact will be the key for new approaches to interrupt transmission [9]. Most molecular research to date has focused on the schistosome parasite Schistosoma mansoni and its New World snail host Biomphalaria glabrata, recently aided by the newly sequenced genomes of both species ([10]; https://www.vectorbase.org/organisms/biomphalaria-glabrata) and transcriptomic studies [11,12]. Several lines of evidence demonstrate that resistance to infection is highly heritable in snails [13–17]. First, infection rates in inbred snail lines are consistent and typically either 0% or close to 100% [13,14]. Second, artificial selection experiments can produce snails with significantly increased or decreased susceptibility in a few generations [15,17]. Third, resistance to infection can be mapped to genetic markers in linkage crosses [16]. However, despite recent advances [18–20], our knowledge of B. glabrata-S. mansoni interactions lags behind that of other host-parasite systems such as mosquito-Plasmodium [21,22]. Expression levels of some genes are known to influence resistance of B. glabrata to S. mansoni (FREPs, [23,24]; Hsp 90, [25]). However, to date there is only one genic locus known at which allelic variation associates with resistance (sod1, [26,27]), and the causality of this association still needs to be proven. Furthermore, there is substantial B. glabrata-strain by S. mansoni-strain (G × G) interaction in compatibility (i.e. one strain of B. glabrata can be highly resistant to one strain of S. mansoni, but highly susceptible to another, and vice versa) [28–32]. This pattern may reflect different per-strain combinations of highly diverse coevolving loci [33] which could conform to one of several models of host-parasite genotype matching [34].
In lab populations of B. glabrata and S. mansoni from natural populations in Guadeloupe, West Indies, only 40–50% of snails can be infected, no matter how many parasite miracidia are used to challenge them [33]. This scenario is consistent with a simple model of host-parasite genotype matching, in which the parasite population lacks the ability to be compatible with certain alleles in the host population [33]. Here we identify and characterize this resistance polymorphism in the snail population at the molecular level.
Results
Selection for resistance produces rapid phenotypic change
We started with a laboratory population of snails approximately ten generations removed from the wild, which had originated from hundreds of wild Guadeloupe snails and had been maintained as a randomly-mating population in the hundreds at the Université de Perpignan (“Guadeloupe laboratory population”) [33]. We selected two independent lines for resistance by challenging individual snails with either 10 (line R10) or 30 (line R30) miracidia and allowing only uninfected snails to found the next generation (S1 Table). Susceptibility (% of snails infected) dropped from approximately 50% (53% +/ - 4) to under 10% (3% +/ - 2 for R10; 6% +/ - 3 for R30) over five generations in the selection lines, but remained at approximately 50% (52% +/ - 7) in the unselected control line GUA (Fig. 1A). Linear regression of generation time on susceptibility (logit transformed) showed no significant effect for GUA (p > 0.1), but a significantly negative slope for both R10 (odds of infection each generation decrease by 45% (95% CI = 36–54%); p < 0.01) and R30 (odds of infection each generation decrease by 42% (95% CI = 28–53%); p < 0.01). A combined regression with both selection lines showed no significant effect of population (R10 vs. R30) on susceptibility (p > 0.1).

The same genomic region responds repeatedly to selection
In samples of 28 individuals each from GUA, R10 and R30, we observed 6573 informative RAD markers that aligned to the reference genome, including 4142 with codominant (two observable alleles) variants (SNPs or small indels) and 2431 null markers. The same null marker, aligning to Scaffold1, Site 1.814Mb, showed the highest allele frequency difference for both the R30-GUA comparison (FST = 0.52) and the R10-GUA comparison (FST = 0.30), with the allele changing frequency in the same direction for both selection lines (Fig. 2). In 100 bootstrap replicates in which 28 samples of each population were chosen randomly with replacement, this marker was the top FST outlier for the R30-GUA comparison 84% of the time (95% CI of FST = 0.38–0.67), and for the R10-GUA comparison 8% of the time (95% CI of FST = 0.15–0.50). We selected this top outlier for further study. We next looked at the additional markers on Scaffold 1 and found two other null markers <250kb away showing relatively high FST (Fig. 2). We supplemented our analysis with Stacks [35], a program that aligns reads to each other rather than to a reference genome, in order to find divergent markers on reads with low similarity to the reference genome. Because the genomic sequence flanking Stacks markers is unknown, they can be difficult to confirm with PCR, so we did not employ Stacks as a stand-alone analysis. However, Stacks revealed an additional RAD marker showing high allele frequency difference (R30-GUA: FST = 0.46; R10-GUA: FST = 0.14), and high linkage disequilibrium (LD) to Scaffold1, Site 1.814Mb (r = 0.94, p < 10-15), which had been too divergent to align unambiguously to the reference genome but which showed sequence similarity to Scaffold4, Site 1.466Mb (Fig. 2).

This genomic region is strongly associated with resistance
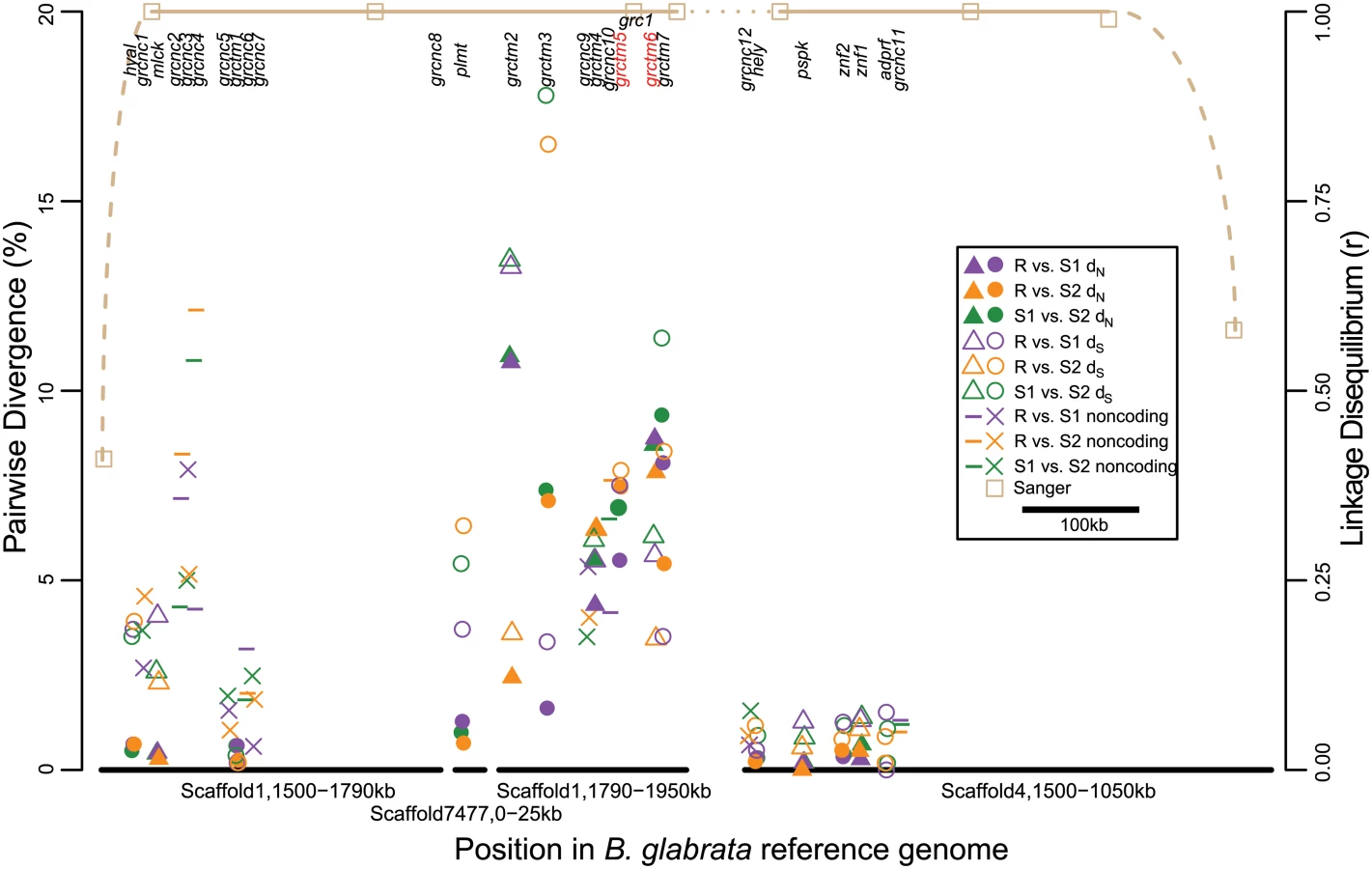
We determined the susceptibility/resistance phenotype of 289 additional snails from the Guadeloupe laboratory population by challenging each with 20 miracidia: 159 were resistant (uninfected) and 130 were susceptible (infected). We genotyped them at one of the high-FST RAD markers on Scaffold1 (“grc1”) using PCR and Sanger sequencing. All Sanger genotypes could be unambiguously phased by eye and were found to comprise three alleles (multi-SNP haplotypes), all of intermediate frequency. Allele R (“resistance”) with a frequency of 32.0% (+/ - 3.4%), showed a strong negative correlation with infection (Fig. 1B) and was dominant to the two S (“susceptibility”) alleles, S1 (42.6% +/ - 3.2% frequency) and S2 (25.4% +/ - 3.6% frequency). Specifically, snails without an R allele (n = 139) had a 70 (± 4) % chance of infection, while snails with at least one R allele (n = 150) had a 22 (± 3) % chance of infection (r2 = 0.23; odds ratio = 8.2; Fisher’s exact test, p < 10-15). There was no significant difference in infection between RR homozygotes and RS heterozygotes (Fisher’s exact test, p > 0.1). Similarly, Sanger sequencing and genotype phasing by eye revealed three alleles (multi-SNP haplotypes) in a PCR-amplified region of Scaffold4 (“grc2”), one of which was in near-perfect LD with allele R: only one individual out of 289 showed recombination between Scaffolds 1 and 4 (r = 0.996, p < 10-15). The remaining two grc2 alleles were in modest LD with S1 and S2 (r = 0.572 among the 139 SS snails, p < 10-12). We genotyped a subset of 94 snails at additional sites on Scaffold1 and Scaffold4 (Table 1; Fig. 3), and found alleles in perfect LD with allele R extending from sites 1.541–1.940Mb on Scaffold1 and sites 1.187–1.468Mb on Scaffold4, with the exception of a single recombinant at Scaffold4_1.187Mb. LD declined rapidly further upstream on both scaffolds, beginning at site 1.501Mb on Scaffold1 and at site 1.081Mb on Scaffold4. The different FST values among the RAD markers on these scaffolds (Fig. 2) is consistent with the imprecision of genotyping with null RAD tags (e.g. null heterozygotes cannot be identified), rather than different allele frequencies in the sample. Thus, the ends of these two scaffolds are tightly linked and probably either adjacent or nearly so, forming a region of > 0.7Mb in near-perfect LD and showing a strong association with resistance. We refer to this region as the Guadeloupe Resistance Complex (GRC).

GRC genes encode proteins with likely immunological roles
We performed RNA-Seq on outbred, parasite-unchallenged individuals of each genotype from an Oregon State University population derived from the original Guadeloupe laboratory population. The purpose of the RNA-Seq analysis was to facilitate gene annotation of the GRC, to obtain coding sequence for each allele, and to identify genes that might be differentially expressed between haplotypes. We sequenced 18 RR homozygotes, 9 S1S1 homozygotes, and 9 S2S2 homozygotes, all showing perfect LD among all three alleles at grc1 and grc2. In order to exhaustively characterize differences in either sequence or expression among haplotypes, we identified 31bp sequences (31-mers; the largest size possible in our analysis pipeline) showing significantly higher counts between RR and SS (8639 31-mers), between S1S1 and RR (5586 31-mers), and between S2S2 and RR (14976 31-mers). The large excess of 31-mers in S2S2 vs. RR, relative to the other comparisons, stems from the fact that LD between these two haplotypes extends ~0.7Mb farther upstream on Scaffold4. We ignored these 31-mers that align upstream of Scaffold4_1.1Mb because recombination between R and S1 indicates that they do not contain the causal variant. We assembled transcripts from 27 genes (defined as a transcribed sequence >500bp), including 15 coding and 12 putative noncoding (no open reading frame >500bp; designated grcnc for “Guadeloupe resistance complex noncoding”) genes that encompassed most of these 31-mers (95% for R, 90% for S1, and 31% for S2; Table 2). Nearly all transcripts aligned to the GRC region on Scaffold1 and Scaffold4, with two exceptions: one transcript (plmt) aligned to Scaffold7477, and another (grctm2) aligned partially to Scaffold1 and partially to Scaffold7477. However, SNPs in these transcripts were in perfect LD with the GRC, indicating that this section of Scaffold7477 (at least 6kb in size) occurs within the GRC.

With one exception (described below), all genes appeared in all three genotypes, although in some cases the orthologous transcript was under 500bp due to apparent truncation, and in some cases multiple isoforms of the same transcript were observed for some genotypes but not others (S1 Fig). The coding genes represented a diversity of protein families, including hyaluronidase, myosin light chain kinase, hemolysin, paraspeckle component, zinc finger, ADP ribosylation factor GTPase-activating protein, and palmitoyltransferase (Table 2). Other coding genes showed low or no similarity to any characterized sequence. Seven genes, all on Scaffold1 between positions 1.6–2.0Mb (middle of the GRC), show similar secondary structure, all encoding single-pass transmembrane proteins (TM1s) with >50 residues on either side of the transmembrane domain, and with a large (>50% of the protein) N-terminal (extracellular) domain in which β-strand residues are prevalent (>25% of residues and >1.5 times as common as α-helix residues). TM1 proteins are a structural class seen in a wide variety of protein families with roles in processes like cell migration, adhesion, and growth, and typically acting as receptors for extracellular signals [36]. Notably, TM1s play an important role in the immune system, and include B - and T-cell receptors, Fc receptors (binding the fragment crystallizable region of antibodies), some major histocompatibility complex (MHC) receptors, and Toll-like receptors, all of which are membrane-bound receptors recognizing foreign molecules [36]. Four of the TM1 genes in the GRC (grctm2, grctm3, grctm4, and grctm5) encode fibronectin III domains (DELTA-BLAST, E < 10-3; Fig. 4). Although the TM1 genes in the GRC do not all share primary sequence similarity, we designated them grctm1-grctm7 for “Guadeloupe resistance complex transmembrane” due to their similar secondary structure.

Low expression variation among GRC alleles
In the parasite-unchallenged snails, although expression varied substantially among genes (per-sample, per-site depth from 1x to >50x), expression differences among the three genotypes were low (S1 Fig, S2 Fig). We performed 90 t-tests for expression depth among genotypes in the unchallenged snails, and thus employed a Bonferroni-corrected α value of 0.0006. There was only a single instance of a gene not observed in all genotypes, and it was a short (605bp), noncoding sequence, grcnc8, observed only in RR snails at relatively low coverage (depth = 1.1x) (t-test between RR and SS, p<10-15). Only one other gene, the non-coding grcnc9, showed a >2-fold difference between RR (4.9x) and both SS (0.8x, 0.9x) genotypes (t-test, p<10-9). To be conservative with respect to identifying candidate genes, we also noted genes with expression differences that would be significant if uncorrected for multiple tests. There were two such genes showing small (<2 fold) differences between RR and both SS genotypes in the same direction: the non-coding grcnc5 (RR = 9.7x, S1S1 = 6.6x, S2S2 = 5.0x; p<10-2), and the TM1 gene grctm6 (RR = 1.2x, S1S1 = 2.2x, S2S2 = 2.4x; p<10-2). A few other genes showed >2-fold expression differences in some genotype comparisons, but not in both RR-S1S1 and RR-S2S2 comparisons.
In order to test whether any expression differences are only apparent during parasite challenge, we also performed RNA-Seq on pooled DNA from six families (3 RR, 1 S1S1, 2 S2S2) at 2 and 6 hour intervals after exposure to miracidia. We performed 62 t-tests for expression depth among genotypes in these challenged snails, and thus employed a Bonferroni-corrected α value of 0.0008. Expression differences among haplotypes appeared slightly greater in challenged snails than in unchallenged snails, but still low overall and not significant. As with unchallenged snails, we conservatively noted all genes with expression differences that would be significant if uncorrected for multiple tests. There were 4 genes with greater expression in SS families: grcnc2 at both intervals (2 hr RR = 2.1x, 2 hr SS = 3.6x, 6 hr RR = 1.6x, 6 hr SS = 4.3x; p < 0.05 for both), grctm6 at 6 hr (RR = 1.6x, SS = 4.5x; p < 0.05); adprf at 6 hr (RR = 13.6x, SS = 17.2x; p < 0.05) and hely at both intervals (2 hr RR = 1.9x, 2 hr SS = 4.2x, 6 hr RR = 1.6x, 6 hr SS = 3.0x; p < 0.05 for both). Only grcnc8, which was absent in both challenged and unchallenged SS snails, was higher in challenged RR families (2 hr RR = 0.8x, 6 hr RR = 0.9x). Additionally, there were 5 genes with >2-fold but nonsignificant (even if uncorrected) challenged expression differences among RR and SS snails: hyal at both intervals (2 hr RR = 2.8x, 2 hr SS = 9.9x, 6 hr RR = 3.3x, 6 hr SS = 8.5x), grcnc4 at 2 hr (RR = 7.9x, SS = 3.7x), grcnc6 at 2 hr (RR = 12.3x; SS = 3.5x), grcnc9 at both intervals (2 hr RR = 1.2x, 2 hr SS = 0.4x, 6 hr RR = 1.0x, 6 hr SS = 0.4x), and grctm6 at 2 hr (RR = 1.8x, SS = 3.8x). Notably, there were no coding genes with increased expression in RR families.
High sequence variation among GRC alleles
In contrast to expression differences, sequence divergence among alleles was often very high (Fig. 3; Fig. 4). Silent (synonymous and noncoding) divergence (dS) was over 5% in at least one pairwise comparison for 12 genes, and over 10% for 4 genes. All coding genes harbored nonsynonymous variants, but for non-TM1 coding genes, pairwise nonsynonymous divergence (dN) was 1% or less. In contrast, six of the TM1 genes had mean dN >5%, and all of these had at least one 75bp window with dN >15% in at least one comparison. The alleles at the remaining TM1 gene, grctm1, showed dN <1% but still encoded very different proteins because the S2 allele had a premature stop codon approximately 30% of the way through its open reading frame. Because dS was also very high among TM1 alleles, dN did not significantly exceed dS. Only two genes showed high nonsynonymous substitution (>3%) specific to the R allele: grctm5 (both R isoforms) and grctm6. Thus, the TM1 genes not only show structural similarity to known immune-relevant receptor genes, they also show the highest dN among alleles of all the genes in the GRC.
The GRC appears to be under balancing selection
In this snail population the GRC harbors three highly divergent haplotypes that have very even allele frequencies (>25%). In order to test for the signature of balancing selection, we compared Tajima's D and nucleotide diversity (π) for the GRC to that in the rest of the genome. Tajima’s D at Sanger-sequenced marker locus grc1 (15 polymorphisms, 289 samples) is 3.99. Tajima’s D in the GRC cannot be directly estimated from the RNA-Seq data because samples were chosen non-randomly based on GRC genotype. However, if we adjust RNA-Seq SNP frequencies based on the population allele frequencies of the three haplotypes, Tajima’s D in the GRC is estimated as 3.85. In order to compare this estimate to genome-wide patterns, we calculated Tajima’s D in two independent datasets: the RNA-Seq data from the 36 unchallenged snails, and the RAD data from the 28 GUA snails. We identified 779 genomic scaffolds for which we could confidently align at least 15 RNA-Seq SNPs with no missing genotypes (15–157 SNPs per scaffold; excludes SNPs in the GRC). Genome-wide, mean Tajima’s D is significantly less than in the GRC (mean = 1.68; 95% CI = -0.98 to 3.62; empirical p for D ≥ 3.85 is 0.01). In the RAD data, only 709 variants had no missing data across all GUA samples, and only a single scaffold contained at least 15 such variants, so we could not estimate a distribution of Tajima’s D as we did with the RNA-Seq data. However, overall Tajima’s D for these RAD variants is 1.76, a very similar estimate to the RNA-Seq data, and again much lower than at the GRC. Both genome-wide estimates are still significantly higher than the neutral expectation of D = 0 (coalescent simulation, p < 0.05).
Similarly, silent and noncoding diversity (πS) in the GRC ranged among genes from 0.16% to 7.40%, with a majority of genes (15 out of 26 universally expressed genes) showing πS > 2%. Nonsynonymous diversity (πN) in the GRC ranged among genes from 0.07% to 5.69%, with six TM1 genes showing πN > 3%. In order to compare π in the GRC to genome-wide patterns, we identified 3061 genomic scaffolds for which we could confidently align at least 100 contiguous bp of RNA-Seq reads at high coverage (length = 100–11,694bp; excludes reads in the GRC). Genome-wide π is significantly lower than for most GRC genes (mean = 0.49%; 95% CI = 0.06–1.75%; empirical p for π ≥ 2% is 0.01; empirical p for π ≥ 3% is 0.001).
BAC sequencing refines the assembly of the GRC
Scaffold1 includes the most interesting candidate genes as well as some obvious assembly errors (e.g. Scaffold7477 must be inserted somewhere). Therefore, in order to verify the assembly of the Scaffold1 portion of the GRC, we sequenced eight BAC clones that putatively tiled across the GRC region, and aligned them to the B. glabrata reference genome (BAC clones and the genome assembly were created using a Brazilian strain, BB02; [37]). All aligned to Scaffold1 as expected and largely confirmed the reference genome assembly between sites 1.471–1.971Mb, with a few exceptions. One large exception is the insertion (relative to the genome assembly) of a 69kb region aligning to Scaffold4587, Scaffold7002, and Scaffold7477. This complete insertion is found in two BACs, which also overlap on Scaffold1 between sites 1.74–1.80 Mb. This result is consistent with our RNA-Seq transcripts, one of which (plmt) is on Scaffold7477 but in perfect LD with the GRC, and one of which (grctm2) is partially on Scaffold7477 and partially on Scaffold1, sites 1.79–1.81 Mb. This insertion is accompanied by a 14kb deletion between sites 1.762–1.776 Mb on Scaffold1. Notably, this deletion contains a putative gene encoding an acetylglucosaminyltransferase. Thus this gene is not part of the GRC, consistent with RNA-Seq results. A second insertion of indefinite size (at least 67kb) occurs after Scaffold1 site 1.971Mb, as observed in two BACs. We did not observe sequence corresponding to the end of Scaffold1 from site 1.971Mb to 2.184Mb, which may occur after this final insertion or may have been incorrectly assembled onto Scaffold1. A small but interesting insertion matching Scaffold866, sites 57.7–58.1kb, is an exon of gene grctm6, corresponding exactly to the region of the gene where allele S1 shows no sequence similarity to the other alleles (Fig. 4).
Discussion
We have identified and characterized a region of the B. glabrata genome, the GRC, that greatly influences immunity to schistosome parasites. Three major lines of evidence demonstrate the functional importance of the GRC. First, this region responded rapidly to selection for resistance in two independent experiments (Fig. 1A, Fig. 2). Second, it shows a strong and significant association with infection status in the unselected population of snails (Fig. 1B), explaining 23% of the variance in resistance in the population. Third, genetic diversity at this locus shows an unusual pattern with three highly divergent alleles in nearly perfect LD over hundreds of kilobases, suggestive of balancing selection (Fig. 3, Fig. 4).
Genes in the GRC
The GRC contains 15 coding genes, some of which are duplicated within some haplotypes. Pinpointing the specific causal gene(s) responsible for resistance in this system will require additional experimental evidence, but the most promising candidates are the TM1 genes, especially those with many nonsynonymous variants unique to the resistant haplotype. Gene regulation in cis is not a strong candidate for the functional mechanism because there were few significant and/or large (>2-fold) expression differences, especially in coding genes (S1 Fig; S2 Fig). Although we cannot rule out a role for subtle expression differences, including in noncoding transcripts, the simplest mechanism of dominant resistance via gene expression would be overexpression of the resistance allele in a coding gene, and such a pattern is never observed. In contrast, the strikingly high amino acid sequence divergence is a much stronger candidate for the functional mechanism. All genes show some nonsynonymous differences, but only the TM1 genes show high dN (Fig. 3). The TM1 alleles also harbor many length polymorphisms, and some alleles with short open reading frames may be nonfunctional (e.g. grctm1_S2, grctm5_S1, grctm7_S1, Fig. 4). We also note that the cluster of TM1 genes is in the center of the region of statistical association with resistance, the most likely position in which to find the causal locus or loci.
That these TM1 genes also code for proteins that share structural similarity to known pathogen-recognition molecules [36] is particularly intriguing. Typical TM1s usually have their transmembrane domain very close to the N - or C-terminal (<50 residues) [38], but in all seven GRC TM1 genes, the transmembrane domain is >50 residues from either end, a feature they share with Toll-like receptors and a minority of other TM1s. Most also contain fibronectin III domains which are often involved in molecular recognition [39]. Thus, although not all the TM1 genes show sequence similarity to each other, they may be functionally or even evolutionarily related. Specifically, we hypothesize that their extracellular (N-terminal) domains recognize foreign substances such as parasite PAMPs (pathogen-associated molecular patterns), while their intracellular (C-terminal) domains transmit this signal to other cellular components, leading to a physiological response. For example, S. mansoni produces polymorphic mucins, SmPoMucs [40], that interact with snail FREP immunity proteins [20,41,42]. It is possible that the TM1 proteins recognize SmPoMucs, SmPoMucs-FREP complexes, or unrelated PAMPs.
The most compelling candidates for controlling resistance to S. mansoni in the TM1 gene cluster are grctm5 and grctm6. Only these two genes show high nonsynonymous substitution specific to the R haplotype (Fig. 4). Gene grctm5 encodes a fibronectin III protein with sequence similarity to chitinase. Intriguingly, it is present in two copies in RR snails but only one copy in the susceptible genotypes. Furthermore, these two R isoforms are substantially divergent from each other (dN = 7.9%, dS = 7.4%), as well as from S1 (dN = 4.2–6.8%, dS = 7.1–7.8%) and S2 (dN = 7.4–7.5%, dS = 6.7–9.1%) alleles. Both grctm5 R sequences show large, significant expression differences between RR (depth = 2.8x, 3.0x) and S1S1 (depth = 1.0x; t-tests, p < 10-3 for both), but not between RR and S2S2 (2.3x; t-tests, p >0.1 for both) (S1 Fig). Gene grctm6 has no strong sequence similarity to any known proteins. It shows high divergence among all three haplotypes, and includes a ~300bp segment where the S1 haplotype shows no sequence similarity to R or S2. The grctm6 gene also shows significantly lower expression in RR than in S1S1 or S2S2.
Balancing selection on the GRC and host-parasite coevolution
The GRC, especially the TM1 genes, shows three patterns of genetic variation consistent with balancing selection. First, with three intermediate frequency alleles, the site-frequency spectrum is highly skewed away from neutral expectation, as evidenced by the extremely high Tajima's D value. Second, high LD across several Mb is an unusual pattern suggestive of selection [43]. Although high LD can also be caused by low recombination rates, this explanation seems unlikely. In most genomes, regions of low recombination are gene-poor [44], unlike the GRC. The main exceptions are chromosomal inversions which can suppress recombination across suites of genes; however, an inversion cannot maintain high divergence among more than two haplotypes, so the three divergent haplotypes of the GRC require another explanation. Both Tajima’s D and LD can be elevated by demographic bottlenecks. Genome-wide values of Tajima’s D in the Guadeloupe laboratory population are higher than the neutral equilibrium expectation, suggesting such a demographic effect. However, Tajima’s D at the GRC is significantly higher still, suggesting that selection has also played a role. Because the Guadeloupe laboratory population has had a census size in the hundreds at collection and ever since, we expect genetic drift in these snails to have been minimal. Thus, both allele frequencies and the extent of LD at the GRC should be similar to values in the wild, which we cannot directly estimate. Guadeloupe B. glabrata undergoes population fluctuations in the wild [45], and these natural demographic dynamics likely contribute to the genome-wide population genetic patterns. In addition, our genome-wide estimates of Tajima’s D are based on samples that did experience minor laboratory bottlenecks (N = 100 over five generations for GUA RAD; N = 30 for the founding of the Oregon State University population from which the RNA-Seq data is derived). Therefore, genome-wide Tajima’s D and LD may be lower in the wild, and the GRC may be an even more extreme outlier.
The third non-neutral pattern, high genetic diversity, cannot be explained by demographics. The GRC is an outlier with respect to nucleotide diversity (π) at both synonymous and nonsynonymous sites, especially in six of the TM1 genes. The high nonsynonymous diversity (πN = 3.2–5.7%) observed at six of the TM1 genes is quite remarkable, as it greatly exceeds the presumably neutral values seen throughout most of the genome (p < 0.001). Of course, reads at highly diverse loci might be less likely to align to the reference genome, so our genome-wide estimate of π is likely biased downward. Still, such high nonsynonymous diversity is rarely seen in any species; for example, 99.9% of Drosophila simulans genes show lower πN values [46]. We did not observe a dN/dS ratio significantly greater than one at any gene. At grctm6, dN is 1.65 times as high as dS (synonymous plus noncoding) divergence (Fig. 3), but only 0.83 times the magnitude of synonymous divergence alone (excluding noncoding sites). Such high diversity at both synonymous and nonsynonymous sites is evidence that alleles have coexisted for an unusually long time at intermediate frequency, presumably because selection has maintained them in balance [47,48]. As with other immunity genes that show similar patterns [49], it may be the case that heterozygotes have a fitness advantage because they can recognize and respond to a greater diversity of parasites, or it may be that parasite community composition changes so quickly that no one allele is advantageous for long enough to fix. Because S. mansoni arrived in the New World only in historical times [50], the selection pressure maintaining these alleles must be due to other native parasites, perhaps including other trematodes which have a very close and ancient relationship with snails [17,51]. As with Tajima’s D and LD, π in the Guadeloupe laboratory population may be slightly different than in the wild, although we expect the effect of genetic drift in the lab to have been minimal. If anything, we would expect even greater diversity in natural populations, as there may be additional unsampled divergent alleles, especially if populations outside of Guadeloupe are considered (indeed, the reference genome provides an example). Additional work is needed to study the relevance of the GRC to phenotypic variation in resistance across the range of B. glabrata, or in other snail species.
Théron et al. [33] showed that the shape of dose-response curves in the Guadeloupe snail-schistosome community is consistent with a simple host-parasite phenotype compatibility model, in which allelic variation in the snails controls matching with the parasite, likely via a complimentary locus (or loci) in the Guadeloupe population of S. mansoni. Such phenotype compatibility could occur by several mechanisms. Under the matching alleles (MA) model, parasites avoid detection by matching host self determinants, whereas under the inverse matching allele (IMA) model, host molecules recognize parasite molecules leading to an immune response [34]. Importantly, heterozygous hosts are susceptible under MA and resistant under IMA [34]. Thus, the genetic dominance of resistance at the GRC favors IMA.
Similar signatures of balancing selection are seen across a wide range of host taxa at immunity loci that interact directly with infectious disease agents [52–54]. In the Anopheles/Plasmodium pair, the best studied system of invertebrate host and eukaryotic parasite, several genomic loci have been associated with host resistance and show patterns paralleling those seen at the GRC [55–57]. For example, the Anopheles APL1 locus also represents a cluster of structurally similar genes that show extraordinarily high nonsynonymous diversity of the same magnitude as the GRC TM1 genes (3–6%) [55,57]. Likewise, nonsynonymous divergence is high among alleles of the Anopheles TEP1 locus, although gene conversion rather than balancing selection appears to be responsible [56]. Nonsynonymous diversity exceeding neutral expectations is also consistently observed in the MHC of vertebrates [49] and plant R genes [58]. As with the GRC, genetic dominance of resistance is common across these systems, consistent with the IMA model. Furthermore, the close physical linkage of the TM1 genes resembles the gene clusters that form many of these resistance genomic regions in other taxa. Such clustering may be a neutral artifact of tandem gene duplication, or it may have adaptive significance through shared gene regulation or the maintenance of beneficial multi-gene haplotypes [59].
Possible applications
The GRC is an obvious target for applications in the control of schistosomiasis. Genetic manipulation of disease vectors is a promising approach that is already underway for mosquitoes and other pest species [9,60,61], and driving resistance genes into B. glabrata populations is under discussion [27]. Gene knockdowns in B. glabrata have shown repeated success [23,24,62,63], and targeted inhibition of GRC genes or their products may pinpoint the specific causal gene. Alternatively, transfection with a multi-gene haplotype could have practical utility even without perfect understanding of each gene’s functional contribution. Although genetic modification of snails is still in its infancy, the CRISPR nuclease system shows promise for fine-scale modification of even non-model species [9]. Importantly, transgenic vectors would not need to have higher fitness than wild-type organisms if transgenes were spread by gene drives [9]. Thus, if only the R allele recognizes S. mansoni while other alleles recognize other parasites of no importance to human health, it would be possible to drive the R allele to fixation even without an adaptive benefit to the snails, if it were coupled to a gene drive conferring preferential non-Mendelian inheritance of the R allele [9]. Conversely, if multiple alleles at this locus are required to recognize all strains of S. mansoni, it might be possible to engineer a single haplotype with duplicated genes that included all relevant sequences. Alternatively, if the GRC genes initiate an immune signal cascade upon recognition of the parasite, future research could seek to manipulate snails such that this signal cascade is constitutively upregulated regardless of genotype or infection status. The matching loci in the parasite, once they are identified, may also be targets for drugs or genetic manipulation.
Materials and Methods
Ethics statement
Mice and hamsters were used to maintain the schistosome parasites and to produce miracidia for challenge experiments. Infection is through contact with inoculated water and involves minimal discomfort. Infected rodents are euthanized with CO2 prior to showing clinical signs of disease and are dissected to recover parasitic worms and their eggs. This research was approved by OSU IACUC and the French veterinary agency.
Experimental evolution
B. glabrata and S. mansoni were collected on the island of Guadeloupe, West Indies, in 2005 as described previously [33]. Selection lines were created at the University of Perpignan beginning in 2008. We selected for resistance by challenging snails with 10 (line R10) or 30 (line R30) S. mansoni miracidia and allowing only uninfected snails in each independent line to contribute to the next generation (N = 50–100 snails challenged per line per generation, and so the numbers used to found each subsequent generation varied each generation and increased from as low as 38 to 90 as the susceptibility of the two lines decreased; S1 Table). This was done for five generations. We measured susceptibility in each line as the percentage of snails infected (Fig. 1A). As a control, we maintained a line of uninfected snails (GUA) at a similar population size for the same time period (N = 100 snails per generation). For GUA, 50–70 snails from each offspring generation were challenged in order to measure the susceptibility of GUA each generation (percentage of snails infected, S1 Table), but these challenged snails did not contribute to subsequent generations. We tested for a correlation between generation and susceptibility by logit transforming susceptibility and using linear regression. We estimated the standard errors of proportions using √((p*(1-p))/N).
RAD genotyping
We used SbfI in RAD genotyping [64] of 28 individuals each from the control population (GUA) and the two selection lines (R10 and R30) (Illumina data at NCBI SRA, Bioproject Accession PRJNA268191). We aligned reads to the B. glabrata reference genome with BWA [65]. All genomic analyses in this study used version 4.3 of the reference genome, and positions were subsequently converted to nearly identical reference genome version BglaB1 (https://www.vectorbase.org/organisms/biomphalaria-glabrata). In order to maximize the density of markers, we considered both markers with observable variants as well as null markers for which a RAD tag aligned in only a subset of individuals. To estimate allele frequencies of SNPs, we counted two alleles per individual with 10x or greater depth, and two half-alleles per individual with 2–9x depth to account for possible nonobserved alleles, while genotypes with <2x depth were counted as missing. We only analyzed sites with a per-population allele count of at least 24 for both GUA and at least one other population, and which did not violate Hardy-Weinberg equilibrium (χ2 > 20) in any population nor show overall heterozygote excess among populations. Allele frequencies for null markers were estimated using the method of Zhivotovsky [66]. For each informative marker we estimated the difference in allele frequency between GUA and each selected population (FST) as 1 –(mean within-population expected heterozygosity)/(total expected heterozygosity). To assess RAD tags with low sequence similarity to the reference genome, we aligned RAD tags to each other with Stacks [35] and tested for any previously undetected high-FST markers. We assessed the robustness of FST results with 100 bootstrap replicates in which 28 samples from each population were chosen randomly with replacement. We chose the single most extreme outlier (highest FST), along with markers showing evidence of close physical linkage to this marker (<1Mb away on same scaffold and/or high LD), for further analysis. For these promising markers, we designed primer pairs to amplify and Sanger sequence the RAD tag in order to obtain the complete codominant genotype for all individuals.
Sanger verification
We phenotyped an independent sample of 289 snails by challenging them with 20 miracidia each and then designating them as either infected or uninfected following Theron et al. [33]. We examined markers showing exceptionally high FST by Sanger sequencing them in these 289 snails and testing for an association with phenotype. We calculated allele frequencies and standard errors of proportions. We calculated the proportion of phenotypic variation explained by the R allele as the square of the correlation coefficient (r2) from a logistic regression of phenotype (infected or not) versus genotype (R/ - or not). We then developed primers to amplify nearby genomic regions in order to find and characterize the genomic region showing the strongest association with resistance. We tested these markers on a subset of 94 phenotyped snails.
RNA-Seq of unchallenged snails
We extracted RNA from whole bodies of randomly-chosen, size-matched juvenile snails that had not been parasite-challenged. We prepared samples using the TruSeqTM RNA v2 kit (Illumina RS-122–2001) following the low-throughput protocol found in the Sample Preparation v2 Guide. We performed RNA-Seq on the following 36 samples: 12 RR homozygotes (single-end), 6 RR homozygotes (paired-end), 6 S1S1 homozygotes (single-end), 3 S1S1 homozygotes (paired-end), 6 S2S2 homozygotes (single-end), and 3 S2S2 homozygotes (paired-end) (we first Sanger genotyped a large number of randomly-chosen snails from tentacle snips, and then we chose the above 36 to use for RNA-Seq). Sequencing was conducted on the Illumina HiSeq 2000 at Oregon State University (Illumina data at NCBI SRA, Bioproject Accession PRJNA264063). We converted FASTQ files to FASTA and used Jellyfish 1.0.2 [67] to count 31-mers in each sample, which is the largest kmer size that Jellyfish can count, but which is long enough to be typically unique in the genome and therefore represent specific transcripts. We then used a custom perlscript (https://github.com/jacobtennessen/HOLDRS) to identify 31-mers that differed significantly among genotypes, defined as showing at least a 2-fold difference in total count and a Welch’s t-statistic of at least 5. These parameters were chosen to encompass both sequence and expression differences: expression differences under 2-fold may not be biologically meaningful, true sequence differences should result in a much larger (all or nothing) count ratio, and this t-statistic typically corresponds to a p-value under 10-4, which will minimize the number of false positives among 31-mers from thousands of transcripts, while still detecting 31-mers that are truly overabundant in one sample (N = 9 or 18) versus a control (N = 18). We identified reads containing these divergent 31-mers and used ABySS v. 1.3.4 [68] to assemble them into contigs. We used grep and manual alignment to extend these contigs and assemble additional contigs from divergent reads. We focused subsequent analysis on contigs >500bp, as well as shorter contigs with putative orthology to a contig >500bp in another genotype (sequences in NCBI Transcriptome Shotgun Assembly Database, Bioproject Accession PRJNA264063). In order to measure sequencing depth and identify polymorphisms, we aligned reads to these long contigs using BWA [65] and converted genotypes to vcf format.
RNA-Seq of challenged snails
We chose six snail lineages homozygous at the GRC (3 RR, 1 S1S1, 2 S2S2). For each family we exposed 6 randomly-chosen, size-matched juvenile individuals to 20 miracidia each, and then we extracted RNA from whole bodies at 2 hr (n = 3) and 6 hr (n = 3) post infection. RNA from each set of 3 snails from the same family and time point was then pooled. We prepared and sequenced samples single-end as described above (Illumina data at NCBI SRA, Bioproject Accession PRJNA264063). We aligned reads to the assembled transcripts from the unchallenged dataset and compared depths as described above.
BAC sequencing
We BLASTed the GRC portion of Scaffold1 (B. glabrata reference genome) against end sequences from the BAC library generated for B. glabrata strain BB02 (ftp://ftp.ncbi.nih.gov/pub/TraceDB/biomphalaria_glabrata/; [37]). We chose BACs from among the hits, ordered them from the Arizona Genomics Institute, extracted DNA, and sequenced them in a Nano run of the Illumina MiSeq at Oregon State University (Illumina data at NCBI SRA Bioproject Accession PRJNA268208). We aligned the reads from eight BACs to the B. glabrata reference genome with BWA [65].
Analysis
We calculated linkage disequilibrium between markers as the correlation coefficient, r, in number of R alleles per genotype (0, 1, or 2) We classified transcripts as “coding” if they contained an open reading frame of at least 500bp, otherwise they were designated “noncoding.” We used DELTA-BLAST [69] to match candidate transcripts with known protein families. Genes were named following the guidelines in Bayne [70]. We characterized secondary structure with Jpred3 [71] and identified transmembrane domains using TMHMM v. 2.0 (http://www.cbs.dtu.dk/services/TMHMM/). We measured pairwise synonymous (dS) and nonsynonymous (dN) divergence, as well as allele-specific nonsynonymous substitution, with custom Perl scripts and DnaSP [72]. For each transcript, we used t-tests to identify significant differences in adjusted per-sample, per-site depth among genotypes in which all values were divided by the ratio of the total count of reads in that individual relative to the mean per-individual read count, and then log-transformed. Depth values of zero were arbitrarily recorded as 1/6, which assumes they are present but too rare (much less than 1x) to have been sampled. For each transcript under each experimental scenario (unchallenged, 2 hours post-challenge, and 6-hours post-challenge) we compared RR vs. SS. For unchallenged snails we also compared RR vs. S1S1 and RR vs. S2S2. If multiple sequences of the same gene were observed for one genotype, these were all tested separately. In order to estimate Tajima’s D [73] in the GRC, we calculated the frequency of all RNA-Seq SNPs on their haplotype (R, S1, or S2), and then multiplied this frequency by the unbiased haplotype frequency estimate from the 289 snails Sanger sequenced at grc1. In order to estimate genome-wide Tajima’s D and π, we aligned RNA-Seq reads to the B. glabrata reference genome with BWA [65]. We identified regions with at least 100 contiguous bp showing high (≥6x) depth in all 36 samples. All of these high-depth regions were used to estimate π, while scaffolds with at least 15 SNPs in such high-depth regions were included in the estimate of Tajima’s D. Significance of Tajima’s D was estimated with standard neutral coalescent simulations in DnaSP [72].
Supporting Information
Zdroje
1. King CH, Dickman K, Tisch DJ (2005) Reassessment of the cost of chronic helmintic infection: a meta-analysis of disability-related outcomes in endemic schistosomiasis. Lancet 365 : 1561–1569. 15866310
2. Steinmann P, Keiser J, Bos R, Tanner M, Utzinger J (2006) Schistosomiasis and water resources development: systematic review, meta-analysis, and estimates of people at risk. Lancet Infect Dis 6 : 411–425. 16790382
3. King CH (2010) Parasites and poverty: The case of schistosomiasis. Acta Tropica 113 : 95–104. doi: 10.1016/j.actatropica.2009.11.012 19962954
4. King CH (2011) Schistosomiasis: challenges and opportunities. In Institute of Medicine (US) Forum on Microbial Threats. The Causes and Impacts of Neglected Tropical and Zoonotic Diseases: Opportunities for Integrated Intervention Strategies National Academies Press (US), Washington (DC). 21977543
5. Doenhoff MJ, Hagan P, Cioli D, Southgate V, Pica-Mattoccia L, et al. (2009) Praziquantel: its use in control of schistosomiasis in sub-Saharan Africa and current research needs. Parasitology 136 : 1825–1835. doi: 10.1017/S0031182009000493 19281637
6. Fallon PG, Doenhoff MJ (1994) Drug resistant schistosomiasis: Resistance to praziquantel and oxaminquine induced in Schistosoma mansoni in mice is drug specific. Am J Trop Med Hyg 51 : 83–88. 8059919
7. Melman SD, Steinauer ML, Cunningham C, Kubatko LS, Mwangi IN, et al. (2009) Reduced susceptibility of Kenyan Schistosoma mansoni to praziquantel following repeated exposures: origin, measurement, and likelihood of persistence. PLoS Negl Trop Dis 3: e504. doi: 10.1371/journal.pntd.0000504 19688043
8. Fenwick A, Webster JP, Bosque-Oliva E, Blair L, Fleming FM, et al. (2009) The Schistosomiasis Control Initiative (SCI): rationale, development and implementation from 2002–2008. Parasitology 136 : 1719–1730. doi: 10.1017/S0031182009990400 19631008
9. Esvelt KM, Smidler AL, Catteruccia F, Church GM (2014) Concerning RNA-guided gene drives for the alteration of wild populations. eLife 3: e03401. doi: 10.7554/eLife.03401 25035423
10. Berriman M, Haas BJ, LoVerde PT, Wilson RA, Dillon GP, et al. (2009) The genome of the blood fluke Schistosoma mansoni. Nature 460 : 352–358. doi: 10.1038/nature08160 19606141
11. Lockyer A, Spinks J, Kane RA, Hoffmann KF, Fitzpatrick JM, et al. (2008) Biomphalaria glabrata transcriptome: cDNA microarray profiling identifies resistant - and susceptible-specific gene expression in haemocytes from snail strains exposed to Schistosoma mansoni. BMC Genomics 9 : 634. doi: 10.1186/1471-2164-9-634 19114004
12. Adema CM, Hanington PC, Lun CM, Rosenberg GH, Aragon AD, et al. (2010) Differential transcriptomic responses of Biomphalaria glabrata (Gastropoda, Mollusca) to bacteria and metazoan parasites, Schistosoma mansoni and Echinostoma paraensei (Digenea, Platyhelminthes). Mol Immunol 47 : 849–860. doi: 10.1016/j.molimm.2009.10.019 19962194
13. Richards CS, Shade PC (1987) The genetic variation of compatibility in Biomphalaria glabrata and Schistosoma mansoni. J Parasitol 73 : 1146–1151. 3437352
14. Richards CS, Knight M, Lewis FA (1992) Genetics of Biomphalaria glabrata and its effect on the outcome of Schistosoma mansoni infection. Parasitol Today 8 : 171–174. 15463608
15. Webster JP, Woolhouse MEJ (1998) Selection and strain specificity of compatibility between snail intermediate hosts and their parasitic schistosomes. Evolution 52 : 1627–1643.
16. Knight M, Miller AN, Patterson CN, Rowe CG, Michaels G, et al. (1999) The identification of markers segregating with resistance to Schistosoma mansoni infection in the snail Biomphalaria glabrata. Proc Natl Acad Sci USA 96 : 1510–1515. 9990054
17. Webster JP, Gower CM, Blair L (2004) Do hosts and parasites coevolve? Empirical support from the Schistosoma system. Am Nat 164: S33–S51. 15540140
18. Bayne CJ (2009) Successful parasitism of vector snail Biomphalaria glabrata by the human blood fluke (trematode) Schistosoma mansoni: A 2009 assessment. Mol Biochem Parasitol 165 : 8–18. doi: 10.1016/j.molbiopara.2009.01.005 19393158
19. Loker ES (2010) Gastropod immunobiology. Adv Exp Med Biol 708 : 17–43. 21528691
20. Mitta G, Adema CM, Gourbal B, Loker ES, Théron A (2012) Compatibility polymorphism in snail/schistosome interactions: From field to theory to molecular mechanisms. Dev Comp Immunol 37 : 1–8. doi: 10.1016/j.dci.2011.09.002 21945832
21. Waterhouse RM, Kriventseva EV, Meister S, Xi Z, Alvarez KS, et al. (2007) Evolutionary dynamics of immune-related genes and pathways in disease-vector mosquitoes. Science 316 : 1738–1743. 17588928
22. Severson DW, Behura SK (2012) Mosquito genomics: progress and challenges. Annu Rev Entomol 57 : 143–166. doi: 10.1146/annurev-ento-120710-100651 21942845
23. Hanington PC, Forys MA, Dragoo JW, Zhang SM, Adema CM, et al. (2010) Role for a somatically diversified lectin in resistance of an invertebrate to parasite infection. Proc Natl Acad Sci USA 107 : 21087–21092. doi: 10.1073/pnas.1011242107 21084634
24. Hanington PC, Forys MA, Loker ES (2012) A somatically diversified defense factor, FREP3, is a determinant of snail resistance to schistosome infection. PLoS Negl Trop Dis 6: e1591. doi: 10.1371/journal.pntd.0001591 22479663
25. Ittiprasert W, Knight M (2012) Reversing the resistance phenotype of the Biomphalaria glabrata snail host Schistosoma mansoni infection by temperature modulation. PLoS Pathog 8: e1002677. doi: 10.1371/journal.ppat.1002677 22577362
26. Goodall CP, Bender RC, Brooks JK, Bayne CJ (2006) Biomphalaria glabrata cytosolic copper/zinc superoxide dismutase (SOD1) gene: Association of SOD1 alleles with resistance/susceptibility to Schistosoma mansoni. Mol Biochem Parasitol 147 : 207–210. 16564582
27. Bonner KM, Bayne CJ, Laurson MK, Blouin MS (2012) Effects of Cu/Zn superoxide dismutase (sod1) genotype and genetic background on growth, reproduction and defense in Biomphalaria glabrata. PLoS Negl Trop Dis 6: e1701. doi: 10.1371/journal.pntd.0001701 22724037
28. Basch PF (1975) An interpretation of snail-trematode infection rates: specificity based on concordance of compatible phenotypes. Int J Parasitol 5 : 449–452. 1140882
29. Basch PF (1976) Intermediate host specificity in Schistosoma mansoni. Exp Parasitol 39 : 159–169.
30. Richards CS (1975) Genetic factors in susceptibility of Biomphalaria glabrata for different strains of Schistosoma mansoni. Parasitology 70 : 231–241. 1128925
31. Webster JP, Woolhouse MEJ (1998) Selection and strain specificity of compatibility between snail intermediate hosts and their parasitic schistosomes. Evolution 52 : 1627–1634.
32. Théron A, Rognon A, Gourbal B, Mitta G (2014) Multi-host parasite infectivity and multi-parasite host susceptibility: a new approach of the Biomphalaria glabrata/Schistosoma mansoni compatibility polymorphism. Infect Genet Evol 26 : 80–88. doi: 10.1016/j.meegid.2014.04.025 24837670
33. Théron A, Coustau C, Rognon A, Gourbiere S, Blouin MS (2008) Effects of laboratory culture on compatibility between snails and schistosomes. Parasitology 135 : 1179–1188. doi: 10.1017/S0031182008004745 18700994
34. Nuismer SL, Otto SP (2004) Host-parasite interactions and the evolution of ploidy. Proc Natl Acad Sci USA 101 : 11036–11039. 15252199
35. Catchen J, Hohenlohe P, Bassham S, Amores A, Cresko W (2013) Stacks: an analysis tool set for population genomics. Mol Ecol 22 : 3124–3140. doi: 10.1111/mec.12354 23701397
36. Pahl MC, Askinazi OL, Hamilton C, Cheng I, Cichewicz K, et al. (2013) Signalling via single-pass transmembrane proteins. In: eLS. John Wiley & Sons, Ltd: Chichester.
37. Adema CM, Luo MZ, Hanelt B, Hertel LA, Marshall JJ, et al. (2006) A bacterial artificial chromosome library for Biomphalaria glabrata, intermediate snail host of Schistosoma mansoni. Mem Inst Oswaldo Cruz 101 : 167–177. 17308766
38. Sawada R, Mitaku S (2011) How are exons encoding transmembrane sequences distributed in the exon-intron structure of genes? Genes Cells 16 : 115–21. doi: 10.1111/j.1365-2443.2010.01468.x 21143351
39. Skerra A (2000) Engineered protein scaffolds for molecular recognition. J Mol Recognit 13 : 167–187. 10931555
40. Roger E, Grunau C, Pierce RJ, Hirai H, Gourbal B, et al. (2008) Controlled chaos of polymorphic mucins in a metazoan parasite (Schistosoma mansoni) interacting with its invertebrate host (Biomphalaria glabrata). PLoS Negl Trop Dis 2: e330. doi: 10.1371/journal.pntd.0000330 19002242
41. Adema CM, Hertel LA, Miller RD, Loker ES (1997) A family of fibrinogen-related proteins that precipitates parasite-derived molecules is produced by an invertebrate after infection. Proc Natl Acad Sci USA 94 : 8691–8696. 9238039
42. Moné Y, Gourbal B, Duval D, Du Pasquier L, Kieffer-Jaquinod S, et al. (2010) A large repertoire of parasite epitopes matched by a large repertoire of host immune receptors in an invertebrate host/parasite model. PLoS Negl Trop Dis 4: e813. doi: 10.1371/journal.pntd.0000813 20838648
43. Charlesworth D (2006) Balancing selection and its effects on sequences in nearby genome regions. PLoS Genet 2: e64. 16683038
44. Nam K, Ellegren H (2012) Recombination drives vertebrate genome contraction. PLoS Genet 8: e1002680. doi: 10.1371/journal.pgen.1002680 22570634
45. Sire C, Durand P, Pointier JP, Théron A (1999) Genetic diversity and recruitment pattern of Schistosoma mansoni in a Biomphalaria glabrata snail population: a field study using random-amplified polymorphic DNA markers. J Parasitol 85 : 436–441. 10386434
46. Begun DJ, Holloway AK, Stevens K, Hillier LW, Poh YP, et al. (2007) Population genomics: whole-genome analysis of polymorphism and divergence in Drosophila simulans. PLoS Biol 5: e310. 17988176
47. Meagher S, Potts WK (1997) A microsatellite-based MHC genotyping system for house mice (Mus domesticus). Hereditas 127 : 75–82. 9420473
48. Roux C, Pauwels M, Ruggiero MV, Charlesworth D, Castric V, et al. (2013) Recent and ancient signature of balancing selection around the S-locus in Arabidopsis halleri and A. lyrata. Mol Biol Evol 30 : 435–447. doi: 10.1093/molbev/mss246 23104079
49. Hedrick PW (2002) Pathogen resistance and genetic variation at MHC loci. Evolution 56 : 1902–1908. 12449477
50. Desprès L, Imbert-Establet D. Monnerot M (1993) Molecular characterisation of mitochondrial DNA provides evidence for the recent introduction of Schistosoma mansoni into America. Mol Biochem Parasitol 60 : 221–230. 7694146
51. Blair D, Davis GM, Wu B (2001) Evolutionary relationships between trematodes and snails emphasizing schistosomes and paragonimids. Parasitology 123 Suppl:S229–S243. 11769286
52. Takahata N, Satta Y, Klein J (1992) Polymorphism and balancing selection at major histocompatibility complex loci. Genetics 130 : 925–938. 1582567
53. Tennessen JA, Blouin MS (2008) Balancing selection at a frog antimicrobial peptide locus: fluctuating immune effector alleles? Mol Biol Evol 25 : 2669–2680. doi: 10.1093/molbev/msn208 18799711
54. Andrés AM, Hubisz MJ, Indap A, Torgerson DG, Degenhardt JD, et al. (2009) Targets of balancing selection in the human genome. Mol Biol Evol 26 : 2755–2764. doi: 10.1093/molbev/msp190 19713326
55. Riehle MM, Markianos K, Niaré O, Xu J, Li J, et al. (2006) Natural malaria infection in Anopheles gambiae is regulated by a single genomic control region. Science. 312 : 577–579. 16645095
56. Obbard DJ, Callister DM, Jiggins FM, Soares DC, Yan G, et al. (2008) The evolution of TEP1, an exceptionally polymorphic immunity gene in Anopheles gambiae. BMC Evol Biol. 8 : 274. doi: 10.1186/1471-2148-8-274 18840262
57. Rottschaefer SM, Riehle MM, Coulibaly B, Sacko M, Niaré O, et al. (2011) Exceptional diversity, maintenance of polymorphism, and recent directional selection on the APL1 malaria resistance genes of Anopheles gambiae. PLoS Biol. 9: e1000600. doi: 10.1371/journal.pbio.1000600 21408087
58. Hulbert SH, Webb CA, Smith SM, Sun Q (2001) Resistance gene complexes: evolution and utilization. Annu Rev Phytopathol 39 : 285–312. 11701867
59. Trowsdale J, Powis SH (1992) The MHC: relationship between linkage and function. Curr Opin Genet Dev 2 : 492–497. 1504627
60. Marrelli MT, Li C, Rasgon JL, Jacobs-Lorena M (2007) Transgenic malaria-resistant mosquitoes have a fitness advantage when feeding on Plasmodium-infected blood. Proc Natl Acad Sci USA 104 : 5580–5583. 17372227
61. Wise de Valdez MR, Nimmo D, Betz J, Gong HF, James AA, et al. (2011) Genetic elimination of dengue vector mosquitoes. Proc Natl Acad Sci USA 108 : 4772–4775. doi: 10.1073/pnas.1019295108 21383140
62. Baeza Garcia A, Pierce RJ, Gourbal B, Werkmeister E, Colinet D, et al. (2010) Involvement of the cytokine MIF in the snail host immune response to the parasite Schistosoma mansoni. PLoS Pathog 6: e1001115. doi: 10.1371/journal.ppat.1001115 20886098
63. Knight M, Miller A, Liu Y, Scaria P, Woodle M, et al. (2011) Polyethyleneimine (PEI) mediated siRNA gene silencing in the Schistosoma mansoni snail host, Biomphalaria glabrata. PLoS Negl Trop Dis 5: e1212. doi: 10.1371/journal.pntd.0001212 21765961
64. Baird NA, Etter PD, Atwood TS, Currey MC, Shiver AL, et al. (2008) Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS ONE 3: e3376. doi: 10.1371/journal.pone.0003376 18852878
65. Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows-Wheeler Transform. Bioinformatics 25 : 1754–1760. doi: 10.1093/bioinformatics/btp324 19451168
66. Zhivotovsky LA (1999) Estimating population structure in diploids with multilocus dominant DNA markers. Mol Ecol. 8 : 907–913. 10434412
67. Marçais G, Kingsford C (2001) A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27 : 764–770.
68. Simpson JT, Wong K, Jackman SD, Schein JE, Jones SJ, et al. (2009) ABySS: a parallel assembler for short read sequence data. Genome Res 19 : 1117–1123. doi: 10.1101/gr.089532.108 19251739
69. Boratyn GM, Schäffer AA, Agarwala R, Altschul SF, Lipman DJ, et al. (2012) Domain enhanced lookup time accelerated BLAST. Biol Direct 7 : 12. doi: 10.1186/1745-6150-7-12 22510480
70. Bayne CJ (2012) A convention for naming molluscan genes. Curr Top in Genet 5 : 45–48.
71. Cole C, Barber JD, Barton GJ (2008) The Jpred 3 secondary structure prediction server. Nucleic Acids Res 36: W197–201. doi: 10.1093/nar/gkn238 18463136
72. Librado P, Rozas J (2009) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25 : 1451–1452. doi: 10.1093/bioinformatics/btp187 19346325
73. Tajima F (1989) Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123 : 585–595. 2513255
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2015 Číslo 3
Nejčtenější v tomto čísle
- Clonality and Evolutionary History of Rhabdomyosarcoma
- Morphological Mutations: Lessons from the Cockscomb
- Maternal Filaggrin Mutations Increase the Risk of Atopic Dermatitis in Children: An Effect Independent of Mutation Inheritance
- Transcriptomic Profiling of Reveals Reprogramming of the Crp Regulon by Temperature and Uncovers Crp as a Master Regulator of Small RNAs