
Global Analysis of Neutrophil Responses to Reveals a Self-Propagating Inflammatory Program
Gonorrhea is a sexually transmitted infection caused by the bacteria Neisseria gonorrhoeae. These bacteria have re-emerged as a public health priority due to its acquisition of resistance to multiple antibiotics, leading to fears of untreatable infection. The symptoms of gonorrhea include an intense inflammatory response that may lead to pus discharged from the infected genital tract and scarring of the reproductive tract caused by neutrophils recruited to the site of infection. Past studies have detailed molecular interactions that lead to neutrophil binding and engulfment of N. gonorrhoeae, yet it remains unclear why N. gonorrhoeae elicits such a pathogenic inflammatory response. In this study, we reveal that N. gonorrhoeae binding to the human innate decoy receptor, CEACAM3, elicits a potent intracellular signaling cascade that leads to neutrophil expression of cytokines that actively recruit other neutrophils to the infected tissues. As they encounter the gonococci, the next wave of neutrophils becomes similarly activated, leading to the progressive expansion in phagocytic cell numbers until they overwhelm the infected tissues. While this process promotes a rapid response to a troubling pathogen early during infection, the unrestrained recruitment of neutrophils and their toxic antimicrobial arsenal also lead to the pathogenic consequences associated with gonorrhea.
Published in the journal:
. PLoS Pathog 10(9): e32767. doi:10.1371/journal.ppat.1004341
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004341
Summary
Gonorrhea is a sexually transmitted infection caused by the bacteria Neisseria gonorrhoeae. These bacteria have re-emerged as a public health priority due to its acquisition of resistance to multiple antibiotics, leading to fears of untreatable infection. The symptoms of gonorrhea include an intense inflammatory response that may lead to pus discharged from the infected genital tract and scarring of the reproductive tract caused by neutrophils recruited to the site of infection. Past studies have detailed molecular interactions that lead to neutrophil binding and engulfment of N. gonorrhoeae, yet it remains unclear why N. gonorrhoeae elicits such a pathogenic inflammatory response. In this study, we reveal that N. gonorrhoeae binding to the human innate decoy receptor, CEACAM3, elicits a potent intracellular signaling cascade that leads to neutrophil expression of cytokines that actively recruit other neutrophils to the infected tissues. As they encounter the gonococci, the next wave of neutrophils becomes similarly activated, leading to the progressive expansion in phagocytic cell numbers until they overwhelm the infected tissues. While this process promotes a rapid response to a troubling pathogen early during infection, the unrestrained recruitment of neutrophils and their toxic antimicrobial arsenal also lead to the pathogenic consequences associated with gonorrhea.
Introduction
Neisseria gonorrhoeae, the causative agent of gonorrhea, is a re-emerging global health concern, with over a hundred million cases diagnosed each year, the recent emergence of multi-drug resistant strains that have led to its ‘superbug’ status, and a lack of success in vaccine development [1], [2], [3]. Symptomatic infection with N. gonorrhoeae results in acute inflammation of the urogenital tract and a purulent urethral discharge consisting almost exclusively of neutrophils. If left untreated, gonococcal infection can lead to serious chronic conditions, such as pelvic inflammatory disease and infertility, which stem from an overzealous response to the infection [2].
N. gonorrhoeae is a Gram-negative diplococcus that is highly adapted to colonization of the human urogenital tract. The initial interaction between the bacteria and epithelia is mediated by type IV pili, which retract to allow tight association with the mucosal epithelia [4]. More intimate interactions are then facilitated by adhesins including the neisserial Opa proteins binding to certain epithelial cell-expressed members of the carcinoembryonic antigen-related adhesion molecule (CEACAM) family: CEACAM1, CEACAM5, and CEACAM6 [5], [6], [7], [8], [9]. CEACAMs represent a subset of the Ig superfamily and consist of a variable number of Ig-like constant domains and an Ig variable domain-like N-terminus that allows Opa binding [10], [11], [12]. Attachment to apically expressed CEACAMs is sufficient to trigger bacterial engulfment and transcytosis across the epithelia to allow entry into the subepithelial space [13], [14]. CEACAM1 is notable among the family in that, in addition to being on epithelial cells, it is also expressed on certain endothelial, lymphocytic and myeloid cells. Bacteria exploit its co-inhibitory function, which depends upon its cytoplasmic immunoreceptor tyrosine-based inhibitory motif (ITIM), to suppress T cell [15], [16], [17], B cell [18], dendritic cell [19] and epithelial cell [20] responses (reviewed in [21]).
While binding to CEACAMs on most cell types tends to facilitate infection, Opa proteins may also bind to neutrophil-expressed CEACAM3. When this occurs, CEACAM3 triggers an efficient opsonin-independent phagocytosis of the bacteria [22], [23], [24]. Ligation of CEACAM3 also promotes a Syk kinase - and phosphatidylinositol 3-kinase-dependent recruitment and downstream activation of the neutrophils' antimicrobial responses, including degranulation and oxidative burst [22], [23], [24], [25], [26], [27], [28]. These effects are driven by the cytoplasmic immunoreceptor tyrosine-based activation motif (ITAM), which distinguishes CEACAM3 from the other CEACAMs that N. gonorrhoeae binds. Considering that CEACAM3 is human-restricted, expressed on neutrophils and lacks cell adhesion function, CEACAM3 is now generally considered to be an innate immune receptor allowing capture and elimination of bacteria that colonize epithelial tissues via other CEACAMs [23], [24], [29], [30], [31].
Neutrophils are specialized for rapid transmigration to sites of infection in response to a variety of stimuli, including chemotactic gradients and presence of bacterial components. Following recruitment to the infected tissue, neutrophils effectively phagocytose opsonized bacteria, activate production of reactive oxygen species [32] and release toxic antimicrobial peptides and proteins from cytoplasmic granules [33], [34]. Conventionally, neutrophils were thought to have little to no controlled expression of new gene products, depending mostly on constitutively-expressed proteins and pre-loaded granules assembled during maturation. In recent years, it has become evident that properly stimulated neutrophils respond by synthesizing new proteins [35], [36], [37], however surprisingly little is known about the control of gene expression.
In this work, we show that heterologous expression of human CEACAMs in transgenic mouse neutrophils permits effective opsonin-independent neisserial binding and neutrophil activation in a manner reflecting that seen with human neutrophils. Moreover, we reveal that Opa-dependent CEACAM3 binding drives a potent neutrophil transcriptional response that elicits production of pro-inflammatory cytokines via a PKCδ and Tak1 serine/threonine kinase-dependent pathway triggered downstream of Syk tyrosine kinase. Furthermore, we observed that infection of human CEACAM-expressing transgenic mice with N. gonorrhoeae results in a dramatically higher neutrophil influx to the infection site when compared to wild-type mice. Together, this study establishes that bacterial binding to CEACAM3 effectively recruits more neutrophils to the infected tissues. While providing an effective strategy to combat the initial infection, this self-propagating cycle of events can also lead to the pathogenic inflammatory response that typifies symptomatic gonorrhea.
Results
CEACAM-humanized transgenic mouse neutrophils respond to N. gonorrhoeae
Neisserial infection is exquisitely human-specific, with major receptors for the bacterial Opa protein adhesins being certain members of the human CEACAM family. While CEACAM homologues can be found in all vertebrates [38], only human CEACAMs have been observed to bind Neisseria. Mouse polymorphonuclear leukocytes (PMNs), which express mouse CEACAM1 on their surface, do not bind N. gonorrhoeae [24], [39], whereas human PMNs specifically bind Opa-expressing but not Opa-deficient N. gonorrhoeae in an opsonin-independent fashion (Figure 1A, C). We have recently established that recombinant human CEACAMs encoded from constitutively expressed cDNA were functionally expressed in a mouse promyelocytic (MPRO) cell line [24]. We considered whether ectopic expression of intact human CEACAM genes in transgenic mouse neutrophils would also confer responsiveness to N. gonorrhoeae. To address this question, we performed experiments with bone marrow-derived neutrophils from human CEACAM-expressing CEABAC2 mice [40]. These mice were engineered using a BAC that encodes human CEACAM3, CEACAM5, CEACAM6 and CEACAM7, none of which have murine homologues. Using flow cytometric analysis and immunoblotting with CEACAM-specific antibodies, we confirmed that human CEACAM3 and CEACAM6 were expressed on the surface of CEABAC neutrophils, in a manner reflecting their expression on human neutrophils (Figure 1A,B). When we exposed CEABAC neutrophils to N. gonorrhoeae expressing either the CEACAM-specific Opa+ or no Opa protein (Opa−), they effectively bound and engulfed the Opa+ but not Opa− bacteria, whereas no such association was apparent with wild type neutrophils regardless of Opa expression (Figure 1C,D). While the number of Opa-expressing bacteria captured by human CEACAM-expressing neutrophils was substantially (∼10-fold) greater than what occurs with WT mouse neutrophils and/or Opa− bacteria (Figure 1D), the bacteria that become engulfed are effectively killed regardless of whether or not human CEACAMs are involved in the uptake (Figure 1E). These results differ from a recent study with human neutrophils which describe increased killing of Opa+ (relative to Opa−) bacteria [41], however it remains unclear whether this is a neutrophil species-dependent effect or result from differences in bacterial strain or methodology used in the two studies.

N. gonorrhoeae infection activates CEABAC neutrophils
Previous work addressing individual CEACAM expression and its effect on neisserial infection was undertaken using transfected promyelocytic cell lines [24]. Because CEABAC transgenic neutrophils can bind and engulf N. gonorrhoeae (Figure 1C), we wondered whether neutrophil-specific responses to N. gonorrhoeae were also reproduced in these cells. Human neutrophils respond to Opa-expressing N. gonorrhoeae by triggering an increased consumption of oxygen, resulting in the production of free oxygen radicals in the cell (the ‘oxidative burst’), as well as by releasing granule components to the cell surface or into the newly formed phagosome (‘degranulation’) [25]. Consistent with this, we observed that Opa-expressing N. gonorrhoeae were able to efficiently stimulate both the oxidative burst and release of primary and secondary granules (as determined by the release of neutrophil elastase and lactoferrin, respectively), in infected human neutrophils (Figure 1F). In stark contrast, WT mouse neutrophils are surprisingly unresponsive to N. gonorrhoeae infection, illustrating the importance of human CEACAMs for these effects. However, in CEABAC neutrophils, we observed that the oxidative burst and degranulation were heightened in response to Opa-expressing bacteria (Figure 1G), consistent with the function of Opa proteins in CEACAM binding.
N. gonorrhoeae drives an acute inflammatory program in neutrophils
While neutrophils were classically considered to be transcriptionally quiet, Fc receptor-mediated phagocytosis has long been known to promote IL-8 mRNA expression in neutrophils [32]. More recently, it has become clear that neutrophils have the capacity to become transcriptionally active in response to certain stimuli [35], [36], [37], yet neutrophil transcriptional responses to specific infections remain poorly understood. Consequently, to investigate whether N. gonorrhoeae might elicit a transcriptional response, we isolated RNA from uninfected and infected WT and CEABAC bone marrow-derived neutrophils and compared their transcriptional profile by full genome gene array 1 hour post-infection. DAVID functional annotation [42] and manual analysis of the results revealed that the general pattern of genes expressed in response to N. gonorrhoeae were similar in the WT and CEABAC animals, however, two categories of transcriptional up-regulation were apparent. In the first group are genes that are induced to a similar level in WT and CEABAC PMNs. Of these, the largest functional classes of genes are those involved in the regulation of inflammation (i.e. IL-10 Receptor α Subunit (il10ra); Suppressor of Cytokine Signaling 3 (SOCS3); Inhibitor of kappa B subunits (IκBδ, IκBζ)) and control of cell cycle and apoptosis (i.e. Bcl2, Gadd34, Gadd45) (Figure 2A). In contrast, the CEABAC neutrophils displayed a marked up-regulation of acute inflammatory cytokine expression, including TNFα (2.1-fold induction over wild type neutrophils), IL-1α (2.7-fold induction) and the neutrophil chemoattractant and activators Gro-α/KC, MIP-1α and MIP-1β (1.5-, 3.3 - and 3.5-fold induction) (Figure 2B). It is pertinent to note that we detected no down-regulation of any gene expression at that time point. Considering that regulatory genes are expressed at similar levels between WT and CEABAC PMNs, while pro-inflammatory mediators are drastically higher in CEABAC cells, we infer that the cumulative effect of these changes in gene expression would be a heightened pro-inflammatory cytokine response when the gonococcal Opa proteins engage the neutrophil-expressed CEACAM3.

Opa-expressing N. gonorrhoeae drive production of pro-inflammatory cytokines in CEACAM-expressing neutrophils
To validate gene array results, and confirm that increases in transcript levels corresponded to PMN secretion of the protein products, we measured the production of MIP-1α, MIP-2, KC and TNF-α protein in infected PMNs from WT and CEABAC mice (Figure 3A). Significant (between 3 - to-10 fold) increases in chemokine protein levels were observed in supernatants from CEABAC PMNs infected with Opa+ bacteria, compared to supernatants from CEABAC PMNs infected with Opa− bacteria. Critically, the WT neutrophil response was not affected by Opa expression, instead reflecting that seen with Opa− bacteria and CEABAC PMNs, demonstrating that both Opa and human CEACAMs are required for this effect. This increased chemokine secretion corresponded with increased levels of chemokine transcripts in these samples (Figure 3B), reflecting the data obtained via the gene array experiments, and establishing that de novo transcription is driving the cytokine response. Collectively, these data provide the first evidence of a neutrophil transcription-based inflammatory response to N. gonorrhoeae infection, and point to the CEACAM-Opa interaction as a critical driver of this inflammation.

CEACAM-Opa interaction drives an inflammatory response in human peripheral blood neutrophils
To confirm that the CEACAM - and Opa-dependent transcriptional response apparent in our transgenic mouse model reflected that occurring in human PMNs, peripheral blood neutrophils isolated from healthy volunteers were infected with Opa− or Opa+ N. gonorrhoeae and then subjected to quantitative RNA analysis. Opa+ N. gonorrhoeae-infected PMNs had substantially higher levels of MIP-1α, MIP-2, TNF-α, and IL-1α transcript in all donors tested, when compared to Opa− infected controls (Figure 3C). We therefore conclude that the CEACAM-Opa interaction potentiates the inflammation observed during human PMN infection, reflecting our findings with the CEACAM-humanized mouse model.
Phagocytosis and production of reactive oxygen species are not essential for inflammatory cytokine production
Unlike WT PMNs, CEABAC neutrophils efficiently bind and phagocytose Opa-expressing N. gonorrhoeae (Figure 1C–D). This prompted us to consider whether uptake alone can account for the cytokine response of human CEACAM-expressing PMNs. To address this question, WT and CEABAC PMNs were pre-treated with cytochalasin D to inhibit phagocytosis prior to infection. While cytochalasin D pre-treatment led to a marked decrease in bacterial internalization, (Figure S1A), it had no effect on MIP-1α secretion (Figure 4A). Furthermore, we used PMNs from a transgenic mouse line expressing human CEACAM1 but no CEACAM3. While the human CEACAM1-expressing neutrophils can efficiently phagocytose Opa-expressing N. gonorrhoeae [43], the neutrophils showed little chemokine response to infection (Figure S1B).

In considering that reactive oxygen species (ROS) have been linked to the activation of various inflammatory signals [44], we also sought to confirm whether ROS formed during the well-characterized oxidative burst response to Opa-expressing N. gonorrhoeae [24], [25], [28], [41] explained the induced cytokine response. To this end, we prevented ROS production using the NADPH oxidase inhibitor diphenylene iodonium (DPI) and then measured cytokine production in WT and CEABAC neutrophils infected with Opa− and Opa+ bacteria. While DPI completely abolished the otherwise high levels of ROS produced upon exposure of CEABAC PMNs to Opa+ N. gonorrhoeae (Figure S1C), this had no affect the chemokine response of CEABAC PMNs (Figure 4A). Taken together our data suggest that Opa protein-dependent engagement of CEACAM3 drives a cytokine response that is independent of its ability to promote bacterial phagocytosis and is not mediated by the ROS produced in response to infection.
CEACAM-Opa interaction drives activation of NF-κB and MAPK signaling
A large proportion of genes identified in our gene array study (MIP-1α, MIP-2, TNF-α, IL-1α) are known to be activated by NF-κB. NF-κB transcription factors are major mediators of inflammation and have been shown to stimulate transcription of pro-inflammatory cytokines in response to LPS in PMNs [45] [46] [47]. NF-κB also governs transcriptional responses downstream of various ITAM receptors in other (non-neutrophil) cell types [48], including the innate immune receptor Dectin-1 [49]. This led us to investigate NF-κB activation downstream of CEACAM3. Consistent with NF-κB being a downstream effector of CEACAM3, IκBα was degraded more rapidly and completely in CEACAM-expressing PMNs than in the WT cells (Figure 4B).
The p38 mitogen associated kinase (MAPK) has been shown to act along side NF-κB in the activation of PMN transcriptional responses to LPS [50] [46]. Consequently, we considered whether p38 might also be involved in the CEACAM-mediated transcriptional response. Infected CEABAC neutrophils exhibit increased levels of p38 phosphorylation when compared to WT (Figure 4C), indicating that they are more effectively activated in response to N. gonorrhoeae infection. In contrast, the Erk1/2 MAPKs were not phosphorylated in a CEACAM-dependent manner (Figure 4D), suggesting selective activation of p38 kinase upon CEACAM ligation. To assess whether p38 activity contributed to the CEACAM-dependent cytokine response, PMNs were exposed to a p38-specific inhibitor (SB203580) prior to infection. This treatment effectively blocked production of MIP-1α and MIP-2 in CEABAC PMNs infected with Opa+ N. gonorrhoeae (Figure 4E), while inhibition of Erk1/2 phosphorylation had no effect on cytokine secretion (Figure S1D). Together, this data supports an essential role for p38 MAPK in the CEACAM-dependent transcriptional response.
It has been reported that the mitogen-activated kinase kinase kinase (MAPKKK) family member TAK1 can activate both p38 MAPKs and NF-κB [51]. To test the involvement of TAK1 in the p38 activation observed during neisserial infection of PMNs, we used the TAK1 inhibitor (5z)-7-oxozeanol. TAK1 inhibition effectively abrogated MIP-1α and MIP-2 secretion by the Opa+ N. gonorrhoeae-infected CEABAC neutrophils (Figure 4F). Importantly, neither bacterial adherence nor phagocytosis by the PMNs were affected by either the p38 or TAK1 inhibitors (Figure 5E, F), confirming that the effect of these two compounds on cytokine expression was not due to their inhibition of these cellular processes.

SFK, Syk, and PKCδ couple CEACAM3 to the downstream transcriptional response
CEABAC neutrophils express both human CEACAM3 and CEACAM6. Both receptors facilitate bacterial uptake, yet previous work has shown that the activation of neutrophil bactericidal processes, including degranulation and oxidative burst, occur via CEACAM3 alone [24], [25]. We sought to confirm whether the pro-inflammatory response observed in CEABAC neutrophils was solely mediated by CEACAM3, and to determine whether the inflammation depended upon the CEACAM3 ITAM-dependent signaling. Since CEACAM3, unlike the GPI-anchored CEACAM6, relies on phosphorylation of its cytoplasmic ITAM by Src family kinases (SFK) for its activation, we exploited the SFK-specific inhibitor PP2 that has previously been shown to block CEACAM3 ITAM-dependent signaling [22], [25], [52], [53]. Inhibition of SFK significantly abrogated cytokine production (Figure 5A), but did not affect bacterial adherence or phagocytosis (Figure 5E,F). Considered together, the data suggests that CEACAM3 ITAM phosphorylation is essential for induction of pro-inflammatory response. This also indicates a divergence in the bacterial engulfment and transcriptional pathways, since the tyrosine phosphorylation-independent CEACAM3 - and CEACAM6-mediated engulfment of N. gonorrhoeae [24] is not, itself, sufficient to elicit the pro-inflammatory response. Work in other systems has shown that Syk kinase is an essential mediator of ITAM-mediated responses in general [54] and CEACAM3-specific neutrophil responses in particular [24], though there is some suggestion that CEACAM3 can signal independent of Syk [55]. Therefore, we tested whether Syk contributed to CEACAM3-dependent inflammatory signaling using the specific inhibitor, piceatannol. Inhibiting Syk function led to a significant reduction in chemokine production by CEABAC neutrophils (Figure 5B), implicating a critical role for this kinase in the CEACAM3-dependent pro-inflammatory cytokine responses.
Recently, the serine/threonine kinase PKCδ was shown to link signaling from the ITAM-containing innate immune receptor Dectin-1 to NF-κB activation in dendritic cells [56]. Consequently, we assessed PKCδ activation during N. gonorrhoeae infection of WT and/or CEABAC PMNs. CEABAC neutrophils showed substantially increased phosphorylation of PKCδ relative to that seen in WT PMNs (Figure 5C), suggesting it is also activated downstream of CEACAM3. Furthermore, pretreatment of CEABAC PMNs with the PKC inhibitor bisindolylmaleimide II (BIS II) led to the inhibition of MIP-1α production in response to Opa+ N. gonorrhoeae (Figure 5D), consistent with PKCδ actively contributing to the CEACAM3-driven inflammatory response. In summary, we have outlined a novel signaling cascade downstream of CEACAM3 that is distinct from CEACAM3-mediated bacterial engulfment and activation of antimicrobial responses (Figure 5G), and potentially contributes to the excessive inflammatory response typical of N. gonorrhoeae infection.
CEACAM-Opa intensifies the inflammatory response in vivo
The heightened expression of pro-inflammatory cytokines by CEACAM-humanized PMNs should result in increased neutrophil chemotaxis to the site of N. gonorrhoeae infection, an outcome consistent with the clinical manifestations of gonorrhea. To test this, we collected supernatants from WT and CEABAC PMNs infected with Opa+ N. gonorrhoeae, and measured their ability to affect the speed and directionality of neutrophils using a Zigmond chamber. Consistent with the increased expression of chemotactic factors when CEABAC neutrophils are infected with N. gonorrhoeae, the chemotaxis of uninfected neutrophils was significantly greater in response to culture supernatants from the N. gonorrhoeae-infected CEABAC neutrophils, as compared to supernatants from infected WT neutrophils (Figure 6A). To understand the implications of these effects during in vivo infection, we measured the relative contribution of neutrophil CEACAM3 on inflammation using a subcutaneous air-pouch model, which allows the effective recovery and analysis of leukocyte recruitment to an otherwise sterile site [57]. Opa expression did not affect leukocyte recruitment into air pouches formed in WT mice. However, in CEABAC mice, the Opa-expressing gonococci elicited a significantly increased infiltration of neutrophils relative to that seen in response to Opa− bacteria (Figure 6B). Giemsa-Wright staining of the CEABAC-Opa+ air pouch infiltrate showed that nearly all of the cells (>95%) present are PMNs (Figure 6C), with many neutrophils containing intracellular gonococci. Notably, the number of PMNs present in CEABAC mice infected with Opa− bacteria reflected that seen in the WT mice, indicating that both Opa and human CEACAMs are required to drive the more intense inflammatory response.

Considering the marked increase in neutrophil response in the CEABAC animals, we considered whether it was possible that cells lining the air pouch could differentially associate with N. gonorrhoeae, which would contribute to the inflammatory milieu. To test this, we administered trypsin/EDTA into an uninfected air pouch from CEABAC mice, harvested the cells that were released, and then seeded them onto glass coverslips. The next day, cells were infected with Opa+ bacteria, and bacterial binding and CEACAM expression were assessed using immunofluorescence microscopy. Cells lining the pouch did not express CEACAM and did not bind or take up the N. gonorrhoeae (Figure 6D, top). When the same experiment was conducted on air pouches infected with Opa+ N. gonorrhoeae, we observed a large number of PMNs had infiltrated the air pouch lining. As expected, these PMNs expressed high levels of the human CEACAMs, and effectively bound the bacteria, unlike the adjacent fibroblast lining (Figure 6D, bottom). These results are consistent with the enhanced inflammation being due to differential association of the gonococci with the CEACAM3-expressing neutrophils, without obvious effect on their interaction with the surrounding tissues.
The air pouch experiments suggest that the CEACAM-dependent association between N. gonorrhoeae and the resident neutrophils promotes subsequent neutrophil recruitment, presumably due to the establishment of a chemotactic gradient. Consistent with this, the pro-inflammatory chemokines MIP-1α, MIP-2, KC, and IL-1β were all increased in the washes from Opa+-N. gonorrhoeae infected air pouches in CEABAC mice relative to that seen in the infected WT littermates (Figure 6E). To address whether neutrophils directly contribute to the higher levels of cytokines observed, we infected mice that had been depleted of neutrophils by administration of the Gr1-specific RB6-8C5 antibody prior to the introduction of N. gonorrhoeae. The levels of MIP-1α and IL-1β were significantly lower in the air pouches from PMN-depleted mice, indicating that neutrophils are the primary source for both of these chemokines (Figure 6E). Interestingly, the levels of KC and MIP-2 were dramatically higher in PMN-depleted mice, indicating that these chemokines are produced by cells other than PMNs under these conditions. While these chemokines are both produced by CEABAC neutrophils in response to N. gonorrhoeae (Figure 3A,B), they can also be produced by a variety of tissues [33], including synovial fibroblasts [58]. Considering that their levels increased upon neutrophil depletion, we interpret the increased response to suggest a delay in bacterial clearance in the absence of PMNs. The increased IL-1β and MIP-1α in CEABAC mice thereby reflect a local neutrophil response whereas MIP-2 and KC levels appear to be the cumulative effect of both neutrophil and underlying tissue responses.
To link the CEACAM3-dependent intracellular response evident from our cell-based experiments with inflammation in vivo, we repeated the air pouch experiment, this time assessing the effect of the TAK1 inhibitor, which was administered to mice 1 h prior to infecting the air pouch. Consistent with our model that the inhibition of CEACAM3 signaling would suppress the inflammatory response to N. gonorrhoeae, we observed a decrease in the number of infiltrating PMNs upon administration of the TAK1-inhibitor relative to the untreated animals (Figure 6F).
Discussion
The picture of neutrophils as ever-ready weapons of defense aiming to achieve efficient pathogen clearance has become an axiom. While still true, recent evidence suggests that they have the ability to nuance their response through de novo gene expression in response to certain microbial cues. In addition to their classical role in direct microbial killing, neutrophils can produce a range of cytokines with the potential to affect inflammation through the activation and induced chemotaxis of various leukocytes [37], [59]. However, their specific contribution to the cytokine milieu and inflammatory response remains underappreciated in vivo. The emerging picture of neutrophils as a dynamic, responsive cell population has important implications for our understanding of the overzealous neutrophil response that typifies gonorrhea. Our findings reveal that the decoy receptor CEACAM3, in addition to facilitating the effective capture and killing of N. gonorrhoeae, also helps drive inflammation. While recruitment of more PMNs to combat infection would seem to be an effective innate immune strategy during early infection, the persistent exposure of CEACAM3-expressing PMNs to Opa-expressing gonococci can promote a self-perpetuating and, ultimately, pathogenic response such as is associated with gonorrhea or pelvic inflammatory disease.
In this work, we demonstrate that human CEACAM-expressing transgenic mouse PMNs respond to N. gonorrhoeae in a manner that parallels those of human PMNs. Unlike neutrophils from WT mice, the CEABAC neutrophils undergo a vigorous oxidative burst and degranulation response to N. gonorrhoeae. Since the pathology associated with gonococcal disease primarily arises due to tissue damage caused by the recruited neutrophils, we considered whether CEACAM-dependent interactions with the bacteria could also contribute to the inflammatory response. We observed that N. gonorrhoeae binding to human CEACAM3 leads to the acute activation of a pro-inflammatory transcriptional program that results in the production of the inflammatory mediators such as MIP-1α, MIP-2, KC and TNF-α. While relatively little is known about signal transduction in PMNs relative to other cell types, Opa binding to CEACAM3 elicits signaling via a pathway closely reminiscent of that triggered downstream of the innate anti-fungal receptor Dectin-1 [30], [60], [61]. As with Dectin-1, SFK and Syk kinase represent the first effectors downstream of CEACAM3, and their activity is required for the PMN oxidative burst and degranulation responses to Opa-expressing gonococci [24]. While Syk was shown to serve a regulatory role in the context of PMNs [62], we have observed that Syk contributes to the inflammatory cytokine response to N. gonorrhoeae by eliciting the PKCδ and TAK1-dependent activation of NF-κB. This CEACAM3-dependent expression of MIP-1α, MIP-2 and KC stimulates chemotaxis of uninfected neutrophils so as to augment their recruitment to the infected tissues, an effect that has the potential to contribute to both innate defense and the massive neutrophil recruitment that typifies gonorrhea (Figure 7).
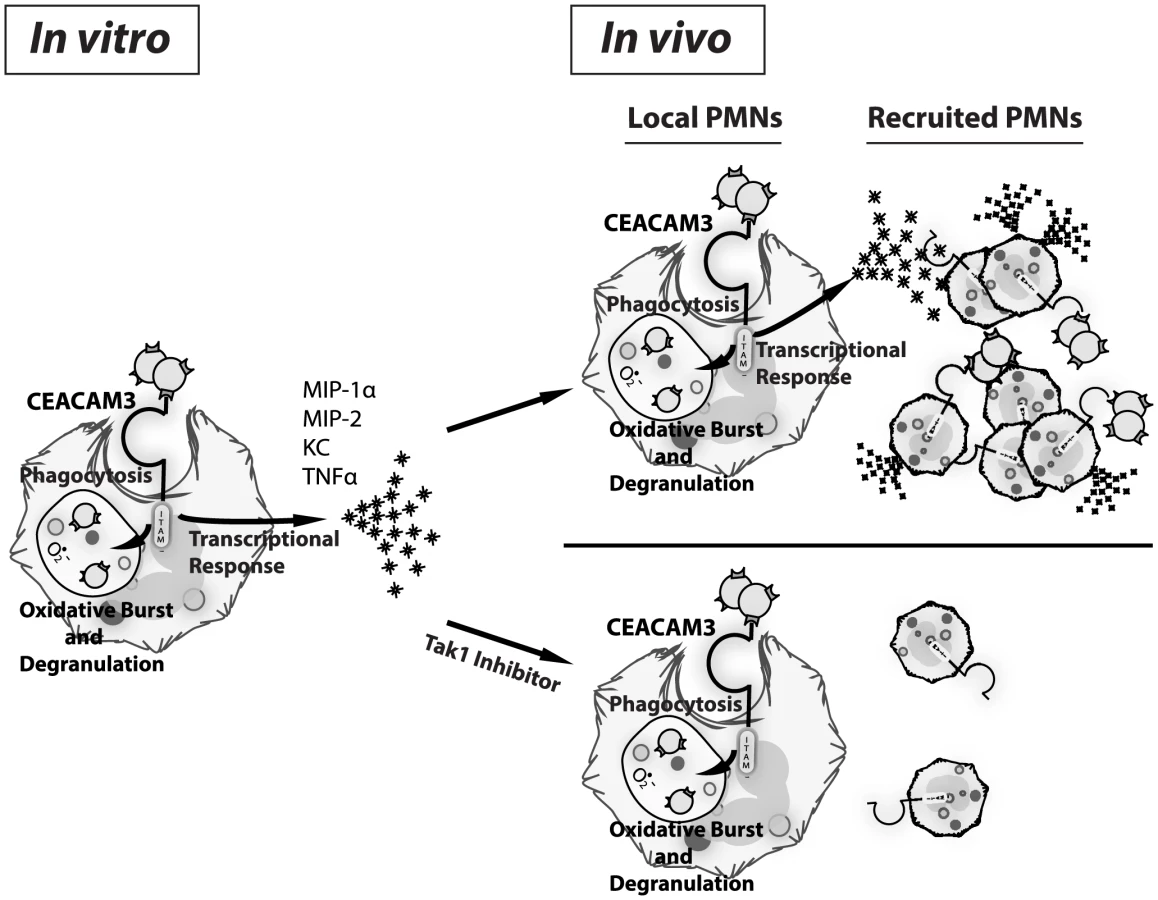
The trade-off of having a direct link between CEACAM3 and inflammation is that, when uncontrolled, this response results in the ongoing infiltration of neutrophils, leading to permanent tissue damage such as is observed with N. gonorrhoeae-associated fallopian tube scarring and pelvic inflammatory disease. Supporting this model, we observed CEACAM-dependent increases in neutrophil recruitment into the gonococcal-infected tissues, brought on by the CEACAM3-dependent production of chemokines with the potential to promote continuous infiltration of neutrophils. Thus, CEACAM3 activation serves as a double-edged sword, promoting the immunopathology of gonorrhea through an over-activated immune response that is meant to clear the bacteria causing the infection. While the complete ablation of neutrophil recruitment would increase the bacterial burden, a better outcome would be to limit the chemokine response without blocking bacterial binding and phagocytosis (Figure 7). As a proof of concept for this, we used a TAK1 inhibitor in vivo to block the pro-inflammatory response and, thereby, the influx of neutrophils without affecting their phagocytic capacity. Satisfyingly, this treatment decreased PMN recruitment to that seen in WT mice, effectively eliminating the CEACAM3-dependent inflammation in response to N. gonorrhoeae.
It has been reported previously that the majority of neisserial isolates from infected patients have the capacity to bind to CEACAMs [5]. This may seem contradictory when considering the negative outcome for bacteria that bind to CEACAM3 on neutrophils. However, it is important to consider that, in contrast to CEACAM3, neisserial binding to other human CEACAMs facilitates attachment to mucosal epithelia [14], [63], [64], [65] and suppression of both innate [20] and adaptive [15], [16], [17] immune responses, activities which are central to the establishment and persistence of infection. In this respect, it is curious to contrast CEACAM1, which is the evolutionary precursor of the CEA family, with the evolutionarily ‘new’ CEACAM3. While CEACAM1 is present in all vertebrates and is broadly expressed on many cell types, CEACAM3 can only be found in humans and is only expressed by neutrophils. When this is considered along with the fact that CEACAM3 possesses no cell adhesion function yet has a sufficiently conserved extracellular domain that can be engaged by the neisserial Opa proteins, we and others have suggested that CEACAM3 functions as a decoy receptor that allows the capture and killing of CEACAM-targeting microbes. The apparent involvement of CEACAM3 in pro-inflammatory signaling establishes a role for neutrophils beyond basic microbial killing, and places CEACAM3 as both a potential contributor to the accelerated response to N. gonorrhoeae infection and, subsequently, to the immunopathology associated with the gonococcal disease. The evolutionary advent of CEACAM3 thus reflects the latest step in the ongoing dance between Neisseria and their only natural host, providing a snare that mobilizes our most potently bactericidal cells against this stealthy invader.
Materials and Methods
Ethics statement
All animal experiment procedures were approved by the Animal Ethics Review Committee of the University of Toronto (Approval #20010054 and #20010055), which is subject to the ethical and legal requirements under the province of Ontario's Animals for Research Act and the federal Council on Animal Care.
Animals
Generation of the CEACAM1-humanized mouse line was previously described [43]. CEABAC2 mice, generated by stable integration of a human-derived bacterial artificial chromosome (BAC) encoding the human CEACAM3, CEACAM5, CEACAM6 and CEACAM7 genes, have been previously described [40]. Wild type (WT) mice used are littermates of the CEABAC2 animals.
Reagents and antibodies
All reagents were from Sigma (Oakville, Ontario, Canada) unless otherwise indicated. The anti-gonococcal polyclonal rabbit antibody (UTR01) was described previously [18]. Rabbit anti - CEACAM polyclonal and normal serum was from Dako (Mississauga, ON). CEACAM pan-specific D14HD11, CEACAM1-specific 4/3/17 and CEACAM6-specific 9A6 antibodies were from Genovac (Freiburg, Germany), and the CEACAM3-specific Col-1 antibody was from Zymed (San Francisco, CA). Phospho-p38, p38, phospho-Erk1/2, Erk1/2, phospho-PKCδ and PKCδ-specific antibodies were form Cell Signaling Technology. The IκBα-specific antibody was from Santa Cruz Biotechnology (Santa Cruz, CA). Fluorescent conjugates were from Jackson ImmunoResearch Laboratories (Mississauga, ON), except Texas red-phalloidin, which was from Molecular Probes (Eugene, OR). The Tak1 inhibitor ((5z)-7-oxozeaenol) was from Millipore (Billerica, MA), and the p38 inhibitor (SB203580) and the Src family kinase-specific PP2 were from Calbiochem (La Jolla, CA). Erk1/2 inhibitor (UT0126) was purchased from Cell Signaling.
Bacterial strains
The isogenic Opa− and Opa+ (Opa57) N. gonorrhoeae MS11 strains (N302 and N313, respectively; [66]) were kindly provided by Dr. T.F. Meyer, and their phenotypes have been described previously [7].
Primary neutrophil isolation
Human neutrophils were isolated from citrated whole blood taken from healthy volunteers by venipuncture using Ficoll-Paque Plus (Amersham Biosciences; Buckinghamshire, England). Contaminating erythrocytes were removed by dextran sedimentation and hypotonic shock, as described previously [29].
Mouse bone marrow neutrophils were taken from 8 to 10-week old mice that were humanely euthanized by CO2 inhalation. Femurs and tibias were removed, and bone marrow was isolated and separated on a discontinuous Percoll gradient (80%/65%/55%) as described previously by others [67]. Neutrophils were recovered at the 80%/65% interface.
Human CEACAM expression in CEABAC neutrophils
WT and CEABAC neutrophils (106 cells) were lysed in boiling SDS buffer and CEACAMs detected using SDS-PAGE and immunoblots probed with indicated CEACAM-specific antibodies. For flow cytometry analysis of cell-surface CEACAM expression, 106 PMNs from CEABAC or WT littermates were spun down and fixed in 1% PFA in Hank's Buffered Saline Solution (HBSS) prior to immunofluorescence staining.
Whole cell phosphorylation assays
106 neutrophils per sample were infected with N. gonorrhoeae at multiplicity of infection [45] of 10 in 250 µl of HBSS. Infections were stopped by centrifugation at 2400 g for 3 min at 4°C, lysed in boiling SDS sample buffer, and boiled for a further 10 min. Samples were resolved by SDS-PAGE and immunoblotted.
Bacterial infections for immunofluorescence microscopy
5×105 WT or CEABAC bone marrow-derived PMNs were centrifuged onto washed mouse serum-coated coverslips at 1500 rpm for 10 min. Cells were infected at MOI of 25 (for binding and internalization studies) in a volume of 500 µl, re-centrifuged for 5 minutes at 500 rpm to facilitate bacterial association with cells, and then incubated at 37°C for indicated durations. Post-infection, samples were washed with HBSS, and fixed using 3.7% paraformaldehyde. Cells were stained for CEACAM, actin and bacteria and observed as described previously [6]. Intracellular bacteria were differentiated from extracellular via exclusion of antibody prior to membrane solubilization, as described [29].
PMN killing assays
Killing assays were adapted from Ball et al. [41]. Briefly, adherent WT and CEABAC neutrophils were infected at an MOI = 1. At indicated time points, cells were washed and incubated with protease inhibitors for 15 minutes prior to lysis with 1% saponin and plated on GC agar. Bacterial survival was evaluated relative to CFUs present at 0 time point.
Oxidative burst and degranulation assays
For chemiluminescence-based oxidative burst assay, 5×105 cells were incubated with 25 µg/ml 5-amino-2,3-dihydro-1,4-phthalazinedione (‘luminol’; Sigma) in a volume of 100 µl, and then treated with agonists in a total volume of 200 µl, in triplicate. Infections proceeded for 60 min at 37°C, after which luminescence was read using a Tecan plate reader with i-control software. For flow cytometry-based degranulation assay (CD11b release), 106 PMNs were treated with agonists in 500 µl of HBSS for 30 minutes at 37°C. Infections were stopped by centrifugation at 2,400 g for 3 min at RT and cell pellets then fixed in 1% PFA before staining with 1.25 µg of PE-conjugated rat anti-mouse CD11b in a total volume of 50 µl. Myeloperoxidase (MPO), elastase, and lactoferrin release assays were performed essentially as described by others. Briefly, 106 PMNs were exposed to agonists in a total volume of 500 µl and then incubated for 30 minutes at 37°C. Cells were then pelleted and supernatants collected. For MPO assays, 50 µl of supernatant was mixed with 150 µl SureBlue tetramethylbenzidine peroxidase substrate (KPL; Gaithersberg, MD), and plates were read spectrophotometrically at 650 nm. For the elastase assay, 50 µl of supernatant was diluted 2-fold in PBS and incubated with 100 µl DQ elastin substrate conjugated to BODIPY FL (from the EnzCheck Elastase kit; Molecular Probes), and then incubated for 24 hours at RT before reading fluorescence with 488 nm excitation and 515 nm emission. For both MPO and elastase assays, a percentage (%) release is shown, calculated as the amount of the protein in the supernatant divided by the total amount in 106 CHAPS-lysed cells.
Lactoferrin release from PMN granules was assayed by ELISA as described by others [48]. To induce release of primary granule components into medium, cells were pre-treated with 5 µM cytochalasin B for 5 minutes at 37°C prior to agonist treatment.
PMN gene array experiments
CEABAC and WT bone marrow neutrophils (107 cells) were either infected with Opa+ N. gonorrhoeae (MOI = 10) or left uninfected for 1 h. The infections were stopped by centrifugation at 2400 g for 5 min at 4°C. RNA was extracted and purified using the Qiagen RNeasy kit. Samples from 3 independent experiments were analyzed using an Illumina Mouse Whole Genome V2R2 array with 45,281 probes. The original data normalization and analysis were provided as a service by the Bioinformatics Department of the University Health Network (UHN) Microarray Centre, Toronto, ON. Data was analyzed using Genespring v11.0.1. 66 genes showed ≥2 fold change (FC) in gene expression in infected PMNs relative to uninfected controls. Gene lists were analyzed using the database for annotation, visualization, and integrated discovery (DAVID) (Huang da et al., 2009) and manual examination. To compare WT vs. CEABAC neutrophil responses, we considered genes ≥1.5 FC in CEABAC over WT.
Cytokine measurements
106 cells were infected with N. gonorrhoeae at MOI of 10 and incubated at 37°C for 3 h. Infections were then stopped by centrifugation at 2400 g for 5 min at 4°C, and supernatants were collected. Quantitative measurements of cytokines were performed using ELISA kits form R&D Systems (MIP-1α, KC and MIP-2) and BD Biosciences (TNF-α). For qRT-PCR, cells were infected as described above. At 3 h post infection, RNA was collected using Qiagen RNeasy kit and converted to cDNA using iScript RT Supermix (Bio-Rad Laboratories). qPCR was carried out using SsoAdvanced SYBR Green Supermix (Bio-Rad Laboratories). All transcript levels are shown as relative to those of GAPDH.
Chemotaxis assay
Bone marrow neutrophils were isolated and suspended in HBSS with 1% gelatin (Sigma, G7041). A neutrophil suspension (1×106/mL) was allowed to attach to bovine serum albumin (BSA, Sigma A7906; 1 mg/ml)-coated glass coverslips (22×40 mm, Fisher 12–543-A) at 37°C for 10 minutes. The coverslip was inverted into a Zigmond chamber (Neuroprobe, z02) and 100 µL HBSS media was added to the right chamber with 100 µL HBSS media mixed with supernatants derived from infected CEABAC or WT neutrophils added to the left chamber. Time-lapse video microscopy was used to examine neutrophil movements in Zigmond chambers. Images were captured at 20-second intervals with a Nikon Eclipse E1000 Microscope using the 40× objective. Cell-tracking software (Retrac version 2.1.01 Freeware) was used to characterize cellular chemotaxis from the captured images. Data comes from three independent experiments.
Air pouch
CEABAC and WT littermate control mice (6–8 wk) were anesthetized with isoflurane, and dorsal air pouches were raised by injecting 3 ml sterile air subcutaneously on days 0 and a further 2 ml on day 3. On day 5, the mice were anesthetized with isoflurane and injected with 1 ml PBS containing 2×106 cfu/ml of Opa− or Opa+ N. gonorrhoeae. Mice were sacrificed 6 h after the injection, and air pouches were then washed with 2 ml PBS. The cells present in the wash were counted with a hemocytometer and analyzed by Diff-Quick staining of the cytospins. The supernatants were analyzed by ELISA, as described above. In some cases, air pouch fibroblasts were obtained by instilling 0.05% trypsin containing 0.5 mM EDTA in DMEM (3 ml/pouch), as previously described [68]. These cells were seeded onto glass coverslips in a 24-well plate, cultured in DMEM-10% FBS with antibiotics overnight, and then infected with Opa+ N. gonorrhoeae the next day for 30 min. Cells were fixed and stained for immunofluorescence microscopy.
Neutrophil depletion
Neutrophil depletion from mice was achieved by a single i.p. injection of 200 µl of sterile saline containing 250 µg of the Gr-1 specific monoclonal antibody RB6-8C5 at 24 h prior to infection. The RB6-8C5 hybridoma was generously provided by Professor Paul Allen, Department of Pathology and Immunology, Washington University School of Medicine, St. Louis.
Supporting Information
Zdroje
1. World Health Organization DoRHaR (2012) Global action plan to control the spread and impact of antimicrobial resistance in Neisseria gonorrhoeae. http://www.who.int/reproductivehealth/publications/rtis/9789241503501/en/
2. World Health Organization DoRHaR (2012) Global incidence and prevalence of selected curable sexually transmitted infections - 2008. http://www.who.int/reproductivehealth/publications/rtis/stisestimates/en/
3. ZhuW, ChenCJ, ThomasCE, AndersonJE, JerseAE, et al. (2011) Vaccines for gonorrhea: can we rise to the challenge? Frontiers in Microbiology 2 : 124.
4. BiaisN, LadouxB, HigashiD, SoM, SheetzM (2008) Cooperative retraction of bundled type IV pili enables nanonewton force generation. PLoS Biology 6: e87.
5. VirjiM, MakepeaceK, FergusonDJ, WattSM (1996) Carcinoembryonic antigens (CD66) on epithelial cells and neutrophils are receptors for Opa proteins of pathogenic Neisseria. Molecular Microbiology 22 : 941–950.
6. Gray-OwenSD, DehioC, HaudeA, GrunertF, MeyerTF (1997) CD66 carcinoembryonic antigens mediate interactions between Opa-expressing Neisseria gonorrhoeae and human polymorphonuclear phagocytes. The EMBO Journal 16 : 3435–3445.
7. Gray-OwenSD, LorenzenDR, HaudeA, MeyerTF, DehioC (1997) Differential Opa specificities for CD66 receptors influence tissue interactions and cellular response to Neisseria gonorrhoeae. Molecular Microbiology 26 : 971–980.
8. ChenT, GrunertF, Medina-MarinoA, GotschlichEC (1997) Several carcinoembryonic antigens (CD66) serve as receptors for gonococcal opacity proteins. The Journal of Experimental Medicine 185 : 1557–1564.
9. BosMP, GrunertF, BellandRJ (1997) Differential recognition of members of the carcinoembryonic antigen family by Opa variants of Neisseria gonorrhoeae. Infection and Immunity 65 : 2353–2361.
10. VirjiM, EvansD, HadfieldA, GrunertF, TeixeiraAM, et al. (1999) Critical determinants of host receptor targeting by Neisseria meningitidis and Neisseria gonorrhoeae: identification of Opa adhesiotopes on the N-domain of CD66 molecules. Molecular Microbiology 34 : 538–551.
11. BillkerO, PoppA, Gray-OwenSD, MeyerTF (2000) The structural basis of CEACAM-receptor targeting by neisserial Opa proteins. Trends in Microbiology 8 : 258–260 discussion 260–251.
12. PoppA, DehioC, GrunertF, MeyerTF, Gray-OwenSD (1999) Molecular analysis of neisserial Opa protein interactions with the CEA family of receptors: identification of determinants contributing to the differential specificities of binding. Cellular Microbiology 1 : 169–181.
13. McGeeZA, StephensDS, HoffmanLH, SchlechWF3rd, HornRG (1983) Mechanisms of mucosal invasion by pathogenic Neisseria. Reviews of Infectious Diseases 5 Suppl 4: S708–714.
14. WangJ, Gray-OwenSD, KnorreA, MeyerTF, DehioC (1998) Opa binding to cellular CD66 receptors mediates the transcellular traversal of Neisseria gonorrhoeae across polarized T84 epithelial cell monolayers. Molecular Microbiology 30 : 657–671.
15. BoultonIC, Gray-OwenSD (2002) Neisserial binding to CEACAM1 arrests the activation and proliferation of CD4+ T lymphocytes. Nature Immunology 3 : 229–236.
16. LeeHS, BoultonIC, ReddinK, WongH, HalliwellD, et al. (2007) Neisserial outer membrane vesicles bind the coinhibitory receptor carcinoembryonic antigen-related cellular adhesion molecule 1 and suppress CD4+ T lymphocyte function. Infection and Immunity 75 : 4449–4455.
17. LeeHS, OstrowskiMA, Gray-OwenSD (2008) CEACAM1 dynamics during Neisseria gonorrhoeae suppression of CD4+ T lymphocyte activation. Journal of Immunology 180 : 6827–6835.
18. PantelicM, KimYJ, BollandS, ChenI, ShivelyJ, et al. (2005) Neisseria gonorrhoeae kills carcinoembryonic antigen-related cellular adhesion molecule 1 (CD66a)-expressing human B cells and inhibits antibody production. Infection and Immunity 73 : 4171–4179.
19. YuQ, ChowEM, McCawSE, HuN, ByrdD, et al. (2013) Association of Neisseria gonorrhoeae Opa(CEA) with dendritic cells suppresses their ability to elicit an HIV-1-specific T cell memory response. PloS One 8: e56705.
20. SlevogtH, ZabelS, OpitzB, HockeA, EitelJ, et al. (2008) CEACAM1 inhibits Toll-like receptor 2-triggered antibacterial responses of human pulmonary epithelial cells. Nature Immunology 9 : 1270–1278.
21. Gray-OwenSD, BlumbergRS (2006) CEACAM1: contact-dependent control of immunity. Nature Reviews Immunology 6 : 433–446.
22. SchmitterT, AgererF, PetersonL, MunznerP, HauckCR (2004) Granulocyte CEACAM3 is a phagocytic receptor of the innate immune system that mediates recognition and elimination of human-specific pathogens. The Journal of Experimental Medicine 199 : 35–46.
23. PilsS, GerrardDT, MeyerA, HauckCR (2008) CEACAM3: an innate immune receptor directed against human-restricted bacterial pathogens. International Journal of Medical Microbiology 298 : 553–560.
24. SarantisH, Gray-OwenSD (2012) Defining the roles of human carcinoembryonic antigen-related cellular adhesion molecules during neutrophil responses to Neisseria gonorrhoeae. Infection and Immunity 80 : 345–358.
25. SarantisH, Gray-OwenSD (2007) The specific innate immune receptor CEACAM3 triggers neutrophil bactericidal activities via a Syk kinase-dependent pathway. Cellular Microbiology 9 : 2167–2180.
26. BoothJW, TelioD, LiaoEH, McCawSE, MatsuoT, et al. (2003) Phosphatidylinositol 3-kinases in carcinoembryonic antigen-related cellular adhesion molecule-mediated internalization of Neisseria gonorrhoeae. The Journal of Biological Chemistry 278 : 14037–14045.
27. BuntruA, KoppK, VogesM, FrankR, BachmannV, et al. (2011) Phosphatidylinositol 3′-kinase activity is critical for initiating the oxidative burst and bacterial destruction during CEACAM3-mediated phagocytosis. The Journal of Biological Chemistry 286 : 9555–9566.
28. SmirnovA, DailyKP, CrissAK (2014) Assembly of NADPH Oxidase in Human Neutrophils Is Modulated by the Opacity-Associated Protein Expression State of Neisseria gonorrhoeae. Infection and Immunity 82 : 1036–1044.
29. McCawSE, SchneiderJ, LiaoEH, ZimmermannW, Gray-OwenSD (2003) Immunoreceptor tyrosine-based activation motif phosphorylation during engulfment of Neisseria gonorrhoeae by the neutrophil-restricted CEACAM3 (CD66d) receptor. Molecular Microbiology 49 : 623–637.
30. BuntruA, RothA, Nyffenegger-JannNJ, HauckCR (2012) HemITAM signaling by CEACAM3, a human granulocyte receptor recognizing bacterial pathogens. Archives of Biochemistry and Biophysics 524 : 77–83.
31. SadaranganiM, PollardAJ, Gray-OwenSD (2011) Opa proteins and CEACAMs: pathways of immune engagement for pathogenic Neisseria. FEMS Microbiology Reviews 35 : 498–514.
32. BazzoniF, CassatellaMA, RossiF, CeskaM, DewaldB, et al. (1991) Phagocytosing neutrophils produce and release high amounts of the neutrophil-activating peptide 1/interleukin 8. The Journal of Experimental Medicine 173 : 771–774.
33. KolaczkowskaE, KubesP (2013) Neutrophil recruitment and function in health and inflammation. Nature Reviews Immunology 13 : 159–175.
34. JaillonS, GaldieroMR, Del PreteD, CassatellaMA, GarlandaC, et al. (2013) Neutrophils in innate and adaptive immunity. Seminars in Immunopathology 35 : 377–394.
35. SubrahmanyamYV, YamagaS, PrasharY, LeeHH, HoeNP, et al. (2001) RNA expression patterns change dramatically in human neutrophils exposed to bacteria. Blood 97 : 2457–2468.
36. KobayashiSD, VoyichJM, BuhlCL, StahlRM, DeLeoFR (2002) Global changes in gene expression by human polymorphonuclear leukocytes during receptor-mediated phagocytosis: cell fate is regulated at the level of gene expression. Proceedings of the National Academy of Sciences of the United States of America 99 : 6901–6906.
37. ScapiniP, Lapinet-VeraJA, GasperiniS, CalzettiF, BazzoniF, et al. (2000) The neutrophil as a cellular source of chemokines. Immunological Reviews 177 : 195–203.
38. KammererR, PoppT, HartleS, SingerBB, ZimmermannW (2007) Species-specific evolution of immune receptor tyrosine based activation motif-containing CEACAM1-related immune receptors in the dog. BMC Evolutionary Biology 7 : 196.
39. VogesM, BachmannV, KammererR, GophnaU, HauckCR (2010) CEACAM1 recognition by bacterial pathogens is species-specific. BMC Microbiology 10 : 117.
40. ChanCH, StannersCP (2004) Novel mouse model for carcinoembryonic antigen-based therapy. Molecular Therapy : the Journal of the American Society of Gene Therapy 9 : 775–785.
41. BallLM, CrissAK (2013) Constitutively Opa-expressing and Opa-deficient Neisseria gonorrhoeae strains differentially stimulate and survive exposure to human neutrophils. Journal of Bacteriology 195 : 2982–2990.
42. Huang daW, ShermanBT, LempickiRA (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature Protocols 4 : 44–57.
43. GuA, ZhangZ, ZhangN, TsarkW, ShivelyJE (2010) Generation of human CEACAM1 transgenic mice and binding of Neisseria Opa protein to their neutrophils. PloS One 5: e10067.
44. TamasD, CeciliaC, Sanae BenM, ThiloMF, SiegfriedW, et al. (2010) Potentiation of Epithelial Innate Host Responses by Intercellular Communication. PLoS Pathogens
45. TamassiaN, Le MoigneV, CalzettiF, DoniniM, GasperiniS, et al. (2007) The MyD88-independent pathway is not mobilized in human neutrophils stimulated via TLR4. Journal of Immunology 178 : 7344–7356.
46. CloutierA, EarT, Blais-CharronE, DuboisCM, McDonaldPP (2007) Differential involvement of NF-kappaB and MAP kinase pathways in the generation of inflammatory cytokines by human neutrophils. Journal of leukocyte biology 81 : 567–577.
47. McDonaldPP, BaldA, CassatellaMA (1997) Activation of the NF-kappaB pathway by inflammatory stimuli in human neutrophils. Blood 89 : 3421–3433.
48. AbramCL, LowellCA (2007) The expanding role for ITAM-based signaling pathways in immune cells. Science's STKE : signal transduction knowledge environment 2007: re2.
49. KingeterLM, LinX (2012) C-type lectin receptor-induced NF-kappaB activation in innate immune and inflammatory responses. Cellular & molecular immunology 9 : 105–112.
50. FesslerMB, MalcolmKC, DuncanMW, WorthenGS (2002) A genomic and proteomic analysis of activation of the human neutrophil by lipopolysaccharide and its mediation by p38 mitogen-activated protein kinase. The Journal of Biological Chemistry 277 : 31291–31302.
51. LandstromM (2010) The TAK1-TRAF6 signalling pathway. The international Journal of Biochemistry & Cell Biology 42 : 585–589.
52. McCawSE, LiaoEH, Gray-OwenSD (2004) Engulfment of Neisseria gonorrhoeae: revealing distinct processes of bacterial entry by individual carcinoembryonic antigen-related cellular adhesion molecule family receptors. Infection and Immunity 72 : 2742–2752.
53. HauckCR, MeyerTF, LangF, GulbinsE (1998) CD66-mediated phagocytosis of Opa52 Neisseria gonorrhoeae requires a Src-like tyrosine kinase - and Rac1-dependent signalling pathway. The EMBO Journal 17 : 443–454.
54. MocsaiA, RulandJ, TybulewiczVL (2010) The SYK tyrosine kinase: a crucial player in diverse biological functions. Nature Reviews Immunology 10 : 387–402.
55. SchmitterT, PilsS, SakkV, FrankR, FischerKD, et al. (2007) The granulocyte receptor carcinoembryonic antigen-related cell adhesion molecule 3 (CEACAM3) directly associates with Vav to promote phagocytosis of human pathogens. Journal of immunology 178 : 3797–3805.
56. StrasserD, NeumannK, BergmannH, MarakalalaMJ, GulerR, et al. (2012) Syk kinase-coupled C-type lectin receptors engage protein kinase C-sigma to elicit Card9 adaptor-mediated innate immunity. Immunity 36 : 32–42.
57. EdwardsJC, SedgwickAD, WilloughbyDA (1981) The formation of a structure with the features of synovial lining by subcutaneous injection of air: an in vivo tissue culture system. The Journal of Pathology 134 : 147–156.
58. PiererM, RethageJ, SeiblR, LauenerR, BrentanoF, et al. (2004) Chemokine secretion of rheumatoid arthritis synovial fibroblasts stimulated by Toll-like receptor 2 ligands. Journal of Immunology 172 : 1256–1265.
59. TatedaK, MooreTA, DengJC, NewsteadMW, ZengX, et al. (2001) Early recruitment of neutrophils determines subsequent T1/T2 host responses in a murine model of Legionella pneumophila pneumonia. Journal of Immunology 166 : 3355–3361.
60. KerriganAM, BrownGD (2010) Syk-coupled C-type lectin receptors that mediate cellular activation via single tyrosine based activation motifs. Immunological Reviews 234 : 335–352.
61. GoodridgeHS, UnderhillDM, TouretN (2012) Mechanisms of Fc receptor and Dectin-1 activation for phagocytosis. Traffic 13 : 1062–1071.
62. ZhangX, MajlessiL, DeriaudE, LeclercC, Lo-ManR (2009) Coactivation of Syk kinase and MyD88 adaptor protein pathways by bacteria promotes regulatory properties of neutrophils. Immunity 31 : 761–771.
63. MuenznerP, BachmannV, ZimmermannW, HentschelJ, HauckCR (2010) Human-restricted bacterial pathogens block shedding of epithelial cells by stimulating integrin activation. Science 329 : 1197–1201.
64. MuenznerP, RohdeM, KneitzS, HauckCR (2005) CEACAM engagement by human pathogens enhances cell adhesion and counteracts bacteria-induced detachment of epithelial cells. The Journal of Cell Biology 170 : 825–836.
65. JohswichKO, McCawSE, IslamE, SintsovaA, GuA, et al. (2013) In vivo adaptation and persistence of Neisseria meningitidis within the nasopharyngeal mucosa. PLoS Pathogens 9: e1003509.
66. KupschEM, KnepperB, KurokiT, HeuerI, MeyerTF (1993) Variable opacity (Opa) outer membrane proteins account for the cell tropisms displayed by Neisseria gonorrhoeae for human leukocytes and epithelial cells. The EMBO Journal 12 : 641–650.
67. SunCX, DowneyGP, ZhuF, KohAL, ThangH, et al. (2004) Rac1 is the small GTPase responsible for regulating the neutrophil chemotaxis compass. Blood 104 : 3758–3765.
68. KukulskiF, Ben YebdriF, BahramiF, LevesqueSA, Martin-SatueM, et al. (2010) The P2 receptor antagonist PPADS abrogates LPS-induced neutrophil migration in the murine air pouch via inhibition of MIP-2 and KC production. Molecular Immunology 47 : 833–839.
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2014 Číslo 9
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Měli bychom postcovidový syndrom léčit antidepresivy?
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- The Secreted Peptide PIP1 Amplifies Immunity through Receptor-Like Kinase 7
- Symbionts Commonly Provide Broad Spectrum Resistance to Viruses in Insects: A Comparative Analysis of Strains
- MIF Contributes to Associated Immunopathogenicity Development
- The Ins and Outs of Rust Haustoria