
Pervasive Variation of Transcription Factor Orthologs Contributes to Regulatory Network Evolution
The phenotypic differences observed between closely related organisms are thought to be due largely to changes in regulatory networks. Changes in transcriptional networks can occur via mutations in cis binding sites, for which there are numerous known examples, as well as via binding specificity variation in transcription factors (TFs), a less studied phenomenon that has been observed primarily in multi-gene families. Though large-scale experimental studies ascertaining the extent to which TFs contribute to regulatory network variation across organisms are lacking and would be time-consuming, computational methods can begin to address this challenge. Here, we present a systematic, large-scale analysis of DNA-binding specificity evolution in TF orthologs by computationally leveraging specific features of Cys2-His2 zinc finger proteins, the largest class of TFs in animals and major components of their regulatory programs. We find not only that divergence of DNA-binding residues in 1-to-1 orthologous C2H2-ZFs is pervasive but also that these changes show evidence of functional constraint and occur in a gradual, evolutionarily viable manner. We conclude that the diversification of orthologous TFs has most likely played a major and largely unstudied role in gene regulatory network evolution in metazoans.
Published in the journal:
. PLoS Genet 11(3): e32767. doi:10.1371/journal.pgen.1005011
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1005011
Summary
The phenotypic differences observed between closely related organisms are thought to be due largely to changes in regulatory networks. Changes in transcriptional networks can occur via mutations in cis binding sites, for which there are numerous known examples, as well as via binding specificity variation in transcription factors (TFs), a less studied phenomenon that has been observed primarily in multi-gene families. Though large-scale experimental studies ascertaining the extent to which TFs contribute to regulatory network variation across organisms are lacking and would be time-consuming, computational methods can begin to address this challenge. Here, we present a systematic, large-scale analysis of DNA-binding specificity evolution in TF orthologs by computationally leveraging specific features of Cys2-His2 zinc finger proteins, the largest class of TFs in animals and major components of their regulatory programs. We find not only that divergence of DNA-binding residues in 1-to-1 orthologous C2H2-ZFs is pervasive but also that these changes show evidence of functional constraint and occur in a gradual, evolutionarily viable manner. We conclude that the diversification of orthologous TFs has most likely played a major and largely unstudied role in gene regulatory network evolution in metazoans.
Introduction
Differences in regulatory networks have been proposed to be one of the major determinants of the phenotypic variations observed across organisms [1]. There are two ways by which regulatory networks evolve: changes in cis or trans. The predominant view is that regulatory evolution results mainly from the gain and loss of binding sites in cis-regulatory regions because incremental, evolutionarily viable steps can occur [2–5]. Mutations in transcription factors (TFs), on the other hand, can affect the expression of multiple genes and are thought therefore to be more likely to have detrimental consequences [6–9]. Nevertheless, case studies of specific biological systems have revealed instances of regulatory divergence stemming from TF variation. These variations include gene loss as well as gene duplication where the subsequent paralogs exhibit gain and loss of effector domains, changes in interactions with other regulatory proteins, or novel TF binding potential [10–15]. Specific cases of variations in non-duplicated TFs are also known; an example of 1-to-1 orthologous plant TFs with differing binding specificities was recently discovered [16], along with a homeodomain TF in animals where the addition of a functionally important transcriptional repressor domain is found in insect orthologs [17, 18]. However, a large-scale experimental study ascertaining the extent to which TF variation may contribute to overall regulatory network evolution is still lacking; it would require determining DNA-binding specificities or genomic occupancies for numerous TFs across a diverse set of organisms. Computational methods can begin to address this challenge by leveraging specific features of TFs.
TFs come in distinct structural classes based upon their incorporation of various DNA-binding domains. For many of these domains, the amino acids conferring DNA-binding specificity are known. This provides a platform to assess TF variation via comparative sequence analysis. The Cys2-His2 zinc finger (C2H2-ZF) TFs in particular are an excellent system to probe for variation, as C2H2-ZF domains have a conserved modular structure with binding specificity conferred largely by four DNA-contacting residues within the domain’s alpha-helix [19]. Further, they constitute the largest group of TFs in higher metazoans [20], making up nearly half of all annotated TFs in human, and are major participants in regulatory programs. A C2H2-ZF domain can specify a wide range of three or four base pair targets, and tandem arrays of these domains bind contiguous DNA sequences, giving C2H2-ZF genes the ability to recognize an incredibly diverse set of motifs [21]. These features of C2H2-ZFs allow us to make binding specificity predictions of reasonably high quality for this TF family [22–26].
Previous evolutionary analyses of C2H2-ZF genes revealed a dichotomy in conservation patterns of this family. Tandemly-duplicated C2H2-ZF paralogs exhibit differences in their C2H2-ZF and effector domain counts and can be highly dynamic across short evolutionary distances [27]. The subset of C2H2-ZF KRAB repressor regulators in particular have undergone recent, rapid expansion and divergence in primates and show evidence of adaptive evolution in their DNA-binding domains in human [13, 28–30]. However, such divergence has been found primarily in extremely recent and often species-specific expansions of C2H2-ZFs [31]. In contrast, examples of single-copy 1-to-1 orthologous C2H2-ZF genes have been shown to be highly conserved across large evolutionary distances [27, 32–35]. Prdm9, a C2H2-ZF gene that mediates homologous recombination but is not known to be a TF, is a notable exception to this trend, and is highly dynamic between and within species despite being single-copy [36–40]. Several other orthologous C2H2-ZF genes have been found to diverge across vertebrates, primarily through the gain and loss of C2H2-ZF domains [27, 31]. More generally, however, it is widely believed that 1-to-1 orthologous TFs tend to maintain their DNA-binding specificities whereas paralogous TFs are free to vary [41].
In this paper, we analyze 1-to-1 orthologous C2H2-ZF TFs across closely related species. We leverage the well-understood binding interface of C2H2-ZFs to evaluate DNA-binding specificity changes resulting from C2H2-ZF variation. We focus on C2H2-ZFs in the 12 sequenced Drosophila species (phylogenetic tree in Fig. 1A), as these species benefit from relatively high-quality assembled genomes [42]. Further, as a result of their ∼45 million years of evolutionary divergence [43], they exhibit extensive regulatory variation [44, 45] and diversity in terms of morphology, physiology and ecology [46]. The flies are an ideal model organism set for our study because they have several hundred C2H2-ZF genes which are found in well-established orthologous relationships. This is in contrast to primate genomes where large-scale species-specific expansions complicate 1-to-1 orthology determination.

To assess change, we consider only C2H2-ZF genes that are in 1-to-1 orthologous relationships between D. melanogaster, which we use as a reference species, and each of the 11 other fly species. We find evidence of functional modifications to DNA-binding potential in a significant proportion of these genes. Furthermore, these changes often result in increasingly diverse predicted DNA-recognition motifs as evolutionary distance from D. melanogaster increases, implying that C2H2-ZF DNA-binding specificities may evolve gradually in evolutionarily viable steps. Our findings challenge the assumption that 1-to-1 orthologous TFs are always highly conserved and provide evidence that binding specificity modifications in single-copy TFs may play an important role in the regulatory evolution of Drosophila and other higher metazoans.
Results
C2H2-ZF Domains and Orthogroup Dataset
The initial step of our framework to assess variation in C2H2-ZFs was to assemble groups of orthologs (orthogroups) of C2H2-ZF genes across the 12 fly species (Fig. 1A). We identified all C2H2-ZF domains and sequences in these species using Pfam [47] and HMMER [48] and determined 1-to-1 orthogroups from existing Flybase [43] annotations. We then augmented this set using the UCSC Genome Browser [49] whole genome fly alignment, resulting in a dataset of all C2H2-ZF sequences in the Drosophila species (Methods M1–M3).
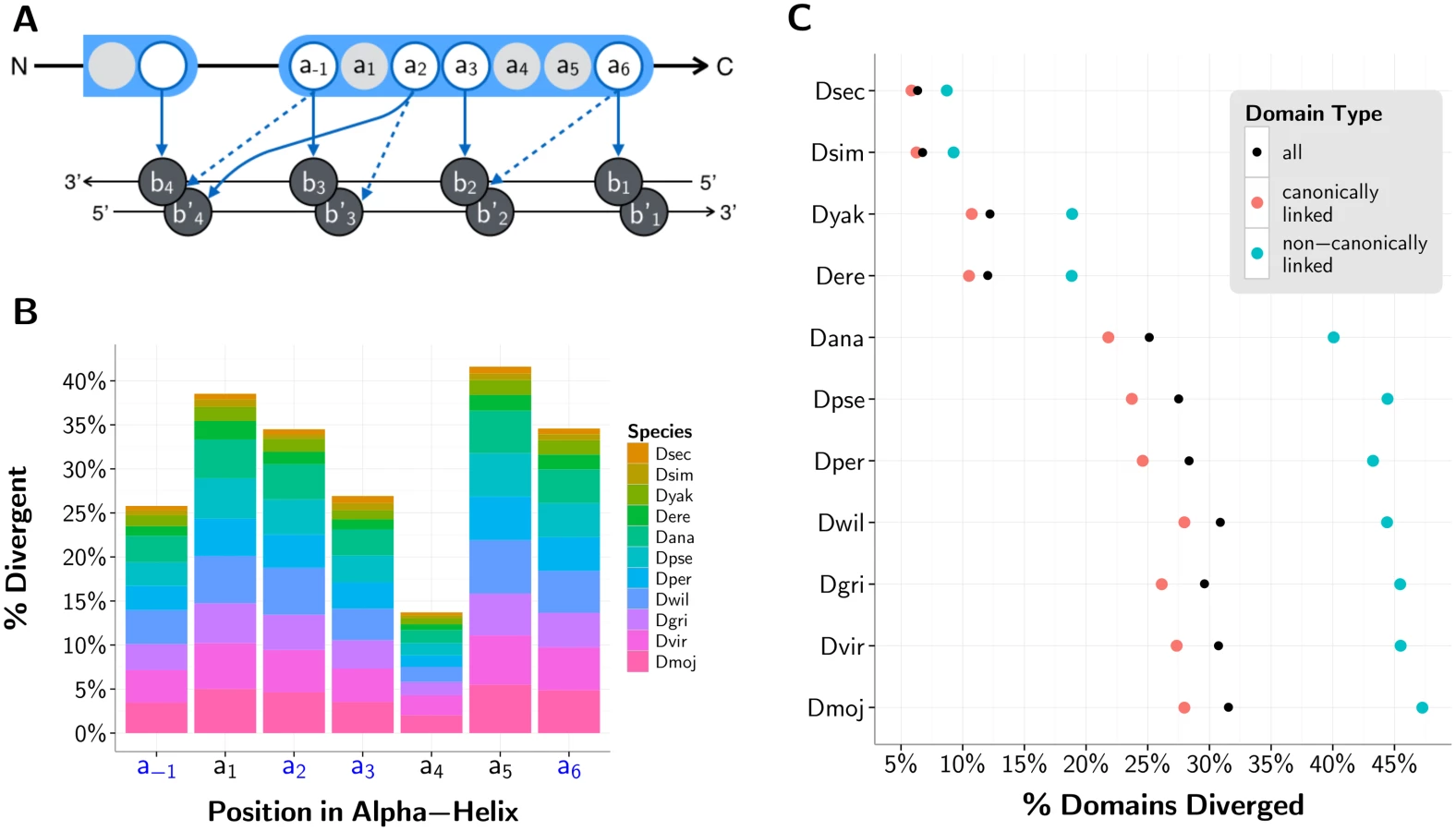
C2H2-ZF domains are known to primarily work in tandem to specify DNA motifs [50] (S1B Fig.), and so we include only those C2H2-ZF genes with 2+ C2H2-ZF domains in our analysis; we refer to these genes as poly-ZF. Tandem C2H2-ZF domains that are separated by canonical linkers—stretches of 5 to 12 amino acids, most often matching the expression TGE[K|R]P[F|Y]X (S1C Fig.)—have the strongest structural evidence for DNA binding [19, 21]. We refer to all domains that are bordered by at least one canonical linker, as defined above, to be “canonically linked.” In D. melanogaster, of the 329 genes with at least one C2H2-ZF domain, 283 have multiple C2H2-ZF domains, and 246 of those contain canonically linked domains.
We found from 319 to 366 genes with at least one C2H2-ZF domain in each of the 12 Drosophila species, 263 to 308 of which were poly-ZF (Fig. 1B, cols. 1–2), in accordance with previous studies’ findings [13, 21]. We found 278 (98.2%) poly-ZF genes in D. melanogaster with a 1-to-1 ortholog in at least one other fly species, and 165 (59.4%) of these were in 1-to-1 relationships across all species. These 278 1-to-1 orthologous poly-ZFs constitute 36.9% of the estimated 753 TFs in D. melanogaster [51]. In each non-melanogaster species, 72.4% to 88.8% of poly-ZF genes had a 1-to-1 ortholog in D. melanogaster (Fig. 1B, col. 3). In the non-melanogaster poly-ZF genes with 1-to-1 orthologs in D. melanogaster, we identified 1000+ C2H2-ZF domains per species (Fig. 1B, col. 4) that are used for comparative analysis in further steps of our framework.
Substantial Loss and Recruitment of C2H2-ZF Domains Relative to D. melanogaster
We first assessed the loss and gain of C2H2-ZF domains across our orthogroups, as the number and arrangement of C2H2-ZF domains likely affects the binding specificity of each poly-ZF gene. D. melanogaster domains are considered “lost” in each non-melanogaster species without a corresponding aligned domain; D. melanogaster domains with no aligned domains in any of the other fly species are ignored because they most likely are species-specific D. melanogaster gains. Conversely, domains from non-melanogaster sequences that did not align back to a D. melanogaster domain are considered “gains” with respect to the reference.
The loss and gain of C2H2-ZF domains was recently identified as the major source of divergence in vertebrate ZF paralogs and orthologs [31]. We quantify this phenomenon in 1-to-1 orthologous TFs in D. melanogaster, where we find that between 2.4% and 10.3% of domains were lost in the other fly species (Fig. 2A), and between 0.8% and 2.9% of domains from non-melanogaster species were gained with respect to the reference (Fig. 2B). A notable 24.8% of all non-reference poly-ZF genes in 1-to-1 orthologous relationships with a D. melanogaster gene have lost or gained a C2H2-ZF domain with respect to the reference. 75.6% of gains or losses occur outside of or at an end of an array of canonically linked domains. The proportion of domains lost and gained in the non-melanogaster species with respect to the reference increases as the phylogenetic distance from D. melanogaster increases. When considering only canonically linked C2H2-ZF domains, we see the same overall phylogenetic trends, albeit at a lower level.

We note that D. melanogaster benefits from more complete sequencing coverage in comparison to the the other fly genomes [42], and relatively poor coverage and subsequent inaccurate sequence assembly would result in a greater number of unidentified or misidentified domains in those genomes. D. sechellia, D. simulans, and D. persimilis, which exhibit the greatest relative C2H2-ZF domain loss (Fig. 2A), also have the lowest relative coverage: 4.9x, 2.1x, and 4.1x, respectively, compared to between 8.4x and 11.0x for the other species. For this reason, the C2H2-ZF domain gains relative to D. melanogaster are especially noteworthy, while some of the apparent domain losses, especially from D. sechellia, D. simulans, and D. persimilis, may be due to incomplete assemblies.
Pervasive Variation in Specificity-Determining Residues in Aligned C2H2-ZF Domains
Binding specificity may also be altered as a result of deviations in the DNA-contacting, specificity conferring residues in positions -1, 2, 3, or 6 of the C2H2-ZF domain [52] (Fig. 3A). As expected, with the exception of structurally constrained position 4, these four functional sites are more conserved than the neighboring, non-DNA-contacting residues within the domain’s alpha-helix. However, these functional sites still show substantial divergence (Fig. 3B). We consider an aligned domain in any non-reference fly species to be “diverged” if at least one of its residues from positions -1, 2, 3, or 6 has diverged from the D. melanogaster reference. Of the > 98% of domains from poly-ZF genes that aligned between the non-reference sequences and their orthologs in D. melanogaster, we observe from 6.3% of domains diverged (in D. sechellia, last common ancester [LCA] with D. melanogaster ∼2 Mya) to a substantial 31.5% of domains diverged (in D. mojavensis, LCA with D. melanogaster ∼45 Mya) (Fig. 3C). These divergent domains are not confined to a small subset of genes: across the 11 non-reference fly species, 19.5% to 62.4% of poly-ZF genes with 1-to-1 orthologs in D. melanogaster contain at least one divergent C2H2-ZF domain. Moreover, as with the proportion of domains lost and gained with respect to the reference, the proportion of domains diverged steadily increases as phylogenetic distance from D. melanogaster increases. The same trend with slightly lower overall divergence is observed in the subset of canonically linked domains. Of the 37.6% of domains situated in the middle of canonically linked arrays, 15.3% contain divergent binding residues. Of the remaining domains outside of or flanking canonically linked arrays, 25.1% contain divergent binding residues. Arrays of canonically linked domains appear to be under stricter constraints than singleton domains are (S2A-C Fig.). Altogether, changes in these DNA-contacting residues are substantially more frequent than the complete gain or loss of C2H2-ZF domains.
Functional and Evolutionary Importance of Divergent Sites
Diverging DNA-binding residues show conservation within phylogenetic clades
We reasoned that changes in these single-copy TFs relative to D. melanogaster that are functionally important are likely to be conserved across phylogenetic clades. To test this, we extracted sequences from the most closely related pairs of species—D. sechellia and D. simulans (LCA < 2 Mya), D. pseudoobscura and D. persimilis (LCA ∼2 Mya), D. yakuba and D. erecta (LCA ∼5 Mya), and D. virilis and D. mojavensis (LCA ∼25 Mya)—and asked how often a particular mutation with respect to the reference in one species was supported by an identical mutation in its partner species. In all cases, divergent DNA-binding residues in poly-ZF genes from each non-melanogaster fly species exhibit clade support more often than background divergent residues in these genes (Fig. 4A-B); these changes are significant (p < 0.001, binomial test) in 4 species, with small sample sizes a limiting factor in the other species (S1 Table). Residues within and between adjacent C2H2-ZFs are also under structural constraints and may be implicated in secondary binding specificities [54], resulting in their high conservation according to the clade support measure. This trend is particularly apparent in the species closest to D. melanogaster with fewer overall divergent residues. Altogether, this analysis suggests that the substantial binding residue variation we see across species is functional rather than random.

Relatively low evolutionary rate of diverging DNA-binding residues
To further support the claim that observed variations in C2H2-ZF binding residues are functionally important, we estimate site-based evolutionary rates using Rate4Site [55] for all divergent residues per sequence per orthogroup (Fig. 4C). For each sequence, we ranked its divergent residues from lowest to highest evolutionary rates, and normalized these ranks to values between 0 and 1. In each non-reference species across all orthogroups, divergent binding residues evolve more slowly than background residues outside of C2H2-ZF domains (p < 0.001 in the 10 species furthest from D. melanogaster, Wilcoxon test; S1 Table). Because we consider only divergent residues in each sequence, this signal is strongest in species with a large number of total divergent residues per sequence, as normalized ranks are more continuous in these cases and therefore differences between the four classes of residues (i.e., specificity-conferring binding residues, background, non-helical C2H2 residues, and linker regions) are apparent with higher resolution. In the flies furthest away from D. melanogaster, where the most variation from D. melanogaster is observed, the divergent binding residues exhibit the slowest evolutionary rate relative to all other classes of divergent residues, including the structurally constrained non-binding regions within domains and the linker regions between domains in poly-ZF genes. In the four species closest to D. melanogaster, non-binding residues in C2H2-ZF domains appear to evolve as slowly as binding residues themselves, as the low number of total divergent residues per sequence (S1 Table) restricts the resolution of differences between these residue classes.
Population analysis suggests positive selection in evolutionary history
We have shown that divergent binding residues are under functional constraints, yet the pervasiveness of such changes in these 1-to-1 orthologs suggests these deviations may confer an evolutionary advantage and were selected for when they arose. The classic approach for detecting positive selection from cross-species sequences is to calculate dN/dS, the ratio of observed nonsynonymous mutations over all possible nonsynonymously mutable sites to observed synonymous mutations over all possible synonymously mutable sites [56]. However, we found that across the Drosophila species, dS naturally saturates and thus impedes a positive selection signal of dN/dS > 1, suggesting that this measure is inappropriate for use across the evolutionary distances considered here [57].
In order to detect positive selection in the evolutionarily dispersed fly species, therefore, we utilize an alternate approach which considers both cross-species sequences and within-species population data. If a nonsynonymous mutation was neutral and accumulates via random genetic drift, it is more likely to persist as a polymorphism within a population, whereas if such a mutation was advantageous and became fixed rapidly through positive selection, finding nonsynonymous mutations in the same location in population data would be highly unlikely [58]. Consequently, for each divergent residue in each non-reference species, we asked whether that same site was or was not polymorphic in a population of 139 D. melanogaster organisms [59] (Fig. 4D). In every species, a greater proportion of divergent binding residues are disjoint from polymorphic sites than divergent background residues are (p < 1e-15 in all species, binomial test; S1 Table), and in 8 of the 11 species these proportions are greater than those for all other types of diverging residues. In four species, linker regions between domains, which may impact overall specificity by affecting flexibility of canonical binding arrays and the positioning of C2H2-ZF domains within them, had residue changes present as polymorphisms as often as the binding residues themselves. In two species, diverging non-helical residues, which may alter the structure of the DNA-binding domains, also overlapped with polymorphic sites in D. melanogaster as rarely as binding residues did. For the set of diverging residues in each of the 11 species separately, we also computed site frequency spectra [60] to analyze polymorphic sites (S3 Fig.). These polymorphic sites are heavily skewed towards smaller minor allele frequencies, with this trend most evident in the diverging DNA-contacting residues. Altogether, these analyses of a D. melanogaster population suggest that a greater proportion of binding residue divergences in each species were likely advantageous rather than neutral as compared with other variations within poly-ZF genes.
Divergent Residues Lead to Distinct Computationally-Predicted Specificities
We next aimed to ascertain whether and how the variation we observe in poly-ZF orthologs changes binding specificity, as it is possible that distinct assignments of binding residues still specify the same overall recognition motif [26, 61]. We predicted the specificity of each C2H2-ZF domain with a predictor [24, 62] that utilizes a linear support vector machine based on an expanded structural model (Fig. 3A); this method is referred to as SVM. Since no method can predict binding specificity perfectly and consensus predictions are more likely to be correct (S1 Text, S2 Table), we compared the SVM predictions to those produced by an independent predictor referred to as ML that uses a probabilistic recognition code generated via maximum likelihood [22], and a random forest based predictor referred to as RF [25]. We calculate the average Pearson correlation coefficients (PCCs) across positions b1 through b4 between SVM predicted position weight matrices (PWMs) and ML and RF PWMs, and consider only the subset of SVM predictions with average PCCs > 0.25 to either of the corresponding ML or RF predictions (S4 Fig.). Of the 17734 aligned binding domains from all 12 fly species, 87.3% passed this confidence threshold; thus, overall there is good agreement between the independent methods on predicted DNA-binding specificities. Results using alternate confidence thresholds of PCC > 0.0, PCC > 0.5 and PCC > 0.75 are found in S3 Table.
We compared the SVM-predicted PWM for each divergent domain in a non-melanogaster species to the predicted PWM for the corresponding, aligned domain in its D. melanogaster ortholog by calculating the average PCC across positions b1 through b4. Overall, 74.2% of divergent domains over the 11 flies exhibit a PCC < 0.25 from their reference domain in at least one predicted position (S5A Fig.). In six non-reference fly species, 100% of all divergent domains exhibit a PCC < 1 from their reference domains in at least one predicted position. Of the remaining five species, < 1% of divergent domains do not show a significant change in predicted specificity in any position compared to their aligned D. melanogaster reference domains. Many domains from non-melanogaster species exhibit a diverged specificity from the reference in more than one predicted position (S5B Fig.). Overall, this analysis suggests that the divergent binding residues within C2H2-ZF domains likely result in changed DNA-binding specificities.
Predicted binding specificities change gradually over evolutionary distance
To establish how specificity may change in relation to phylogenetic distance, we compared the predicted PWM for each D. melanogaster domain to those PWMs predicted for every corresponding aligned domain from the other flies (example domain multiple alignment in Fig. 5A-B). In the majority of such domain alignments between D. melanogaster and the other fly species, we note that as the phylogenetic (species) distance from D. melanogaster increases, the corresponding domain’s predicted specificity change from D. melanogaster also tends to increase. Specifically, we measure change between D. melanogaster and non-melanogaster predicted specificities using PCC, where a lower PCC implies greater change, and so we see that an increase in phylogenetic distance is correlated with a decrease in PCC (Fig. 5C-E). As such, Spearman correlations relating phylogenetic distance to change in predicted specificity (as measured by PCC) were < 0 for 88.7% of domain alignments and < −0.5 for 65.2% of domain alignments (see S3 Table). When we group divergent non-melanogaster domains by species rather than by orthology to a particular D. melanogaster domain, we still observe this same trend, where an increase in phylogenetic distance between D. melanogaster and non-reference species correlates with a decrease in the PCCs between predicted D. melanogaster specificities and non-reference predicted specificities (Fig. 5F). Overall, our analysis of predicted specificities is consistent with a model where DNA-binding specificities diverge gradually over evolutionary time in non-duplicated, 1-to-1 poly-ZF orthologs.

Binding Landscape Divergences Across Species
We next set out to determine whether the variation we observe in poly-ZF DNA-binding residues may result in changes in regulatory network topology. To experimentally test this, we would need experimentally-determined binding specificities and/or genomic occupancies for many poly-ZF genes across the fly species. Although we do not have TF binding data for non-melanogaster flies, there are poly-ZF TFs for which binding specificities or genomic binding locations have been experimentally determined in D. melanogaster.
We first sought to use chromatin-immunoprecipitation (ChIP) data. Of the 12 D. melanogaster poly-ZFs with associated ChIP data from modENCODE [64], five poly-ZFs—three of which exhibit divergences in their DNA-contacting residues and two of which are completely conserved—did not have associated PWMs representing their binding specificities available in the Fly Factor Survey [65], JASPAR [66], or public Transfac [67] databases, thereby precluding any efforts to determine whether these TFs bind in the other fly genomes. The remaining seven poly-ZFs are conserved TFs involved in development; thus, we would not be able to compare how the diverged and conserved poly-ZF genes in this set differ with respect to the loss of binding sites in the non-reference fly genomes. Because most ChIP studies have been carried out at various developmental stages in D. melanogaster and because, as we show in the next section, conserved poly-ZFs are enriched for developmental functions whereas diverged poly-ZFs are not, it is not surprising that few divergent poly-ZFs have associated ChIP data or specific binding at these developmental stages.
We next compiled experimentally-determined binding specificities for 52 fly poly-ZF TFs from the Fly Factor Survey, JASPAR, and public Transfac databases (Fig. 6A) and computationally mapped their binding sites using fimo [68] in the 2000 base pair promoter regions upstream of known genes in D. melanogaster. To obtain a subset of high-confidence binding site predictions in D. melanogaster, we required that the sites be conserved in the four most closely related species—D. sechellia, D. simulans, D. yakuba, and D. erecta. For each TF, we next examined whether high-confidence D. melanogaster binding sites are lost in the remaining seven fly species, and whether orthologous promoter regions are no longer bound in these species. In each species, we compare the fraction of binding sites lost for those TFs with completely conserved DNA-contacting residues across their 1-to-1 orthologs with the fraction lost for those TFs exhibiting some divergence in their DNA-contacting residues as compared to their D. melanogaster orthologs (Methods M4). We note that various features of a TF (e.g., its function) influence the extent to which its binding sites and targets vary across organisms; thus, we compare the conserved and divergent groups of TFs in aggregate.

We find that single-copy poly-ZF orthologs with divergent DNA-contacting residues are significantly more associated with a loss of bound promoter regions than are completely conserved poly-ZF orthologs (p < 1e-9 across all species, Wilcoxon test; Fig. 6B). Changes between the sets of genes predicted to be regulated by D. melanogaster poly-ZFs and the sets of genes predicted to be regulated by their orthologs in other species, therefore, are more common and pronounced when those orthologs show divergences in their DNA-binding domains. When examining individual binding sites that were predicted to be bound by a given D. melanogaster poly-ZF gene, we find that divergent poly-ZFs are significantly more associated with a loss of binding sites than are conserved single-copy poly-ZFs (p < 1e-6 across all species, Wilcoxon test; Fig. 6C). We note that relaxing our criterion for making high-confidence binding site predictions in D. melanogaster by requiring conservation in fewer species does not substantially alter our findings at either the level of promoters or binding sites (S6 Fig.). Altogether, these results suggest that the binding landscapes of divergent poly-ZFs are more different from their D. melanogaster orthologs than are those of conserved poly-ZFs, and subsequently that regulatory network topologies have most likely been affected by variation in 1-to-1 orthologous poly-ZFs.
Diverged Poly-ZFs are Functionally Varied; Conserved Poly-ZFs are Developmentally Enriched
Do divergent poly-ZF genes exhibit distinct biological functions from the set of conserved poly-ZF genes? To answer this question, we divided the genes from our analysis into two main sets: conserved and diverged. The first set contained 82 poly-ZF genes from D. melanogaster with completely conserved DNA-contacting residues across all its orthologs; 28 (34.1%) had orthologs in all other fly species, and 64 (78.0%) contained canonically linked domains. The second set contained 181 D. melanogaster poly-ZF genes with a diverged C2H2-ZF domain in 2+ orthologs; 81 (44.8%) had orthologs in all 11 other fly species, 155 (85.6%) contained canonically linked domains, and 144 (79.6%) contained a divergent canonically linked domain.
Divergent poly-ZFs have limited functional annotations
We ran GO Term Finder [69] on these two gene sets to find enrichment of Gene Ontology terms from the biological process, molecular function, and cellular component association categories, excluding annotations inferred from sequence models, as the presence of C2H2-ZF domains would likely have automatically inferred transcriptional regulation and DNA-binding for all poly-ZFs. Both the conserved and divergent sets are separately enriched for DNA-templated regulation of transcription, positive or negative regulation of gene expression, and regulation of RNA metabolic process and are localized in the nucleus (p < 0.001, Bonferroni-corrected hypergeometric test; S4 Table). Unsurprisingly, those poly-ZF genes with conserved binding specificities are also enriched for such developmental functions as segmentation, morphogenesis, and organ development. The poly-ZF genes with divergent C2H2-ZF domains, on the other hand, exhibit no additional functional enrichments, even when considering only genes with orthologs in every species, genes with canonically linked domains, or the gene set augmented with functional protein partners from STRING [70] (version 9.1, interaction scores > 0.9).
Although no functions beyond transcriptional regulation were significantly enriched across the entire set of divergent poly-ZF genes, certain genes within this set were annotated with functions such as organ development (muscle, respiratory system, axon, wing disc), dorsal/ventral pattern formation, and neurogenesis. Indeed, several known TFs are found in the set of divergent genes. For instance, hermaphrodite (her), a regulator required for sexual differentiation [71], has four C2H2-ZFs, the first of which has a mutation in position 2 in D. yakuba and D. erecta, the fourth of which has a mutation in position 2 in D. pseudoobscura and D. persimilis, and the second and third of which both have mutations in position 2 in D. willistoni. Matotopetli (topi), a testis-specific regulator of meiosis and terminal differentiation [72], has 11 C2H2-ZF domains in D. melanogaster, of which five were mutated in the six species furthest from D. melanogaster, four were mutated in D. ananassae, two were mutated in D. yakuba, and one was mutated in D. sechellia and D. erecta. Tiptop (tio), a repressor of the teashirt TF and regulator of clypeolabrum patterning [73], has five C2H2-ZF domains, the first, third, and fifth of which have diverged from the D. melanogaster ortholog in seven other species. Overall, however, functional analysis reveals a clear study bias toward conserved, developmentally involved TFs.
Co-domain presence suggests transcriptional regulation activity
Because we excluded GO terms inferred from sequence models when looking for functional enrichment, we separately analyzed the co-domains present in the complete conserved and diverged poly-ZF gene sets to get a better sense of these genes’ functions. We downloaded domain annotations from InterPro [74]. Both of these gene sets contain the regulation-related effector domain BTB/POZ, which mediates homomeric dimerization, and additional DNA-binding AT-hook and homeobox domains. Remarkably, a third of poly-ZFs in D. melanogaster contain the ZAD domain; this is largely an insect-specific domain, and its prevalence in fly proteins containing C2H2-ZF domains has been noted before [75–77]. While a few proteins with ZAD domains are completely conserved across the flies, nearly 90% exhibit some divergence, with 78% falling into the divergent set as defined above. Altogether they constitute ∼40% of the divergent set. The domains found uniquely in the conserved set are DZF, a nucleotidyltransferase; SET, a histone methyltransferase found predominantly in enhancer TFs; Ovo, which plays a role in germline sex determination; SANT/Myb, another DNA-binding domain; and ELM2, a domain of unknown function. Divergent C2H2-ZF genes uniquely contain several domains implicating their regulatory activity—PHD, responsible for chromatin-mediated transcriptional regulation; PWWP, ING, WD40, and bromodomain, all important for chromatin remodeling, genome stability maintenance, protein-histone association, and cell cycle progression regulation; EPL1, involved in transcriptional activation; and BESS, TRAF, and SWR1, which direct a variety of protein-protein interactions.
Divergent poly-ZFs are less essential and more widespread
Additional phenotypic information derived from gene knockout experiments are available via FlyBase and modENCODE [64] for 97.3% of conserved poly-ZF genes and 96.2% of divergent poly-ZF genes. Conserved poly-ZF genes are more often essential than divergent poly-ZF genes are: gene knockouts were lethal for 23.3% of conserved and only 15.8% of diverged genes. An additional 43.8% and 21.5% of conserved and divergent genes respectively had semi-lethal, recessive lethal, or larval lethal knockouts. In concurrence with the GO term enrichment, we found that 37.0% of conserved poly-ZF gene knockouts affected phenotypes in the embryonic or larval stages, whereas only 12.0% of diverged poly-ZF knockouts had a phenotypic effect during development.
To further determine where and when poly-ZF genes affect phenotype, we looked at expression locale and levels derived from FlyAtlas [78], available for 95.8% of conserved poly-ZFs and 97.5% of diverged poly-ZFs. We considered adult, larval, and germline tissues separately (S7A Fig.). Interestingly, we found that larger proportions of divergent poly-ZFs were found in each tissue than the proportions of conserved poly-ZFs. Although divergent poly-ZFs tended to be present in a larger number of distinct tissues than conserved poly-ZFs were (S7B Fig.), their expression was consistently lower than the expression of conserved poly-ZFs in corresponding tissues (S7C Fig.).
Discussion
Previously, binding site turnover has been shown via ChIP experiments to be an essential component in regulatory network variation across closely-related organisms [79–83] and even across individuals of the same species [84, 85]. Here we present an analysis suggesting that divergence of orthologous TFs also plays a role in regulatory variation.
Over half of the single-copy, poly-ZF 1-to-1 gene orthogroups in Drosophila exhibit variation with respect to the number and arrangement of DNA-binding C2H2-ZF domains and the composition of specificity-conferring residues within these domains. Variations within these specificity-determining positions are known via structural studies to influence the binding specificities of the proteins in which they are found. These mutations’ conservation across phylogenetic clades, low rate of evolution, and rapid fixation as determined by their lack of overlap with population polymorphisms further demonstrate their functional importance. Additionally, predicted specificities of C2H2-ZF domains increasingly diverge as evolutionary distance from the reference D. melanogaster increases, offering evidence that specificity-altering trans changes are feasible and occur in evolutionarily viable steps even in non-duplicated orthologs.
Though C2H2-ZF binding to RNA [86] or protein [87] rather than or in addition to DNA has been observed, several lines of evidence suggest that a large fraction of the domains in our study bind DNA. We focus on only those genes with multiple C2H2-ZF domains, a requirement for specific DNA recognition. Even when we limit our analysis to canonically linked domains, which have the strongest structural evidence for DNA-binding, we observe the same overall divergence trends. Some DNA-binding C2H2-ZFs may regulate processes other than transcription; however, GO term enrichment analysis and co-domain presence suggests that many of these poly-ZFs are regulating transcription and gene expression and are likely interacting with other protein co-factors. Altogether, this suggests that a substantial set of the divergent poly-ZF genes included in our analysis are DNA-binding TFs. However, it is also possible that the likely specificity-altering mutations we see in these DNA-binding TFs may leave overall gene expression unaffected. There are cases of divergent cis-regulatory sequences that do not confer a change in gene expression [88–93], review by [94], as sometimes these binding site changes are accompanied by complementary TF changes [95]. Compensatory change may occur for some of the diverging poly-ZF TFs we observe. For those poly-ZFs with experimentally-derived PWMs in D. melanogaster, however, we see that TF orthologs across the other fly species with diverged DNA-contacting residues are associated with significantly fewer conserved binding sites and bound promoter regions than are TF orthologs with completely conserved DNA-binding domains. This suggests that the substantial trans variations must result in, at minimum, modulated expression changes, as multiple cis mutations co-occurring with and counteracting each trans specificity change would be extremely unlikely.
Poly-ZFs in D. melanogaster that diverge across the flies appear to have several notable characteristics. They tend to have limited functional annotations and are less essential than conserved poly-ZF genes. Further, they tend to be more broadly expressed, albeit at lower levels, than poly-ZF genes whose binding specificities are conserved. Intriguingly, a substantial fraction of diverging poly-ZF genes contain ZAD domains, and the vast majority of all ZAD-containing poly-ZFs diverge in their DNA-contacting residues. Uncovering the functional roles of diverging poly-ZFs, especially those containing ZAD domains, may be a particularly promising avenue for future work.
Earlier work on C2H2-ZF genes in vertebrates has established the plasticity of this class of DNA-binding domains and the potential role these genes may play in shaping species-specific regulatory networks. In particular, the human C2H2-ZF genes that contain KRAB repressor domains have been studied in depth [28, 32, 96, 97]. The KRAB C2H2-ZF family of proteins are unique to tetrapods and have undergone major species-specific segmental and tandem duplications in mammals and primates [98]. Paralogous KRAB-ZF genes residing in these clusters exhibit frequent pseudogenization, loss and gain of binding domains, and evidence of positive selection acting on the DNA-contacting residues within these domains [13, 29, 32, 97]. These findings on paralogous genes are consistent with the long-standing belief that gene duplication followed by subsequent diversification is the primary means by which otherwise conserved genes can accrue functional divergences [99]. Where attempts have been made to identify and evaluate orthologs across species containing these expansions of KRAB-ZFs, orthologs have been found to either be deeply conserved or to exhibit differences in C2H2-ZF domain count rather than in the identities of DNA-binding residues, though a few cases of variation in DNA-binding residues have been previously reported [27, 28, 31]. We note that the plasticity of domains within these expanded C2H2-ZF gene families in vertebrates does not necessarily imply that C2H2-ZF domains in other organisms will have similar properties. Indeed, we see far fewer losses and gains of domains in 1-to-1 C2H2-ZF orthologs in flies as compared to what has been observed in C2H2-ZF gene expansions in primates, and we observe a relatively higher rate of divergence in specificity-conferring residues. It remains to be seen if divergences within DNA-contacting residues are also prevalent in single-copy orthologs of other TF families.
Although prior research has recognized the possibility of TF variation occurring in multi-gene families, it has long been thought that single-copy TFs are under stringent conservation, as loss or change of function mutations in these genes could not be masked by the functional gene products of paralogs and would thus have catastrophic effects. We cannot, of course, rule out the possibility that ancient transient gene duplications and losses have complicated the detection of 1-to-1 orthologs in Drosophila. However, our large-scale results on 1-to-1 C2H2-ZF orthogroups in flies are consistent with a recent experimental case study of specificity divergence of a single-copy TF in plants [16]. Here, binding specificities of 1-to-1 orthologs of the plant TF LEAFY (lfy) were analyzed across algal, moss, and plant species, and three distinct binding preferences were found. The lfy ortholog in hornworts was dubbed a “promiscuous intermediate” as it recognizes all three binding motifs with various preferences. This intermediate, which is not accompanied by a definitive ancestral gene duplication event [100, 101], highlights a means by which TF binding specificity can evolve in single-copy genes. The gradual TF variation we observe may also give rise to such analogous TF intermediates.
In conclusion, we propose that variation in 1-to-1 orthologous TFs can shape regulatory network evolution. Changes in TFs need not be catastrophic. Rather, single amino acid mutations in DNA-contacting positions may result in overall TF binding of similar targets with varying affinities. Such variations provide the opportunity for gradual evolution of binding specificity. We propose that these changes in single-copy TFs may be substantial contributors to overall regulatory evolution in Drosophila and in other metazoans in general.
Materials and Methods
M1. Sequence Collection
Translated protein sequences for the 12 sequenced fly species—D. melanogaster (build r6.01), D. sechellia (r1.3), D. simulans (r1.4), D. yakuba (r1.3), D. erecta (r1.3), D. ananassae (r1.3), D. pseudoobscura (r3.2), D. persimilis (r1.3), D. willistoni (r1.3), D. mojavensis (r1.3), D. virillis (r1.2), and D. grimshawi (r1.3)—were downloaded from FlyBase [43], version FB2014_04. Additional D. simulans sequences were downloaded from the Andolfatto Lab site [102]. To identify C2H2-ZF genes, HMMER’s hmmsearch (versions 2.3.2 [48] and 3.0 [103]) was run on each translated protein file using 12 Pfam HMMs [47], which were selected based upon their similarity to and presence in the same clan as the consensus C2H2-ZF profile (S1B Fig.), zf-C2H2 (PF00096)—zf-C2H2 (PF00096), zf-C2H2_2 (PF12756), zf-C2H2_6 (PF13912), zf-C2H2_jaz (PF12171), zf-C2HC_2 (PF13913), zf-H2C2_5 (PF13909), zf-met (PF12874), zf-met2 (PF12907), zf-BED (PF02892), zf-U1 (PF06220), GAGA (PF09237), DUF3449 (PF11931). Any protein sequence containing at least one HMMER hit with a bit score above the specified gathering domain threshold for that HMM was considered.
C2H2-ZF domains themselves were identified from these proteins as any HMMER hit matching the regular expression CX2, CX8, ΨX2HX3, [H|C], where Ψ is a large, hydrophobic amino acid. Hits that did not match this expression and thus no longer have the structure necessary to bind DNA are considered degenerate, and are not identified as domains. HMMER hits below the corresponding bitscore thresholds but which matched this regular expression were retained in these proteins because C2H2-ZFs are known to occur in tandem, and therefore we are more confident about all C2H2-ZF domains which co-occur with at least one high scoring domain. All C2H2-ZF domains can be found in S5 Table.
Where possible, the longest protein splice form per gene containing all C2H2-ZF domains was selected to represent each gene. If no single protein isoform contained all domains present in the gene, a minimal set of proteins which together include all unique C2H2-ZF domains was selected to represent the gene.
M2. Orthogroup Collection & Augmentation
A list of pairwise orthologs to D. melanogaster was downloaded from FlyBase and from the Andolfatto Lab build of D. simulans [102], and orthogroups were constructed from overlaps of these orthologs. Those orthogroups containing at least one D. melanogaster poly-ZF gene were selected. Of 13273 total original orthogroups, 272 had at least one D. melanogaster poly-ZF gene.
D. melanogaster poly-ZF orthogroups with sequences missing from one or more species were augmented according to the 15 insect whole genome alignment (WGA) from the UCSC Genome Browser [49]. A missing species is defined as any species not present in the orthogroup but present in the phylogenetic subtree rooted at the most recent common ancestor of those species that are present in the orthogroup. For each of the 52 orthogroups containing at least one missing species, known protein sequences were aligned to the UCSC 15-insect WGA using BLAT [104]. Where possible, sequence(s) from the missing species were extracted from the section of the alignment with the best hits and aligned back to their corresponding translated protein files using BLAT again. Gene IDs of proteins with BLAT hits with an e-value cutoff of 0.001 were extracted and, when they were not present in pseudogene lists, were added to the corresponding orthogroups. Through this process, 13 of the orthogroups with missing species were augmented with at least one new gene.
M3. Orthogroup Reconciliation
All 1-to-many (i.e., one gene from D. melanogaster but more than one gene from at least one other species) orthogroups were truncated such that only those species with a single gene in the original orthogroup were included in the new orthogroup. In this manner, our analysis was restricted to variation in 1-to-1 orthologs.
A gene tree was constructed from a multiple alignment for each many-to-many orthogroup using T-Coffee, version 10 [105]. Each of these gene trees was then reconciled with the phylogenetic species tree for the 12 Drosophila species using Notung, version 2.8 [106]. For each input pair of gene and species trees, the reconciled tree output by Notung is marked with the most parsimonious duplication and loss events along ancestral branches, such that branches of the gene tree now coincide with speciation events of the species tree. Each subtree of the reconciled Notung tree was considered separately as a new potential orthogroup.
Potential orthogroups that contained fewer or greater than one D. melanogaster gene were discarded. All remaining potential orthogroups were truncated as before where necessary, such that only genes that were found to be 1-to-1 with a single D. melanogaster gene were retained. Potential orthogroups containing sequences from at least two species were extracted as new 1-to-1 orthogroups. Six original orthogroups were reconciled using Notung in this manner. All augmented and reconciled orthogroups can be found in S6 Table.
M4. Binding Landscapes Across Species
We initially obtained binding specificity motifs, represented as PWMs, for 62 D. melanogaster poly-ZF genes from the FlyFactorSurvey, JASPAR, and public Transfac databases. There are 96 binding specificity motifs for these 62 genes, as different isoforms or subsets of binding domains may correspond to distinct motifs (e.g., peb_Z1-3 and peb_Z5-7). For cases of duplicate binding motifs, we preferentially selected the PWM generated from SOLEXA sequencing over SANGER sequencing, and the longer PWM over the shorter. To exclude binding motifs that are non-specific, we discarded PWMs with fewer than six columns exhibiting information content (IC) > 0.5. To exclude binding motifs of low complexity (e.g. poly-A motifs), we discarded PWMs where > 80% of columns with IC > 0.5 correspond to the same consensus nucleotide, where consensus is defined as the most common nucleotide in a position, or ‘N’ in the case of a tie. Slight variations to these thresholds do not affect our findings. To exclude TFs which cannot be compared across species, we discarded binding motifs corresponding to TFs with 1-to-1 orthologs in fewer than two non-reference species. This filtering process resulted in 64 binding specificity motifs for 52 genes. These motifs were properly formatted for use by fimo with jaspar2meme, available from the MEME suite [68].
The 2000 basepair regions upstream of all genes in D. melanogaster and their alignments to orthologous regions across the other 11 fly species were obtained from the UCSC Genome Browser 15-fly promoter region alignments [49]. For each binding specificity motif, fimo was run on these aligned upstream regions from all 12 fly species to find all predicted TF binding site occurrences.
To obtain a set of high-confidence predicted binding sites in D. melanogaster, we required that each predicted binding site in D melanogaster be found within 25 basepairs in the UCSC genome alignments to binding sites in D. sechellia, D. simulans, D. yakuba, and D. erecta; this allows detection of conserved sites while allowing for slight variations in the genomes and/or slight error in the genome alignment [107, 108]. We note that restricting D. melanogaster binding sites to those found within 15 or 50 basepairs to binding sites in these other four species did not affect results nor significance. Considering alternate definitions of confident binding sites by restricting D. melanogaster binding sites to those found within 25 basepairs in only D. sechellia, only D. sechellia and D. simulans, or only D. sechellia, D. simulans, and D. yakuba also did not affect results nor significance (S6 Fig.).
For each PWM, the set of “bound” promoter regions, or those containing one or more high-confidence binding sites, was obtained in D. melanogaster. For each of these bound promoter regions, the orthologous promoter region in a non-reference species was also considered bound if it contained one or more binding sites within 25 basepairs of a high-confidence D. melanogaster binding site. For each PWM, we were thus able to determine the percent of bound promoter regions in D. melanogaster that were also bound across each other fly species. Similarly, each high-confidence binding site in D. melanogaster was considered conserved in another species if a binding site was found in that species within 25 basepairs of the D. melanogaster binding site. If another binding site was not found in that species within this window, the high-confidence D. melanogaster binding site was considered lost. The proportion of orthologous promoter regions bound and proportion of binding sites conserved were calculated for each binding motif in each species that contained a 1-to-1 ortholog of the corresponding TF (Fig. 6B-C).
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. King M, Wilson A (1975) Evolution at two levels in humans and chimpanzees. Science 188 : 107–116. doi: 10.1126/science.1090005 1090005
2. Wray GA, Hahn MW, Abouheif E, Balhoff JP, Pizer M, et al. (2003) The evolution of transcriptional regulation in eukaryotes. Mol Biol Evol 20 : 1377–1419. doi: 10.1093/molbev/msg140 12777501
3. Prud’homme B, Gompel N, Carroll SB (2007) Emerging principles of regulatory evolution. PNAS 104 : 8605–8612. doi: 10.1073/pnas.0700488104 17494759
4. Stern DL, Orgogozo V (2008) The loci of evolution: How predictable is genetic evolution? Evolution 62 : 2155–2177. doi: 10.1111/j.1558-5646.2008.00450.x 18616572
5. Liao BY, Weng MP, Zhang J (2010) Contrasting genetic paths to morphological and physiological evolution. PNAS 107 : 7353–7358. doi: 10.1073/pnas.0910339107 20368429
6. Britten RJ, Davidson EH (1969) Gene regulation for higher cells: A theory. Science 165 : 349–357. doi: 10.1126/science.165.3891.349 5789433
7. Stern DL (2000) Perspective: Evolutionary developmental biology and the problem of variation. Evolution 54 : 1079–1091. doi: 10.1554/0014-3820(2000)054%5B1079:PEDBAT%5D2.0.CO;2 11005278
8. Carroll SB (2005) Evolution at two levels: On genes and form. PLoS Biol 3: e245. doi: 10.1371/journal.pbio.0030245 16000021
9. Wray GA (2007) The evolutionary significance of cis-regulatory mutations. Nat Rev Genet 8 : 206–216. doi: 10.1038/nrg2063 17304246
10. Vlad D, Kierzkowski D, Rast MI, Vuolo F, Dello Ioio R, et al. (2014) Leaf shape evolution through duplication, regulatory diversification, and loss of a homeobox gene. Science 343 : 780–783. doi: 10.1126/science.1248384 24531971
11. Wagner GP, Lynch VJ (2008) The gene regulatory logic of transcription factor evolution. Trends Ecol Evol 23 : 377–385. doi: 10.1016/j.tree.2008.03.006 18501470
12. Singh LN, Hannenhalli S (2008) Functional diversification of paralogous transcription factors via divergence in DNA binding site motif and in expression. PLoS ONE 3: e2345. doi: 10.1371/journal.pone.0002345 18523562
13. Emerson RO, Thomas JH (2009) Adaptive evolution in zinc finger transcription factors. PLoS Genet 5: e1000325. doi: 10.1371/journal.pgen.1000325 19119423
14. Baker CR, Tuch BB, Johnson AD (2011) Extensive DNA-binding specificity divergence of a conserved transcription regulator. PNAS 108 : 7493–7498. doi: 10.1073/pnas.1019177108 21498688
15. Nakagawa S, Gisselbrecht SS, Rogers JM, Hartl DL, Bulyk ML (2013) DNA-binding specificity changes in the evolution of forkhead transcription factors. PNAS 110 : 12349–12354. doi: 10.1073/pnas.1310430110 23836653
16. Sayou C, Monniaux M, Nanao MH, Moyroud E, Brockington SF, et al. (2014) A promiscuous intermediate underlies the evolution of LEAFY DNA-binding specificity. Science 343 : 645–648. doi: 10.1126/science.1248229 24436181
17. Galant R, Carroll SB (2002) Evolution of a transcriptional repression domain in an insect Hox protein. Nature 415 : 910–913. doi: 10.1038/nature717 11859369
18. Ronshaugen M, McGinnis N, McGinnis W (2002) Hox protein mutation and macroevolution of the insect body plan. Nature 415 : 914–917. doi: 10.1038/nature716 11859370
19. Pabo CO, Peisach E, Grant RA (2001) Design and selection of novel Cys2His2 zinc finger proteins. Ann Rev Biochem 70 : 313–340. doi: 10.1146/annurev.biochem.70.1.313 11395410
20. Vaquerizas JM, Kummerfeld SK, Teichmann SA, Luscombe NM (2009) A census of human transcription factors: Function, expression and evolution. Nat Rev Genet 10 : 252–263. doi: 10.1038/nrg2538 19274049
21. Enuameh MS, Asriyan Y, Richards A, Christensen RG, Hall VL, et al. (2013) Global analysis of Drosophila Cys2-His2 zinc finger proteins reveals a multitude of novel recognition motifs and binding determinants. Genome Res 23 : 928–940. doi: 10.1101/gr.151472.112 23471540
22. Benos PV, Lapedes AS, Stormo GD (2002) Probabilistic code for DNA recognition by proteins of the EGR family. J Mol Biol 323 : 701–727. doi: 10.1016/S0022-2836(02)00917-8 12419259
23. Kaplan T, Friedman N, Margalit H (2005) Ab initio prediction of transcription factor targets using structural knowledge. PLoS Comput Biol 1: e1. doi: 10.1371/journal.pcbi.0010001 16103898
24. Persikov AV, Singh M (2014) De novo prediction of DNA-binding specificities for Cys2His2 zinc finger proteins. Nucleic Acids Res 42 : 97–108. doi: 10.1093/nar/gkt890 24097433
25. Gupta A, Christensen RG, Bell HA, Goodwin M, Patel RY, et al. (2014) An improved predictive recognition model for Cys2-His2 zinc finger proteins. Nucleic Acids Res 42 : 4800–4812. doi: 10.1093/nar/gku132 24523353
26. Persikov AV, Wetzel JL, Rowland EF, Oakes BL, Xu DJ, et al. (2015) A systematic survey of the Cys2His2 zinc finger DNA-binding landscape. Nucleic Acids Res In press: doi: 10.1093/nar/gku1395 25593323
27. Nowick K, Fields C, Gernat T, Caetano-Anolles D, Kholina N, et al. (2011) Gain, loss and divergence in primate zinc-finger genes: A rich resource for evolution of gene regulatory differences between species. PLoS ONE 6: e21553. doi: 10.1371/journal.pone.0021553 21738707
28. Shannon M, Hamilton AT, Gordon L, Branscomb E, Stubbs L (2003) Differential expansion of zinc-finger transcription factor loci in homologous human and mouse gene clusters. Genome Res 13 : 1097–1110. doi: 10.1101/gr.963903 12743021
29. Nowick K, Hamilton AT, Zhang H, Stubbs L (2010) Rapid sequence and expression divergence suggest selection for novel function in primate-specific KRAB-ZNF genes. Mol Biol Evol 27 : 2606–2617. doi: 10.1093/molbev/msq157 20573777
30. Stubbs L, Sun Y, Caetano-Anolles D (2011) Function and evolution of C2H2 zinc finger arrays, Houten, Netherlands: Springer Publishing. In A Handbook of Transcription Factors (ed. Hughes TR), pp. 75–94.
31. Liu H, Chang LH, Sun Y, Lu X, Stubbs L (2014) Deep vertebrate roots for mammalian zinc finger transcription factor subfamilies. Genome Biol Evol 6 : 510–525. doi: 10.1093/gbe/evu030 24534434
32. Looman C, Åbrink M, Mark C, Hellman L (2002) KRAB zinc finger proteins: An analysis of the molecular mechanisms governing their increase in numbers and complexity during evolution. Mol Biol Evol 19 : 2118–2130. doi: 10.1093/oxfordjournals.molbev.a004037 12446804
33. Knight R, Shimeld S (2001) Identification of conserved C2H2 zinc-finger gene families in the Bilateria. Genome Biol 2: R16.1–R16.8. doi: 10.1186/gb-2001-2-5-research0016
34. Seetharam A, Bai Y, Stuart G (2010) A survey of well conserved families of C2H2 zinc-finger genes in Daphnia. BMC Genomics 11 : 276–295. doi: 10.1186/1471-2164-11-276 20433734
35. Seetharam A, Stuart GW (2013) A study on the distribution of 37 well conserved families of C2H2 zinc finger genes in eukaryotes. BMC Genomics 14 : 420–426. doi: 10.1186/1471-2164-14-420 23800006
36. Oliver PL, Goodstadt L, Bayes JJ, Birtle Z, Roach KC, et al. (2009) Accelerated evolution of the PRDM9 speciation gene across diverse metazoan taxa. PLoS Genet 5: e1000753. doi: 10.1371/journal.pgen.1000753 19997497
37. Myers S, Bowden R, Tumian A, Bontrop RE, Freeman C, et al. (2010) Drive against hotspot motifs in primates implicates the PRDM9 gene in meiotic recombination. Science 327 : 876–879. doi: 10.1126/science.1182363 20044541
38. Berg IL, Neumann R, Sarbajna S, Odenthal-Hesse L, Butler NJ, et al. (2011) Variants of the protein PRDM9 differentially regulate a set of human meiotic recombination hotspots highly active in African populations. PNAS 108 : 12378–12383. doi: 10.1073/pnas.1109531108 21750151
39. Ségurel L, Leffler EM, Przeworski M (2011) The case of the fickle fingers: How the PRDM9 zinc finger protein specifies meiotic recombination hotspots in humans. PLoS Biol 9: e1001211. doi: 10.1371/journal.pbio.1001211 22162947
40. Groeneveld LF, Atencia R, Garriga RM, Vigilant L (2012) High diversity at PRDM9 in chimpanzees and bonobos. PLoS ONE 7: e39064. doi: 10.1371/journal.pone.0039064 22768294
41. Hoekstra HE, Coyne JA (2007) The locus of evolution: Evo devo and the genetics of adaptation. Evolution 61 : 995–1016. doi: 10.1111/j.1558-5646.2007.00105.x 17492956
42. Drosophila 12 Genomes Consortium (2007) Evolution of genes and genomes on the Drosophila phylogeny. Nature 450 : 203–218. doi: 10.1038/nature06341 17994087
43. Marygold SJ, Leyland PC, Seal RL, Goodman JL, Thurmond J, et al. (2013) FlyBase: Improvements to the bibliography. Nucleic Acids Res 41 : 751–757. doi: 10.1093/nar/gks1024
44. Gompel N, Carroll SB (2003) Genetic mechanisms and constraints governing the evolution of correlated traits in Drosophilid flies. Nature 424 : 931–935. doi: 10.1038/nature01787 12931186
45. Jeong S, Rokas A, Carroll SB (2006) Regulation of body pigmentation by the Abdominal-B Hox protein and its gain and loss in Drosophila evolution. Cell 125 : 1387–1399. doi: 10.1016/j.cell.2006.04.043 16814723
46. Markow TA, O’Grady PM (2007) Drosophila biology in the genomic age. Genetics 177 : 1269–1276. doi: 10.1534/genetics.107.074112 18039866
47. Punta M, Coggill PC, Eberhardt RY, Mistry J, Tate J, et al. (2012) The Pfam protein families database. Nucleic Acids Res 40 : 290–301. doi: 10.1093/nar/gkr1065
48. Finn RD, Clements J, Eddy SR (2011) HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res 39: W29–W37. doi: 10.1093/nar/gkr367 21593126
49. Meyer LR, Zweig AS, Hinrichs AS, Karolchik D, Kuhn RM, et al. (2013) The UCSC Genome Browser database: Extensions and updates 2013. Nucleic Acids Res 41 : 64–69. doi: 10.1093/nar/gks1048
50. Iuchi S (2001) Three classes of C2H2 zinc finger proteins. Cell Mol Life Sci 58 : 625–635. 11361095
51. Adryan B, Teichmann SA (2006) Flytf: A systematic review of site-specific transcription factors in the fruit fly Drosophila melanogaster. Bioinformatics 22 : 1532–1533. doi: 10.1093/bioinformatics/btl143 16613907
52. Wolfe SA, Nekludova L, Pabo CO (2000) DNA recognition by Cys2His2 zinc finger proteins. Ann Rev Bioph Biom 29 : 183–212. doi: 10.1146/annurev.biophys.29.1.183
53. Persikov AV, Singh M (2011) An expanded binding model for Cys2 His2 zinc finger protein-DNA interfaces. Phys Biol 8: e035010. doi: 10.1088/1478-3975/8/3/035010
54. Siggers T, Reddy J, Barron B, Bulyk ML (2014) Diversification of transcription factor paralogs via noncanonical modularity in C2H2 zinc finger DNA binding. Mol Cell 55 : 1–9. doi: 10.1016/j.molcel.2014.06.019
55. Mayrose I, Graur D, Ben-Tal N, Pupko T (2004) Comparison of site-specific rate-inference methods for protein sequences: Empirical Bayesian methods are superior. Mol Biol Evol 21 : 1781–1791. doi: 10.1093/molbev/msh194 15201400
56. Kimura M (1977) Preponderance of synonymous changes as evidence for the neutral theory of molecular evolution. Nature 267 : 275–276. doi: 10.1038/267275a0 865622
57. Yang Z (1997) PAML: A program package for phylogenetic analysis by maximum likelihood. Comput Appl Biosci 13 : 555–556. 9367129
58. McDonald JH, Kreitman M (1991) Adaptive protein evolution at the Adh locus in Drosophila. Nature 351 : 652–654. doi: 10.1038/351652a0 1904993
59. Pool JE, Corbett-Detig RB, Sugino RP, Stevens KA, Cardeno CM, et al. (2012) Population genomics of sub-Saharan Drosophila melanogaster: African diversity and non-African admixture. PLoS Genet 8: e1003080. doi: 10.1371/journal.pgen.1003080 23284287
60. Bustamante CD, Wakeley J, Sawyer S, Hartl DL (2001) Directional selection and the site-frequency spectrum. Genetics 159 : 1779–1788. 11779814
61. Persikov AV, Rowland EF, Oakes BL, Singh M, Noyes MB (2014) Deep sequencing of large library selections allows computational discovery of diverse sets of zinc fingers that bind common targets. Nucleic Acids Res 42 : 1497–1508. doi: 10.1093/nar/gkt1034 24214968
62. Persikov AV, Osada R, Singh M (2009) Predicting DNA recognition by Cys2His2 zinc finger proteins. Bioinformatics 25 : 22–29. doi: 10.1093/bioinformatics/btn580 19008249
63. Crooks GE, Hon G, Chandonia JM, Brenner SE (2004) WebLogo: A sequence logo generator. Genome Res 14 : 1188–1190. doi: 10.1101/gr.849004 15173120
64. modENCODE Consortium (2010) Identification of functional elements and regulatory circuits by Drosophila modENCODE. Science 330 : 1787–1797. doi: 10.1126/science.1198374 21177974
65. Zhu LJ, Christensen RG, Kazemian M, Hull CJ, Enuameh MS, et al. (2011) FlyFactorSurvey: A database of Drosophila transcription factor binding specificities determined using the bacterial one-hybrid system. Nucleic Acids Res 39: D111–D117. doi: 10.1093/nar/gkq858 21097781
66. Mathelier A, Zhao X, Zhang AW, Parcy F, Worsley-Hunt R, et al. (2014) JASPAR 2014: An extensively expanded and updated open-access database of transcription factor binding profiles. Nucleic Acids Res 42: D142–D147. doi: 10.1093/nar/gkt997 24194598
67. Matys V, Fricke E, Geffers R, Gößling E, Haubrock M, et al. (2003) TRANSFAC: transcriptional regulation, from patterns to profiles. Nucleic Acids Res 31 : 374–378. doi: 10.1093/nar/gkg108 12520026
68. Bailey TL, Boden M, Buske FA, Frith M, Grant CE, et al. (2009) MEME Suite: Tools for motif discovery and searching. Nucleic Acids Res 37: W202–W208. doi: 10.1093/nar/gkp335 19458158
69. Boyle EI, Weng S, Gollub J, Jin H, Botstein D, et al. (2004) GO∷TermFinder—open source software for accessing Gene Ontology information and finding significantly enriched Gene Ontology terms associated with a list of genes. Bioinformatics 20 : 3710–3715. doi: 10.1093/bioinformatics/bth456 15297299
70. Franceschini A, Szklarczyk D, Frankild S, Kuhn M, Simonovic M, et al. (2013) STRING v9.1: Protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res 41: D808–D815. doi: 10.1093/nar/gks1094 23203871
71. Li H, Baker B (1998) Her, a gene required for sexual differentiation in Drosophila, encodes a zinc finger protein with characteristics of ZFY-like proteins and is expressed independently of the sex determination hierarchy. Development 125 : 225–235. 9486796
72. Perezgasga L, Jiang J, Bolival B, Hiller M, Benson E, et al. (2004) Regulation of transcription of meiotic cell cycle and terminal differentiation genes by the testis-specific Zn-finger protein matotopetli. Development 131 : 1691–1702. doi: 10.1242/dev.01032 15084455
73. Laugier E, Yang Z, Fasano L, Kerridge S, Vola C (2005) A critical role of teashirt for patterning the ventral epidermis is masked by ectopic expression of tiptop, a paralog of teashirt in Drosophila. Dev Biol 283 : 446–458. doi: 10.1016/j.ydbio.2005.05.005 15936749
74. Hunter S, Jones P, Mitchell A, Apweiler R, Attwood TK, et al. (2012) InterPro in 2011: new developments in the family and domain prediction database. Nucleic Acids Res 40: D306–D312. doi: 10.1093/nar/gkr948 22096229
75. Chung HR, Schäfer U, Jäckle H, Böhm S (2002) Genomic expansion and clustering of ZAD-containing C2H2 zinc-finger genes in Drosophila. EMBO reports 3 : 1158–1162. doi: 10.1093/embo-reports/kvf243 12446571
76. Jauch R, Bourenkov GP, Chung HR, Urlaub H, Reidt U, et al. (2003) The zinc finger-associated domain of the Drosophila transcription factor Grauzone is a novel zinc-coordinating protein-protein interaction module. Structure 11 : 1393–1402. doi: 10.1016/j.str.2003.09.015 14604529
77. Chung HR, Löhr U, Jäckle H (2007) Lineage-specific expansion of the zinc finger associated domain ZAD. Mol Biol Evol 24 : 1934–1943. doi: 10.1093/molbev/msm121 17569752
78. Robinson SW, Herzyk P, Dow JAT, Leader DP (2013) FlyAtlas: database of gene expression in the tissues of Drosophila melanogaster. Nucleic Acids Res 41: D744–D750. doi: 10.1093/nar/gks1141 23203866
79. Tuch BB, Galgoczy DJ, Hernday AD, Li H, Johnson AD (2008) The evolution of combinatorial gene regulation in fungi. PLoS Biol 6: e38. doi: 10.1371/journal.pbio.0060038 18303948
80. Borneman AR, Gianoulis TA, Zhang ZD, Yu H, Rozowsky J, et al. (2007) Divergence of transcription factor binding sites across related yeast species. Science 317 : 815–819. doi: 10.1126/science.1140748 17690298
81. Bradley RK, Li XY, Trapnell C, Davidson S, Pachter L, et al. (2010) Binding site turnover produces pervasive quantitative changes in transcription factor binding between closely related Drosophila species. PLoS Biol 8: e1000343. doi: 10.1371/journal.pbio.1000343 20351773
82. Odom DT, Dowell RD, Jacobsen ES, Gordon W, Danford TW, et al. (2007) Tissue-specific transcriptional regulation has diverged significantly between human and mouse. Nat Genet 39 : 730–732. doi: 10.1038/ng2047 17529977
83. Schmidt D, Wilson MD, Ballester B, Schwalie PC, Brown GD, et al. (2010) Five-vertebrate ChIP-seq reveals the evolutionary dynamics of transcription factor binding. Science 328 : 1036–1040. doi: 10.1126/science.1186176 20378774
84. Kasowski M, Grubert F, Heffelfinger C, Hariharan M, Asabere A, et al. (2010) Variation in transcription factor binding among humans. Science 328 : 232–235. doi: 10.1126/science.1183621 20299548
85. Zheng W, Zhao H, Mancera E, Steinmetz LM, Snyder M (2010) Genetic analysis of variation in transcription factor binding in yeast. Nature 464 : 1187–1191. doi: 10.1038/nature08934 20237471
86. Pelham HRB, Brown DD (1980) A specific transcription factor that can bind either the 5S RNA gene or 5S RNA. PNAS 77 : 4170–4174. doi: 10.1073/pnas.77.7.4170 7001457
87. Brayer KJ, Segal DJ (2008) Keep your fingers off my DNA: Protein—protein interactions mediated by C2H2 zinc finger domains. Cell Biochem Biophys 50 : 111–131. doi: 10.1007/s12013-008-9008-5 18253864
88. Dermitzakis ET, Clark AG (2002) Evolution of transcription factor binding sites in mammalian gene regulatory regions: Conservation and turnover. Mol Biol Evol 19 : 1114–1121. doi: 10.1093/oxfordjournals.molbev.a004169 12082130
89. Costas J, Casares F, Vieira J (2003) Turnover of binding sites for transcription factors involved in early Drosophila development. Gene 310 : 215–220. doi: 10.1016/S0378-1119(03)00556-0 12801649
90. Moses AM, Pollard DA, Nix DA, Iyer VN, Li XY, et al. (2006) Large-scale turnover of functional transcription factor binding sites in Drosophila. PLoS Comput Biol 2: e130. doi: 10.1371/journal.pcbi.0020130 17040121
91. Doniger SW, Fay JC (2007) Frequent gain and loss of functional transcription factor binding sites. PLoS Comput Biol 3: e99. doi: 10.1371/journal.pcbi.0030099 17530920
92. Kim J, He X, Sinha S (2009) Evolution of regulatory sequences in 12 Drosophila species. PLoS Genet 5: e1000330. doi: 10.1371/journal.pgen.1000330 19132088
93. Venkataram S, Fay JC (2010) Is transcription factor binding site turnover a sufficient explanation for cis-regulatory sequence divergence? Genome Biol Evol 2 : 851–858. doi: 10.1093/gbe/evq066 21068212
94. Weirauch MT, Hughes TR (2010) Conserved expression without conserved regulatory sequence: The more things change, the more they stay the same. Trends Genet 26 : 66–74. doi: 10.1016/j.tig.2009.12.002 20083321
95. Tan K, Feizi H, Luo C, Fan SH, Ravasi T, et al. (2008) A systems approach to delineate functions of paralogous transcription factors: Role of the Yap family in the DNA damage response. PNAS 105 : 2934–2939. doi: 10.1073/pnas.0708670105 18287073
96. Lespinet O, Wolf YI, Koonin EV, Aravind L (2002) The role of lineage-specific gene family expansion in the evolution of eukaryotes. Genome Res 12 : 1048–1059. doi: 10.1101/gr.174302 12097341
97. Hamilton AT, Huntley S, Kim J, Branscomb E, Stubbs L (2003) Lineage-specific expansion of KRAB zinc-finger transcription factor genes: Implications for the evolution of vertebrate regulatory networks. CSHL Symposia on Quant Biol 68 : 131–140. doi: 10.1101/sqb.2003.68.131
98. Urrutia R (2003) KRAB-containing zinc-finger repressor proteins. Genome Biol 4 : 231. doi: 10.1186/gb-2003-4-10-231 14519192
99. Taylor JS, Raes J (2004) Duplication and divergence: The evolution of new genes and old ideas. Annu Rev Genet 38 : 615–643. doi: 10.1146/annurev.genet.38.072902.092831 15568988
100. Brunkard JO, Runkel AM, Zambryski PC (2015) Comment on “A promiscuous intermediate underlies the evolution of LEAFY DNA binding specificity.” Science 347 : 621. doi: 10.1126/science.1256011 25657240
101. Brockington SF, Moyroud E, Sayou C, Monniaux M, Nanao MH, et al. (2015) Response to Comment on “A promiscuous intermediate underlies the evolution of LEAFY DNA binding specificity.” Science 347 : 621. doi: 10.1126/science.1256011 25657241
102. Hu TT, Eisen MB, Thornton KR, Andolfatto P (2013) A second-generation assembly of the Drosophila simulans genome provides new insights into patterns of lineage-specific divergence. Genome Res 23 : 89–98. doi: 10.1101/gr.141689.112 22936249
103. Eddy SR (2011) Accelerated profile HMM searches. PLoS Comput Biol 7: e1002195. doi: 10.1371/journal.pcbi.1002195 22039361
104. Kent WJ (2002) Blat-the BLAST-Like Alignment Tool. Genome Res 12 : 656–664. doi: 10.1101/gr.229202 11932250
105. Notredame C, Higgins DG, Heringa J (2000) T-coffee: A novel method for fast and accurate multiple sequence alignment. J Mol Biol 302 : 205–217. doi: 10.1006/jmbi.2000.4042 10964570
106. Vernot B, Stolzer M, Goldman A, Durand D (2008) Reconciliation with non-binary species trees. J Comput Biol 15 : 981–1006. doi: 10.1089/cmb.2008.0092 18808330
107. Kheradpour P, Stark A, Roy S, Kellis M (2007) Reliable prediction of regulator targets using 12 Drosophila genomes. Genome Res 17 : 1919–1931. doi: 10.1101/gr.7090407 17989251
108. Jiang P, Singh M (2014) CCAT: Combinatorial Code Analysis Tool for transcriptional regulation. Nucleic Acids Res 42 : 2833–2847. doi: 10.1093/nar/gkt1302 24366875
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2015 Číslo 3
Nejčtenější v tomto čísle
- Clonality and Evolutionary History of Rhabdomyosarcoma
- Morphological Mutations: Lessons from the Cockscomb
- Maternal Filaggrin Mutations Increase the Risk of Atopic Dermatitis in Children: An Effect Independent of Mutation Inheritance
- Transcriptomic Profiling of Reveals Reprogramming of the Crp Regulon by Temperature and Uncovers Crp as a Master Regulator of Small RNAs