
Hematopoietic but Not Endothelial Cell MyD88 Contributes to Host Defense during Gram-negative Pneumonia Derived Sepsis
Klebsiella pneumoniae is an important causative pathogen in hospital acquired or health care associated pneumonia and sepsis. Toll-like receptors recognize conserved motifs expressed by pathogens and thereby initiate the innate immune response. Myeloid differentiation primary response gene (MyD)88 is a common adapter for multiple Toll-like receptors that is important for protective immunity during Klebsiella infections. The contribution of different cell types to MyD88 mediated protection is not known. We used a model of Klebsiella pneumonia and secondary sepsis in mice that were selectively deficient for MyD88 in specific cell-types that are implicated to be important for host defense mechanisms by use of a tissue specific gene recombination system and bone marrow transfer. We demonstrate that MyD88 in myeloid cells, but not in endothelial cells, is important for host defense during pneumonia derived sepsis caused by Klebsiella pneumoniae.
Published in the journal:
. PLoS Pathog 10(9): e32767. doi:10.1371/journal.ppat.1004368
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004368
Summary
Klebsiella pneumoniae is an important causative pathogen in hospital acquired or health care associated pneumonia and sepsis. Toll-like receptors recognize conserved motifs expressed by pathogens and thereby initiate the innate immune response. Myeloid differentiation primary response gene (MyD)88 is a common adapter for multiple Toll-like receptors that is important for protective immunity during Klebsiella infections. The contribution of different cell types to MyD88 mediated protection is not known. We used a model of Klebsiella pneumonia and secondary sepsis in mice that were selectively deficient for MyD88 in specific cell-types that are implicated to be important for host defense mechanisms by use of a tissue specific gene recombination system and bone marrow transfer. We demonstrate that MyD88 in myeloid cells, but not in endothelial cells, is important for host defense during pneumonia derived sepsis caused by Klebsiella pneumoniae.
Introduction
Globally, lower respiratory tract infections are in the top ten causes of death, both in high - and low-income countries [1]. Pneumonia is the most common cause of sepsis and frequently caused by gram-negative pathogens from the family of Enterobacteriaceae, including Klebsiella (K.) pneumoniae [2]–[4]. Increasing rates of extended-spectrum β-lactamases producing Enterobacteriaceae are a major health concern and make the development of new therapies urgent, since infection with such pathogens is associated with increased mortality [5]–[7].
Infection is detected by sensors of the innate immune system collectively called pattern recognition receptors [8], [9]. Toll-like receptors (TLRs) prominently feature herein, able to detect a variety of conserved microbial patterns as well as “danger signals” released from host cells as a consequence of injurious inflammation. As such, TLRs play an important role in the initiation and amplification of the host response [8], [9]. The universal adaptor for all TLRs except TLR3 is myeloid differentiation primary response gene (MyD)88, that propagates the signal of activated TLRs intracellularly, leading to NFκB and MAP kinase activation. In addition, MyD88 mediates IL-1β and IL-18 receptor signaling [10]. We and others recently demonstrated the importance of MyD88 dependent signaling for survival and antibacterial defense during K. pneumoniae infection [3], [11], [12]. During respiratory tract infection different MyD88 expressing cells may contribute to host defense, including innate immune cells, such as alveolar macrophages, intraepithelial dendritic cells and migrated leukocytes, and parenchymal cells, such as lung epithelium and endothelium [13]–[15]. By creating chimeric mice using bone marrow (BM) transplantation, we reported the importance of MyD88 in both radiosensitive (hematopoietic) cells and radioresistant (parenchymal) cells for antibacterial defense and survival during Klebsiella pneumonia derived sepsis [12].
Whereas the role of hematopoietic cells in host defense against bacteria is undisputed, there are only few reports about the specific contribution of the vascular endothelium to the pathophysiology of infection and sepsis. Some evidence points to an attenuation of tissue and organ injury during polymicrobial sepsis when endothelial NFκB signaling was specifically targeted, without an effect on bacterial clearance [16]–[19]. However on the other hand the specific expression of endothelial TLR4 was reported to be sufficient for adequate bacterial clearance in a model of gram-negative infection [20]. Therefore, we here aimed to study the role of MyD88 dependent signaling in myeloid and endothelial cells during K. pneumoniae pneumosepsis by using mice with cell-specific targeted deletion of Myd88 and BM transfer. We demonstrate that myeloid, but nor endothelial cell MyD88 is important for host defense during pneumonia derived sepsis caused by Klebsiella.
Results
Genetic and functional characterization of primary cells from LysM-Myd88−/− and Tie2-Myd88−/− mice
To investigate the relative contribution of MyD88 dependent signaling in myeloid and endothelial cells to protective immunity during gram-negative pneumosepsis we crossed mice homozygous for the conditional Myd88 flox allele (Myd88fl/fl mice) [21] with mice expressing Cre under control of the myeloid cell LysM promoter (to generate LysM-Myd88−/− mice) [22] or the myeloid plus endothelial cell Tie2 promoter (to generate Tie2-Myd88−/− mice) [23]. To determine the efficiency of Cre-induced Myd88 deletion in specific cell types, we performed qPCR to quantify the remaining Myd88fl/fl in blood total leukocytes, granulocytes, monocytes and lymphocytes, in alveolar and peritoneal macrophages, in splenocytes and in lung endothelial and epithelial cells (figure 1A). As expected, the deletion efficiency of Cre in LysM-Myd88−/− was very high in the myeloid compartment, especially in macrophages, granulocytes and to a lesser extent monocytes; lymphocytes and endothelial cells were unaffected. As anticipated, the Myd88fl/fl allele was almost completely absent in endothelial cells of Tie2-Myd88−/− mice. In addition, excision of the Myd88fl/fl allele was also virtually complete in all hematopoietic cell types of Tie2-Myd88−/− mice, as well as in lymphocytes and (accordingly) in splenocytes. Next, to determine the functional consequences of these Cre-mediated cell-specific Myd88 deletions, we incubated whole blood leukocytes, alveolar and peritoneal macrophages and splenocytes obtained from LysM-Myd88−/−, Tie2-Myd88−/− and control mice with K. pneumoniae LPS or heat-killed K. pneumoniae, using TNFα release as readout; we focused on these cell types since they confer protective functions during infection and sepsis [24]–[26]. In agreement with the genetic characterization of cells from LysM-Myd88−/− and Tie2-Myd88−/− mice, whole blood leukocytes from both genotypes showed a clearly reduced responsiveness to Klebsiella and Klebsiella LPS, with Tie2-Myd88−/− leukocytes showing the largest defect (figure 1B). In addition, LysM-Myd88−/− and Tie2-Myd88−/− alveolar and peritoneal macrophages displayed strongly reduced TNFα release upon stimulation (figure 1C,D), while the strongest defect in splenocyte responsiveness was seen in Tie2-Myd88−/− cell cultures (figure 1E). Together these results indicate that Tie2-Myd88−/− mice are MyD88 deficient in hematopoietic, lymphoid and endothelial cells, while in LysM-Myd88−/− mice MyD88 deficiency is restricted to hematopoietic cells.
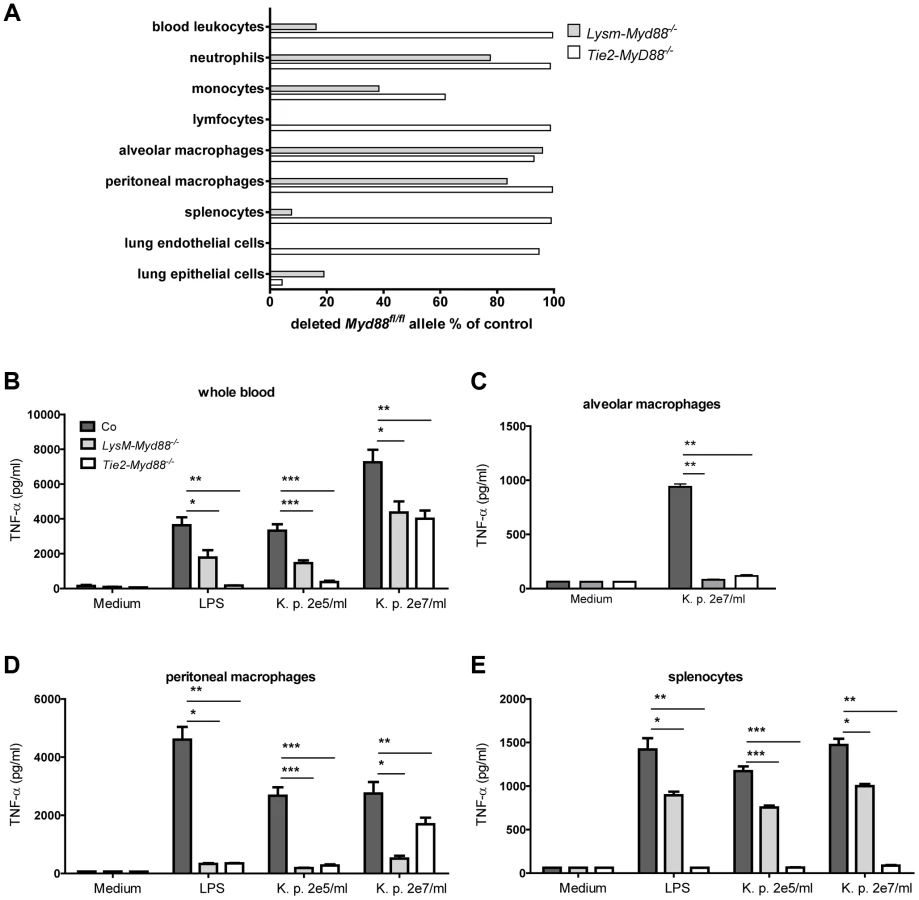
LysM-Myd88−/− and Tie2-Myd88−/− mice demonstrate a strongly impaired host defense during gram-negative pneumosepsis
Next, we infected LysM-Myd88−/−, Tie2-Myd88−/− and Myd88fl/fl Cre negative control mice with K. pneumoniae via the airways and monitored mortality during a 5-day follow up (figure 2A). LysM-Myd88−/− and Tie2-Myd88−/− mice displayed massive mortality within the first 2 days after infection with median survival times of 1.8 and 1.5 days respectively, while control mice had a median survival time of 2.9 days (both p<0.001 versus control mice). Notably, Tie2-Myd88−/− mice showed an accelerated mortality relative to LysM-Myd88−/− mice (p<0.01 for the difference between groups). To obtain insight in the cause of early lethality of LysM-Myd88−/− and Tie2-Myd88−/− mice we next infected mice with Klebsiella in a separate experiment and harvested lungs, blood, spleen and liver for quantitative cultures 24 hours post infection (i.e. shortly before the first deaths were expected to occur), seeking to collect data representative for host defense at the primary site of infection and bacterial dissemination. At this time point, both LysM-Myd88−/− and Tie2-Myd88−/− mice had ≥2-log more bacteria in their lungs relative to control mice (p<0.01 and 0.001 respectively compared to controls, figure 2B). Moreover, bacterial counts were significantly higher in blood and spleen of LysM-Myd88−/− and Tie2-Myd88−/− mice (both p<0.05 compared to control mice, figure 2C and D). In addition, Tie2-Myd88−/− mice had significantly higher amounts of bacteria in their livers (p<0.01 compared to control mice, figure 2E). Tie2-Myd88−/− mice had higher bacterial counts when compared with LysM-Myd88−/− mice in all body sites, although these differences did not reach statistical significance. Together these data indicate that LysM-Myd88−/− and Tie2-Myd88−/− mice demonstrate a strongly enhanced bacterial growth and dissemination during gram-negative pneumonia derived sepsis, resulting in accelerated mortality.

LysM-Myd88−/− and Tie2-Myd88−/− mice show modest alterations in the inflammatory and injurious response during gram-negative pneumosepsis
To obtain insight in local inflammation at the primary site of infection we harvested lungs from LysM-Myd88−/−, Tie2-Myd88−/− and control mice 24 hours post infection for semi-quantitative histopathology, focusing on key histological features characteristic for severe pneumonia (figure 3). The extent of lung pathology did not differ between groups. LysM-Myd88−/− mice had lower myeloperoxidase (MPO) concentrations in whole lung homogenates, indicative of a reduced neutrophil content. In accordance, the number of Ly6+ cells was lower in LysM-Myd88−/− mice relative to controls. To obtain further insight in the role of MyD88 in cells targeted by LysM - and Tie2-driven Cre recombinase in lung inflammation during Klebsiella pneumonia, we measured the levels of the proinflammatory cytokines IL-1β, TNF-α, IL-6, the anti-inflammatory cytokine IL-10 and the neutrophil attracting chemokines CXCL-1 and CXCL-2 in lung homogenates (table 1). The pulmonary concentrations of all mediators were similar in LysM-Myd88−/−, Tie2-Myd88−/− and control mice, with the exception of TNF-α levels which were significantly lower in Tie2-Myd88−/− mice (p<0.01 compared to controls). Plasma IL-6 levels were significantly increased in LysM-Myd88−/− and Tie2-Myd88−/− mice (p<0.05 to 0.01 respectively compared to controls, table 1) likely as a result of higher bacterial loads. In addition, we determined E-selectin levels in both lung homogenates and plasma as a reflection of endothelial cell activation [27] and observed that lung levels of E-selectin were significantly increased in Tie2-Myd88−/− mice, probably as a result of the higher bacterial burden (p<0.05 compared to control mice) (figure S1).


The model of Klebsiella pneumonia and sepsis used here is associated with rises in the plasma concentrations of LDH (indicative for cellular injury in general) and AST (reflecting hepatocellular injury) in the late stage of infection [28]. To study if the absence of MyD88 in myeloid and/or endothelial cells affected the degree of liver and cellular injury we determined the plasma levels of these parameters but observed no differences (figure S2).
Together these data suggest that the increased mortality in LysM-Myd88−/− and Tie2-Myd88−/− mice occurred as a result of overwhelming bacterial growth rather than as a result of pulmonary or distant organ injury.
MyD88 dependent signaling in the hematopoietic compartment is crucial for antibacterial defense while MyD88 in endothelial cells is not important
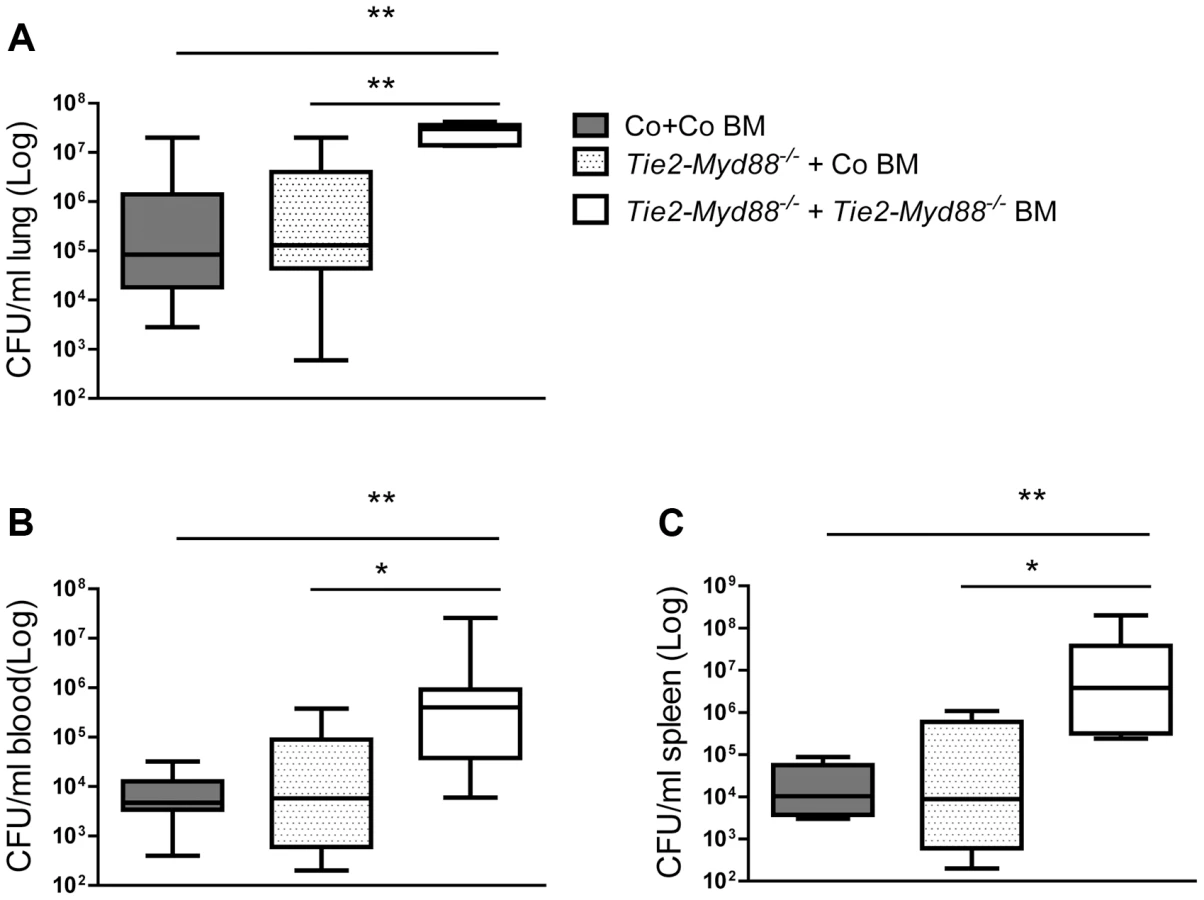
Considering that Tie2-Myd88−/− mice have strongly impaired MyD88 signaling in hematopoietic and endothelial cells, we decided to restore the hematopoietic compartment of Tie2-Myd88−/− mice with BM of Myd88fl/fl control mice after lethal irradiation, thereby creating mice with a more exclusive MyD88 deficiency in endothelial cells. In order to adequately estimate the effect size, we created two control groups: Tie2-Myd88−/− mice transplanted with Tie2-Myd88−/− BM and control mice transplanted with control BM. After 6 weeks of recovery, we infected mice with K. pneumoniae intranasally and sacrificed them 24 hours later. In addition, to check the efficiency of the BM transplantation to restore the responsiveness of relevant cell types from Tie2-Myd88−/− mice to Klebsiella, we euthanized 2–3 uninfected mice of each recipient group and repeated cell stimulation experiments as described above. These experiments revealed that transfer of control BM in Tie2-Myd88−/− mice fully restored the capacity of blood leukocytes, and alveolar and peritoneal macrophages, and partially that of splenocytes, to produce TNFα upon exposure to Klebsiella in vitro (figure 4A–D). The response of cells obtained from the two control groups transplanted with isogenic BM (control mice+control BM and Tie2-Myd88−/− mice+Tie2-Myd88−/− BM) replicated the impaired response of untransplanted Tie2-Myd88−/− mice relative to control mice. Importantly, after 24 hours of infection, lung bacterial loads of Tie2-Myd88−/− +control BM mice were indistinguishable from control+control BM mice, while the difference between Tie2-Myd88−/−+Tie2-Myd88−/− BM mice and control+control BM mice phenocopied the difference between Tie2-Myd88−/− and control mice observed in untransplanted mice (p<0.001, figure 5A). In line, lung bacterial levels were significantly lower in Tie2-Myd88−/−+control BM mice compared to Tie2-Myd88−/−+Tie2-Myd88−/− BM mice (p<0.01). Bacterial numbers in blood and spleen confirmed the protective effect of reconstitution of the hematopoietic compartment of Tie2-Myd88−/− mice with MyD88 sufficient BM (p<0.05 versus Tie2-Myd88−/−+Tie2-Myd88−/− BM mice, figure 5B,C). The extent of lung pathology, lung MPO levels and the number of Ly6+ cells in lung tissue were not different between groups (figure S3). Moreover, lung and plasma cytokine/chemokine and E selectin concentrations were not affected by the selective absence of endothelial MyD88 in Tie2-Myd88−/−+control BM mice, except for slightly lower lung levels of IL-10 compared to control+control BM mice (table S1; figure S4)). Also, the plasma levels of AST and LDH did not differ between groups (figure S4). Together, these data indicate that endothelial cell MyD88 has no role in antibacterial defense or in lung or distant organ injury after infection with Klebsiella via the airways.

LysM-Myd88−/− mice demonstrate an attenuated early inflammatory response
Mice with a complete MyD88 deficiency show a strongly impaired antibacterial defense after infection with Klebsiella via the airways caused by a mitigated neutrophil recruitment into the airways associated with strongly reduced local levels of neutrophil attracting mediators [11]. We wished to determine whether a similar mechanism is at play in LysM-Myd88−/− mice. Thus, LysM-Myd88−/− and control mice were infected with K. pneumoniae intranasally and lungs and bronchoalveolar lavage (BAL) fluid was harvested 6 hours later. LysM-Myd88−/− mice showed higher bacterial loads in whole lung homogenates, but not in BAL fluid (figure 6A). Importantly, LysM-Myd88−/− mice displayed a strongly attenuated influx of neutrophils into the bronchoalveolar compartment (figure 6B), which was associated with markedly reduced levels of TNFα, CXCL-1 and CXCL-2 in BAL fluid; IL-6 concentrations in BAL fluid did not differ between groups (figure 6D). Hence, these data suggest that LysM-Myd88−/− mice replicate the phenotype of Myd88−/− mice with regard to impaired neutrophil influx in the airways during early Klebsiella pneumonia at least in part caused by a reduced chemotactic gradient due to impaired chemoattractant production.

Discussion
Several MyD88 dependent TLRs are known to be important for the innate immune response to respiratory tract infection with K. pneumoniae, particularly TLR4 and TLR9, and during late stage infection or in the presence of high bacterial numbers, TLR2 [29]–[32]. Since TLRs and other innate immune sensors are widely distributed among different cell types in the airways, comprising both hematopoietic and non-hematopoietic cells, our laboratory engaged in several studies seeking to dissect the cell-specific contribution of TLR and MyD88 signaling in host defense during Klebsiella pneumonia derived sepsis [12], [32]. Using BM chimeras we reported that TLR2 and TLR4 expression in hematopoietic cells are crucial for antibacterial defense, while MyD88 in hematopoietic and parenchymal cells is equally important [12], [32]. BM transplantation can introduce artefacts caused by the irradiation and/or incomplete replacement of recipient hematopoietic cells, and cannot provide detailed information about the specific cell type that is affected [33]. In the present study we used the Cre-lox system combined with BM transfer to study the role of myeloid and endothelial cell specific MyD88 signalling in the host response during Klebsiella induced pneumosepsis. We demonstrate that while myeloid MyD88 contributes significantly to host defense, endothelial cell MyD88 has no role herein.
Endothelial cells are resident cells implicated in sepsis pathogenesis and the induction of organ injury [34]. Earlier investigations examined the contribution of TLR and NFκB signaling within the vascular endothelium to the host response during experimental sepsis. Inhibition of endothelial NFκB signaling by overexpression of a degradation-resistant form of the NF-κB inhibitor I-κBα under the control of the endothelial cell specific VE-cadherin-5 promoter attenuated tissue inflammation and organ injury during endotoxemia and abdominal sepsis [16]–[19]. In addition, these mice displayed strongly reduced coagulation activation upon administration of endotoxin [19]. Endothelial cell specific NFκB inhibition did not influence the clearance of Listeria monocytogenesis, Streptococcus pneumoniae or Salmonella enterica after intravenous infection [16]–[19]. However, transgenic Tie2 driven expression of TLR4 in Tlr4−/− mice, resulting in mice with TLR4 expression restricted to endothelial cells, was sufficient for adequate bacterial clearance after intraperitoneal infection with Escherichia coli [20]. We used mice with Tie2 driven expression of Cre recombinase to delete hematopoietic and endothelial MyD88 in Myd88fl/fl mice and observed a strongly impaired host defense as reflected by very high bacterial loads and increased mortality. Previous studies support Tie2 expression in hematopoietic cells and the lack of specificity for endothelial cells [23], [35]. Similarly, the VE-cadherin-5 promoter is reported to drive Cre recombinase gene expression not only in endothelial cells but also in a subset of hematopoietic cells [36]. As such, the Cre-lox system seems less suitable to specifically study the function of genes in endothelial cells. Therefore, to generate mice with endothelial cell specific MyD88 deficiency, we reconstituted Tie2-Myd88−/− mice with BM of control mice and confirmed functional recovery of their hematopoietic cells with regard to responsiveness to Klebsiella. These mice were indistinguishable from control mice with regard to antibacterial defense, inflammation and distant organ injury, strongly suggesting that endothelial cell MyD88 does not play an important role in the host response during Klebsiella induced pneumosepsis. Although this “negative” finding may seem to contrast with previous studies on the role of endothelial cells in severe infection [16]–[20], our approach clearly differs from these earlier investigations, both with regard to the target of genetic manipulation (deletion of MyD88 versus inhibition of NFκB [16]–[19] and endothelial cell TLR4 expression on an otherwise TLR4 deficient background [20]) and the sepsis model used (pneumonia versus abdominal or intravenous infection [16]–[20]). Importantly, mice with TLR4 exclusively on endothelial cells were unable to recruit neutrophils into the lungs upon intratracheal LPS administration [20] and, similarly, studies in TLR4 BM chimeras have indicated that neutrophil influx after airway exposure to LPS occurs by mechanisms that do not rely on TLR4 expression by radioresistant (including endothelial) cells [37], which is completely consistent with our present data. Of note, findings in TLR4 BM chimeras have suggested that neutrophil accumulation in lungs upon intravenous LPS challenge does largely dependent on TLR4 in radioresistant cells [38], indicating that the role of cell-specific TLR signaling in neutrophil recruitment likely depends on the route by which the bacterial stimulus is administered.
LysM-Cre mediated deletion of the floxed Myd88 allele resulted in MyD88 deficiency especially in macrophages and neutrophils, and to a lesser extent monocytes [22], [39]. Clearly, these myeloid cell MyD88 deficient mice showed a strongly compromised host defense after infection with Klebsiella, as reflected by enhanced mortality, increased bacterial numbers at the primary site of infection and an impaired early neutrophil influx and cytokine/chemokine release in the airways. Thus, MyD88 expressed by alveolar macrophages and neutrophils is essential for initiation of an adequate early innate immune response in the lung after infection with Klebsiella via the airways and the absence thereof results in uncontrolled bacterial growth and death. The phenotype of LysM-Myd88−/− mice was very similar to the previously documented phenotype of Myd88−/− mice during Klebsiella pneumonia [3], [11], [12], underlining the importance of myeloid cell MyD88 during respiratory tract infection. The Klebsiella strain used here cannot be killed by macrophages or neutrophils in vitro, illustrating its high virulence and precluding analysis of a possible direct role of MyD88 in killing. Previous studies have reported a role for MyD88 in killing of commensal and attenuated pathogenic Gram-negative bacteria [40], but not in killing of Listeria by macrophages [41].
While innate immunity is important for antibacterial defense, it can also cause harm by hyperinflammation induced organ injury [42], [43]. Deficiency of MyD88 has been shown to be protective in polymicrobial sepsis, in which especially liver injury was found to be associated with MyD88 dependent signaling [43], [44]. A recent study demonstrated that mice with selective expression of MyD88 in myeloid cells displayed enhanced hepatocellular injury during abdominal sepsis induced by cecal ligation and puncture [45]. Here we found no evidence for a role of either myeloid or endothelial cell MyD88 in hepatocellular damage during pneumonia derived sepsis caused by K. pneumoniae. Hence, although MyD88 may contribute to organ injury during sepsis, its role likely depends on the type and primary source of the infection.
Using MyD88 BM chimeras, we recently reported a role for both hematopoietic and parenchymal MyD88 in host defense in this model [12]. Since we could not demonstrate a role for endothelial cell MyD88 in the present investigation, MyD88 expressed in the respiratory epithelium may be involved. Indeed, lung epithelial cells have been implicated in host defense during respiratory tract infection [15]. The importance of MyD88 dependent signaling in lung epithelial cells was recently elegantly demonstrated in a model of Pseudomonas pneumonia in epithelial specific MyD88 knock-in mice [46], [47]. Selective expression of MyD88 in the airway epithelium was sufficient for neutrophil recruitment to the site of infection and bacterial clearance [47]. In addition, transgenic overexpression of IκB-α in alveolar and bronchial epithelium in mice resulted in a reduced neutrophil influx into BAL fluid upon intrapulmonary delivery of LPS [48], [49] and an increased growth of the gram-positive pathogen Streptococcus pneumoniae upon intratracheal infection [50]. Studies using mice in which Myd88 is deleted specifically in respiratory epithelium are warranted to establish the role of epithelial MyD88 in host defense against Klebsiella pneumonia derived sepsis. However, our first preliminary results with mice generated from intercrossings of Myd88fl/fl mice and mice with Cre recombinase controlled by the surfactant protein C promoter [51], resulting in mice with a targeted deletion of Myd88 in distal airway epithelium, suggest that epithelial cell MyD88 does not contribute to protective immunity during Klebsiella pneumonia. Therefore, our earlier data using MyD88 BM chimeras [12] may have been confounded by incomplete replacement of recipient (MyD88 sufficient) hematopoietic cells.
LysM-Myd88−/− mice showed a strongly impaired neutrophil influx into the bronchoalveolar space 6 hours after infection with Klebsiella, together with markedly reduced local concentrations of neutrophil attracting mediators such as TNF-α, CXCL1 and CXCL2. Notably, global MyD88 deficiency similarly results in an early impairment of neutrophil chemoattractant release and neutrophil migration into the airways in mouse models of pneumonia caused by a variety of bacterial and viral species [11], [52]–[57], as well as during sterile lung inflammation [58], [59]. These data suggest that hematopoietic and global MyD88 deficiency impair host defense during pneumonia by a largely similar mechanism that involves an inability to produce a chemotactic gradient that would normally attract neutrophils to the site of the infection. Notably, MyD88 deficient mice showed extensive lung inflammation, including high E-selectin levels, at 24 hours after infection, suggesting that these late responses can be induced by Klebsiella via MyD88-independent mechanisms (e.g., via the TRIF pathway) in the presence of the (by then) very high bacterial loads. Similarly, global Myd88−/− mice were previously reported to show profound lung inflammation during late stage bacterial pneumonia in the presence of high bacterial loads [54], [56], [60]. E-selectin, while implicated in the rolling of neutrophils along the vascular endothelium [61], does not seem to play a role in neutrophil recruitment to the lungs elicited by bacterial stimuli [62], [63].
In conclusion, to our knowledge, we here report for the first time on the role of MyD88 in myeloid and endothelial cells in severe bacterial infection, using a clinically relevant model of gram-negative pneumonia derived sepsis characterized by gradual growth of bacteria at the primary site of infection followed by dissemination, tissue injury and death. While myeloid MyD88 was crucial for protective immunity, endothelial MyD88 played no role herein. Our results suggest that myeloid MyD88 deficiency results in enhanced lethality during Klebsiella pneumonia by a mechanism that involves a strongly attenuated early inflammatory response at the primary site of infection and as a consequence thereof uncontrolled bacterial growth. These data provide new insights in the pathophysiology of gram-negative sepsis and may be helpful for the development of therapeutics aimed at specific cell types.
Materials and Methods
Ethics statement
Experiments were carried out in accordance with the Dutch Experiment on Animals Act and approved by the Animal Care and Use Committee of the University of Amsterdam (Permit number: DIX 100121, sub-protocols DIX102300 and DIX101613).
Animals
Homozygous Myd88fl/fl mice [21] were crossed with LysM-Cre [22] or Tie2-Cre mice [23], both obtained from the Jackson Laboratory (Bar Harbor, Maine), to generate myeloid (LysM-Myd88−/−) and myeloid plus endothelial cell (Tie2-Myd88−/) specific MyD88 deficient mice. Myd88fl/fl Cre negative littermates were used as controls. All mice were backrossed at least 8 times to a C57Bl/6 background and age - and sex matched when used in experiments.
Harvest of primary cells for genetic and functional characterization of LysM-Myd88−/− and Tie2-Myd88−/− mice
Peritoneal lavage was performed with 5 ml sterile PBS under isoflurane anesthesia and lavage fluid was collected in PBS containing a final concentration of 10% FBS, 1% antibiotics (penicillin - streptomycin - amphotericin B (Gibco, Paisley, United Kingdom); heart puncture was performed and blood was collected in EDTA or heparin containing tubes; BAL was performed with 10 ml PBS in portions to obtain alveolar macrophages and spleens were harvested. For whole blood stimulation, 100 µl of heparinized blood was pipetted in a 96 wells U-bottom cell culture plate (Greiner bio-one, Alphen a/d Rijn, Netherlands). Spleens were crushed through a 40 µm mesh and after lysis of erythrocytes with an ammoniumchloride containing lysis buffer, splenocytes were seeded in RPMI complete (containing 10% FBS, 1% antibiotics, 10 mM L-glutamine, Gibco) at a density of 500.000 cells per well in 96 wells U-bottom culture plate (Greiner bio-one). Peritoneal and alveolar macrophages were seeded in flat bottom 96 wells cell culture plates (Greiner Bio-one) at a density of approximately 50.000 and 30.000 respectively per well in RPMI complete and left to adhere overnight. Cells were stimulated for 20 hours with the indicated concentrations of heat-killed K. pneumoniae or LPS derived from Klebsiella pneumoniae (Sigma) diluted in RPMI complete medium in a final volume of 200 microliter.
Whole blood leukocyte genomic DNA was isolated from fresh EDTA blood and primary cells using the Nucleospin Blood Kit (Machery Nagel, Düren, Germany) and in addition, from FACS purified monocytes, neutrophils and lymphocytes. For this, erythrocyte lysis of EDTA blood with ammoniumchloride containing lysis buffer was performed and cells were stained for cell surface molecules using FITC-conjugated anti-mouse Ly6-C &Ly6-G (Gr-1), PE-conjugated anti-mouse CD11b (BD Biosciences) and biotinylated anti-mouse CD115 (eBioscience), secondary staining was performed with streptavidin-APC (BD Biosciences). Monocytes were identified as Gr-1dim/CD-115+ and neutrophils as Gr-1high/Cd115− within the fraction of CD11b+ cells, the fraction of Cd11b− cells with a low Side Scatter and Forward Scatter pattern were identified as lymphoid cells [64].
Real-time PCR
Total RNA was reverse transcribed using oligo (dT) primer and Moloney murine leukemia virus reverse transcriptase (Invitrogen, Breda, The Netherlands).
We quantified the residual amount of the “floxed” region of MyD88 in LysM-Myd88−/− and Tie2-Myd88−/− mice in blood and particular cell types using the primer sequences 5′-ACGCCGGAACTTTTCGAT-3′ (forward); 5′-TTTTCTCAATTAGCTCGCTGG-3′ relative to the unaffected Socs-3 gene with primer sequences 5′ - ACCTTTCTTATCCGCGACAG - 3′ (forward) and 5′ - TGCACCAGCTTGAGTACACAG-3′ (reverse) in a SybrGreen reaction on an LightCycler system (LC480, Roche Applied Science, Mannheim, Germany). The amount of remaining “floxed” MyD88 region in LysM-MyD88−/− and Tie2-MyD88−/− mice was calculated using the 2-deltaCt (ΔΔCt) method using the amount of genomic DNA from Myd88fl/fl mice for the no-deletion control [21]. The deletion efficiency was calculated as (1 - residual Myd88fl) ×100.
Induction of pneumonia and sampling of organs
Pneumonia was induced by intranasal inoculation with ∼6×103 colony forming units (CFU) of K. pneumoniae serotype 2 (ATCC 43816; American Type Culture Collection, Manassas, VA) and survival was monitored or in separate experiments mice were euthanized after 6 or 24 hours of infection when organs were harvested and processed exactly as described [12], [32].
Measurements of inflammatory proteins and clinical chemistry
Lung (and cell supernatant) levels of IL-1β, TNF-α, IL-6, IL-10, CXCL-1 and CXCL-2 were measured by ELISA (R&D Systems, Minneapolis, MN). Plasma levels of TNF-α, IL-6, and IL-10 were measured by using a cytometric bead array multiplex assay (BD Biosciences). MPO was measured by ELISA from HyCult Biotechnology (Uden, the Netherlands).Lactate dehydrogenase (LDH) and aspartate aminotransferase (AST were measured using kits from Sigma and a Hittachi analyzer (Boehringer Mannheim).
Histopathology
Histologic examination of lungs was performed exactly as described [32]. For granulocyte immunohistochemic stainings lung tissue slides were deparaffinized and rehydrated. Endogenous peroxidase activity was quenched by a solution of 0.3% H2O2 (Merck). Slides were then digested by a solution of pepsin 0.025% (Sigma, St. Louis, MO, USA) in 0.1 M HCl. After being rinsed, the sections were incubated in Ultra V Block (Thermo Scientific, Fremont, CA) and then exposed to a FITC-labeled anti-mouse Ly6-G and Ly6-C monoclonal antibody (BD Pharmingen, San Diego, CA). After washes, slides were incubated with a rabbit anti-FITC antibody (Nuclilab, Ede, The Netherlands) followed by further incubation with Brightvision poly-horseradish peroxidase anti Rabbit IgG (Immunologic, Duiven, The Netherlands), rinsed again and developed using Bright DAB (Immunologic, Duiven, the Netherlands). The sections were counterstained with methyl green and mounted in Pertex mounting medium (Histolab, Gothenburg, Sweden). The Ly-6G and Ly-6C+ percentage of total lung surface was determined with imageJ software (Rasband, W.S., ImageJ, U. S. National Institutes of Health, Bethesda, Maryland, USA, http://rsb.info.nih.gov/ij/, 1997–2011).
Bone marrow transplantation
BM transplantation was done as described previously [12]. Three groups were generated: Tie2-Myd88−/− (recipient) +control BM (donor), Tie2-Myd88−/−+Tie2-Myd88−/− BM and control+control BM mice. Myd88fl/fl mice and BM were used as control.
Statistical analysis
Data are expressed as box-and-whisker diagrams depicting the smallest observation, lower quartile, median, upper quartile, and largest observation (in vivo experiments) or as means ± standard error of the mean (tables, cell stimulation experiments); Comparison of these data was done by Mann Whitney U test. Differences in the proportion of positive cultures were analyzed by Fisher's exact test. Survival curves are depicted as Kaplan-Meier plots and compared using log-rank test. These analyses were done using GraphPad Prism (San Diego, CA). P<0.05 was considered statistically significant.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. World Health Organisation (2012) WHO causes of death 2008, Global health Observatory, World Health Organisation.
2. KollefMH, ShorrA, TabakYP, GuptaV, LiuLZ, et al. (2005) Epidemiology and outcomes of health-care-associated pneumonia: results from a large US database of culture-positive pneumonia. Chest 128 : 3854–3862.
3. WelteT, TorresA, NathwaniD (2012) Clinical and economic burden of community-acquired pneumonia among adults in Europe. Thorax 67 : 71–79 thx.2009.129502 [pii];10.1136/thx.2009.129502 [doi]
4. ZaharJR, TimsitJF, Garrouste-Org, FrancaisA, VesinA, et al. (2011) Outcomes in severe sepsis and patients with septic shock: pathogen species and infection sites are not associated with mortality. Crit Care Med 39 : 1886–1895 10.1097/CCM.0b013e31821b827c [doi]
5. CoqueTM, BaqueroF, CantonR (2008) Increasing prevalence of ESBL-producing Enterobacteriaceae in Europe. Euro Surveill 13 19044.
6. GiamarellouH (2005) Multidrug resistance in Gram-negative bacteria that produce extended-spectrum beta-lactamases (ESBLs). Clin Microbiol Infect 11 Suppl 4 : 1–16.
7. SchwaberMJ, CarmeliY (2007) Mortality and delay in effective therapy associated with extended-spectrum beta-lactamase production in Enterobacteriaceae bacteraemia: a systematic review and meta-analysis. J Antimicrob Chemother 60 : 913–920 dkm318 [pii];10.1093/jac/dkm318 [doi]
8. BeutlerBA (2009) TLRs and innate immunity. Blood 113 : 1399–1407 blood-2008-07-019307 [pii];10.1182/blood-2008-07-019307 [doi]
9. KawaiT, AkiraS (2011) Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 34 : 637–650 S1074-7613(11)00190-7 [pii];10.1016/j.immuni.2011.05.006 [doi]
10. AdachiO, KawaiT, TakedaK, MatsumotoM, TsutsuiH, et al. (1998) Targeted disruption of the MyD88 gene results in loss of IL-1 - and IL-18-mediated function. Immunity 9 : 143–150 S1074-7613(00)80596-8 [pii].
11. CaiS, BatraS, ShenL, WakamatsuN, JeyaseelanS (2009) Both TRIF - and MyD88-dependent signaling contribute to host defense against pulmonary Klebsiella infection. J Immunol 183 : 6629–6638.
12. van LieshoutMH, BlokDC, WielandCW, de VosAF, van 't VeerC, et al. (2012) Differential roles of MyD88 and TRIF in hematopoietic and resident cells during murine gram-negative pneumonia. J Infect Dis 206 : 1415–1423 jis505 [pii];10.1093/infdis/jis505 [doi]
13. KawaiT, AkiraS (2010) The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol 11 : 373–384 ni.1863 [pii];10.1038/ni.1863 [doi]
14. OpitzB, van LaakV, EitelJ, SuttorpN (2010) Innate immune recognition in infectious and noninfectious diseases of the lung. Am J Respir Crit Care Med 181 : 1294–1309 200909-1427SO [pii];10.1164/rccm.200909-1427SO [doi]
15. ParkerD, PrinceA (2011) Innate immunity in the respiratory epithelium. Am J Respir Cell Mol Biol 45 : 189–201 2011-0011RT [pii];10.1165/rcmb.2011-0011RT [doi]
16. DingJ, SongD, YeX, LiuSF (2009) A pivotal role of endothelial-specific NF-kappaB signaling in the pathogenesis of septic shock and septic vascular dysfunction. J Immunol 183 : 4031–4038.
17. XuH, YeX, SteinbergH, LiuSF (2010) Selective blockade of endothelial NF-kappaB pathway differentially affects systemic inflammation and multiple organ dysfunction and injury in septic mice. J Pathol 220 : 490–498.
18. YeX, DingJ, ZhouX, ChenG, LiuSF (2008) Divergent roles of endothelial NF-kappaB in multiple organ injury and bacterial clearance in mouse models of sepsis. J Exp Med 205 : 1303–1315.
19. SongD, YeX, XuH, LiuSF (2009) Activation of endothelial intrinsic NF-{kappa}B pathway impairs protein C anticoagulation mechanism and promotes coagulation in endotoxemic mice. Blood 114 : 2521–2529 blood-2009-02-205914 [pii];10.1182/blood-2009-02-205914 [doi]
20. AndoneguiG, ZhouH, BullardD, KellyMM, MullalySC, et al. (2009) Mice that exclusively express TLR4 on endothelial cells can efficiently clear a lethal systemic Gram-negative bacterial infection. J Clin Invest 119 : 1921–1930.
21. HouB, ReizisB, DeFrancoAL (2008) Toll-like receptors activate innate and adaptive immunity by using dendritic cell-intrinsic and -extrinsic mechanisms. Immunity 29 : 272–282 S1074-7613(08)00315-4 [pii];10.1016/j.immuni.2008.05.016 [doi]
22. ClausenBE, BurkhardtC, ReithW, RenkawitzR, ForsterI (1999) Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res 8 : 265–277.
23. KoniPA, JoshiSK, TemannUA, OlsonD, BurklyL, et al. (2001) Conditional vascular cell adhesion molecule 1 deletion in mice: impaired lymphocyte migration to bone marrow. J Exp Med 193 : 741–754.
24. EddensT, KollsJK (2012) Host defenses against bacterial lower respiratory tract infection. Curr Opin Immunol 24 : 424–430 S0952-7915(12)00102-1 [pii];10.1016/j.coi.2012.07.005 [doi]
25. TakabayshiK, CorrM, HayashiT, RedeckeV, BeckL, et al. (2006) Induction of a homeostatic circuit in lung tissue by microbial compounds. Immunity 24 : 475–487 S1074-7613(06)00173-7 [pii];10.1016/j.immuni.2006.02.008 [doi]
26. MebiusRE, KraalG (2005) Structure and function of the spleen. Nat Rev Immunol 5 : 606–616 nri1669 [pii];10.1038/nri1669 [doi]
27. AchouitiA, de VosAF, de BeerR, FlorquinS, van 't Veer, et al. (2013) Limited Role of the Receptor for Advanced Glycation End Products during Streptococcus pneumoniae Bacteremia. J Innate Immun 5 : 603–612 000348739 [pii];10.1159/000348739 [doi]
28. RenckensR, RoelofsJJ, BontaPI, FlorquinS, de VriesCJ, et al. (2007) Plasminogen activator inhibitor type 1 is protective during severe Gram-negative pneumonia. Blood 109 : 1593–1601.
29. BhanU, LukacsNW, OsterholzerJJ, NewsteadMW, ZengX, et al. (2007) TLR9 is required for protective innate immunity in Gram-negative bacterial pneumonia: role of dendritic cells. J Immunol 179 : 3937–3946.
30. BrangerJ, KnappS, WeijerS, LeemansJC, PaterJM, et al. (2004) Role of Toll-like receptor 4 in gram-positive and gram-negative pneumonia in mice. Infect Immun 72 : 788–794.
31. SchurrJR, YoungE, ByrneP, SteeleC, ShellitoJE, et al. (2005) Central role of toll-like receptor 4 signaling and host defense in experimental pneumonia caused by Gram-negative bacteria. Infect Immun 73 : 532–545.
32. WielandCW, van LieshoutMH, HoogendijkAJ, van der PollT (2011) Host defence during Klebsiella pneumonia relies on haematopoietic-expressed Toll-like receptors 4 and 2. Eur Respir J 37 : 848–857 09031936.00076510 [pii];10.1183/09031936.00076510 [doi]
33. Duran-StruuckR, DyskoRC (2009) Principles of bone marrow transplantation (BMT): providing optimal veterinary and husbandry care to irradiated mice in BMT studies. J Am Assoc Lab Anim Sci 48 : 11–22.
34. SchoutenM, WiersingaWJ, LeviM, van der PollT (2008) Inflammation, endothelium, and coagulation in sepsis. J Leukoc Biol 83 : 536–545 jlb.0607373 [pii];10.1189/jlb.0607373 [doi]
35. TangY, HarringtonA, YangX, FrieselRE, LiawL (2010) The contribution of the Tie2+ lineage to primitive and definitive hematopoietic cells. Genesis 48 : 563–567 10.1002/dvg.20654 [doi]
36. AlvaJA, ZoveinAC, MonvoisinA, MurphyT, SalazarA, et al. (2006) VE-Cadherin-Cre-recombinase transgenic mouse: a tool for lineage analysis and gene deletion in endothelial cells. Dev Dyn 235 : 759–767 10.1002/dvdy.20643 [doi]
37. HollingsworthJW, ChenBJ, BrassDM, BermanK, GunnMD, et al. (2005) The critical role of hematopoietic cells in lipopolysaccharide-induced airway inflammation. Am J Respir Crit Care Med 171 : 806–813 200407-953OC [pii];10.1164/rccm.200407-953OC [doi]
38. AndoneguiG, BonderCS, GreenF, MullalySC, ZbytnuikL, et al. (2003) Endothelium-derived Toll-like receptor-4 is the key molecule in LPS-induced neutrophil sequestration into lungs. J Clin Invest 111 : 1011–1020 10.1172/JCI16510 [doi]
39. KongX, ThimmulappaR, CraciunF, HarveyC, SinghA, et al. (2011) Enhancing Nrf2 pathway by disruption of Keap1 in myeloid leukocytes protects against sepsis. Am J Respir Crit Care Med 184 : 928–938 201102-0271OC [pii];10.1164/rccm.201102-0271OC [doi]
40. LarouxFS, RomeroX, WetzlerL, EngelP, TerhorstC (2005) Cutting edge: MyD88 controls phagocyte NADPH oxidase function and killing of gram-negative bacteria. J Immunol 175 : 5596–5600 175/9/5596 [pii].
41. EdelsonBT, UnanueER (2002) MyD88-dependent but Toll-like receptor 2-independent innate immunity to Listeria: no role for either in macrophage listericidal activity. J Immunol 169 : 3869–3875.
42. MancusoG, MidiriA, BeninatiC, BiondoC, GalboR, et al. (2004) Dual role of TLR2 and myeloid differentiation factor 88 in a mouse model of invasive group B streptococcal disease. J Immunol 172 : 6324–6329.
43. WeighardtH, Kaiser-MooreS, VabulasRM, KirschningCJ, WagnerH, et al. (2002) Cutting edge: myeloid differentiation factor 88 deficiency improves resistance against sepsis caused by polymicrobial infection. J Immunol 169 : 2823–2827.
44. WeighardtH, MagesJ, JusekG, Kaiser-MooreS, LangR, et al. (2006) Organ-specific role of MyD88 for gene regulation during polymicrobial peritonitis. Infect Immun 74 : 3618–3632 74/6/3618 [pii];10.1128/IAI.01681-05 [doi]
45. GaisP, ReimD, JusekG, Rossmann-BloeckT, WeighardtH, et al. (2012) Cutting edge: Divergent cell-specific functions of MyD88 for inflammatory responses and organ injury in septic peritonitis. J Immunol 188 : 5833–5837 jimmunol.1200038 [pii];10.4049/jimmunol.1200038 [doi]
46. HajjarAM, HarowiczH, LiggittHD, FinkPJ, WilsonCB, et al. (2005) An essential role for non-bone marrow-derived cells in control of Pseudomonas aeruginosa pneumonia. Am J Respir Cell Mol Biol 33 : 470–475 2005-0199OC [pii];10.1165/rcmb.2005-0199OC [doi]
47. MijaresLA, WangdiT, SokolC, HomerR, MedzhitovR, et al. (2011) Airway Epithelial MyD88 Restores Control of Pseudomonas aeruginosa Murine Infection via an IL-1-Dependent Pathway. J Immunol 186 : 7080–7088 jimmunol.1003687 [pii];10.4049/jimmunol.1003687 [doi]
48. PoynterME, ClootsR, vanWT, ButnorKJ, VacekP, et al. (2004) NF-kappa B activation in airways modulates allergic inflammation but not hyperresponsiveness. J Immunol 173 : 7003–7009 173/11/7003 [pii].
49. SkerrettSJ, LiggittHD, HajjarAM, ErnstRK, MillerSI, et al. (2004) Respiratory epithelial cells regulate lung inflammation in response to inhaled endotoxin. Am J Physiol Lung Cell Mol Physiol 287: L143–L152 10.1152/ajplung.00030.2004 [doi];00030.2004 [pii]
50. QuintonLJ, JonesMR, SimmsBT, KoganMS, RobsonBE, et al. (2007) Functions and regulation of NF-kappaB RelA during pneumococcal pneumonia. J Immunol 178 : 1896–1903 178/3/1896 [pii].
51. KorfhagenTR, GlasserSW, WertSE, BrunoMD, DaughertyCC, et al. (1990) Cis-acting sequences from a human surfactant protein gene confer pulmonary-specific gene expression in transgenic mice. Proc Natl Acad Sci U S A 87 : 6122–6126.
52. AlbigerB, SandgrenA, KatsuragiH, Meyer-HoffertU, BeiterK, et al. (2005) Myeloid differentiation factor 88-dependent signalling controls bacterial growth during colonization and systemic pneumococcal disease in mice. Cell Microbiol 7 : 1603–1615 CMI578 [pii];10.1111/j.1462-5822.2005.00578.x [doi]
53. BradleyLM, DouglassMF, ChatterjeeD, AkiraS, BaatenBJ (2012) Matrix metalloprotease 9 mediates neutrophil migration into the airways in response to influenza virus-induced toll-like receptor signaling. PLoS Pathog 8: e1002641 10.1371/journal.ppat.1002641 [doi];PPATHOGENS-D-11-01153 [pii]
54. HawnTR, SmithKD, AderemA, SkerrettSJ (2006) Myeloid differentiation primary response gene (88) - and toll-like receptor 2-deficient mice are susceptible to infection with aerosolized Legionella pneumophila. J Infect Dis 193 : 1693–1702 JID35426 [pii];10.1086/504525 [doi]
55. NaikiY, MichelsenKS, SchroderNW, AlsabehR, SlepenkinA, et al. (2005) MyD88 is pivotal for the early inflammatory response and subsequent bacterial clearance and survival in a mouse model of Chlamydia pneumoniae pneumonia. J Biol Chem 280 : 29242–29249 M503225200 [pii];10.1074/jbc.M503225200 [doi]
56. SkerrettSJ, LiggittHD, HajjarAM, WilsonCB (2004) Cutting edge: myeloid differentiation factor 88 is essential for pulmonary host defense against Pseudomonas aeruginosa but not Staphylococcus aureus. J Immunol 172 : 3377–3381.
57. WiersingaWJ, WielandCW, RoelofsJJ, van der PollT (2008) MyD88 dependent signaling contributes to protective host defense against Burkholderia pseudomallei. PLoS ONE 3: e3494.
58. DozE, NoulinN, BoichotE, GuenonI, FickL, et al. (2008) Cigarette smoke-induced pulmonary inflammation is TLR4/MyD88 and IL-1R1/MyD88 signaling dependent. J Immunol 180 : 1169–1178 180/2/1169 [pii].
59. GasseP, MaryC, GuenonI, NoulinN, CharronS, et al. (2007) IL-1R1/MyD88 signaling and the inflammasome are essential in pulmonary inflammation and fibrosis in mice. J Clin Invest 117 : 3786–3799 10.1172/JCI32285 [doi]
60. WielandCW, FlorquinS, MarisNA, HoebeK, BeutlerB, et al. (2005) The MyD88-dependent, but not the MyD88-independent, pathway of TLR4 signaling is important in clearing nontypeable haemophilus influenzae from the mouse lung. J Immunol 175 : 6042–6049 175/9/6042 [pii].
61. KolaczkowskaE, KubesP (2013) Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol 13 : 159–175 nri3399 [pii];10.1038/nri3399 [doi]
62. BurnsJA, IssekutzTB, YagitaH, IssekutzAC (2001) The alpha 4 beta 1 (very late antigen (VLA)-4, CD49d/CD29) and alpha 5 beta 1 (VLA-5, CD49e/CD29) integrins mediate beta 2 (CD11/CD18) integrin-independent neutrophil recruitment to endotoxin-induced lung inflammation. J Immunol 166 : 4644–4649.
63. MizgerdJP, MeekBB, KutkoskiGJ, BullardDC, BeaudetAL, et al. (1996) Selectins and neutrophil traffic: margination and Streptococcus pneumoniae-induced emigration in murine lungs. J Exp Med 184 : 639–645.
64. de BruinAM, LibregtsSF, ValkhofM, BoonL, TouwIP, et al. (2012) IFNgamma induces monopoiesis and inhibits neutrophil development during inflammation. Blood 119 : 1543–1554 blood-2011-07-367706 [pii];10.1182/blood-2011-07-367706 [doi]
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2014 Číslo 9
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Měli bychom postcovidový syndrom léčit antidepresivy?
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- The Secreted Peptide PIP1 Amplifies Immunity through Receptor-Like Kinase 7
- Symbionts Commonly Provide Broad Spectrum Resistance to Viruses in Insects: A Comparative Analysis of Strains
- MIF Contributes to Associated Immunopathogenicity Development
- The Ins and Outs of Rust Haustoria