
The Class Homeodomain Factors and Cooperate in . Embryonic Progenitor Cells to Regulate Robust Development
Animals develop as one initial cell, the fertilized egg, repeatedly divides and its progeny differentiate, ultimately producing diverse cell types. This occurs in large part by the expression of unique combinations of regulatory genes, such as transcription factors, in precursors of each cell type. These early factors are typically reused in precursors of different cell types. The nematode worm Caenorhabditis elegans is a powerful system in which to identify developmental regulators because it has a rapid and reproducible development, yet it shares most of its developmental regulators with more complex organisms such as humans. We used state-of-the-art microscopy and computer-aided cell tracking methods to identify the developmental role of worm homologs of the OTX and PITX genes, whose human homologs play a role in the development of the brain, eye, and pituitary among other tissues. We identified broad roles for OTX in regulating development for many distinct cell types including muscles, neurons and skin, and found a redundant role for both OTX and PITX in a subset of cells. Future studies of these genes should address whether these genes also act redundantly in mammals.
Published in the journal:
. PLoS Genet 11(3): e32767. doi:10.1371/journal.pgen.1005003
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1005003
Summary
Animals develop as one initial cell, the fertilized egg, repeatedly divides and its progeny differentiate, ultimately producing diverse cell types. This occurs in large part by the expression of unique combinations of regulatory genes, such as transcription factors, in precursors of each cell type. These early factors are typically reused in precursors of different cell types. The nematode worm Caenorhabditis elegans is a powerful system in which to identify developmental regulators because it has a rapid and reproducible development, yet it shares most of its developmental regulators with more complex organisms such as humans. We used state-of-the-art microscopy and computer-aided cell tracking methods to identify the developmental role of worm homologs of the OTX and PITX genes, whose human homologs play a role in the development of the brain, eye, and pituitary among other tissues. We identified broad roles for OTX in regulating development for many distinct cell types including muscles, neurons and skin, and found a redundant role for both OTX and PITX in a subset of cells. Future studies of these genes should address whether these genes also act redundantly in mammals.
Introduction
Identifying regulators of the intermediate steps that link pluripotency and terminal differentiation is a fundamental challenge in developmental biology. These regulators are comparatively poorly understood for most tissues due to the difficulty of recognizing and isolating cells in these transient intermediate states (“progenitors”) and their complex combinatorial logic. Individual transcription factors (TFs) acting at these stages often have broad and diverse expression domains that don’t correlate well with specific tissue or cell types [1], with multiple TFs typically acting together to specify any given intermediate progenitor. Therefore, loss of function can lead to pleiotropic phenotypes, while partial redundancy between regulators can lead to reduced penetrance, making it hard to determine the relationship between expression and biological function. Large-scale screens for gene pairs with synthetic phenotypes, as has been done for yeast [2] can identify genes acting in parallel, but screening at that scale is not feasible in animals. We are overcoming these challenges with a systematic approach to define pleiotropic and redundant progenitor TFs in Caenorhabditis elegans, a simple model organism where lineage relationships are already understood, large-scale gene expression resources allow rapid identify patterns of TF overlap, and powerful tools exist for characterizing mutant phenotypes across all embryonic cells. Previous studies of genetic redundancy in C. elegans have prioritized gene pairs for synthetic lethality testing based on similar functional interactions [3,4], expression patterns [5] and homology or conservation [6,7].
Progenitor cells are easily identified in C. elegans because the relationship between cell lineage and fate is known and invariant[8,9]. The first several embryonic divisions give rise to founder cells, some of which have clonal or partially clonal cell fates. Most cells, however, retain a multipotent state until the final round of embryonic cell divisions, when two daughters adopt such different fates as a neuron and an epithelial tube or neuron and hypodermal (skin) cell. Thus, any TF expressed in a non-clonal progenitor cell or group of lineally related cells (i.e. lineage) at any time after the earliest cell divisions but prior to the final round could play a role in progenitor identity. Despite this potential, genetic studies have identified numerous regulators of both early founder cell identity [10–16] and of terminal fate[17–19], but fewer regulators of intermediate progenitor identity. Automated methods to track cell lineages from confocal microscopy image series have allowed quantitative expression measurements for over 200 transcription factors across every cell of C. elegans embryos [1,20–22], and this EPIC (Expression Patterns In Caenorhabditis) dataset suggests many candidate regulators of progenitor identity [1,23]. Computer-aided cell tracking of mutant embryos can confirm these regulators by identifying a wide range of pleiotropic defects, from wholesale fate transformations to subtle defects in cell migration or division timing [10,14,24–27].
Many previous studies of TF function relied on reporter gene expression to infer developmental defects. We reasoned that the complex patterns of cell cycle length asynchrony and cell migration that occur in later embryos may allow identification of defects at single cell resolution without such reporters. We used this approach to characterize the developmental role of the candidate progenitor regulator ceh-36, which encodes an orthodenticle/OTX homeodomain family transcription factor orthologous to mammalian OTX1, OTX2 and CRX proteins. A ceh-36 reporter is expressed in multiple progenitor cells, encompassing the precursors of 248 terminal cells with diverse fates including neurons, glia, the excretory (renal) system, visceral and body muscles, epidermal and rectal epithelial cells[1]. Vertebrate OTX factors are similarly expressed and required in precursors of diverse tissues [28–37], suggesting these factors could be conserved regulators of progenitor identity. However, previous studies of ceh-36 mutants identified defects only in the embryonic specification of four neurons [38–40]. The large number of expressing cells combined with the small number of cells known to require ceh-36 raises the question of whether ceh-36 is required across most expressing cells or only a minority of these cells.
We found that ceh-36 null mutants are viable embryonically, with partially penetrant larval lethality and superficially normal morphology. Cell lineage tracing of ceh-36(-) embryos revealed variably penetrant defects in cell division patterns or cell migration in over 100 cells that normally express ceh-36. Double mutants lacking both ceh-36 and the coexpressed PITX-family homeobox gene unc-30 exhibited 100% synthetic lethality and severe morphological defects. These double mutants have dramatically increased rates of defective cell division and migration in coexpressing cells, indicating ceh-36 and unc-30 act in parallel to regulate the development of these cells. This provides the first evidence for genetic redundancy between OTX and PITX homeodomain factors, two bicoid class TFs that are predicted to bind similar sequences, yet diverged prior to the radiation of metazoan species.
Results
ceh-36 is required for larval progression but not morphogenesis
A ceh-36 deletion allele that removes the majority of coding regions, including the homeodomain, was annotated as embryonic lethal in WormBase based on limited previous characterization [40,41]. After outcrossing, we found that nearly all embryos homozygous for this allele hatched, while ~60% of animals arrest as larvae (Table 1, Fig. 1A, B, S1 Table). The remainder of ceh-36(ok795) animals survived to adulthood and were fertile. Most arrested larvae had normal body morphology, with 5.6% of L1s containing a small bubble-like “vacuole” at the tip of the head (Table 2, Fig. 1C). Two other ceh-36 alleles predicted to eliminate or alter the homeodomain displayed similar rates of larval arrest (Table 1), suggesting this is the null phenotype. A fourth allele, ky640, which truncates the protein but is predicted to encode a complete homeodomain, displayed lower lethality rates, suggesting it leads to partial loss of function. An extrachromosomal genomic fosmid transgene containing CEH-36::GFP (+) rescued ceh-36(ok795) larval lethality; the 85% survival in this strain corresponds to nearly 100% after accounting for the 25% rate of transgene loss (Table 1). Consistent with this, 95% of CEH-36::GFP-positive L1s survived. Ectopic expression of CEH-36::GFP under the control of a heat-shock promoter caused extensive lethality when induced prior to the 50-cell stage, while later induction had little effect (Fig. 1D), indicating that CEH-36 is toxic when expressed in these early embryonic cells, but not in later cells. We conclude that ceh-36 is required for robust larval viability but not for gross morphology or embryonic viability.



ceh-36 is expressed in bilateral lineages that produce diverse tissues
We previously analyzed expression of a 5-kb ceh-36 promoter fusion reporter and identified expression in several major lineages (Fig. 2, S1 Fig) [1]. Since this reporter may not contain all relevant regulatory sequences, we generated transgenic strains using a fosmid clone from the “Transgeneome” project [22] where CEH-36 protein is fused to GFP in the context of the endogenous locus (Fig. 2A). This transgene rescues the higher-penetrance ceh-36 mutant lineage defects and larval arrest phenotype described below (Table 1). Using lineage analysis, we identified all CEH-36::GFP expressing cells through the comma stage, at which point the embryo starts to move. CEH-36::GFP is expressed in progenitors of 248 terminal cells from six lineages that together produce a mix of diverse cell types including pharyngeal cells, muscles, neurons, glia and specialized cell types, and programmed cell deaths (Fig. 2B, C). CEH-36::GFP is predominantly (>90%) expressed symmetrically between left and right symmetric lineages, despite left-right asymmetric expression and function for two of the four neurons previously shown to require ceh-36 [38,40]. The spatial expression pattern is similar to the previously analyzed ceh-36 promoter fusion (Fig. 2B, S2 Fig), but includes additional expression in the ABara lineage. We also analyzed a previously published 2-kb promoter fusion reporter [38,40,42] that we found is expressed in the MSa, MSp and ABalpa lineages but not ABara, ABplp or ABprp, indicating the existence of multiple regulatory elements for ceh-36 in different lineages (S3 Fig).

The CEH-36 protein fusion reporter exhibits complex dynamics that we confirmed by single molecule RNA-FISH (smFISH) [43] of endogenous ceh-36 mRNA (see below). Expression in the ABpxp, MSaa, and MSpa lineages begins between the 50-cell and 100-cell stages and decreases in most cells after 2–3 cell cycles, prior to morphogenesis (Fig. 2B). However, a few cells maintain stable expression much longer, up to at least comma stage. The CEH-36::GFP expressing cells include progenitors of three neurons previously shown to require ceh-36 (MI, AWCL and AWCR), with additional stronger expression in AWCL, AWCR and the fourth ceh-36-requiring cell, ASEL, beginning after the worm begins to elongate and twitch [38–40]. In total, we found expression of ceh-36 in progenitors of over 30% of embryonic cells suggesting it could play a broad role in embryonic patterning. Its early and transient expression in progenitor cells suggested that ceh-36 might be an important regulator of progenitor identity or function.
Quantitative analysis identifies broad roles for ceh-36 in regulating cell cycle timing and cell position
The lack of obvious morphological defects in ceh-36 mutants suggests that ceh-36 might play a minimal role in the development of most expressing cells. To test this, we searched for defects in lineage patterns and cell migrations in mutant embryos using automated cell tracking. We examined quantitative features of embryonic development, including timing and patterns of cell division, division orientation, and positions in eight ceh-36(ok795) embryos through the comma stage (~400 minutes after fertilization, when nearly all cell divisions have occurred) and compared these phenotypes to a wild-type reference set [27] (see Materials and Methods) and to three embryos expressing a rescuing CEH-36::GFP transgene. We also examined one embryo carrying a second predicted ceh-36 null mutation (ky646). As detailed below, we found that many cells in ceh-36(-) embryos have partially penetrant defects in both cell cycle timing and cell position (Figs. 3,4, S2 Table). In total, 5.1% (495/9636) of cells in ceh-36(ok795) embryos were defective in cell division or position, compared with 0.3% (85/26171) of cells in wild-type control embryos (p < 10-220; chi-squared test). This suggests that ceh-36 is broadly important for robust development across its expressing cells.
Loss of ceh-36 disrupts robust control of cell division and cell death
The C. elegans lineage is composed of an invariant pattern of cell divisions and deaths. In wild-type embryos, the division timing is highly stereotyped, with most cells having variability in cell cycle length of less than 5% [27] [44]. We identified 49 cells with cell cycle or lineage timing defects in at least one ceh-36 mutant embryo (Figs. 3A, D, 4, S2 Table), defined as cells dividing both three standard deviations and at least five minutes earlier or later than expected, not dividing at all, or dividing inappropriately. In addition, three cells failed to undergo programmed cell death when expected, as recognized by the characteristic pattern of chromatin compaction observed for histone-mCherry. For example, in three embryos, MSpaapp, which normally is the first embryonic cell to undergo apoptosis, instead survived and divided, with both sisters migrating into the pharynx to adopt unknown fates (Fig. 3C). In some cases, cells not passing our threshold for defect calling appeared to have different mean cell cycles or positions. For example 35 of 49 cells with cell cycle defects in one or more embryos also had a nominally significant difference in mean cell cycles (p < 0.1; FDR < 0.15; S2 Table). The CEH-36::GFP fusion protein is expressed in precursors of 86%(12/14) of cells with cell division timing defects in two or more ceh-36(-) embryos, and 60% (21/35) of cells with defects in one embryo. This is significantly more than the 30% of all cells that express CEH-36::GFP (chi squared p < 2 × 10-6). CEH-36::GFP is also expressed in all of the cells with supernumerary divisions or failed cell death.

ceh-36 is required for cell migration and bilateral symmetry
Cell positions are also highly consistent between wild-type embryos, allowing us to identify cell migration defects by comparing cell positions between ceh-36 mutant and wild-type embryos. We identified 124 cells whose deviation from expected position was at least 3.5 standard deviations greater than in the wild-type set and that had aberrant neighbors as defined by an empirical neighbor-distance score (S2 Table; see Methods). Position defects were strongly enriched in expressing cells; 81% (55/68) of cells with position defects in two or more embryos normally express CEH-36::GFP. By comparison, in 22 wild-type embryos examined, only 13 cells had defective positions, in one embryo each.
A cell could be misplaced because of a defective migration, in which case it would have both different position and different neighbors than in the wild type. Alternatively, a cell could be misplaced because its normal position is occupied by another cell that migrated inappropriately, in which case its position relative to its normal neighbors would be unchanged. We used these criteria to classify 50 cells with position defects by examining their position and neighbors in 3D visualizations (Fig. 3E, F). We scored 82% (41/50) of cells as likely defective migrations, while 9/50 (18%) defects could be explained by defective migration of other cells (S3 Table). 100% (18/18) of higher-penetrance (seen in at least three of eight ok795 embryos) position defects examined were scored as likely migration defects. The migration defects include both cells that undergo novel migrations in the mutant (Fig. 3E) as well as cells that fail to undergo their expected migrations (Fig. 3F). The cells scored as possible secondary defects were less penetrant, with each identified as defective in one or two embryos. Still, most low-penetrance defects (23/32) were scored as likely migration defects
We observed dramatic defects in eight laterally positioned cells that were born in the correct position but subsequently migrated across the midline to the opposite lateral side of the embryo, sometimes displacing the position of their bilateral counterpart (e.g. Fig. 3E). These lateral migration defects occurred on both sides of the embryo (3 L→R, 5 R→L) and include diverse cell types: neurons (I1R and I2L), pharyngeal cells (pm3R and mc1DR), rectal cells (left intestinal muscle and anal depressor muscle), and tail cells (Hyp10 and tail spike). These defects were all low penetrance (seen in one or two of eight ok795 embryos), but we saw no defects of this class in the 22 wild-type control embryos, and all eight of these cells normally express ceh-36. This indicates that C. elegans cells’ lateral position is not merely a result of their birth position but is regulated by factors that include ceh-36.
We determined that lateralization defects are maintained through embryonic elongation and not corrected by subsequent cell movements by examining worms expressing FKH-4::GFP, a marker of three visceral muscles (left and right intestinal muscles and anal depressor; Fig. 3E, S4 Fig). 100% of both wild-type and ceh-36 mutant elongated (pretzel-stage) embryos have three FKH-4(+) cells, indicating that ceh-36 is not necessary for FKH-4 expression. However, one FKH-4(+) cell is laterally mispositioned in 14% of ceh-36(-) embryos (Table 3; Fig. 3E). This is consistent with the left-right migration phenotype and low penetrance observed in our lineage data (1/8 ok795 embryos, 12.5%), increasing confidence in the low-penetrance defects identified by lineage analysis.

Pharyngeal gland defects correlate with ceh-36(-) larval lethality
Multiple pharyngeal gland cell precursors had cell cycle and position defects in ceh-36 mutants. For example, the daughters of the MSaapapa cell normally produce a pharyngeal gland cell and a programmed cell death and the early division of this cell was the largest division-timing defect we observed in ceh-36 mutants (Fig. 3A). Precursors of four of the five pharyngeal gland cells express CEH-36::GFP and all four of these had partial penetrance defects in cell cycle or position (Figs. 3A, 4). Since pharyngeal gland cells are known to be required for feeding and viability [45], we examined them for additional defects by examining expression of the pharyngeal gland marker hlh-6::GFP in elongated ceh-36(ok795) embryos. We observed altered pharyngeal gland morphology in 20% of ceh-36(ok795) elongated embryos. An additional 9% of embryos were missing one or more hlh-6::GFP-positive cells (Fig. 3B, Table 2), suggesting that ceh-36 regulates not only gland cell cycle patterns and morphology but also terminal fate. While only 41% (23/56) of larvae with normal gland morphology arrested prior to the L4 stage, 92% (46/50) of larvae with abnormal gland morphology arrested. Thus, defects in pharyngeal gland morphology predict larval arrest in ceh-36 mutants.
ceh-36 mutant defects are partially penetrant
Defects occurred in 223 unique cells, typically with low penetrance; only 82/223 (37%) cells were defective in two or more (of eight) ok795 embryos. Most of the defective cells normally express CEH-36::GFP (77%), significantly more than the 30% fraction of all cells that express ceh-36 (p < 10-90, chi-squared test). Most of the defective cells that do not normally express ceh-36 were only called as defective in one embryo. Still, even defects seen in a single embryo were enriched in expressing cells (59% of such cells express CEH-36::GFP). While cells with prior cell cycle defects were 2.9-fold more likely to have position defects (p < 10-9), 90% of cells with position defects had no detectable cell cycle defect. Only 22 expressing cells had defects in at least 50% of analyzed embryos (e.g. Fig. 4, S2 Table). The low penetrance of most individual defects may explain the viability of ceh-36(-) embryos.

We determined whether cells with low penetrance defects have noticeable defective terminal positions or numbers by examining several fluorescent markers expressed in these cells (Table 2). Reporters for two cells previously reported as requiring ceh-36 (MI(sams-5) and ASEL(gcy-5)) showed the expected terminal defect frequencies in the ceh-36 deletion. As described above, the visceral muscle reporter FKH-4::GFP and the pharyngeal gland reporter hlh-6p::GFP also showed terminal position defect frequencies consistent with the observed embryonic defects. Finally, a FLP-1::GFP reporter reported as expressed in the AVK neuron (which expresses ceh-36 but was not identified as defective in our analysis) showed little or no terminal defects (~2%). Given that the mutant embryos hatch without major morphological defects despite an average 40 cells with position defects and 10 cells with altered division timing, development of C. elegans embryos must be robust to a substantial amount of developmental error.
ceh-36 mutant defects are clustered in related cells
The rescuing CEH-36::GFP transgene expression is typically strongest several divisions before the birth of the terminal cells where most defects were identified, suggesting that defects in ceh-36(-) may result from regulatory events occurring in mitotic progenitor cells. If this is true, partially penetrant defects should preferentially co-occur in closely related cells within a given embryo. We identified 71 examples of defective sister cell pairs in ceh-36-expressing cells. We found preferential co-occurrence of defects in sisters for seven embryos (p < 0.001) by using a bootstrap evaluation, and this co-occurrence was only significant in cells expressing ceh-36. This along with the early and dynamic CEH-36::GFP expression suggests that ceh-36 regulates development in part through its activity in progenitor cells, rather than the terminal cells that exhibit the defects.
Lineage defects are observed with multiple ceh-36 alleles and are rescued by CEH-36::GFP
To confirm that most defects identified in ceh-36(ok795) embryos result from loss of ceh-36, we specifically examined high-penetrance (≥6 of 8 ok795 embryos) position defects in an embryo carrying a second predicted ceh-36 null mutation (ky646). We found four of the five cells examined had similar defects in this embryo. We examined these cells in two ceh-36(ok795) embryos expressing CEH-36::GFP, and one embryo with mosaic CEH-36::GFP expression, and found that these defects were rescued in all CEH-36::GFP expressing cells. Taken together, these results show that ceh-36 regulates the robustness of cell cycle and migration patterns in many cells. Our analysis did not explicitly test for changes in cell fate, but given the known role of ceh-36 in fate specification [38–40], there may be additional unidentified cells with defects in fate, but not position or cell cycle timing.
Expression of ceh-36 is distinct from its OTX paralogs ceh-37 and ttx-1 but overlaps substantially with unc-30/PITX in early embryos
Most defects in ceh-36(ok795) have low penetrance, so other transcriptional regulators likely function in parallel with ceh-36 to ensure robust development. Therefore, we searched for transcription factors that might act redundantly with ceh-36 (Fig. 5). Previous work demonstrated that the three OTX family members ceh-36, ceh-37, and ttx-1 can rescue the others’ mutant phenotypes when expressed in the appropriate cells [39]. We asked if these genes’ early embryonic expression overlaps with that of ceh-36 by lineage analysis of fluorescent reporters and single molecule (sm)RNA-FISH [43]. Lineage analysis of a ceh-37 promoter-fusion reporter[46] identified ten cells where its expression overlaps spatially but not temporally with ceh-36; the ceh-37 reporter is expressed after CEH-36::GFP in these cells (Fig. 5A). The ceh-37 reporter is also expressed in several lineages that do not express CEH-36::GFP. ceh-37 transcripts identified by smRNA-FISH did not overlap with positions of ceh-36 transcripts prior to the 50-cell stage and there was only a small amount of overlap between the 50 and 200 cell stages (Fig. 5B, S5 Fig). We could detect no embryonic expression of a ttx-1 promoter reporter prior to morphogenesis and little or no overlap between ttx-1 and ceh-36 transcripts by smRNA-FISH (Fig. 5B). We examined these genes’ expression in ceh-36(ok795) by smRNA-FISH and observed no ceh-36 transcripts and no changes in ceh-37 or ttx-1 expression. We also observed no substantial increase in ceh-36(ok795) lethality after ttx-1 or ceh-37 RNAi. This indicates that most ceh-36-expressing cells do not express other OTX homologs in wild-type or ceh-36 mutant embryos, and redundancy with these factors is unlikely to explain the low penetrance of most ceh-36 mutant defects.

We mined the EPIC database of embryonic expression patterns [1,23] for additional factors coexpressed with ceh-36, and identified substantial coexpression with the PITX homolog unc-30. An UNC-30::GFP fosmid “Transgeneome” reporter [22] was transiently expressed at the same time as CEH-36::GFP in the descendants of the ABplp and ABprp progenitor cells (together “ABpxp”), which give rise to diverse cell types, but not in other CEH-36-expressing lineages. We confirmed this expression overlap between endogenous ceh-36 and unc-30 transcripts in ABpxp-derived cells by lineage tracing (Fig. 5C) and observed significant overlap of these genes’ endogenous transcripts by smRNA-FISH between the 28-cell and 50-cell stages (Fig. 5D). ceh-36/OTX and unc-30/PITX both encode bicoid-type homeodomains that are predicted to bind similar target sequences [47,48]. In addition, the combined frequency of position and cell cycle defects in ceh-36(ok795) embryos was lower (3%) in the ABpxp lineages than in other CEH-36::GFP-expressing cells (12%) suggesting that ceh-36 may have more redundancy in ABpxp than in other lineages. This suggested the possibility that these two factors might act redundantly to regulate the development of the ABpxp lineages.
unc-30 and ceh-36 are redundantly required for embryonic and larval viability
In addition to its early ABpxp expression, we observed UNC-30::GFP in the six embryonic type D GABA-ergic motorneurons as well as a few other neurons (PVP, AWA, ASG, AIB, ASI and GLR) at morphogenesis (bean stage), consistent with the known role of unc-30 in the terminal differentiation of type D neurons [49] (S6 Fig). Consistent with the phenotypes of other unc-30 alleles, the deletion allele unc-30(ok613) is uncoordinated yet fully viable, with no embryonic or larval arrest (Table 1).
We tested for redundancy between unc-30 and ceh-36 by examining the progeny of a strain homozygous for both unc-30(ok613); ceh-36(ok795) and carrying the rescuing extrachromosomal CEH-36::GFP fosmid. Animals that had lost the rescuing transgene displayed 100% lethality (54% embryonic, 46% larval), while embryos expressing CEH-36::GFP had no embryonic lethality and low larval arrest rates (Table 1), with 75% progressing to L4. The residual larval arrest rate could result from transgene mosaicism or incomplete rescue by the CEH-36::GFP transgene. This indicates that ceh-36 and unc-30 are redundantly required for viability.
The unc-30(ok613); ceh-36(ok795) double mutants displayed visible phenotypes characteristic of defects in ABpxp-derived cells not observed in either single mutant (Table 3, Fig. 6). These included variable abnormalities in body morphology (Vab) defects, which are also seen when ABpxp-derived cells fail to act as a substrate for hypodermal enclosure [50,51], “no backend” (Nob) tail defects characteristic of severe defects in patterning posterior cells including many derived from ABpxp [52], and a “rod-like” arrest posture and large edemas near the pharynx characteristic of defects in the excretory system [53], which is formed by descendants of ABpxp cells. Double mutants did not contain the more anterior head “vacuoles” we saw in ceh-36(ok795) single mutants; however this phenotype could be masked by the more severe Vab and excretory phenotypes. Taken together, ceh-36 and unc-30 are redundantly required for viability and for aspects of normal development associated with cells produced by ABpxp.

ceh-36 and unc-30 co-regulate lineage patterning in ABpxp
The highly penetrant viability and morphological phenotypes of unc-30(ok613); ceh-36(ok795) double mutants led us to hypothesize that these animals would have more frequent cell lineage and position defects in the cells that normally coexpress both factors. We tested this by automated lineage analysis of six unc-30(ok613); ceh-36(ok795) embryos that had lost the rescuing CEH-36::GFP transgene (Fig. 7, S2 Table). We observed a significant increase in cell cycle and cell position defects in the ABpxp lineages of unc-30; ceh-36 double mutants as compared to ceh-36 alone. Double mutant embryos averaged 15.5 cell cycle defects and 94.7 position defects per embryo in ABpxp compared with 2.25 and 11 in ceh-36 single mutants. We also saw a smaller increase in position defects for cells that do not normally express either CEH-36::GFP or UNC-30::GFP (64 vs 26.1), consistent for a role of the ABpxp cells in migration of cells from other lineages. In contrast, we saw no corresponding increase in cell cycle defects in double mutants for nonexpressing lineages (Fig. 7), and no corresponding defects in UNC-30 single mutants (S7 Fig).
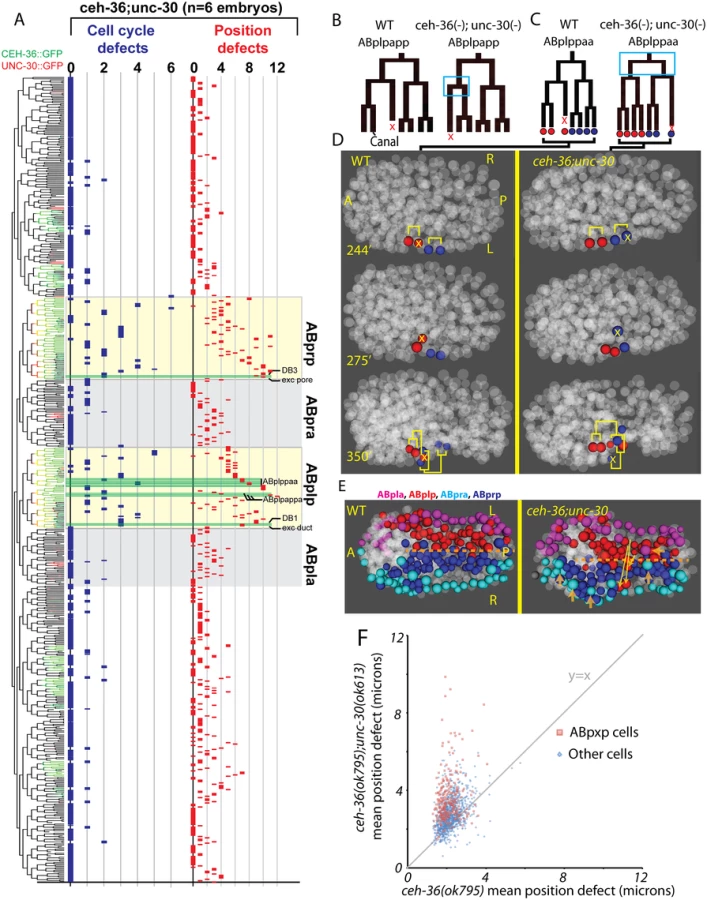
We observed an increased co-occurrence of defects in sister cell pairs in the ABpxp lineages of double mutants (27.2 sisters pairs per embryo) compared with ceh-36(ok795) alone (3.8 sister pairs per embryo). Because unc-30 expression in the ABpxp lineage is even more transient than that of ceh-36, the increased co-occurrence of defective sister cells likely results from primary defects in progenitor cells. We individually examined ceh-36(ok795) single mutant and unc-30(ok613); ceh-36(ok795) embryos for “defect trios” of a defective mother with two defective daughter cells. While ceh-36(ok795) embryos have on average 1.9 of these defect trios (none in ABpxp), double mutant embryos average 13.5 defect trios (10.8 in ABpxp). We observed only one defect trio in three unc-30(ok613); ceh-36(ok795) embryos expressing rescuing CEH-36::GFP, indicating that these defects do not result from unc-30(ok613). Together this suggests that unc-30 and ceh-36 cooperate to regulate robust lineage patterning of ABpxp-derived progenitor cells.
Two cell divisions (ABplpappa and ABplppaa) each displayed a novel type of defect we term “anterior-posterior reversals” in 2 of 6 double mutant embryos. In this class of defect, the anterior daughter of a division adopts the division pattern of the posterior daughter and vice versa. This was evident in the patterns of asymmetric division timing, cell death and migration (Fig. 7B-D). For example, ABplpappap, which undergoes cell death in the wild type, survives and divides in the double mutant. Meanwhile, that cell’s anterior sister ABplpappaa, which should generate RMEV and the excretory canal cell, instead undergoes programmed cell death. Consistent with this being a fate reversal, the division occurs with normal orientation and the daughters of the cell that should have died go on to adopt positions characteristic of RMEV and the excretory canal cell. Defects in these fate-reversed cells or their failure to function correctly in their new location could explain some of the excretory system edema observed in the double mutants.
We identified numerous defects in the organization of the ventral midline in the double mutants. Several ABpxp-derived cells failed to respect the midline and crossed to the opposite side; these were distinct from those seen in ceh-36 single mutants (Fig. 7E). Also, in contrast with ceh-36 single mutants, the double mutants had a much larger number of (presumably nonautonomous) defects in cells that normally express neither ceh-36 nor unc-30. Most of these defects (43/65) were in cells derived from the ABpxa lineages in cells that should form the ventral epidermis. Previous work showed that in the process of ventral enclosure, the epidermal cells migrate over ABpxp-derived substrate cells, some of which are mispositioned in the double mutants. The “leading cells” hyp6/ABpxaappap and hyp7/ABpxaappaa, which initiate ventral enclosure, along with adjacent migrating epidermal cells hyp4, G2, and W, had the largest magnitude defects in cell position of nonexpressing cells (Fig. 7E). This suggests that ceh-36 and unc-30 regulate development of the ABpxp-derived substrate for normal epidermal migration and morphogenesis.
ceh-36 and unc-30 regulate ABpxp-specific expression of mls-2 and excretory system development
Several ABpxp-derived cells had position defects of much larger magnitude than we observed in ceh-36(-) alone (Fig. 7F). Among the cells with the largest defects (average 8.9 micron (>2 cell diameters) deviation from expected position compared with 1.9 microns in ceh-36 alone and 1.6 microns in wild type) were two sister pairs that normally produce two DB neurons and the excretory duct and G1 pore cells (Fig. 8A, B). In wild-type embryos, these cells migrate from anterior lateral positions to the ventral midline where the duct and pore cells connect with the excretory canal cell to form a continuous three-celled tube (Fig. 8C) [54].

Previous work showed that the HMX homeodomain transcription factor mls-2 is required for robust development of the excretory duct and pore [55]. MLS-2::GFP is expressed in several lineages including the precursors of the excretory duct and pore (Fig. 8A, C). To determine if loss of mls-2 leads to cell migration defects similar to those seen in unc-30;ceh-36 double mutants, we traced the lineages and cell positions of the excretory system cells in 23 mls-2 mutant embryos (Fig. 8D). We found migration failure or inappropriate migration into the head in 43% (10/23) of duct cells and 57% (13/23) of pore cells indicating that mls-2 is required for robust migration of these cells. Consistent with this, previous work [55] found that 5 of 25 mls-2 mutant larvae were missing an excretory tube cell; the difference in rates suggests that in some cases the misplaced cells may eventually migrate to the correct position, or that another cell may sometimes adopt a duct or pore fate.
We tested whether mls-2 expression depends on ceh-36 and unc-30 by measuring the expression of a genomically integrated rescuing MLS-2::GFP reporter [55] in unc-30; ceh-36 double mutant embryos. MLS-2::GFP was expressed normally in ceh-36(ok795) single mutant embryos (n = 6). Since MLS-2::GFP is expressed later and ~10-fold more strongly than CEH-36::GFP in the excretory duct and pore lineages (Fig. 8A), we were able to compare MLS-2::GFP expression between double-mutant embryos carrying the rescuing CEH-36::GFP with unrescued embryos. We found that in all (7/7) embryos carrying CEH-36::GFP, MLS-2::GFP was robustly expressed in both the duct and pore precursors, and the duct and pore cells migrated normally. In contrast in six embryos (12 duct/pore lineages) that had lost the rescuing transgene and expressed no CEH-36::GFP, 58% of duct/pore lineages (7/12) had no MLS-2::GFP expression, with the remaining lineages expressing MLS-2::GFP at lower levels than in wild-type or rescued embryos. Absence of MLS-2::GFP expression predicted migration defects; all seven duct or pore cells with no MLS-2::GFP expression had severe migration defects, while three of five MLS-2::GFP-expressing duct/pore cells migrated normally, sometimes with moderate delays. MLS-2::GFP expression in other lineages that don’t normally express ceh-36 or unc-30 was unaffected.
Discontinuities in the excretory tube are associated with formation of edemas and eventual lethality with a characteristic rod-like posture [53–56]. Thus the edemas (Table 3) and rod-like lethality we see in unc-30; ceh-36 double mutants could be explained by the duct and pore migration defects or defects in specification of these cells or the canal cell. We conclude that ceh-36 and unc-30 are required for robust mls-2 expression in ABpxp descendants that give rise to the excretory system, and that misregulation of mls-2 may account for the observed phenotypes in those cells.
Discussion
Our analysis of ceh-36 and unc-30 function across all embryonic cells highlights the complex biology of transcriptional regulation during development that would not have been discovered using traditional approaches. We showed these factors regulate distinct processes including the cell cycle, lineage patterning, cell position, and cell fate specification in many embryonic cells that go on to adopt diverse fates. These factors likely function together to regulate progenitor identity in the ABpxp lineage and ceh-36 likely works with other unknown factors in progenitors from other lineages.
Combinatorial lineage-specific regulatory networks
The lineage-specific cellular phenotypes and defect penetrance in ceh-36(-) and the ABpxp-specific functional interaction between unc-30 and ceh-36 are consistent with context-dependent roles for these factors. Each factor has distinct expression outside of the early ABpxp coexpression, suggesting that each may work with other factors in these other lineages; indeed, unc-30 is a well-established regulator of motor neuron differentiation later in development [49], and ceh-36 mutants have partially penetrant defects in lineages where unc-30 is not expressed. Even within ABpxp, most defects were still partially penetrant even in unc-30; ceh-36 double mutants, consistent with the existence of additional redundant factors. One role of these factors is to directly or indirectly regulate the expression of mls-2. Intriguingly, mls-2 may itself act as a progenitor identity factor, as it regulates the development of lineally-related embryonic cells including glial, excretory and neuronal cells [42,55,57] and is expressed in these cells’ progenitors. In fact, mls-2 is required for expression of ceh-36 in the AWC neurons [42], suggesting that ceh-36 (with unc-30) indirectly regulates its own expression later in development. Similarly, ceh-36 and unc-30 can bind to the unc-30 promoter [4], which is intriguing given that the later expression of unc-30 in GABA-ergic motor neurons occurs in ABpxp-derived cells. We suggest a model in which C. elegans develops robustly with an invariant lineage because each of many lineage-specific TFs [1], provides a small amount of information to each cell about its lineage history. Combining this information from many TFs allows cells to robustly adopt a fate appropriate to their lineage history (Fig. 9). This model suggests that the expression of each individual factor could be regulated by lineage mechanisms (e.g. [1]) in parallel rather than hierarchically. Another intriguing possibility is raised by our observation of cell cycle and migration defects in cells that nonetheless express appropriate terminal fate markers. This suggests that distinct regulators may modularly control different aspects of each cell’s developmental phenotype (i.e. one factor regulates fate, another, cell cycle, and yet another, migration).

ceh-36 and unc-30 are regulators of ABpxp progenitor identity
Our data suggest that ceh-36 and unc-30 act in embryonic progenitor cells to regulate development, which is distinct from their previously characterized role in neuronal terminal differentiation. They are expressed early and transiently in progenitor cells from multiple lineages and these progenitors give rise to varied cell types, similar to multipotent progenitors in other organisms. The migration and cell division defects that we observe occur across these distinct cell types, and while most defects were observed in terminal cells, they were clustered in the lineage suggesting an underlying defect in the common ancestor. Together this strongly suggests that defects occurred in progenitor cells, although it does not rule out additional roles in the subset of terminal cells where ceh-36 expression persists. Early progenitor factors such as ceh-36 and unc-30 may regulate factors important in later progenitor cells, but they could also directly regulate genes expressed in terminal cells by creating stable chromatin alterations, as was recently demonstrated for another factor [58]. Cell division and migration patterns in unc-30; ceh-36 double mutant embryos do not, however, suggest a switch in fate from ABpxp to its sister ABpxa or any other recognizable sublineage. Thus, other ABpxp factors remain to be discovered or other factors are required to specify alternative progenitor fates.
Mechanisms ensuring developmental robustness
Gene regulatory networks are generally robust against biological noise and often employ transcription factors (TFs) with overlapping or redundant functions to decrease transcriptional and phenotypic variability [59]. For example, in C. elegans, redundant pairs of GATA factors regulate intestine development [60], and similar redundancy exists for T-box factors [6,61] and HLH factors [62]. Despite the superficial redundancy of these factors, in some cases the single mutants exhibit decreased robustness in fate determination and partial penetrance phenotypes [60,63]. Our finding of similar redundancy between the more divergent homeodomain factors from the PITX and OTX classes indicates that redundancy can occur between factors with ancient divergence. Worms, insects, and vertebrates all have PITX and OTX homologs, indicating these factors diverged prior to the common ancestor of these phyla. This is the first demonstration of a genetic interaction between these factors that could reflect functional redundancy. Since PITX and OTX factors can bind the same sequence motif this redundancy could reflect regulation of shared targets; consistent with this, a large-scale study of TF binding by yeast 1-hybrid analysis identified binding of ceh-36 and unc-30 to highly overlapping sets of promoters [4]. However it is also possible that they work through independent parallel mechanisms. Intriguingly, vertebrate PITX and OTX homologs have some expression overlap in the pituitary and nervous system, and it will be interesting to determine whether they act together in vertebrates. Our approach of studying robustness across an entire organism at a single-cell level provides the opportunity to sensitively identify cells where each factor or combination of factors plays a role. For example, the overlapping functions of ceh-36 and unc-30 in the ABpxp sublineage allowed us to identify their role in regulating mls-2 expression in the developing excretory system.
Regulation of lateralization
Previous studies identified ceh-36 as a regulator of lateral asymmetry for the MI [40] and ASE [38] neurons. The pharyngeal MI neuron is derived from a right lineage, and the left equivalent lineage produces seemingly equivalent cells except for an epithelial cell, e3D, in place of the MI neuron. Mutations in ceh-36 transform MI into an e3D-like cell, and this asymmetry is driven by asymmetric ceh-36 expression in the MI progenitors and not those of e3D [40]. Surprisingly, the same phenotype occurs in a truncating mutant in histone H3, likely acting downstream of ceh-36 [64]. The fact that loss of either an asymmetrically expressed factor (ceh-36) or a symmetrically expressed factor (histone H3) leads to the same phenotype underscores that asymmetry in regulatory networks can influence which cells have phenotypes. While we do observe asymmetric CEH-36::GFP expression in MI, we found that most expression in other lineages is L-R symmetric and most penetrant defects were seen in both symmetric pairs. However we did identify defects in lateral identity, such as the migrations of the left intestinal muscle and anal depressor, in cells where ceh-36 expression is normally L-R symmetric. This suggests that ceh-36 contributes to the regulation of lateral identity even in cells where it is symmetrically expressed.
Prospects for comprehensive phenotyping of progenitor TF mutants
Although our approach improved the sensitivity for detection of cellular phenotypes compared to previous studies and methods, it is likely that additional cellular defects remain unidentified. For example we identified many defects in only one or two embryos; further improvements to automated cell tracking methods to increase accuracy and reduce curation time would allow analysis of higher numbers of embryos and more sensitive and reliable identification of lower penetrance defects, In the absence of markers for terminal differentiation, a cell with normal migration and division patterns but altered terminal fate cannot be detected. Repeating lineage tracing with a panel of strains expressing distinct fate markers can increase the power to detect lineage transformations [25], but this approach is labor-intensive. On the other hand, some of the cell position defects we identified were apparent only by lineage tracing and not when scored using a terminal fluorescent marker in larvae, which reflects the high sensitivity of the quantitative methods and possibly the correction of some position defects later in development. The power of future applications of lineage-based phenotyping methods would be increased by new methods to directly assay fate transformation while maintaining throughput; such as by analyzing multiple fate markers simultaneously in different colors.
Materials and Methods
Alleles
ceh-36(ks86) X
ceh-36(ky640) X
ceh-36(ky646) X
ceh-36(ok795) X
ceh-37(ok272) X
ceh-37(ok642) X
unc-30(ok613) IV
unc-119(tm4063) III
mls-2(cs71) X
Reporters
bwIs2[flp-1::GFP + pRF4(rol-6(su1006))][65]
nsIs396[sams-5 3′::4xNLS-GFP + lin-15(+)] V [40]
ntIs1[lin-15(+); gcy-5::GFP][38]
sEx14784[ceh-37::GFP][46]
ujEx173[CEH-36::GFP + unc-119(+)]
ujEx130[CEH-36::GFP + myo-2::mCherry + myo-3::mCherry]
oyIs48[ceh-36 2KB promoter::GFP][39]
stIs10501[ceh-36 5KB promoter::HIS-24-mCherry][1]
ujIs113[pie-1::mCherry::H2B + unc-119(+); Pnhr-2::mCherry::histone + unc-119(+)] II
wgIs108[FKH-4::GFP+ unc-119(+)] I [22]
wwIs19[hlh-6::GFP + unc-119(+)] X [66]
csIs55[MLS-2::GFP] X [55]
wgIs395[UNC-30::GFP+unc-119(+)]
Growth conditions and genetics
All strains were grown as previously described [67]. N2 was used as the wild-type reference strain. All manipulations were performed at room temperature (21°C).
Knockout consortium alleles ceh-36(ok795) and unc-30(ok613) were outcrossed three times. VC579 ceh-36(ok795)/szT1 hermaphrodites were mated with males carrying an extrachromosomal copy of ceh-36(+)::GFP (ujEx173), and F2 progeny were tested for ok795, which deletes 406 base pairs of ceh-36, by PCR. Additional outcrossing of ceh-36(ok795) was with N2 males. unc-30(ok613) was outcrossed by mating unc-30(ok613) hermaphrodites with N2 males and picking F2 Unc progeny. Combinations of reporters with ceh-36(-) were created using a mating strategy that did not produce heterozygous ceh-36(-) hermaphrodites at any step or else were verified using PCR.
Combinations of unc-30(ok613) and ceh-36(ok795) were created using nT1[qIs51](IV;V) to balance unc-30(ok613) while testing for ceh-36(ok795) by PCR. unc-30(ok613)/nT1[qIs51](IV;V); ceh-36(ok795) males were mated with unc-119(tm4063); ceh-36(ok795); ujEx173[ceh-36::GFP + unc-119(+)] hermaphrodites, and F2 Unc progeny with the genotype unc-30(ok613); ceh-36(ok795); ujEx173 were isolated. ujEx173 was generated by microparticle bombardment of the CEH-36::GFP Transgeneome fosmid [22] into unc-119(tm4063) using methods previously described [22,68]. ujIs113 was generated by co-bombardment of pAA64H2B (pie-1::mCherry-H2B::pie-1UTR)[69] and pJIM20_nhr-2 (nhr-2promoter::HIS-24-mCherry::let-858YTR) into unc-119(tm4063). ujEx130 was generated by injection of the CEH-36::GFP transgeneome fosmid into ceh-36(ok795) worms.
Lethality checks
All strains were grown at 20°C for over two generations before scoring. Young adult hermaphrodites were dissected at room temperature in egg buffer (118mM NaCl, 48mM KCl, 2mM CaCl2, 2mM MgCl2, 25mM HEPES) [70], and embryos with four or more cells were transferred onto NGM plates. Embryos were counted and replaced in the 20°C incubator. Embryonic lethality was determined by counting unhatched embryos on the subsequent two days. Due to a variable rate of larval development for ceh-36(-) mutants, L4 hermaphrodites were picked off the NGM plates and counted as survivors for one week following dissection. We observed no L4 lethality or adult sterility. Similar rates of lethality for ceh-36(ok795) were obtained by counting eggs laid by free moving ceh-36(ok795) hermaphrodites and following their progeny to the L4 stage. To track the presence of the fosmid in rescued animals, we generated unc-119(tm4063); ceh-36(ok795) worms that were doubly rescued by the presence of the fosmid and reduced the larval lethality of ceh-36(ok795). This allowed us to score absence of the fosmid by the presence of the Unc phenotype.
Lethality checks of unc-30(ok613); ceh-36(ok795) double mutants followed a similar protocol. unc-30(ok613); ceh-36(ok795); ujEx173[ceh-36::GFP + unc-119(+)] young adult hermaphrodites were dissected and embryos counted as described above. Embryonic lethality was scored the next morning. Unhatched embryos were mounted in 20μm beads in egg buffer/methyl cellulose [71] and scored for CEH-36::GFP expression in ASE and AWC neurons. All hatched L1s were examined using a fluorescent dissecting microscope for CEH-36::GFP expression in ASE and AWC neurons (Leica M205FA, Leica Microsystems). CEH-36::GFP expressing and non-expressing L1s were transferred to separate plates, and several larvae were found and transferred the following day. L4 survivors were picked off the NGM plates and counted as survivors for one week following dissection. A similar procedure was used to score survival of ceh-36(ok795) worms carrying wwIs19(hlh-6::GFP).
Examination of L1 phenotypes
All strains were grown at 20°C for over two generations before young adult hermaphrodites were dissected at room temperature in egg buffer and embryos with four or more cells were mounted into a solution of 20μm beads in egg buffer/methyl cellulose. Sealed slides containing 10–15 embryos were incubated overnight at 20°C and scored the following morning.
Examination of unc-30(ok613); ceh-36(ok795) double mutant phenotypes followed the above protocol except that embryos were also scored for CEH-36::GFP expression in ASE and AWC neurons following DIC examination to exclude rescued animals.
Live imaging and lineage tracing
We acquired confocal images with a Leica TCS SP5 resonance scanning confocal microscope (67 z planes at 0.5 μm spacing and 1.5 minute time spacing) and generated lineages using StarryNite and AceTree as previously described [20,21,72–75]. Embryos were mounted in egg buffer/methyl cellulose with 20μm beads as spacers [71] and imaged at 22°C using a stage temperature controller (Brook Industries, Lake Villa, IL).
Detection of deviation in mutant embryos
We updated the 4D reference model of wild-type C. elegans embryogenesis through the 600-cell stage using eighteen embryos expressing fluorescently tagged histone by tracing four embryos to the comma stage. Deviation of cell-cycle length, division orientation, and anterior-posterior position for eight ujIs113; ceh-36(ok795) embryos, one ujIs113; ceh-36(ky646) and five ujIs113; unc-30(ok613); ceh-36(ok795); ujEx173(CEH-36(+)::GFP) embryos was calculated as previously described [27]. Deviant cell cycle length was defined as beyond three standard deviations and five minutes of the average wild-type cell-cycle length. For position defects, we calculated the expected position of each cell in the embryo based on the overall rotation of the embryo and the wild-type model and scored the distance from the expected position. Cells were considered mispositioned if their mean or maximum distance was more than 3.5 standard deviations greater than the wild-type mean. We also developed a heuristic “neighbor distance” score, consisting of the mean distance of the cell to the 10 cells that are closest to that cell in wild-type embryos, and required 3.5 standard deviation defects in this score as well. Deviant cell position was confirmed by comparison of time-lapse 3D-models for both mutant and wildtype embryos. Defects in all cells identified through statistical analysis mentioned in the text were confirmed by manual retracing of curated lineages. For bootstrap analysis of defective sister pairs, the number of total defective cells (X) and defective sister pairs (Y) were separately counted for each embryo as well as for defined subgroups (e.g. the ABpxp lineage or ceh-36 expressing versus non-expressing). Cells born before the onset of ceh-36 expression were not considered. The number of defective sister cells expected by chance was determined by 100,000 iterations of counting sister pairs from (X) randomly picked cells from a defined subgroup. A p-value was calculated by dividing the total number of iterations equal to or greater than the observed value (Y) with 100,000.
Expression analysis
Mixed-stage embryos were picked into a solution of 10mM sodium azide and 1% methyl cellulose in egg buffer with 25μm beads on top of a glass slide. Coverslips were sealed using petroleum jelly, and embryos became immobilized due to azide and hypoxia. All fluorescent reporters were scored by analyzing confocal GFP and DIC z-stacks of pretzel-stage embryos, which provided a more discrete developmental stage than possible in larvae due to the larval arrest of ceh-36(-) mutants. Positional defects and wild-type variation of fluorescent reporters were measured using LASAF software. Single-molecule RNA FISH was performed as previously described [43,76].
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Murray JI, Boyle TJ, Preston E, Vafeados D, Mericle B, et al. (2012) Multidimensional regulation of gene expression in the C. elegans embryo. Genome research.
2. Tong AH, Evangelista M, Parsons AB, Xu H, Bader GD, et al. (2001) Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science 294 : 2364–2368. 11743205
3. Ow MC, Martinez NJ, Olsen PH, Silverman HS, Barrasa MI, et al. (2008) The FLYWCH transcription factors FLH-1, FLH-2, and FLH-3 repress embryonic expression of microRNA genes in C. elegans. Genes Dev 22 : 2520–2534. doi: 10.1101/gad.1678808 18794349
4. Reece-Hoyes JS, Pons C, Diallo A, Mori A, Shrestha S, et al. (2013) Extensive rewiring and complex evolutionary dynamics in a C. elegans multiparameter transcription factor network. Mol Cell 51 : 116–127. doi: 10.1016/j.molcel.2013.05.018 23791784
5. Ritter AD, Shen Y, Fuxman Bass J, Jeyaraj S, Deplancke B, et al. (2013) Complex expression dynamics and robustness in C. elegans insulin networks. Genome Res 23 : 954–965. doi: 10.1101/gr.150466.112 23539137
6. Good K, Ciosk R, Nance J, Neves A, Hill RJ, et al. (2004) The T-box transcription factors TBX-37 and TBX-38 link GLP-1/Notch signaling to mesoderm induction in C. elegans embryos. Development 131 : 1967–1978. 15056620
7. Lehner B, Crombie C, Tischler J, Fortunato A, Fraser AG (2006) Systematic mapping of genetic interactions in Caenorhabditis elegans identifies common modifiers of diverse signaling pathways. Nat Genet 38 : 896–903. 16845399
8. Sulston JE, Horvitz HR (1977) Post-embryonic cell lineages of the nematode, Caenorhabditis elegans. Developmental Biology 56 : 110–156. 838129
9. Sulston JE, Schierenberg E, White JG, Thomson JN (1983) The embryonic cell lineage of the nematode Caenorhabditis elegans. Developmental Biology 100 : 64–119. 6684600
10. Kaletta T, Schnabel H, Schnabel R (1997) Binary specification of the embryonic lineage in Caenorhabditis elegans. Nature 390 : 294–298. 9384382
11. Mello CC, Draper BW, Krause M, Weintraub H, Priess JR (1992) The pie-1 and mex-1 genes and maternal control of blastomere identity in early C. elegans embryos. Cell 70 : 163–176. 1623520
12. Bowerman B, Draper BW, Mello CC, Priess JR (1993) The maternal gene skn-1 encodes a protein that is distributed unequally in early C. elegans embryos. Cell 74 : 443–452. 8348611
13. Mello CC, Draper BW, Priess JR (1994) The maternal genes apx-1 and glp-1 and establishment of dorsal-ventral polarity in the early C. elegans embryo. Cell 77 : 95–106. 8156602
14. Lin R, Thompson S, Priess JR (1995) pop-1 encodes an HMG box protein required for the specification of a mesoderm precursor in early C. elegans embryos. Cell 83 : 599–609. 7585963
15. Zhu J, Hill RJ, Heid PJ, Fukuyama M, Sugimoto A, et al. (1997) end-1 encodes an apparent GATA factor that specifies the endoderm precursor in Caenorhabditis elegans embryos. Genes Dev 11 : 2883–2896. 9353257
16. Tenenhaus C, Schubert C, Seydoux G (1998) Genetic requirements for PIE-1 localization and inhibition of gene expression in the embryonic germ lineage of Caenorhabditis elegans. Developmental Biology 200 : 212–224. 9705228
17. Azzaria M, Goszczynski B, Chung MA, Kalb JM, McGhee JD (1996) A fork head/HNF-3 homolog expressed in the pharynx and intestine of the Caenorhabditis elegans embryo. Dev Biol 178 : 289–303. 8812130
18. Fukushige T, Hawkins MG, McGhee JD (1998) The GATA-factor elt-2 is essential for formation of the Caenorhabditis elegans intestine. Dev Biol 198 : 286–302. 9659934
19. Fukushige T, Krause M (2005) The myogenic potency of HLH-1 reveals wide-spread developmental plasticity in early C. elegans embryos. Development 132 : 1795–1805. 15772130
20. Bao Z, Murray JI, Boyle T, Ooi SL, Sandel MJ, et al. (2006) Automated cell lineage tracing in Caenorhabditis elegans. Proc Natl Acad Sci U S A.
21. Murray JI, Bao Z, Boyle TJ, Boeck ME, Mericle BL, et al. (2008) Automated analysis of embryonic gene expression with cellular resolution in C. elegans. Nat Methods 5 : 703–709. doi: 10.1038/nmeth.1228 18587405
22. Sarov M, Murray JI, Schanze K, Pozniakovski A, Niu W, et al. (2012) A genome-scale resource for in vivo tag-based protein function exploration in C. elegans. Cell 150 : 855–866. doi: 10.1016/j.cell.2012.08.001 22901814
23. Mace DL, Weisdepp P, Gevirtzman L, Boyle T, Waterston RH (2013) A High-Fidelity Cell Lineage Tracing Method for Obtaining Systematic Spatiotemporal Gene Expression Patterns in Caenorhabditis elegans. G3 3 : 851–863. doi: 10.1534/g3.113.005918 23550142
24. Hutter H, Schnabel R (1994) glp-1 and inductions establishing embryonic axes in C. elegans. Development 120 : 2051–2064. 7925009
25. Du Z, Santella A, He F, Tiongson M, Bao Z (2014) De novo inference of systems-level mechanistic models of development from live-imaging-based phenotype analysis. Cell 156 : 359–372. doi: 10.1016/j.cell.2013.11.046 24439388
26. Giurumescu CA, Kang S, Planchon TA, Betzig E, Bloomekatz J, et al. (2012) Quantitative semi-automated analysis of morphogenesis with single-cell resolution in complex embryos. Development.
27. Richards JL, Zacharias AL, Walton T, Burdick JT, Murray JI (2013) A quantitative model of normal Caenorhabditis elegans embryogenesis and its disruption after stress. Developmental biology 374 : 12–23. doi: 10.1016/j.ydbio.2012.11.034 23220655
28. Simeone A, Acampora D, Mallamaci A, Stornaiuolo A, D’Apice MR, et al. (1993) A vertebrate gene related to orthodenticle contains a homeodomain of the bicoid class and demarcates anterior neuroectoderm in the gastrulating mouse embryo. EMBO J 12 : 2735–2747. 8101484
29. Chen S, Wang QL, Nie Z, Sun H, Lennon G, et al. (1997) Crx, a novel Otx-like paired-homeodomain protein, binds to and transactivates photoreceptor cell-specific genes. Neuron 19 : 1017–1030. 9390516
30. Matsuo I, Kuratani S, Kimura C, Takeda N, Aizawa S (1995) Mouse Otx2 functions in the formation and patterning of rostral head. Genes Dev 9 : 2646–2658. 7590242
31. Acampora D, Mazan S, Avantaggiato V, Barone P, Tuorto F, et al. (1996) Epilepsy and brain abnormalities in mice lacking the Otx1 gene. Nat Genet 14 : 218–222. 8841200
32. Kablar B, Vignali R, Menotti L, Pannese M, Andreazzoli M, et al. (1996) Xotx genes in the developing brain of Xenopus laevis. Mech Dev 55 : 145–158. 8861095
33. Andreazzoli M, Pannese M, Boncinelli E (1997) Activating and repressing signals in head development: the role of Xotx1 and Xotx2. Development 124 : 1733–1743. 9165121
34. Furukawa T, Morrow EM, Cepko CL (1997) Crx, a novel otx-like homeobox gene, shows photoreceptor-specific expression and regulates photoreceptor differentiation. Cell 91 : 531–541. 9390562
35. Zhang Y, Miki T, Iwanaga T, Koseki Y, Okuno M, et al. (2002) Identification, tissue expression, and functional characterization of Otx3, a novel member of the Otx family. J Biol Chem 277 : 28065–28069. 12055180
36. Viczian AS, Vignali R, Zuber ME, Barsacchi G, Harris WA (2003) XOtx5b and XOtx2 regulate photoreceptor and bipolar fates in the Xenopus retina. Development 130 : 1281–1294. 12588845
37. Ohtoshi A, Behringer RR (2004) Neonatal lethality, dwarfism, and abnormal brain development in Dmbx1 mutant mice. Mol Cell Biol 24 : 7548–7558. 15314164
38. Chang S, Johnston RJ Jr., Hobert O (2003) A transcriptional regulatory cascade that controls left/right asymmetry in chemosensory neurons of C. elegans. Genes & Development 17 : 2123–2137.
39. Lanjuin A, VanHoven MK, Bargmann CI, Thompson JK, Sengupta P (2003) Otx/otd homeobox genes specify distinct sensory neuron identities in C. elegans. Dev Cell 5 : 621–633. 14536063
40. Nakano S, Ellis RE, Horvitz HR (2010) Otx-dependent expression of proneural bHLH genes establishes a neuronal bilateral asymmetry in C. elegans. Development 137 : 4017–4027. doi: 10.1242/dev.058834 21041366
41. Sarin S, O’Meara MM, Flowers EB, Antonio C, Poole RJ, et al. (2007) Genetic screens for Caenorhabditis elegans mutants defective in left/right asymmetric neuronal fate specification. Genetics 176 : 2109–2130. 17717195
42. Kim K, Kim R, Sengupta P (2010) The HMX/NKX homeodomain protein MLS-2 specifies the identity of the AWC sensory neuron type via regulation of the ceh-36 Otx gene in C. elegans. Development 137 : 963–974. doi: 10.1242/dev.044719 20150279
43. Raj A, van den Bogaard P, Rifkin SA, van Oudenaarden A, Tyagi S (2008) Imaging individual mRNA molecules using multiple singly labeled probes. Nat Methods 5 : 877–879. doi: 10.1038/nmeth.1253 18806792
44. Bao Z, Zhao Z, Boyle TJ, Murray JI, Waterston RH (2008) Control of cell cycle timing during C. elegans embryogenesis. Dev Biol 318 : 65–72. doi: 10.1016/j.ydbio.2008.02.054 18430415
45. Smit RB, Schnabel R, Gaudet J (2008) The HLH-6 transcription factor regulates C. elegans pharyngeal gland development and function. PLoS Genet 4: e1000222. doi: 10.1371/journal.pgen.1000222 18927627
46. Hunt-Newbury R, Viveiros R, Johnsen R, Mah A, Anastas D, et al. (2007) High-throughput in vivo analysis of gene expression in Caenorhabditis elegans. PLoS Biol 5: e237. 17850180
47. Berger MF, Badis G, Gehrke AR, Talukder S, Philippakis AA, et al. (2008) Variation in homeodomain DNA binding revealed by high-resolution analysis of sequence preferences. Cell 133 : 1266–1276. doi: 10.1016/j.cell.2008.05.024 18585359
48. Jolma A, Yan J, Whitington T, Toivonen J, Nitta KR, et al. (2013) DNA-binding specificities of human transcription factors. Cell 152 : 327–339. doi: 10.1016/j.cell.2012.12.009 23332764
49. Jin Y, Hoskins R, Horvitz HR (1994) Control of type-D GABAergic neuron differentiation by C. elegans UNC-30 homeodomain protein. Nature 372 : 780–783. 7997265
50. Williams-Masson EM, Malik AN, Hardin J (1997) An actin-mediated two-step mechanism is required for ventral enclosure of the C. elegans hypodermis. Development 124 : 2889–2901. 9247332
51. Chin-Sang ID, George SE, Ding M, Moseley SL, Lynch AS, et al. (1999) The ephrin VAB-2/EFN-1 functions in neuronal signaling to regulate epidermal morphogenesis in C. elegans. Cell 99 : 781–790. 10619431
52. Van Auken K, Weaver DC, Edgar LG, Wood WB (2000) Caenorhabditis elegans embryonic axial patterning requires two recently discovered posterior-group Hox genes. Proceedings of the National Academy of Sciences of the United States of America 97 : 4499–4503. 10781051
53. Stone CE, Hall DH, Sundaram MV (2009) Lipocalin signaling controls unicellular tube development in the Caenorhabditis elegans excretory system. Developmental biology 329 : 201–211. doi: 10.1016/j.ydbio.2009.02.030 19269285
54. Abdus-Saboor I, Mancuso VP, Murray JI, Palozola K, Norris C, et al. (2011) Notch and Ras promote sequential steps of excretory tube development in C. elegans. Development 138 : 3545–3555. doi: 10.1242/dev.068148 21771815
55. Abdus-Saboor I, Stone CE, Murray JI, Sundaram MV (2012) The Nkx5/HMX homeodomain protein MLS-2 is required for proper tube cell shape in the C. elegans excretory system. Developmental biology.
56. Mancuso VP, Parry JM, Storer L, Poggioli C, Nguyen KC, et al. (2012) Extracellular leucine-rich repeat proteins are required to organize the apical extracellular matrix and maintain epithelial junction integrity in C. elegans. Development 139 : 979–990. doi: 10.1242/dev.075135 22278925
57. Yoshimura S, Murray JI, Lu Y, Waterston RH, Shaham S (2008) mls-2 and vab-3 Control glia development, hlh-17/Olig expression and glia-dependent neurite extension in C. elegans. Development 135 : 2263–2275. doi: 10.1242/dev.019547 18508862
58. Cochella L, Hobert O (2012) Embryonic priming of a miRNA locus predetermines postmitotic neuronal left/right asymmetry in C. elegans. Cell 151 : 1229–1242. doi: 10.1016/j.cell.2012.10.049 23201143
59. Raj A, Rifkin SA, Andersen E, van Oudenaarden A (2010) Variability in gene expression underlies incomplete penetrance. Nature 463 : 913–918. doi: 10.1038/nature08781 20164922
60. Maduro MF, Hill RJ, Heid PJ, Newman-Smith ED, Zhu J, et al. (2005) Genetic redundancy in endoderm specification within the genus Caenorhabditis. Dev Biol 284 : 509–522. 15979606
61. Andachi Y (2004) Caenorhabditis elegans T-box genes tbx-9 and tbx-8 are required for formation of hypodermis and body-wall muscle in embryogenesis. Genes to cells: devoted to molecular & cellular mechanisms 9 : 331–344.
62. Neves A, Priess JR (2005) The REF-1 family of bHLH transcription factors pattern C. elegans embryos through Notch-dependent and Notch-independent pathways. Dev Cell 8 : 867–879. 15935776
63. Boeck ME, Boyle T, Bao Z, Murray J, Mericle B, et al. (2011) Specific roles for the GATA transcription factors end-1 and end-3 during C. elegans E-lineage development. Developmental Biology 358 : 245–255.
64. Nakano S, Stillman B, Horvitz HR (2011) Replication-coupled chromatin assembly generates a neuronal bilateral asymmetry in C. elegans. Cell 147 : 1525–1536. doi: 10.1016/j.cell.2011.11.053 22177093
65. Wightman B, Ebert B, Carmean N, Weber K, Clever S (2005) The C. elegans nuclear receptor gene fax-1 and homeobox gene unc-42 coordinate interneuron identity by regulating the expression of glutamate receptor subunits and other neuron-specific genes. Dev Biol 287 : 74–85. 16183052
66. Grove CA, De Masi F, Barrasa MI, Newburger DE, Alkema MJ, et al. (2009) A multiparameter network reveals extensive divergence between C. elegans bHLH transcription factors. Cell 138 : 314–327. doi: 10.1016/j.cell.2009.04.058 19632181
67. Brenner S (1974) The genetics of Caenorhabditis elegans. Genetics 77 : 71–94. 4366476
68. Praitis V, Casey E, Collar D, Austin J (2001) Creation of low-copy integrated transgenic lines in Caenorhabditis elegans. Genetics 157 : 1217–1226. 11238406
69. McNally K, Audhya A, Oegema K, McNally FJ (2006) Katanin controls mitotic and meiotic spindle length. J Cell Biol 175 : 881–891. 17178907
70. Shaham S (2006) WormBook: Methods in Cell Biology. In: Community TCeR, editor. Wormbook.
71. Bao Z, Murray JI (2011) Mounting Caenorhabditis elegans embryos for live imaging of embryogenesis. Cold Spring Harbor protocols 2011. doi: 10.1101/pdb.prot065599 21880814
72. Boyle TJ, Bao Z, Murray JI, Araya CL, Waterston RH (2006) AceTree: a tool for visual analysis of Caenorhabditis elegans embryogenesis. BMC Bioinformatics 7 : 275. 16740163
73. Murray JI, Bao Z, Boyle T, Waterston RH (2006) The lineaging of fluorescently-labeled Caenorhabditis elegans embryos with StarryNite and AceTree. Nature Protocols 1 : 1468–1476. 17406437
74. Santella A, Du Z, Nowotschin S, Hadjantonakis AK, Bao Z (2010) A hybrid blob-slice model for accurate and efficient detection of fluorescence labeled nuclei in 3D. BMC Bioinformatics 11 : 580. doi: 10.1186/1471-2105-11-580 21114815
75. Murray JI, Bao Z (2012) Automated Lineage and Expression Profiling in Live Caenorhabditis elegans Embryos. Cold Spring Harbor protocols 2012. doi: 10.1101/pdb.prot070615 22854571
76. Raj A, Tyagi S (2010) Detection of individual endogenous RNA transcripts in situ using multiple singly labeled probes. Methods Enzymol 472 : 365–386. doi: 10.1016/S0076-6879(10)72004-8 20580972
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2015 Číslo 3
Nejčtenější v tomto čísle
- Clonality and Evolutionary History of Rhabdomyosarcoma
- Morphological Mutations: Lessons from the Cockscomb
- Maternal Filaggrin Mutations Increase the Risk of Atopic Dermatitis in Children: An Effect Independent of Mutation Inheritance
- Transcriptomic Profiling of Reveals Reprogramming of the Crp Regulon by Temperature and Uncovers Crp as a Master Regulator of Small RNAs