
Coronary Artery Disease Associated Transcription Factor TCF21 Regulates Smooth Muscle Precursor Cells That Contribute to the Fibrous Cap
Coronary artery disease (CAD) is responsible for the majority of deaths in the Western world, and is due in part to environmental and metabolic factors. However, half of the risk for developing heart disease is genetically predetermined. Genome-wide association studies in human populations have identified over 100 sites in the genome that appear to be associated with CAD, however, the mechanisms by which variation in these regions are responsible for predisposition to CAD remain largely unknown. We have begun to study a gene that contributes to CAD risk, the TCF21 gene. Through genomic studies we show that this gene is involved in processes related to alterations in vascular gene expression, and in particular those related to the smooth muscle cell biology. With cell culture models, we show that TCF21 regulates the differentiation state of this cell type, which is believed critical for vascular disease. Using mouse genetic models of atherosclerotic vascular disease we provide evidence that this gene is expressed in precursor cells that migrate into the disease lesions and contribute to the formation of the fibrous cap that is believed to stabilize these lesions and prevent heart attacks.
Published in the journal:
. PLoS Genet 11(5): e32767. doi:10.1371/journal.pgen.1005155
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1005155
Summary
Coronary artery disease (CAD) is responsible for the majority of deaths in the Western world, and is due in part to environmental and metabolic factors. However, half of the risk for developing heart disease is genetically predetermined. Genome-wide association studies in human populations have identified over 100 sites in the genome that appear to be associated with CAD, however, the mechanisms by which variation in these regions are responsible for predisposition to CAD remain largely unknown. We have begun to study a gene that contributes to CAD risk, the TCF21 gene. Through genomic studies we show that this gene is involved in processes related to alterations in vascular gene expression, and in particular those related to the smooth muscle cell biology. With cell culture models, we show that TCF21 regulates the differentiation state of this cell type, which is believed critical for vascular disease. Using mouse genetic models of atherosclerotic vascular disease we provide evidence that this gene is expressed in precursor cells that migrate into the disease lesions and contribute to the formation of the fibrous cap that is believed to stabilize these lesions and prevent heart attacks.
Introduction
Atherosclerotic coronary heart disease (CAD) continues to be the dominant medical problem in the Western world and is growing in minority groups in this country and in developing populations [1–4]. Despite extensive investigation of epidemiological and cellular features of the disease, there are still fundamental questions related to the mechanism of etiology that remain to be answered. For instance, the resident SMC that constitute the majority of the native vessel wall undergo dramatic phenotypic changes in the disease setting, with the loss of SMC gene expression in the medial vessel layer and appearance of a new layer of SMC marker expressing cells in a “fibrous cap” that covers the lipid core of the plaque. While the risk of plaque rupture appears to be inversely correlated with the number of SMC-like cells in the fibrous cap, there is very little understanding of their in vivo origin, and the molecular pathways that regulate their expansion and terminal phenotype determination [5]
Significant new insights into the fundamental cellular processes that drive CAD and other complex human diseases have recently been achieved through genome wide association (GWA) studies employing large cohorts of patients and healthy controls. These studies have provided the first incontrovertible identification of genes and disease pathways that affect risk for many complex human diseases (www.genome.gov/gwastudies), including CAD. A recent GWA meta-analysis has identified 46 loci that associate with CAD at the genome-wide significance level of 5 x 10-8, and another 104 independent variants that associate at a false discovery rate less than 0.05 [6]. Of these CAD associated loci, more than two-thirds act independently of traditional risk factors, and are likely to modulate risk of CAD through regulation of cellular processes in the blood vessel wall. Further study of this latter group of genes is expected to provide insights into novel atherosclerosis pathways.
One CAD associated gene that has not been linked to known environmental or metabolic risk factors is the basic-helix-loop-helix transcription factor TCF21 (capsulin, POD1, epicardin) [6–8]. This gene was initially cloned in four independent laboratories and shown to mark expression of mesodermal cells that give rise to kidney, lung, spleen, gonads, and a restricted group of cranial muscle cells [9–16]. Recent studies have shown that Tcf21 regulates fundamental cell fate decisions in the developing epicardium, serving as a determining factor for divergence between coronary vascular smooth muscle cell and cardiac fibroblast lineages [17,18]. In this setting, Tcf21 is downregulated in cells that are fated to become differentiated coronary SMC and has sustained expression in cells that become interstitial and adventitial fibroblasts [18]. However, its role in molecular and cellular processes related to CAD has not been investigated.
To examine the possible role of TCF21 in coronary artery disease, we assessed its expression pattern in normal and diseased tissues, investigated its role in fundamental cellular processes, and employed next generation sequencing based methods in SMC to identify genes and pathways regulated by TCF21 at the transcriptional level. We have also identified the temporal and cell-specific expression of Tcf21 in reporter gene and lineage tracing mouse models to better understand how this gene might regulate vascular wall cellular processes in the setting of atherosclerotic disease.
Results
TCF21 is expressed in human SMC and other mesodermal lineages in vitro and in human and mouse coronary arteries
To better understand the expression pattern of TCF21 in adult tissues, we studied expression in a number of adult human cells by quantitative real-time polymerase chain reaction (qPCR) (S1A Fig). TCF21 was expressed in human coronary artery smooth muscle cells (HCASMC), but not aortic smooth muscle cells (HAoSMC) or endothelial cells isolated from coronary (HCAEC) or aortic vessels (HAoEC). Because of the reported similarity between pericytes, which have been developmentally linked to Tcf21 [10], and mesenchymal stem cells (MSC) [19], we investigated expression in MSC derived from bone marrow (bmMSC), embryonic-stem cells (eMSC) and induced pluripotent stem cells (iMSC). Results showed that all of these cells expressed TCF21. Cardiac fibroblasts (HCF) and adventitial aortic fibroblasts (HAoAF) were also found to express TCF21.
Immunocytochemistry of cultured HCASMC revealed nuclear expression of TCF21 in cells that were also expressing the SMC marker ACTA2, although a number of prominent cells expressing high levels of ACTA2 appeared to have low-level expression of TCF21 (S1B Fig). In situ hybridization of mouse and human coronary artery specimens with species-specific cRNA probes revealed prominent labeling of adventitial cells with no staining of medial SMC or intimal endothelial cells (S1C Fig).
In preparation for studies in mouse genetic disease models, we investigated Tcf21 expression in normal adult mice with Xgal staining of tissues containing a lacZ reporter gene integrated into the murine Tcf21 locus, and compared this staining with that seen in C57BL/6 mice [12,20]. As with in situ hybridization studies in mouse and human tissues, results showed staining of cells in the adventitia of both the coronary arteries and aorta, with primary localization in those adventitial cells juxtaposed to the vascular media (S1D Fig). Unique to the mouse, there was a patchy distribution of lacZ expressing cells in the proximal aortic wall (S1E Fig). When β-galactosidase activity was pseudocolored green and merged with the Acta2 immunoreactivity (red) there appeared to be minimal overlap in either the coronary arteries or aorta.
Genomic studies identify disease and developmental pathways downstream of TCF21
To provide functional insights into the role of TCF21 in adult SMC in the disease setting, we employed whole transcriptome RNA sequencing (RNA-Seq) studies on cells transfected with either non-silencing control siRNA (siCTRL) or silencing TCF21 siRNA (siTCF21). Quantitative RT-PCR of HCASMC transfected with siTCF21 compared to siCTRL showed a significant decrease in mRNA levels for TCF21 (0.99±0.02 control vs. 0.25±0.02 siTCF21, P<0.0001) (S2A Fig), and quantitation of western blots of protein extracts showed a similar reduction of TCF21 protein levels to 26% of baseline (S2B Fig).
We employed two commonly used algorithms, DESeq and edgeR, to identify genes that were differentially expressed (FDR ≤ 0.05) between the HCASMC treated with siTCF21 or siCTRL reagents [21,22]. To eliminate outliers that might be identified by each analysis, we intersected the 466 genes obtained with DESeq and 430 genes found with edgeR to obtain 380 genes that were common to the two approaches [23]. The intersected genelist was mapped to the Ingenuity IPA Knowledge Base, generating algorithmically computed pathways or networks that were then investigated for over-representation in disease and biological function categories. “Connective Tissue Development and Function, Tissue Morphology, Cardiovascular Disease” was identified as the top network. “Cardiovascular Disease” was ranked third in the disease category, with 20 subcategories including “Vascular disease” (Q = 3.26E-17, right-tailed Fisher’s Exact Test, Benjamini-Hochberg corrected), “Disorder of artery” (Q = 4.04E-12), and “Occlusion of artery” (Q = 2.03E-14) as the top three functional groups (Tables 1 and S1). “Cardiovascular System Development and Function” (Q = 1.88E-16 - 5.30E-04) was the most highly ranked in the “Physiological System Development and Function” category, with a number of subcategories that were related to embryonic development, including “Development of the cardiovascular system” (Q = 1.88E-16), “Angiogenesis” (Q = 4.58E-14), and “Development of blood vessel” (Q = 4.58E-14) being the top three functional groups (Tables 1 and S2). Among the Molecular and Cellular Functions category of traits, the top three functional groups were “Cellular movement” (Q = 4.23E-23), followed by “Cellular growth and proliferation” (Q = 4.23E-23), and “Cell death and survival” (Q = 1.06E-18) (Tables 1 and S3).

Taking the first subcategory in the “Cardiovascular Disease” category, “Vascular disease,” we used well-curated molecular interactions in the IPA Knowledge Base to build a gene network. The initial group included 73 genes, and 18 additional genes were added with the “build” function in IPA, this group functionally related with the “connect” function, and with elimination of non-connected nodes resulted in a final TCF21 Vascular Disease Network containing 83 genes (Fig 1). Also, content was added to the network with annotation from Gene Ontology classifications (S3 Fig). One of the strongest signals in the network was the upregulation of matrix components with TCF21 knockdown, including a large number of collagens, SPARC, LOX, vitronectin (VTN), and components of the matrix that are known to affect vascular cell functions, including thrombospondin 1 (THBS1) and fibrillin 1 (FBN1), as well as vascular cell surface proteins that interact with the matrix including alpha V integrin (ITGAV). Although the matrix degrading enzymes MMP7, MMP10 and MMP25 are upregulated with TCF21 knockdown, the downregulation of the much more highly expressed MMP1 and MMP3 combined with the upregulation of MMP inhibitor TIMP3 are consistent with an anti-matrix overall profile of a mobile cell. Also upregulated are SMC markers such as smooth muscle actin (ACTA2) and myosin light chain kinase (MYLK). Among the genes downregulated with TCF21 knockdown are those involved in vascular development, including neuropilin 1 (NRP1), VEGF receptors II (KDR), angiopoietin 1 (ANGPT1), TGFβ receptor 2 (TGFBR2), and semaphorin 3D (SEMA3D), and cytokines and chemokines such as IL1A, IL1B, CXCL2 and CXCL3. Taken together these data suggest that TCF21 positive cells express low levels of many matrix proteins while expressing high levels of matrix degrading enzymes, consistent with a pro-migratory phenotype, express low levels of SMC lineage markers, and are highly pro-angiogenic and pro-inflammatory, a phenotype quite different from vascular wall SMC.

TCF21 directly regulates cell fate decisions in cultured SMC
To investigate a functional role for TCF21 in fundamental cell fate decisions in vascular SMC, we employed lentiviral shRNA as well as siRNA mediated knockdown and lentiviral over-expression studies. Control lentiviral vectors (pWPI) and lentiviral over-expression vectors (pWPI-TCF21), and control (pLVTHM) and lentiviral shRNA mediated knockdown vectors (pLVTHM-shTCF21) were used to transduce primary cultured HCASMC. pWPI-TCF21 increased TCF21 mRNA levels (1.0±0.04 pWPI vs. 32.5±0.02 pWPI-TCF21, P<0.0001), and pLVTHM-shTCF21 decreased expression (1.0±0.06 pLVTHM vs. 0.34±0.04 pLVTHM-shTCF21 2, P<0.001) (S4A Fig). Western blot of protein extracts from HCASMC that were transduced with over-expression and knockdown lentiviruses showed a 4.5-fold increase, and reduction of TCF21 protein levels to 8% (shTCF21 1, shTCF21 2) of baseline respectively (S4B Fig).
To assess the effect of TCF21 on rate of cell division in vitro, proliferation rates of pools of stably transduced and non-transduced cells were measured over time, where increased or decreased proliferation rates in transduced cells would lead to a change in percentages of this GFP positive cell population as compared to the non-transduced, GFP negative population. Flow cytometry analysis of stable overexpression of TCF21 in HCASMC showed an increase in TCF21 overexpressing cells from 48 to 82 percent of the culture within 28 days (S5A Fig). In the same time period the percentage of cells with stable knockdown of TCF21 decreased from 53 to 16 for construct 1 (shTCF21 1 vs. empty control virus pLVTHM P<0.0001) and from 59 to 2 percent for construct 2 (shTCF21 2 vs. empty control virus pLVTHM P<0.0001) (S5B Fig). The percentage of GFP positive cells did not change when transduced with the empty vector control construct. To provide additional support for these findings we conducted EdU (5-ethynyl-2’-deoxyuridine) labeling proliferation assays. HCASMC transduced with the control, overexpression or knockdown lentiviruses were treated with EdU and imaged for nuclear fluorescence. These data showed an increase in the percentage of TCF21 overexpressing cells compared to DAPI stained cells (33.1% ± 2.3 control vs. 51% ± 4.1 overexpressing cells, P<0.001), and significant decreased percentage of dividing cells transduced with knockdown lentiviruses (43.4% ± 4.4 control vs. 30.8% ± 2.4 knockdown cells, P<0.01) (Fig 2A and 2B). These data suggest that TCF21 positively affects SMC proliferation.

We also evaluated the effect of TCF21 on apoptosis and migration in cultured HCASMC. Apoptosis was evaluated in HCASMC transfected with siControl or siTCF21 RNAs and serum starved for 48 hrs. By caspase assay there was a significant decrease in apoptosis of cells transfected with the siTCF21 reagent (41,824±1872 vs. 18,837±1302 for siCTRL compared to siTCF21, P<0.0001) (Fig 2C). For migration, HCASMC transduced with TCF21 overexpression lentivirus were evaluated with a gap closure assay. Here, TCF21 overexpressing cells covered a significantly larger surface area after 12 hours of incubation (27.8 ± 2.7 vs. 15.8 ± 1.16 control cells, P<0.001) (Fig 2D). With an average doubling time of 71 hours for TCF21 overexpressing HCASMC this effect could not be due to the pro-proliferative effect of TCF21.
Given that TCF21 is downregulated in epicardial cells as they differentiate into coronary SMC, and a number of SMC lineage marker genes were upregulated in the siTCF21 RNA-Seq study, including ACTA2 and MYLK, we specifically investigated the expression of SMC marker genes in additional siRNA knockdown experiments. These studies showed that ACTA2, TAGLN and MYH11 transcript levels were significantly upregulated after 70% TCF21 knockdown (0.96 ± 0.023 vs. 1.38 ± 0.16, P<0.05, 1.0 ± 0.03 vs. 2.59 ± 0.27, P<0.01, and 1.05 ± 0.09 vs. 2.67 ± 0.51, P<0.05, respectively) (Fig 2E), suggesting their direct suppression by TCF21 at the transcriptional level.
To investigate whether TCF21 directly binds and regulates SMC marker genes, a series of studies were conducted with the ACTA2 gene as a representative SMC marker. It is well known that the regulatory regions of the ACTA2 gene are localized in the upstream promoter and first intronic regions of the gene [24], and this information in conjunction with available ENCODE histone modification data, ENCODE DNase hypersensitivity data from human aortic smooth muscle cells, and localization of E-box binding motif sequences provided the approach for choosing a reporter construct for these studies (Fig 2F). Chromatin immunoprecipitation was employed to verify the in vivo binding of TCF21 to a region of the first intron enhancer identified through this analysis (Fig 2G). The luciferase reporter gene construct employed for these studies contained the human ACTA2 promoter and first intron. Obligate heterodimer partners of TCF21, TCF3 and TCF12, were included in these experiments to facilitate transcriptional activation [25]. Also, based on the high degree of conservation of sequence in the bHLH domain between TCF21 and Twist, and the similar inhibitory developmental actions of this related transcription factor, an expression construct for Twist was included to investigate possible interactions with TCF21 in the context of ACTA2 reporter gene expression [26]. When this reporter was transfected into rat aortic smooth muscle cells (RASMC) in conjunction with a TCF21 expression construct, it was found to decrease expression (1.0 ± 0.01 vs. 0.1 ± 0.009, P<0.05), and even greater reductions were observed when the TCF21 expression vector was combined with another vector expressing one of the class II bHLH factors TCF3 (protein isoforms E12 or E47) (Fig 2H). TCF21 was also shown to inhibit expression of a minimal luciferase reporter containing 3 E-box sequences based on the known TCF12 motif (CAGCTG) in conjunction with a minimal promoter. There was significant repression with TCF21 expression alone (1.0 ± 0.29 vs. 0.21 ± 0.02, P<0.05), and additional reductions were noted when bHLH class II factors were included (Fig 2I). For both constructs, the developmental factor Twist produced a modest increase in TCF21 transrepression.
TCF21 expressing cells are found in atherosclerotic lesions and associate with the fibrous cap
To investigate disease-related expression of Tcf21 in the vascular wall, we performed time-course studies in Tcf21lacZ/+, ApoE-/- hyperlipidemic atherosclerotic mice, and compared Xgal staining in these tissues with that observed in negative control ApoE-/- animals. After four weeks of high fat diet (HFD) there were no β-galactosidase expressing cells in the forming lesions, although there was apparent clustering of β-galactosidase positive cells on the luminal side of the media immediately below the areas where lesions had initiated growth (Fig 3). By eight weeks of HFD, lesions in most animals showed abundant β-galactosidase positive cells accumulating toward the luminal side of the atheromatous lesion, with streaks extending from media through the lesion toward the fibrous cap. β-galactosidase positive cells observed in the lesion appeared to be aligned orthogonal to the axis of orientation for cells in the media, suggestive of migratory behavior. By twelve weeks of HFD feeding there was extensive staining in lesions of most animals, with the β-galactosidase positive cells in the vicinity of the forming fibrous cap, or in a cap-like structure that was situated medial to and extended below the actual fibrous cap (Figs 3 and 4). This time interval from 4 to 12 weeks was associated with an apparent decrease in the number of medial cells expressing SMC markers and a disruption of the lamellar medial structure. Tcf21 expressing cells in the media at 8 to 12 weeks were disorganized and had lost their characteristic lamellar structure. By 20 weeks, almost all of the Tcf21 expressing cells in the lesions were associated with the fibrous cap or a subcapsular structure (Fig 3).


To determine whether results in the ApoE model were consistent with TCF21 expression in the diseased human coronary vessel wall, immunostaining of human coronary artery tissues was performed with antibodies for TCF21 and ACTA2. In those vessels with only modest disease there was adventitial TCF21 staining with some staining of the minimal neointima, but no TCF21 staining in the media, which was positive for ACTA2 (S6A–S6C Fig). In vessels with significant disease there was robust TCF21 staining in the adventitia and neointima and some patchy staining in the fibrous cap, but very little staining in the media (S6D and S6E Fig). ACTA2 staining was prominent in the fibrous cap and considerably weaker staining in the media. These studies confirm what was observed in the mouse diseased vasculature.
To assign TCF21 expressing cells to a known cellular lineage we evaluated combined lacZ expression with immunohistochemistry staining for well-known lineage markers for different vascular cell types. Specifically, we worked to identify co-staining for murine adventitial and vascular lesion lineage markers, focusing on lacZ reporter gene expression in the ApoE-/- mice. Xgal and immunohistochemistry staining was performed of adventitial and lesion areas with antibodies specific for endothelial cells (VWF), SMC (ACTA2), neuronal cells (TUBB3), adipocytes (PLIN1), fibroblasts (THY1), stem cells (nestin), macrophages (LAMP2), hematopoietic cells (PTPRC), and dendritic cells (Itgax) (S7 Fig). These studies failed to identify co-localization with known cellular lineage markers, and when combined with published negative data for pericyte markers argue that Tcf21 is not expressed by a common terminally differentiated vascular cell type [17].
Further reporter gene and immunohistochemistry studies focused on the fibrous cap in lesions of mice fed HFD for 4 to 20 weeks. Studies with SMC marker Tagln again revealed that the majority of Tcf21 expressing cells in the adventitia, media and plaque of diseased vessels did not express SMC markers, which were easily identified in association with medial and fibrous cap SMC (Fig 4). Although individual Tcf21 expressing cells and those in subcapsular structures did not express SMC markers, there were Tcf21 expressing cells identified immediately below or within the fibrous cap that appeared to also express Tagln. In addition to SMC lineage markers, we investigated expression of growth factor receptor Tgfbr2, which is involved in SMC development and structural vascular wall disease [27,28] and was identified as one of the differentially expressed genes in the TCF21 knockdown RNA-Seq studies, as well as Pdgfrb which has an important role in coronary SMC development and atherosclerotic disease [5,29]. Fibrous cap cells were shown to express growth factor receptors Tgfbr2 and Pdgfrb, and there appeared to be co-staining for Tcf21 expression and growth factor expression by cells in the vicinity of the fibrous cap. Whether the observed co-staining identifies a unique transitional cell that expresses Tcf21 and SMC marker genes, or is due simply to sustained enzymatic activity of the β-galactosidase protein after Tcf21 expression is downregulated was not clear from these studies. However, these data suggest that Tcf21 expressing cells can adopt an SMC phenotype and contribute to the fibrous cap.
Tcf21 expressing cells give rise to smooth muscle cells in the fibrous cap of ApoE-/- mice
In the next series of experiments we sought to more clearly identify the possible origins and cellular fate of Tcf21 expressing cells found in the vascular disease lesions. For these studies we employed a lineage tracing approach with an inducible Cre recombinase that provided permanent fluorophore marking of cells that expressed Tcf21 at the time of activation of recombinase expression. We used a mouse line expressing a Cre recombinase protein fused to two mutant estrogen-receptor ligand-binding domains (MerCreMer) [30], under the control of the endogenous Tcf21 locus (Tcf21iCre/+). This line has been well characterized and used extensively to investigate the cellular fate of Tcf21 expressing cells in the developing embryo [17,31]. For reporter alleles, we employed both a tandem dimer tomato (tdT) fluorescent reporter line (B6.Cg-Gt(ROSA)26Sortm14(CAGtdTomato)Hze/J) as well as a lacZ reporter line (B6.129S4-Gt(ROSA)26Sortm1Sor/J) that was tracked with histochemical β-galactosidase activity. ApoE-/- mice carrying the Cre and reporter alleles were administered tamoxifen before HFD and sacrificed after 16 weeks of the diet, or administered tamoxifen at 6–8 weeks and harvested after 8, 12 or 16 weeks of HFD before sacrifice.
Lineage tracing studies in ApoE-/- mice were primarily conducted in animals on HFD for 12 weeks, with tamoxifen being administered from 6 to 8 weeks of the diet. At baseline, Tcf21 expression using this lineage tracing system was consistent with that seen for the lacZ knockin reporter mouse line, being restricted to the adventitial cells associated with coronary arteries, cardiac fibroblasts, and some aortic root cells (Fig 5A). Aortic root tissues from animals treated with HFD revealed tdT-expressing cells in the lesions. For early lesions without a defined fibrous cap, Tcf21 lineage traced cells were distributed through the lesion and did not express the SMC marker Tagln (Fig 5B). In those lesions with a mature fibrous cap, Tcf21 lineage traced cells were noted to contribute to this cap and many of these cells shown to express Tagln (Fig 5C and 5D). These data are consistent with the results described above for Tcf21 reporter gene and SMC marker immunohistochemistry studies, and suggest that Tcf21 expressing cells may give rise to SMC in the fibrous cap.
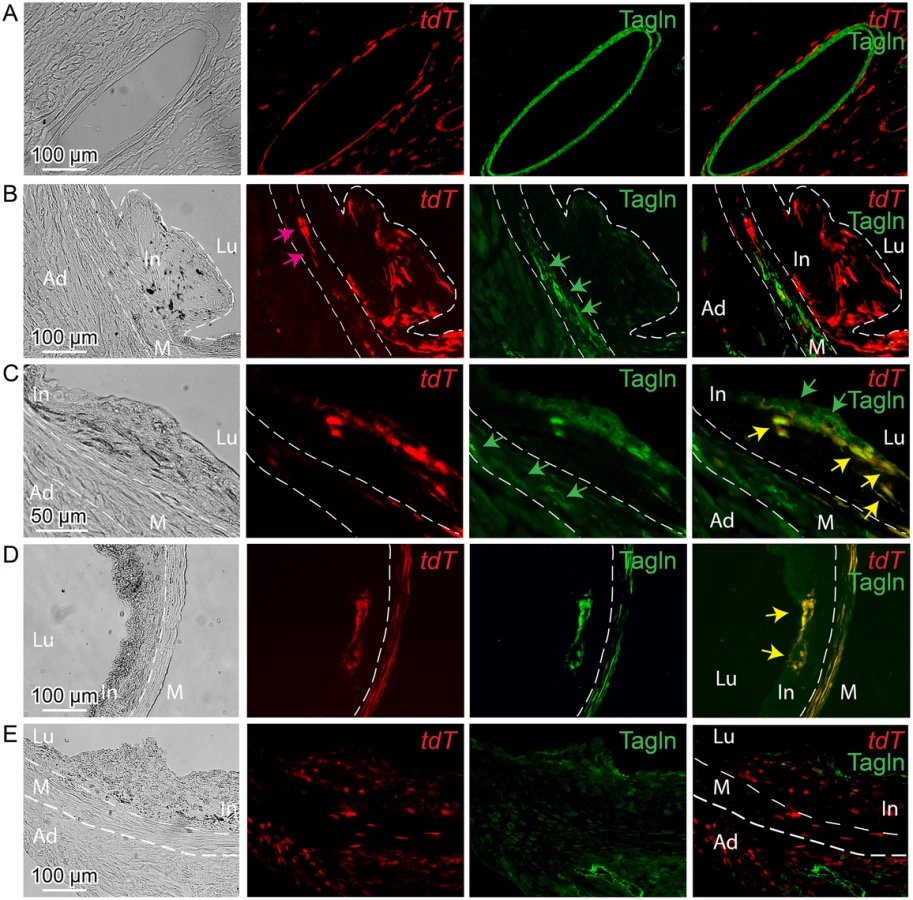
By activating recombinase activity before HFD feeding and lesion development it was possible to investigate the cellular origin of the Tcf21 expressing cells identified in the vascular lesions of the ApoE-/- model. For mice that were administered tamoxifen before HFD, and then maintained on the diet for 12 weeks, lineage traced cells were commonly identified in lesions (Fig 5E). These data are thus consistent with their derivation from Tcf21 expressing cells in the vascular wall adventitia or media, and suggest that Tcf21 lineage traced cells were able to migrate into the forming lesions and contribute to lesion formation.
Finally, to verify the results obtained with the fluorescent tdT reporter, we conducted identical experiments with the Cre-activated lacZ reporter mouse. These studies also found that Tcf21 lineage traced cells in the neointima did not express the SMC marker Tagln, whereas lineage traced cells contributing to the fibrous cap cells were found to express this SMC marker (S8 Fig).
Lineage tracing in Ldlr-/- mice also shows recruitment of Tcf21 expressing cells that contribute to the fibrous cap
To generalize the findings obtained in the ApoE-/- model, we conducted lineage tracing studies in Ldlr-/- mice. Findings from these experiments also showed that activation of the reporter gene at 3 to 5 weeks of age, prior to HFD feeding for 16 to 20 weeks, marked cells that migrated into the atherosclerotic lesion (Fig 6A). As in the ApoE-/- model, some of the Tcf21 traced cells activated expression of SMC markers Tagln and Acta2 and localized to the fibrous cap (Fig 6B). In addition, co-labeling with an antibody specific for the matrix protein periostin showed some general co-localization of Tcf21 traced cells and periostin in the adventitia while there was extensive colocalization in the fibrous cap and co-staining in a subcap region located abluminal to the fibrous cap (Fig 6C). Also, Tcf21 traced cells in the adventitia and fibrous cap showed extensive colocalization for Pdgfra expression (Fig 6D).

The rate of cell division in vascular lesions is related to Tcf21 expression
To investigate whether Tcf21 plays a role in regulating vascular cell proliferation in the disease setting in vivo, we used two different approaches, one employing early stage disease in the Ldlr-/- model, and the other a later stage ApoE-/- murine model. In experiments with Ldlr-/- mice we investigated whether there was a time-dependent increase in the relative number of Tcf21 expressing cells that were undergoing cell division. In these studies, Tcf21iCre/+, ROSAtdT/+, Ldlr-/- mice were treated with tamoxifen at 1 week before isolation, and EdU was injected 24 hours prior to tissue harvest at relatively early disease time points for this model, at 0, 9, 12, and 15 weeks of feeding with HFD. EdU positive cells were found in the adventitia, media and intima of the aortic wall and quantified in sections of the aorta through the region of the sinus. The percentage of proliferating Tcf21 expressing cells was identified by both tdT and EdU fluorescence (Fig 7A). Comparison of the number of Tcf21+EdU+ cells versus all Tcf21+ cells at different time points showed a significantly greater percentage of proliferating cells at 12 weeks of diet compared to time zero (1.94±0.70 vs. 6.42±1.10, P<0.05) (Fig 7B). There was also an increase in the mean percentage of EdU+ Tcf21+ cells to EdU+ cells between the 9 and 12 week time points, although this difference did not reach statistical significance (P = 0.053). Studies also investigated whether there was a difference in the relative rate of cell division in lesions of Tcf21lacZ/+, ApoE-/- mice compared to ApoE-/- animals, after 20 weeks of HFD. All mice were injected with EdU 24 hours prior to tissue harvest and the number of EdU fluorescent labeled cells was compared to the total number of cells as identified by DAPI fluorescence for each genotype. This analysis was restricted to the plaque area where the majority of EdU+ cells were located in these more mature lesions (Fig 7C and 7D). The Tcf21lacZ/+, ApoE-/- mice showed a significantly lower number of EdU+ cells compared to ApoE-/- animals (2.25±0.18 vs. 3.27±0.25, P<0.05) (Fig 7E). Taken together, these data suggest that Tcf21 expressing cells show a time-dependent increase in proliferation with disease progression, possibly greater than other cells not expressing Tcf21, and that this increase may depend on Tcf21 expression.

TCF21 is differentially expressed in the fibrous plaque of ruptured human atherosclerotic lesions
To begin to investigate the possible association of TCF21 expression and function in the fibrous cap of human atherosclerotic lesions, we have investigated the TCF21 expression pattern in fibrous plaque of carotid atherosclerotic lesions collected from patients at time of endarterectomy surgery. Micro-dissection laser capture was used to obtain tissue from the fibrous cap of 10 vessels that showed stable plaque morphology and 10 vessels that had undergone plaque rupture (Fig 8). RNA was isolated from these tissues and employed to assess TCF21 mRNA levels by qPCR. This analysis showed significantly decreased levels of TCF21 expression in the fibrous cap tissue harvested from the ruptured plaque (5.0±0.3 vs 6.9±0.3, ruptured vs stable plaque, P<0.0001).

Discussion
SMC are critical contributors to atherosclerosis pathophysiology, and relevant to the findings reported here is the dramatic alteration in gene expression and basic cellular functions that occur with phenotypic modulation of this cell type in the disease setting [5,32,33]. In the classic response to injury hypothesis, fully differentiated medial SMC express contractile marker genes and have limited proliferative capacity, while under disease conditions they downregulate contractile gene expression, proliferate, and migrate into the neointimal space. Data presented here suggest that upregulation of TCF21 may be directly responsible for at least some aspects of the phenotypic modulation that is believed to characterize the medial SMC response to vascular injury. We have shown that TCF21 binds the ACTA2 locus, transrepresses ACTA2 reporter genes in cultured SMC, and that knockdown of TCF21 is associated with increased ACTA2 expression. Although we have not studied other SMC markers in such detail, both TAGLN and MYH11 are upregulated in siTCF21 treated cells. While additional studies are required to characterize a possible dedifferentiation function for TCF21, also consistent with such a hypothesis are in vitro studies in cultured HCASMC presented here which show that TCF21 promotes proliferation and migration. A differentiation inhibiting function has previously been proposed for TCF21 in skeletal muscle cells [34].
Tcf21 is expressed in epicardial progenitor cells that give rise to coronary SMC, and its expression in the adult might be characteristic of a precursor cell that is resident in the vascular wall and is capable of giving rise to SMC in the setting of vascular injury. A number of previous studies have identified such precursor cells in distinct compartments of the vessel wall, all with some capacity to give rise to differentiated SMC. It is generally accepted that SMC accumulation in the fibrous cap is monoclonal or oligoclonal, and this is consistent with the existence of a resident arterial subpopulation of SMC precursor cells that contribute to healing in the response to disease stresses [35]. In bovine and canine vessels, subsets of medial SMC have prominent proliferation with reduced SMC marker expression, and this population has been estimated to be 6–15% of cells in the murine aorta [36–38]. These observations are consistent with data presented here showing that Tcf21 expressing cells with low SMC marker expression are distributed in the proximal aorta in mouse. Cells with SMC precursor function have also been identified in the adventitial layer [35]. In mouse, Sca1+ cells with SMC differentiation potential have been identified in the aortic adventitia [39], and in humans, a population of cells with SMC potential has been identified in the space between the media and adventitia of large and medium sized vessels [40]. While there are reported differences in the features and markers of these and other progenitor-like cells in the vascular wall, here we demonstrate an overlap of expression pattern and other features in Tcf21 expressing cells. It seems likely that TCF21 may be expressed in one or more of these cell populations and may contribute to an undifferentiated SMC precursor phenotype.
The studies described herein provide important new information regarding the biology of the fibrous cap. While the risk of plaque rupture appears to be inversely correlated with the number of SMC in the fibrous cap, there is limited understanding of their in vivo origin, and the molecular pathways that regulate their expansion and terminal phenotype determination [5,41]. With these studies we have shown Tcf21 to be a marker for precursor cells that give rise to at least a portion of fibrous cap cells, and genomic studies in conjunction with in situ protein marker studies identified a number of signaling molecules that relate to the phenotype of this important cell type. While Pdgfra and Pdgfrb have been previously identified on fibrous cap cells [42,43], we show here that they are not expressed by cells that are migrating toward the cap and expressing Tcf21 but initiate expression in conjunction with upregulation of SMC marker genes when they are juxtaposed to the endothelium. Tgfbr2 has not previously been identified on fibrous cap cells, and its biology in this context not studied, but it has recently been shown to support the differentiated SMC phenotype and homeostasis of this cellular lineage [27,28]. TGFBR2 mRNA levels were found to be regulated by TCF21 in the siRNA knockdown RNA-Seq studies. Another protein expressed by Tcf21 lineage-traced cells in the fibrous cap, periostin, is known to promote SMC differentiation and migration [44–46], and interestingly has recently been associated with the atherosclerotic phenotype in young humans [47]. Additional follow-up of genomic studies are expected to provide insights into other genes and pathways that are present in TCF21 marked fibrous cap precursor cells.
Lineage tracing provides a unique and informative approach to address questions related to cellular origin, movement and differentiation of lesion cells in the vascular wall, however a number of questions remain unanswered. Given the capacity of Tcf21 to promote autonomous cellular proliferation and migration, and the appearance of lineage traced cells in lesions of animals where recombination preceded HFD feeding, it seems likely that lineage traced cells can migrate from the adventitia or media to contribute to lesion formation. Although there is some evidence that bone marrow—derived SMC-like cells may contribute to neointima formation, such cells appear to represent a very small fraction of the total neointimal cells [48,49]. Given the variability in disease progression in different ApoE-/- and Ldlr-/- animals, and the possibility that Tcf21 is upregulated in adventitial or medial cells throughout disease progression, lineage tracing does not allow assessment of how much of the fibrous cap derives from Tcf21 expressing cells. Results from the Tcf21lacZ/+, ApoE-/- studies suggest that Tcf21 expressing cells constitute a significant portion of the lesion SMC but this cannot be readily evaluated with current methods. Also, evidence that some fibrous cap SMC derive from lineage traced cells does not eliminate the possibility that a portion of the fibrous cap cells continue to express Tcf21 and maintain a fibroblast or myofibroblast phenotype, and that the fibrous cap is actually a combination of such cells along with those that have downregulated Tcf21 and adopted a true SMC phenotype. This possibility is consistent with sustained Tcf21 expression in some of the fibrous cap cells at the 20-week disease timepoint (Fig 3).
While the work reported here has provided important initial details of how this gene is expressed during atherosclerotic vascular disease and the types of processes that it might regulate to affect disease pathophysiology, the causal mechanisms of disease risk remain unclear. The direction of effect has not been fully established by current available human genetic and genomic data, with both directions being suggested by different approaches and possible underlying gene regulatory mechanisms characterized for each [8,50,51]. Based on the animal model findings reported here, greater TCF21 expression could inhibit disease risk by promoting the expansion of SMC lineage and contributing to plaque stabilization by supporting fibrous cap development. Alternatively, greater TCF21 expression in SMC precursor cells at the fibrous cap might inhibit their differentiation to the SMC lineage, thus destabilizing the plaque. Similar alternative hypotheses could be generated for the risk allele producing decreased TCF21 expression. Nonetheless, further in vivo gain - and loss-of-function studies are required to determine how changes in Tcf21 gene expression affects the size and architecture of the fibrous cap and how such changes correlate to specific human disease traits linked to plaque vulnerability and rupture. These questions could be addressed using the various plaque rupture models established in mice [52,53]. Finally, it will be important to investigate the phenotype of the TCF21 expressing cells at baseline, and how the phenotype changes as these cells migrate into the lesion. Such studies could be performed with captured cells using modern genomic methods, and would provide extremely valuable insights into the cellular phenotype as these cells respond to the vascular signals of the disease process.
Methods
Ethics statement
All procedures described in this study were approved by the Institutional Animal Care and Use Committees of Stanford University and University of Hawaii and conformed to NIH guidelines for care and use of laboratory animals.
Cell culture
Human primary Coronary Artery Smooth Muscle Cells (HCASMC, #CC-2583) were obtained from Lonza (Allendale, NJ, USA) at passage 4 and cultured in Smooth Muscle Growth Medium-2 including hEGF, insulin, hFGF-B and FBS, but without antibiotics (Lonza, #CC-3182). Human embryonic kidney cells (HEK, ATCC #CRL-11268) and rat aortic smooth muscle cells (A7R5, ATCC #CRL-1444) were cultured in DMEM High Glucose (Life Technologies #311995–065) with 10% FBS (Life Technologies #26140–79). Human cardiac fibroblast (HCF), bone-marrow derived mesenchymal stem cells (bmMSC), embryonic stem cell derived mesenchymal stem cells (eMSC), induced pluripotent stem cell-derived mesenchymal stem cells (iMSC), human aortic adventitial fibroblasts (HAoAF), human aortic endothelial cells (HAoEC), human coronary artery endothelial cells (HCAEC), and human aortic smooth muscle cells (HAoSMC) were obtained from commercial suppliers, maintained in the recommended media, and evaluated with at least 3 technical replicates.
For RNA-Seq studies and individual gene quantitative PCR, HCASMC were transfected with 300 nM TCF21 Trilencer-27 Human siRNA (OriGene #SR304753C) or Trilencer-27 Universal Scrambled Negative Control siRNA (OriGene #SR30004) at 80% confluence using the Amaxa Basic Nucleofector Kit for Primary Mammalian Smooth Muscle Cells (Lonza #VPI-1004) at a density of 1 x 106 cells per 100 μL sample using Nucleofector Program U-025. Cells were changed to medium with supplements at 18 hours post-transfection and cultured for an additional 48 hours.
Lentivirus construction and in vitro cell culture assays
For TCF21 overexpression, a cDNA clone (OriGene #SC125048) representing the protein coding sequence of human TCF21 was cloned into pWPI vector (Addgene #12254) which also contains the GFP reporter gene. For shRNA knockdown the following sequences were cloned into pLVTHM (Addgene #12247):
shTCF21 construct 1
1:GaattcgaacgctgacgtcatcaacccgctccaaggaatcgcgggcccagtgtcactaggcgggaacacccagcgcgcgtgcgccctggcaggaagatggctgtgagggacaggggagtggcgccctgcaatatttgcatgtcgctatgtgttctgggaaatcaccataaacgtgaaatgtctttggatttgggaatcttataagttctgtatgagaccacagatctCCCTGGATTCGAACAAGGAATTTGTGACTTTTCAAGAGAAAGTCACAAATTCCTTGTTCGAATCCATTTTTGGAAaagcttATCGAT
shTCF21 construct 2
GaattcgaacgctgacgtcatcaacccgctccaaggaatcgcgggcccagtgtcactaggcgggaacacccagcgcgcgtgcgccctggcaggaagatggctgtgagggacaggggagtggcgccctgcaatatttgcatgtcgctatgtgttctgggaaatcaccataaacgtgaaatgtctttggatttgggaatcttataagttctgtatgagaccacagatctCCCTGGAGATGTTGGAATGTGACGGGTTGATTCAAGAGATCAACCCGTCACATTCCAACATCTCCATTTTTGGAAaagcttATCGAT
Lentiviruses were produced by polyethylenimine (Polysciences cat #23966–2)-mediated transfection of 293T cells (American Type Culture Collection #CRL-11268). Per 225 cm2 flask, 3.6 x 107 cells were plated 24 hours before transfection in DMEM (Invitrogen cat #11995), 10% fetal bovine serum (Hyclone cat #SH30070.03). For transfection, 30 mg of lentivirus vector, 30 mg of a HIV-1 gag-pol-rev-tat helper plasmid (pCMV DR8.9, Addgene #8455), and 15 mg of a plasmid encoding the VSV-G envelope protein (pMD2.G, Addgene #12259) were used per T-225 flask of cells transfected. The plasmids (75 mg total) were added to OptiMEM (Invitrogen cat # 31985) such that the total volume was 700 ml and then 300 ml of 1 mg/ml polyethylenimine (25 kDa linear, pH 4.5 in 1 x PBS) was added. The PEI/DNA mixture was mixed, incubated 10 min at 20°C, and then added to the cells. The cells were incubated at 37°C in a 5% CO2 atmosphere. Then 24 and 48 hours after transfection the media was harvested and replaced with fresh media. After harvesting the media, dead cells were removed by centrifugation at 3,000 x g for 15 min. The virus was filtered through a 0.45 μm Steri-flip filter (Millipore cat #SE1M003M00) and then aliquoted and stored at -80°C until use.
HCASMC were transduced with lentiviral particles at a MOI of 22.5 at passage 7 to 9. The transduction efficiency was assessed by quantifying the percentage of GFP-positive cells by flow cytometry on a BD FACSCalibur. Transduction efficiencies were between 50 and 95 percent.
Cell migration (gap closure) assay
The gap closure assay (Cell Biolabs #CBA-126) was conducted according to the manufacturer’s protocol. Briefly, 10,000 lentivirus transduced cells were seeded per well and let attach overnight. After gel removal 3 wells per condition were directly stained with crystal violet and imaged using a Zeiss Axioplan 2 instrument using the NIS-Elements F v4.0 software while the remaining 9 wells per condition were incubated for 12h before crystal violet staining. The covered area per well was quantified using ImageJ v1.47.
In vitro proliferation assay (cell counting)
Lentivirally transduced HCASMC were plated into 6 well plates at low density (10,000 cells per well, 3 wells per condition) and harvested at given time points before reaching confluence. Viable cell numbers were then counted manually and percentage of GFP positive cells measured by flow cytometry.
In vitro proliferation assay (EdU labeling)
Lentivirus transduced cells were plated to a 12 well plate at sub-confluence density and allowed to recover overnight. The cells were then serum starved for 24 hours. Following starvation, the cells were exposed to serum for 24 hrs, with treatment during the last 3 hrs of this period with EdU from the Click-iT EdU Alexa Fluor 488 Imaging Kit (Life technologies, Carlsbad, CA; Cat# C10377) at a concentration of 20uM. The cells were incubated with EdU for 3 hours, then fixed and permeabilized using 4% PFA and 0.5% Triton-X in PBS, respectively. This was followed by incubation with Click-iT reaction cocktail, including CuSO4 and Alexa Fluor Azide, and then with nuclear staining with DAPI solution. Using a Leica inverted microscope, the number of total nuclei, and the number of co-stained nuclei were counted using the ImageJ (NIH) software on ten consecutive 10x fields for each condition.
In vitro apoptosis assay
Cells were transfected with siControl or siTCF21 reagents exactly as described above for the RNA-Seq studies, at a density of 1E6 cells per 100 μL sample using Nucleofector Program U-025. Cells were changed to medium without supplements at 18 hours post-transfection and cultured for an additional 48 hours. Caspase activity in these cells was detected using the Caspase-Glo 3/7 Assay (Promega #G8093).
RNA isolation and quantitative real-time RT-PCR
For qPCR RNA was isolated using QIAzol (Qiagen #79306) and the Trizol RNA isolation protocol. DNA was removed with the DNA-free kit from Ambion (Life Technologies #AM1906) followed by first strand cDNA synthesis and subsequent RNA digestion according to the SuperScript III protocol (Life Technologies #18080–051). Relative gene expression was measured using the ViiA 7 Real-Time PCR System from Applied Biosystems (Life Technologies). cDNA was amplified as follows: 95°C 15 min, 40x (95°C 1s, 60° 20s). Locus enrichment in chromatin immunoprecipitation samples was measured using the ABgene SYBR mastermix (AB-1166) and the primers ACTA2chipfor 5’-AGGGAGATGCAAACCAGATATCC-3’, ACTA2chiprev 5’-GCAGGTGACCTGCTGAATTTTTC-3’, PROCRchipfor 5’-GACTCCTGCTTACCTCCTCATA-3’ and PROCRchiprev 5’-GGGTGAAGAAGGTACAAAAGAA-3’. Genomic DNA cycling conditions were 95°C 15min, 40x (95°C 15s, 60°C 1 min). Absolute quantification of expression levels was performed using a standard dilution series and normalization to 18S rRNA. Relative expression levels were then calculated relative to TCF21 expression levels in HEK cells (S1A Fig) or in SiCTRL treated HCASMC (Fig 2E) by division.
RNA sequencing
Total RNA from either siTCF21 or siCTRL treated samples was depleted for ribosomal RNA with the Ribo-Zero magnetic kit from Epicentre (Illumina #MRZH116), libraries generated with the Epicentre ScriptSeq v2 RNA-Seq library preparation kit (Illumina #SSV21106) and thereafter sequenced as 100bp paired-end reads on an Illumina HiSeq 2000 instrument. The resulting data has been deposited at GEO under accession number GSE44461.
Reads resulting from RNA-Sequencing of siCTRL and siTCF21 treated HCASMC were mapped using software tools TopHat+Bowtie2. Differential expression level between samples was analyzed using the software tools DESeq and edgeR at an FDR ≤ 0.05, with intersection of the 466 and 430 respective identified genes providing a group of 380 common genes. This genelist was used to interrogate the Ingenuity Knowledge Base, identifying over-representation of Cardiovascular Disease annotation terms, including the top functional category “Vascular disease.” Gene members of this group were expanded by adding 18 genes with the “build” function in IPA to create the TCF21 Vascular Disease Network. Visualization of the network was performed using Cytoscape open source software. Node color was mapped to log fold change (green-yellow-red palette), node size to absolute expression value in wild type cells, and font size to enrichment Q-value. GO categories were assigned to genes using the Bingo application for Cytoscape and coloring the GO categories was performed using GOlorize application for Cytoscape.
Mouse models
A Tcf21 functional knockout and reporter line, Tcf21lacZ/+, was constructed by substituting the lacZ gene into the first exon of the murine gene employing homologous recombination in embryonic stem cells, as described previously [12]. Fidelity of expression of the lacZ reporter has been described [12], and methods for using Xgal staining to track Tcf21 expression were identical to those published previously from this laboratory [54]. For studies of reporter gene expression in the ApoE-/- model, animals were weaned onto Western high fat diet (HFD, 21% anhydrous milk fat, 19% casein and 0.15% cholesterol, Dyets no. 101511). The animals were euthanized and perfused with PBS followed by 0.4% PFA, the aortic roots excised and subjected to Xgal staining followed by post-fixation for 30 minutes in 4% PFA. After incubation in an ascending sucrose solution series and embedding in OCT, tissue was processed into 7 μm thick frozen sections for immunohistochemistry.
Lineage tracing studies employed a recently described inducible Tcf21-Cre line (Tcf21iCre/+) [31], and tomato (B6.Cg-Gt(ROSA)26Sortm14(CAGtdTomato)Hze/J) Jax #007914 and lacZ (B6.129S4-Gt(ROSA)26Sortm1Sor/J) Jax # 003474 reporter lines. The Tcf21iCre/+ line was obtained on a mixed C57Bl/6 x 129SV mixed background and was bred back onto the C57Bl/6 background for 6 generations before being mated with the ApoE-/- line. The Tcf21iCre/+ allele was bred directly onto the Ldlr-/- line in C57Bl/6 background. All Ldlr-/- mice were treated with a modified Western high fat diet (HFD, 15.8% milk fat and 1.25% cholesterol (diet 94059; Harlan Teklad) for the time periods indicated. Tcf21iCre/+, ROSAtdT/+, ApoE-/-; Tcf21iCre/+, ROSAtdT/+, Ldlr-/-; and Tcf21iCre/+, ROSAlacZ/+, ApoE-/- and Ldlr-/- and ApoE-/- control animals were treated as described with HFD, tissues harvested and sectioned as described above, and analyzed for expression of the reporter genes as well as for other cellular markers as described below. For ApoE-/- animals, Cre recombination was activated with a series of ten 1 mg tamoxifen (Sigma Aldrich) intraperitoneal injections for a total of 10 mg of tamoxifen. Animals were induced at 0, 6, 10 weeks and sacrificed after 20, 8, and 12 weeks respectively of the HFD as described above. For Ldlr-/- mice, Cre induction was accomplished by feeding of tamoxifen chow at 3–5 weeks of age (Harlan Laboratories, Inc).
Immunostaining
For immunocytochemistry, HCASMC were seeded into culture slides (BD Biosciences #35641) in supplemented medium for 24 hours. For immunohistochemistry of human tissues, paraformaldehyde-fixed human coronary arteries were frozen, embedded and sectioned at a thickness of 7 μm and tissue slides were postfixed with 4%PFA in PBS for 10 minutes. Staining for TCF21 expression was conducted on frozen tissue sections with a polyclonal anti-TCF21 antibody (Sigma-Aldrich #HPA013189) at a dilution of 1 : 100, with overnight primary incubation.
Mouse tissues were collected and processed as described above. After postfixation, nonspecific binding sites were blocked for 30 minutes with Rodent Block M (Biocare Medical #RBM961). Primary antibody incubation was performed overnight at 4°C using the following antibodies and dilutions in Da Vinci Green antibody diluent (Biocare Medical #PD900), anti-Acta2 1 : 300 (Abcam #ab5694), anti-Tagln 1 : 300 (Abcam ab14106), anti-Pdgfrb 1 : 500 (Abcam #ab32570), anti-Tgfbr2 1 : 100 (Abcam #ab61213), anti-periostin 1 : 200 (Santa Cruz #sc49480), anti-Pdgfra (R&D Systems #AF1062), anti-vWF 1 : 250 dilution (Abcam ab11713), anti-TUBB3 1 : 1000 dilution (Abcam ab78078), anti-PLIN1 1 : 500 dilution (Abcam ab61682), anti-THY1 1 : 200 dilution (Dianova DIA-100-M), anti-NES 1 : 2000 dilution (Abcam ab134017), anti-LAMP2 1 : 150 dilution (Abcam ab37024), anti-PTPRC 1 : 300 dilution (Abcam ab), and anti-ITGAX 1 : 100 dilution (Abcam ab33483). The slides were incubated for 1h at room temperature with Rabbit-on-Rodent AP polymer (Biocare Medical #RMR625) and 10–15 minutes with the substrate Vulcan Fast Red (Biocare Medical #FR805). Between steps slides were washed 2 x 5 minutes with TBS (Sigma-Aldrich #T6664). The slides were then dehydrated in an ascending alcohol dilution series and mounted in Cytoseal XYL (Thermo Scientific #8312–4). Images were taken with a Zeiss Axioplan 2 using the NIS-Elements F v4.0 software.
EdU staining was employed to quantify in vivo rates of vascular wall cell proliferation in Tcf21iCre/+, ROSAtdT/+, Ldlr-/- mice. These mice were weaned onto HFD at 4 weeks of age, administered tamoxifen by a single gavage of 10 μg/40g body weight 1 week before tissue harvest, and sacrificed and evaluated at T0, 9wks, 12 wks or 15 wks of diet. 5-Ethynyl-2-deoxyuridine (Santa Cruz, 50μg/g body weight) was injected intraperitoneal 24 hours before sacrifice. Aortic root tissues were harvested, processed and sectioned, and Tcf21+EdU+ positive cells were quantified and expressed as the % of Tcf21 lineage cells. This percentage was evaluated for significant differences among the various time points evaluated. These data are based on three 40x images at each time point in each animal.
Similar experiments were performed to compare the relative rates of cell division in Tcf21lacZ/+, ApoE-/- compared to ApoE-/- mice. Animals in each group were injected with 1mg of EdU (Life Technologies, cat#A10044) 24 hours before sacrifice and EdU incorporation was imaged according to the manufacturer's instructions with the Click-iT EdU Alexa Fluor 488 Imaging Kit (Life Technologies) on OCT (Tissue-Tek) frozen sections. Eight sections were assessed from each group with ImageJ software (NIH) to determine the ratio of EdU positive cells to the total number of DAPI positive cells within plaque areas.
Micro-dissection laser capture and TCF21 expression analysis in human atherosclerotic lesions
Human atherosclerotic carotid artery lesions were obtained from patients undergoing endarterectomy surgery for stable (asymptomatic; n = 10) or vulnerable (symptomatic; n = 10) carotid stenosis, as part of the Biobank of Karolinska Endarterectomies (BiKE). Samples were collected with informed consent procedure and the study approved by the Local Ethical Committee. Patient demographics, symptom definition and sampling routines were described previously [55]. Micro-dissection laser capture of fibrous cap structures from both subgroups of patient samples was performed using a PALM Microlaser system (Zeiss, Germany) according to manufacturer’s instruction. Atherosclerotic lesions, stable and ruptured (vulnerable), were paraffin-embedded, sectioned, and stained with hematoxylin & eosin on RNAse-free glass slides. Sections were pre-treated with UV light at 254nm for 30 minutes to overcome the hydrophobic nature of the membranes and to enhance adhesion of the paraffin-embedded sections. Ten consecutive slides per individual patient were micro-dissected and then pooled for gene expression analysis.
In preparation for laser pressure catapulting, sections were first de-paraffinized with Xylene (2 x 2 minutes) and decreasing C2H6O (Ethanol) concentrations (100%, 96%, 70% each for 1 minute). Sections were then rinsed in RNAse-free water, and stained for 10 minutes with Mayer’s hematoxylin (Sigma, USA), again rinsed for 3 minutes in RNAse-free water, and then consecutively stained for 3 minutes with Eosin (Sigma, USA). Finally, samples were dehydrated with increasing concentrations of C2H6O, before briefly being air-dried at room temperature. After micro-dissection and catapulting, the fibrous cap sample was collected into AdhesiveCaps (Zeiss, Germany) and 350μl of RLT buffer (Qiagen, Germany) was added, and mixed by inversion after closure. The lysate was spun down for 5 minutes at 13 000 rpm and stored at -80°C.
RNA extraction from catapulted, micro-dissected samples was performed using the RNeasy Micro Kit (Qiagen, Germany) following the manufacturer’s protocol. RNA was quantified by Nanodrop (Agilent Technologies), and RNA quality was verified using an Agilent 2100 Bioanalyzer (Agilent Technologies). Samples required 260/280 ratios >1.8, and sample RNA integrity numbers >9 for inclusion. The TaqMan High Capacity cDNA Transcription Kit was used for cDNA synthesis, and primer assays for TCF21 and 18S (for housekeeping/endogenous control) were utilized to detect changes in mRNA expression levels. The mean TCF21 mRNA level (run in triplicate) was normalized to the mean 18S mRNA level (run in triplicate) to calculate the change in expression for each condition.
Statistical analysis
Results are expressed as means ± standard deviation (SD) with number (n) of replicates. Statistical comparisons of two groups were performed by two-tailed t-test using Prism if not stated otherwise. Welch’s Correction for unequal variance was applied.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Daviglus ML, Talavera GA, Aviles-Santa ML, Allison M, Cai J, et al. (2012) Prevalence of major cardiovascular risk factors and cardiovascular diseases among Hispanic/Latino individuals of diverse backgrounds in the United States. JAMA: the journal of the American Medical Association 308 : 1775–1784. doi: 10.1001/jama.2012.14517 23117778
2. Gupta R, Gupta KD (2009) Coronary heart disease in low socioeconomic status subjects in India: "an evolving epidemic". Indian heart journal 61 : 358–367. 20635739
3. Ikem I, Sumpio BE (2011) Cardiovascular disease: the new epidemic in sub-Saharan Africa. Vascular 19 : 301–307. doi: 10.1258/vasc.2011.ra0049 21940758
4. Almahmeed W, Arnaout MS, Chettaoui R, Ibrahim M, Kurdi MI, et al. (2012) Coronary artery disease in Africa and the Middle East. Therapeutics and clinical risk management 8 : 65–72. doi: 10.2147/TCRM.S26414 22368447
5. Gomez D, Owens GK (2012) Smooth muscle cell phenotypic switching in atherosclerosis. Cardiovasc Res 95 : 156–164. doi: 10.1093/cvr/cvs115 22406749
6. Deloukas P, Kanoni S, Willenborg C, Farrall M, Assimes TL, et al. (2012) Large-scale association analysis identifies new risk loci for coronary artery disease. Nat Genet 45 : 25–33. doi: 10.1038/ng.2480 23202125
7. Lu X, Wang L, Chen S, He L, Yang X, et al. (2012) Genome-wide association study in Han Chinese identifies four new susceptibility loci for coronary artery disease. Nat Genet 44 : 890–894. doi: 10.1038/ng.2337 22751097
8. Schunkert H, Konig IR, Kathiresan S, Reilly MP, Assimes TL, et al. (2011) Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nat Genet 43 : 333–338. doi: 10.1038/ng.784 21378990
9. Cui S, Ross A, Stallings N, Parker KL, Capel B, et al. (2004) Disrupted gonadogenesis and male-to-female sex reversal in Pod1 knockout mice. Development 131 : 4095–4105. 15289436
10. Cui S, Schwartz L, Quaggin SE (2003) Pod1 is required in stromal cells for glomerulogenesis. Dev Dyn 226 : 512–522. 12619136
11. Hidai H, Bardales R, Goodwin R, Quertermous T, Quertermous EE (1998) Cloning of capsulin, a basic helix-loop-helix factor expressed in progenitor cells of the pericardium and the coronary arteries. Mech Dev 73 : 33–43. 9545526
12. Lu J, Chang P, Richardson JA, Gan L, Weiler H, et al. (2000) The basic helix-loop-helix transcription factor capsulin controls spleen organogenesis. Proc Natl Acad Sci U S A 97 : 9525–9530. 10944221
13. Lu J, Richardson JA, Olson EN (1998) Capsulin: a novel bHLH transcription factor expressed in epicardial progenitors and mesenchyme of visceral organs. Mech Dev 73 : 23–32. 9545521
14. Lu JR, Bassel-Duby R, Hawkins A, Chang P, Valdez R, et al. (2002) Control of facial muscle development by MyoR and capsulin. Science 298 : 2378–2381. 12493912
15. Quaggin SE, Schwartz L, Cui S, Igarashi P, Deimling J, et al. (1999) The basic-helix-loop-helix protein pod1 is critically important for kidney and lung organogenesis. Development 126 : 5771–5783. 10572052
16. Robb L, Mifsud L, Hartley L, Biben C, Copeland NG, et al. (1998) epicardin: A novel basic helix-loop-helix transcription factor gene expressed in epicardium, branchial arch myoblasts, and mesenchyme of developing lung, gut, kidney, and gonads. Dev Dyn 213 : 105–113. 9733105
17. Acharya A, Baek ST, Huang G, Eskiocak B, Goetsch S, et al. (2012) The bHLH transcription factor Tcf21 is required for lineage-specific EMT of cardiac fibroblast progenitors. Development 139 : 2139–2149. doi: 10.1242/dev.079970 22573622
18. Braitsch CM, Combs MD, Quaggin SE, Yutzey KE (2012) Pod1/Tcf21 is regulated by retinoic acid signaling and inhibits differentiation of epicardium-derived cells into smooth muscle in the developing heart. Dev Biol 368 : 345–357. doi: 10.1016/j.ydbio.2012.06.002 22687751
19. Feng J, Mantesso A, Sharpe PT (2010) Perivascular cells as mesenchymal stem cells. Expert opinion on biological therapy 10 : 1441–1451. doi: 10.1517/14712598.2010.517191 20836622
20. Sheikh AY, Chun HJ, Glassford AJ, Kundu RK, Kutschka I, et al. (2008) In vivo genetic profiling and cellular localization of apelin reveals a hypoxia-sensitive, endothelial-centered pathway activated in ischemic heart failure. Am J Physiol Heart Circ Physiol 294: H88–98. 17906101
21. Anders S, Huber W (2010) Differential expression analysis for sequence count data. Genome Biol 11: R106. doi: 10.1186/gb-2010-11-10-r106 20979621
22. Robinson MD, McCarthy DJ, Smyth GK (2010) edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26 : 139–140. doi: 10.1093/bioinformatics/btp616 19910308
23. Soneson C, Delorenzi M (2013) A comparison of methods for differential expression analysis of RNA-seq data. BMC Bioinformatics 14 : 91. doi: 10.1186/1471-2105-14-91 23497356
24. Mack CP, Owens GK (1999) Regulation of smooth muscle alpha-actin expression in vivo is dependent on CArG elements within the 5' and first intron promoter regions. Circ Res 84 : 852–861. 10205154
25. Arab K, Smith LT, Gast A, Weichenhan D, Huang JP, et al. (2011) Epigenetic deregulation of TCF21 inhibits metastasis suppressor KISS1 in metastatic melanoma. Carcinogenesis 32 : 1467–1473. doi: 10.1093/carcin/bgr138 21771727
26. Barnes RM, Firulli AB (2009) A twist of insight—the role of Twist-family bHLH factors in development. Int J Dev Biol 53 : 909–924. doi: 10.1387/ijdb.082747rb 19378251
27. Li W, Li Q, Jiao Y, Qin L, Ali R, et al. (2014) Tgfbr2 disruption in postnatal smooth muscle impairs aortic wall homeostasis. J Clin Invest 124 : 755–767. doi: 10.1172/JCI69942 24401272
28. Inamoto S, Kwartler CS, Lafont AL, Liang YY, Fadulu VT, et al. (2010) TGFBR2 mutations alter smooth muscle cell phenotype and predispose to thoracic aortic aneurysms and dissections. Cardiovasc Res 88 : 520–529. doi: 10.1093/cvr/cvq230 20628007
29. Mellgren AM, Smith CL, Olsen GS, Eskiocak B, Zhou B, et al. (2008) Platelet-derived growth factor receptor beta signaling is required for efficient epicardial cell migration and development of two distinct coronary vascular smooth muscle cell populations. Circ Res 103 : 1393–1401. doi: 10.1161/CIRCRESAHA.108.176768 18948621
30. Zhang Y, Riesterer C, Ayrall AM, Sablitzky F, Littlewood TD, et al. (1996) Inducible site-directed recombination in mouse embryonic stem cells. Nucleic Acids Res 24 : 543–548. 8604292
31. Acharya A, Baek ST, Banfi S, Eskiocak B, Tallquist MD (2011) Efficient inducible Cre-mediated recombination in Tcf21 cell lineages in the heart and kidney. Genesis 49 : 870–877. doi: 10.1002/dvg.20750 21432986
32. Alexander MR, Owens GK (2012) Epigenetic control of smooth muscle cell differentiation and phenotypic switching in vascular development and disease. Annu Rev Physiol 74 : 13–40. doi: 10.1146/annurev-physiol-012110-142315 22017177
33. Owens GK, Kumar MS, Wamhoff BR (2004) Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev 84 : 767–801. 15269336
34. Funato N, Ohyama K, Kuroda T, Nakamura M (2003) Basic helix-loop-helix transcription factor epicardin/capsulin/Pod-1 suppresses differentiation by negative regulation of transcription. J Biol Chem 278 : 7486–7493. 12493738
35. Orlandi A, Bennett M (2010) Progenitor cell-derived smooth muscle cells in vascular disease. Biochemical pharmacology 79 : 1706–1713. doi: 10.1016/j.bcp.2010.01.027 20117099
36. Frid MG, Aldashev AA, Nemenoff RA, Higashito R, Westcott JY, et al. (1999) Subendothelial cells from normal bovine arteries exhibit autonomous growth and constitutively activated intracellular signaling. Arterioscler Thromb Vasc Biol 19 : 2884–2893. 10591665
37. Holifield B, Helgason T, Jemelka S, Taylor A, Navran S, et al. (1996) Differentiated vascular myocytes: are they involved in neointimal formation? J Clin Invest 97 : 814–825. 8609239
38. Sainz J, Al Haj Zen A, Caligiuri G, Demerens C, Urbain D, et al. (2006) Isolation of "side population" progenitor cells from healthy arteries of adult mice. Arterioscler Thromb Vasc Biol 26 : 281–286. 16306431
39. Majesky MW, Dong XR, Hoglund V, Mahoney WM Jr., Daum G (2011) The adventitia: a dynamic interface containing resident progenitor cells. Arterioscler Thromb Vasc Biol 31 : 1530–1539. doi: 10.1161/ATVBAHA.110.221549 21677296
40. Zengin E, Chalajour F, Gehling UM, Ito WD, Treede H, et al. (2006) Vascular wall resident progenitor cells: a source for postnatal vasculogenesis. Development 133 : 1543–1551. 16524930
41. Virmani R, Burke AP, Farb A, Kolodgie FD (2006) Pathology of the vulnerable plaque. J Am Coll Cardiol 47: C13–18. 16631505
42. Kozaki K, Kaminski WE, Tang J, Hollenbach S, Lindahl P, et al. (2002) Blockade of platelet-derived growth factor or its receptors transiently delays but does not prevent fibrous cap formation in ApoE null mice. Am J Pathol 161 : 1395–1407. 12368212
43. Sano H, Sudo T, Yokode M, Murayama T, Kataoka H, et al. (2001) Functional blockade of platelet-derived growth factor receptor-beta but not of receptor-alpha prevents vascular smooth muscle cell accumulation in fibrous cap lesions in apolipoprotein E-deficient mice. Circulation 103 : 2955–2960. 11413086
44. Li G, Jin R, Norris RA, Zhang L, Yu S, et al. (2010) Periostin mediates vascular smooth muscle cell migration through the integrins alphavbeta3 and alphavbeta5 and focal adhesion kinase (FAK) pathway. Atherosclerosis 208 : 358–365. doi: 10.1016/j.atherosclerosis.2009.07.046 19695571
45. Litvin J, Chen X, Keleman S, Zhu S, Autieri M (2007) Expression and function of periostin-like factor in vascular smooth muscle cells. Am J Physiol Cell Physiol 292: C1672–1680. 17092992
46. Lindner V, Wang Q, Conley BA, Friesel RE, Vary CP (2005) Vascular injury induces expression of periostin: implications for vascular cell differentiation and migration. Arterioscler Thromb Vasc Biol 25 : 77–83. 15514205
47. Hixson JE, Shimmin LC, Montasser ME, Kim DK, Zhong Y, et al. (2011) Common variants in the periostin gene influence development of atherosclerosis in young persons. Arterioscler Thromb Vasc Biol 31 : 1661–1667. doi: 10.1161/ATVBAHA.111.224352 21474826
48. Bentzon JF, Falk E (2010) Circulating smooth muscle progenitor cells in atherosclerosis and plaque rupture: current perspective and methods of analysis. Vascular pharmacology 52 : 11–20. doi: 10.1016/j.vph.2009.11.005 19951745
49. Yu H, Stoneman V, Clarke M, Figg N, Xin HB, et al. (2011) Bone marrow-derived smooth muscle-like cells are infrequent in advanced primary atherosclerotic plaques but promote atherosclerosis. Arterioscler Thromb Vasc Biol 31 : 1291–1299. doi: 10.1161/ATVBAHA.110.218578 21372299
50. Miller CL, Anderson DR, Kundu RK, Raiesdana A, Nurnberg ST, et al. (2013) Disease-Related Growth Factor and Embryonic Signaling Pathways Modulate an Enhancer of TCF21 Expression at the 6q23.2 Coronary Heart Disease Locus. PLoS Genet 9: e1003652. doi: 10.1371/journal.pgen.1003652 23874238
51. Miller CL, Haas U, Diaz R, Leeper NJ, Kundu RK, et al. (2014) Coronary Heart Disease-Associated Variation in TCF21 Disrupts a miR-224 Binding Site and miRNA-Mediated Regulation. PLoS Genet 10: e1004263. doi: 10.1371/journal.pgen.1004263 24676100
52. Chen YC, Bui AV, Diesch J, Manasseh R, Hausding C, et al. (2013) A novel mouse model of atherosclerotic plaque instability for drug testing and mechanistic/therapeutic discoveries using gene and microRNA expression profiling. Circ Res 113 : 252–265. doi: 10.1161/CIRCRESAHA.113.301562 23748430
53. Aono J, Suzuki J, Iwai M, Horiuchi M, Nagai T, et al. (2012) Deletion of the angiotensin II type 1a receptor prevents atherosclerotic plaque rupture in apolipoprotein E-/ - mice. Arterioscler Thromb Vasc Biol 32 : 1453–1459. doi: 10.1161/ATVBAHA.112.249516 22460554
54. Hidai C, Zupancic T, Penta K, Mikhail A, Kawana M, et al. (1998) Cloning and characterization of developmental endothelial locus-1: an embryonic endothelial cell protein that binds the alphavbeta3 integrin receptor. Genes Dev 12 : 21–33. 9420328
55. Perisic L, Hedin E, Razuvaev A, Lengquist M, Osterholm C, et al. (2013) Profiling of atherosclerotic lesions by gene and tissue microarrays reveals PCSK6 as a novel protease in unstable carotid atherosclerosis. Arterioscler Thromb Vasc Biol 33 : 2432–2443. doi: 10.1161/ATVBAHA.113.301743 23908247
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2015 Číslo 5
Nejčtenější v tomto čísle
- Drosophila Spaghetti and Doubletime Link the Circadian Clock and Light to Caspases, Apoptosis and Tauopathy
- Autoselection of Cytoplasmic Yeast Virus Like Elements Encoding Toxin/Antitoxin Systems Involves a Nuclear Barrier for Immunity Gene Expression
- Parp3 Negatively Regulates Immunoglobulin Class Switch Recombination
- PERK Limits Lifespan by Promoting Intestinal Stem Cell Proliferation in Response to ER Stress