
RNF26 Temporally Regulates Virus-Triggered Type I Interferon Induction by Two Distinct Mechanisms
Virus infection induces the host cells to produce type I interferons, which are secreted proteins important for the host to clear viruses. Previously, we identified a cellular protein called MITA, which is essential for virus-triggered induction of interferons. In this study, we found an enzyme called RNF26 could covalently modify MITA with one type of polypeptide, called polyubiquitin. This modification caused increased stability of MITA after viral infection. RNF26 also caused disability of IRF3, another important component required for virus-triggered interferon induction. Thus, RNF26 could temporally regulate virus-triggered interferon induction by two distinct mechanisms. This discovery helps to understand how the antiviral response is delicately regulated.
Published in the journal:
. PLoS Pathog 10(9): e32767. doi:10.1371/journal.ppat.1004358
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004358
Summary
Virus infection induces the host cells to produce type I interferons, which are secreted proteins important for the host to clear viruses. Previously, we identified a cellular protein called MITA, which is essential for virus-triggered induction of interferons. In this study, we found an enzyme called RNF26 could covalently modify MITA with one type of polypeptide, called polyubiquitin. This modification caused increased stability of MITA after viral infection. RNF26 also caused disability of IRF3, another important component required for virus-triggered interferon induction. Thus, RNF26 could temporally regulate virus-triggered interferon induction by two distinct mechanisms. This discovery helps to understand how the antiviral response is delicately regulated.
Introduction
Host pattern-recognition receptors (PRRs) detect nucleic acid from invading viruses or necrotic cells and trigger a series of signaling events that lead to the induction of type I interferons (IFNs), which plays a central role in autoimmune diseases as well as protective immune responses against viruses, respectively [1], [2]. Much progress has been made to characterize viral nucleic acid-triggered signaling pathways that result in transcriptional activation of type I IFN genes. A family of DExD/H box RNA helicases consisting of retinoic acid inducible gene I (RIG-I), melanoma differentiation-associated gene 5 (MDA5), and LGP2 are RNA sensors and recruit the adaptors VISA (also called MAVS, IPS-1 and Cardif) [3]–[6] and MITA (also known as STING, MPYS and ERIS) [7]–[10]to activate the transcription factors NF-κB and interferon regulatory factor (IRF) 3 or IRF7, leading to transcriptional induction of the genes encoding type I IFNs and other antiviral effectors [2]. A number of DNA sensors have been identified, including DAI, IFI16, DDX41 and MRE11 which may function in a ligand - and/or cell-type-specific manner [11]–[14]. In addition, cyclic GMP-AMP synthase (cGAS) has been recently characterized as a viral DNA sensor in almost all types of cells [15]. These sensors depend exclusively on the adaptor MITA to activate NF-κB and IRF3/7 and lead to subsequent induction of type I IFNs [16].
MITA is localized to the endoplasmic reticulum (ER), mitochondria-associated membrane and mitochondria [7], [8], [10], [17]. Upon viral infection, MITA translocates to intracellular membrane-containing compartments to form punctate aggregates and acts as a scaffold protein to facilitate the phosphorylation of IRF3 and STAT6 by the kinases TBK1 and IKKε [18]–[21]. Recent studies also suggest that MITA is a direct sensor that recognizes cyclic dinucleotides such as c-di-AMP, c-di-GMP and cGAMP generated from self and viral DNA infection [22]–[24].
It has been demonstrated that MITA undergoes various post-translational modifications and such modifications are key to the activity and stability of MITA [7], [21], [25]–[27]. MITA is phosphorylated at Ser358 by TBK1 which is critical for phosphorylation and activation of IRF3 [7], while UNC-51-like kinase (ULK1) phosphorylates MITA at Ser366 which impairs MITA-IRF3 interaction and subsequent activation of IRF3 [21]. In addition, RNF5 catalyzes K48-linked polyubiquitination of MITA and targets MITA for degradation [25], whereas TRIM56 and TRIM32 promote K63-linked polyubiquitination of MITA and positively regulates virus-triggered type I IFN induction [26], [27]. Whether and how additional proteins mediate other types of modifications of MITA is unknown.
In the present study, we identified an E3 ubiquitin ligase, RING finger protein 26 (RNF26) that targeted MITA for K11-linked polyubiquitination upon viral infection. MITA with K11-linked polyubiquitin chains remained as reservoir of MITA and was protected from RNF5-mediated K48-linked polyubiquitination and degradation. However, overexpression of RNF26 indirectly induced degradation of IRF3 through an autophagy pathway. As a result, knockdown of RNF26 promoted degradation of MITA after viral infection and prevented degradation of IRF3. Virus-triggered phosphorylation of IRF3 and induction of IFN-β were inhibited at the early time points and potentiated at late time points in the absence of RNF26, respectively. Our findings suggest that RNF26 temporally regulates virus-triggered induction of type I IFNs by two distinct mechanisms.
Results
RNF26 is an E3 ubiquitin ligase for MITA
Because post-translational modifications of MITA are critical for mediating viral nucleic acid-triggered type I IFN induction, we assumed there are additional proteins that interact with and target MITA for various modifications and thereby regulate innate antiviral signaling. We attempted to unambiguously identify E3 ubiquitin ligases that regulate MITA ubiquitination and function [27]. This effort led to the identification of RNF26 which could promote polyubiquitination of MITA [27]. Analysis of the NCBI EST profile database indicates that RNF26 is ubiquitously expressed in most examined cells and tissues. In overexpression experiments, RNF26 dose-dependently promoted polyubiquitination of MITA (Figure 1A). In contrast, the enzymatic inactive mutants RNF26(C395S), RNF26(C399S) or RNF26(C401S) failed to mediate polyubiquitination of MITA (Figure 1B). Results from in vitro ubiquitination assays demonstrated that RNF26 catalyzed polyubiquitination of MITA in vitro, which depended on the enzymatic activity of RNF26 (Figure 1C–D). These data suggest that RNF26 is an E3 ubiquitin ligase targeting MITA for polyubiquitination.

RNF26 interacts with MITA
Since RNF26 caused polyubiquitination of MITA, we examined whether RNF26 interacted with MITA. Transient transfection and coimmunoprecipitation experiments indicated that RNF26 was associated with MITA in 293 cells (Figure 2A). In untransfected THP-1 cells, endogenous RNF26 constitutively interacted with MITA. This interaction was enhanced at 6 hours and decreased at 12–24 hours after SeV or HSV-1 infection (Figure 2B). It has been reported that MITA is localized to the ER, mitochondria-associated membrane and mitochondria [7], [8], [10], [17]. The subcellular localization of RNF26 was examined. Fluorescent confocal microscopy and cellular fractionation analysis suggested that RNF26 was mainly localized to the ER and a minor fraction of RNF26 was found to be colocalized with the mitochondria marker (Figure 2C and D). In contrast, RNF26 was not colocalized with the Golgi marker (Figure 2C). RNF26 was colocalized with MITA mostly at the ER and formed punctate dots with MITA after SeV or HSV-1 infection (Figure 2E). In this context, MITA was reported to translocate to microsomes to form punctate aggregates after viral infection or transfection of poly(dA:dT) or interferon stimulatory DNA (ISD) [17], [18], [20], [21]. These data suggest that RNF26 is physically associated with MITA.

Previously, we have demonstrated that the N-terminal transmembrane domains of MITA are critical for its subcellular localization and function [7]. Interestingly, sequence analysis indicated that RNF26 contained five transmembrane domains at the N-terminus and a RING domain at the C-terminus (Figure S1A). Domain mapping experiments indicated that the association of RNF26 and MITA depended on their respective transmembrane domains (Figure S1A and B). These data collectively suggest that RNF26 is physically associated with MITA and the interaction is dependent on their transmembrane domains.
RNF26 promotes polyubiquitination of MITA at lysine 150 (K150)
To map the residue(s) of MITA that are targeted by RNF26, we examined RNF26-mediated polyubiquitination of MITA mutants in which all the lysine residues of MITA were individually substituted by arginine. As shown in Figure 3A, mutation of K150 to arginine impaired its polyubiquitination by RNF26. In addition, RNF26 could not induce polyubiquitination of MITA(K150R) in in vitro ubiquitination assays (Figure 3B). These data suggest that RNF26 targets K150 of MITA for polyubiquitination.
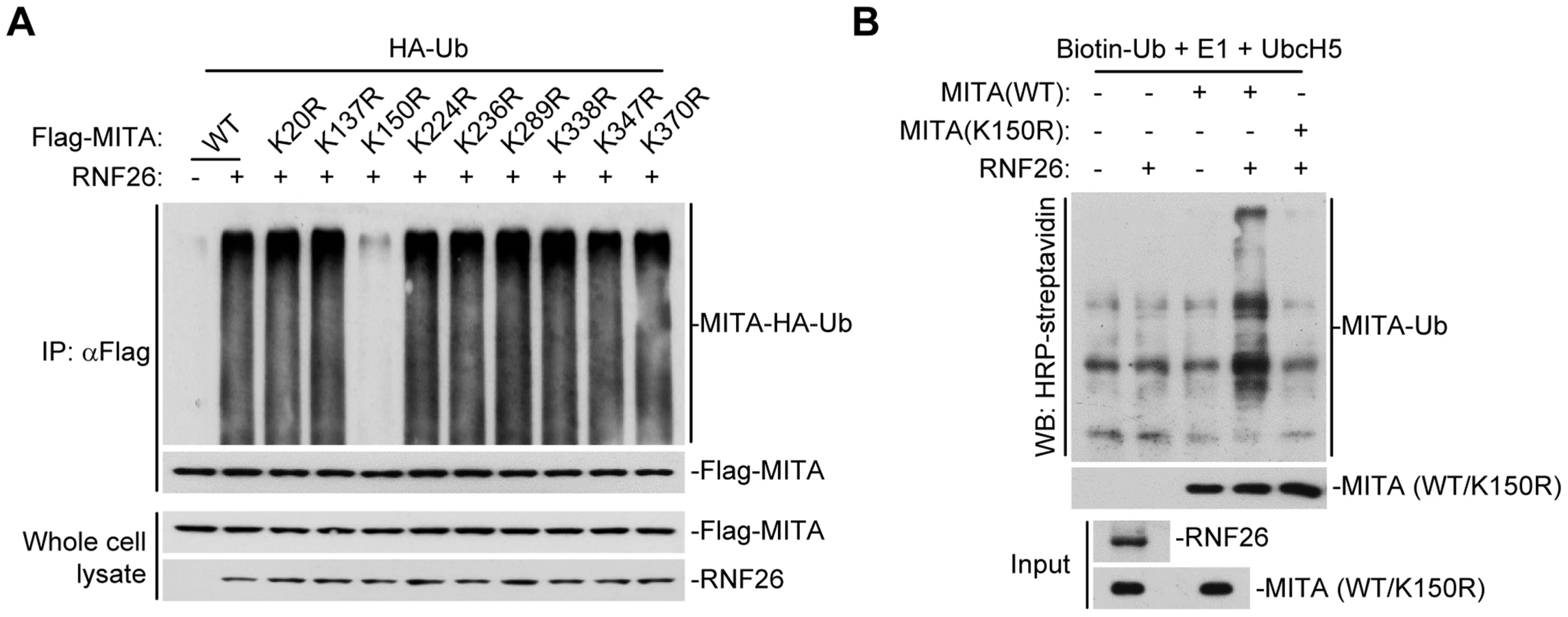
RNF26 catalyzes K11-linked polyubiquitination of MITA
Having demonstrated that RNF26 is a MITA-interacting E3 ubiquitin ligase targeting MITA for polyubiquitination, the types of polyubiquitin chains conjugated to MITA by RNF26 were next examined. Ubiquitin mutants were constructed in which all but one lysine residues were simultaneously mutated to arginines (K-O) or all seven lysine residues were individually mutated to arginine (K-R). These mutants were examined for their abilities to be conjugated to MITA by RNF26. As shown in Figure 4A, the ubiquitin mutant retaining only lysine 11 (Ub-K11O) but not other lysine residues could be conjugated to MITA. Conversely, mutation of lysine 11 to arginine (Ub-K11R) markedly reduced its ability to be conjugated to MITA by RNF26 (Figure 4B). These data indicate that RNF26 induces K11-linked polyubiquitination of MITA.

Since an antibody against K11-linkage of polyubiquitin chains was not available to us, THP-1 cells stably transfected with Flag-Ub-K11O (THP-1-Flag-Ub-K11O) together with a control or RNF26-RNAi plasmids (Figure 4C and D) were established to examine viral infection-induced K11-linked polyubiquitination of MITA and the role of RNF26 in this ubiquitination. As shown in Figure 4E, MITA was modified with K11-linked polyubiquitin chains at 6–12 hours after SeV or HSV-1 infection, and such polyubiquitination was substantially impaired by knockdown of RNF26. Similar results were obtained in 293 cells stably transfected with HA-Ub-K11O plasmids (293-HA-Ub-K11O) together with a control or RNF26-RNAi plasmids (Figure 4F). In similar experiments, K11-linked polyubiquitination of VISA after viral infection was not affected by RNF26 knockdown (Figure S2). These data suggest that RNF26 mediates K11-linked polyubiquitination of MITA upon viral infection.
RNF26 protects MITA from K48-linked polyubiquitination and degradation
Because multiple E3s have been reported to target polyubiquitin chains of distinct linkages (K48-linked or K63-linked) to K150 of MITA [25]–[27], we speculated that RNF26-mediated K11-linked polyubiquitination at K150 might compete with K48 - or K63-linked polyubiquitination at the same lysine residue. Thus, virus-induced K48 - or K63-linked polyubiquitination of MITA in THP-1-RNF26-RNAi and control cells was examined. As shown in Figure 5A, SeV or HSV-1 infection triggered K48 - and K63-linked polyubiquitination of MITA. Knockdown of RNF26 potentiated K48 - but not K63-linked polyubiquitination of MITA after viral infection (Figure 5A). RNF5 has been shown to induce K48-linked polyubiquitination of MITA at K150 [25]. Interestingly, we found that knockdown of RNF5 greatly enhanced SeV-induced K11-linked polyubiquitination of MITA in 293-HA-Ub-K11O cells (Figure 5B). In addition, RNF26-mediated K11-linked polyubiquitination of MITA was diminished by co-expression of RNF5, and RNF5-mediated K48-linked polyubiquitination and degradation of MITA was partially inhibited by co-expression of RNF26 (Figure 5C). Consistent with these observations, SeV - or HSV-1-induced degradation of MITA was accelerated by knockdown of RNF26 (Figure 5D). These data suggest that RNF26 catalyzes K11-linked polyubiquitination of MITA which protects MITA from RNF5-mediated K48-linked polyubiquitination.

RNF26 modulates virus-triggered induction of type I IFNs
Since RNF26-mediated K11-linked polyubiquitination of MITA protected its degradation after viral infection, whether RNF26 regulates virus-triggered induction of type I IFNs was determined. Luciferase reporter assays suggested that knockdown of RNF26 inhibited SeV-induced activation of NF-κB, ISRE and IFN-β promoter but had no marked effects on TNFα - or IL-1β-induced activation of NF-κB (Figure 6A and S3A). The expression of IFNB1 gene was impaired at 6 hours after SeV infection by knockdown of RNF26 in 293 cells (Figure S3B). However, we unexpectedly observed that SeV-induced expression of IFNB1 was potentiated in RNF26 knockdown cells compared to control cells at 12–24 hours after SeV infection (Figure S3B). In a mouse macrophage cell line Raw264.7 cells, SeV - or HSV-1-induced expression of Ifnb1 gene was also inhibited and potentiated at the early and late time points by knockdown of murine Rnf26, respectively (Figure S3C and D). It should be noted that the degrees of inhibition or potentiation of expression of IFNB1 or Ifnb1 genes were correlated with the knockdown efficiencies of the RNF26-RNAi or Rnf26-RNAi plasmids, indicating that RNF26 is involved in regulating RNA and DNA virus-triggered induction of type I IFNs.

To further confirm this notion, RNAi-transduced stable THP-1 cell lines were established and the expression of IFNB1 in these cells was examined after stimulation with SeV or HSV-1. As shown in Figure 6B and C, the mRNA and protein levels of IFN-β were impaired at 6 hours and potentiated at 12–24 hours in THP-1-RNF26-RNAi compared to control cells after viral infection, respectively. Similar results were obtained with various RNA (VSV and EMCV) or DNA (ECTV) viruses (Figure 6D) as well as virus-induced expression of CCL5 (Figure S3E). Interestingly, SeV - or HSV-1-induced expression of the proinflammatory cytokines TNFα and IL-6 was inhibited by RNF26 knockdown at all examined time points after viral infection (Figure 6E, 6F, S4A and B). In similar experiments, RNF26 knockdown did not affect IFN-β-induced expression of ISG15 or ISG56 genes (Figure S4C). Consistent with the gene induction experiments, we found that although SeV - or HSV-1-induced phosphorylation of TBK1 and IκBα was inhibited in THP-1-RNF26-RNAi cells, phosphorylation of IRF3 was inhibited at the early time points and increased at the late time points in THP-1-RNF26-RNAi stable cells compared to that in control cells after viral infection (Figure 6G). Thus, we conclude that RNF26 temporally regulates virus-triggered induction of type I IFNs by two distinct mechanisms.
RNF26 regulates IRF3 stability
When examining virus-triggered activation of IRF3 in RNF26 knockdown and control cells, we observed that the level of IRF3 protein was raised in THP-1-RNF26-RNAi compared to control cells (Figure 6G and 7A), and the mRNA levels of IRF3 were comparable in these cells (Figure 7A), indicating that RNF26 regulates IRF3 at the protein level. In support of this notion, we found that overexpression of RNF26 but not RNF26(C395S) promoted degradation of IRF3 and inhibited SeV-induced activation of the IFN-β promoter (Figure 7B and C). However, we failed to observe an interaction between IRF3 and RNF26 (Figure 2A) or polyubiquitination of IRF3 by RNF26 (Figure 7D). Interestingly, RNF26-mediated degradation of IRF3 was blocked by the autophagy inhibitor 3-methyladenine (3-MA) but not the lysosome inhibitor ammonium chloride (NH4Cl) or the proteasome inhibitor MG132 (Figure 7E). To further determine whether autophagic degradation system is responsible for RNF26-mediated degradation of IRF3, we determined the effect of knockdown of ATG12, an important component of the autophagic degradation system [28], on RNF26-mediated IRF3 degradation. The results indicated that RNF26-mediated IRF3 degradation was markedly inhibited in ATG12 knockdown cells (Figure 7F). Thus, RNF26 might temporally regulate virus-triggered type I IFN induction through regulating K11-linked polyubiquitination of MITA at the early phase and autophagy-dependent degradation of IRF3 at late phase, respectively.

RNF26 functions as a negative regulator in cellular antiviral response
Since RNF26 regulates virus-induced expression of IFN-β and other downstream genes, we examined its roles in cellular antiviral response. We found that knockdown of RNF26 inhibited VSV and HSV-1 replication in plaque assays (Figure S5), suggesting that RNF26 functions as a negative regulator in cellular antiviral response.
Discussion
MITA plays critical roles in virus-triggered type I IFN induction and innate antiviral immune response [7], [8], [10], [15], [17], [22]–[24]. Various studies have shown that post-translational modifications of MITA are essential for its function [7], [21], [25]–[27]. In this study, we demonstrated that RNF26 but not the enzymatic inactive mutants induced polyubiquitination of MITA. RNF26 mediated K11-linked polyubiquitination of MITA and modulated expression of type I IFN triggered by viral infection. RNF26 was localized mainly at the ER and constitutively interacted with MITA through their respective transmembrane domains. Viral infection potentiated this association and induced RNF26 to form punctate dots with MITA. Our studies suggest that RNF26 is a MITA-interacting E3 ubiquitin ligase which targets MITA for K11-linked polyubiquitination.
Polyubiquitination of MITA catalyzed by RNF26 was mapped to K150, which is also targeted by RNF5 for K48-linked polyubiquitination and by TRIM56 or TRIM32 for K63-linked polyubiquitination [25]–[27]. These observations prompted us to hypothesize that polyubiquitin chains of distinct linkages might compete with each other at the same residue of MITA. Interestingly, virus-triggered K48 - but not K63-linked polyubiquitination of MITA was enhanced by knockdown of RNF26. Previously, it has been demonstrated that TRIM32 targets not only K150 but also K20, 224 and 236, whereas RNF5 targets only K150 of MITA [27]. This is consistent with our observations that RNF26 impaired K48 - but not K63-linked polyubiquitination of MITA. RNF26-mediated K11-linked polyubiquitination of MITA protected it from RNF5-mediated K48-linked polyubiquitination and degradation. Consistently, degradation of MITA was accelerated in RNF26 knockdown cells compared to control cells after viral infection. These results suggest that RNF26-meidated K11-linked polyubiquitination competes with RNF5-mediated K48-linked polyubiquitination of MITA at K150. These results are consistent with our observations that knockdown of RNF26 inhibited induction of type I IFNs at early phase of viral infection. The functions of K11-linked polyubiquitination are not well understood so far [29]–[33]. To our knowledge, our study represents the first report on the function of K11-linked polyubiquitination in virus-triggered signaling and innate antiviral response.
Unexpected, although knockdown of RNF26 inhibited virus-triggered induction of type I IFNs at the early phase of viral infection, it had opposite effect at the late phase of viral infection. This led us to hypothesize that RNF26 temporally regulates virus-triggered type I IFN induction by distinct mechanisms. In this context, we observed that overexpression of RNF26 promoted degradation of IRF3, whereas knockdown of RNF26 increased the level of IRF3. Interestingly, RNF26-mediated degradation of IRF3 was blocked by the autophagy inhibitor 3-MA but not the lysosome inhibitor NH4Cl or the proteasome inhibitor MG132. In addition, RNF26-induced degradation of IRF3 was markedly inhibited by knockdown of ATG12, an essential component in the autophagic degradation pathway. These results indicate that RNF26 indirectly regulates stability of IRF3 protein in an autophagy-dependent manner. The exact mechanism on how RNF26 mediates autophagy-dependent degradation of IRF3 is currently unknown. Based on our findings, we come to a working model on how RNF26 temporally regulates virus-triggered type I IFN induction (Figure 8). At the early phase of infection, RNF26 mediates K11-linked polyubiquitination of MITA, which protects it from K48-linked polyubiquitination and degradation to facilitate the fast induction of type I IFN genes. In addition to its early phase function through K11-linked polyubiquitination of MITA, RNF26 constitutively down-regulates IRF3 level by autophagic degradation. This may contribute to the termination of type I IFN induction at the late phase of viral infection. Interestingly, since IRF3 activation is not required for virus-triggered induction of the proinflammatory cytokines such as TNFα and IL-6, RNF26 positively regulates virus-triggered induction of the proinflammatory cytokines in a constitutive but not temporal manner. Therefore, our findings not only reveal the mechanisms on how RNF26 temporally modulate virus-triggered type I IFN induction, but also provide an explanation on how virus-triggered induction of type I IFNs and proinflammatory cytokines can be distinctly regulated.

Materials and Methods
Reagents and antibodies
Recombinant IFN-β, TNFα and IL-1β (R&D Systems); mouse monoclonal antibodies against FLAG (Sigma), HA (Covance), β-actin (Sigma), AIF, KDEL (Santa Cruz Biotechnology), β-tubulin (Invitrogen), HSV-1 ICP27, ATG12 (Abcam), TBK1, p-TBK1 and p-IκBα (CST); rabbit polyclonal antibodies against ubiquitin, IRF3, p-IRF3 (Santa Cruz Biotechnology), polyubiquitin K48-linkage and K63-linkage (Millipore) were purchased from the indicated manufacturers. SeV, HSV-1, VSV, EMCV, ECTV, anti-SeV, anti-RIG-I, anti-VISA, anti-MITA anti-RNF5 and anti-IκBα sera were previously described [7], [25], [27], [34]. Rabbit anti-RNF26 was raised against recombinant human RNF26 (241–433).
Constructs
IFN-β, ISRE and NF-κB luciferase reporter plasmids, mammalian expression plasmids for HA-, Flag-, or GFP-tagged MITA and its mutants, ubiquitin, RIG-I, VISA, TRAF3, TRAF6, TBK1, IRF3, IRF7, Sec61-β, GALT, RNF5 and β-actin were previously described [7], [25], [27], [34], [35]. Mammalian expression plasmids for human Flag-, GFP-, Cherry - or CFP-tagged RNF26 and its mutants were constructed by standard molecular biology techniques.
Transfection and reporter assays
The cells were seeded and transfected the following day by standard calcium phosphate precipitation method or by FuGENE (Roche). Empty control plasmids were added to ensure that each transfection receives the same amount of total DNA. To normalize for transfection efficiency, pRL-TK Renilla luciferase reporter plasmids were added to each transfection. Luciferase assays were performed using a dual-specific luciferase assay kit (Promega). Firefly luciferase activities were normalized on the basis of Renilla luciferase activities.
Immunoprecipitation under denatured conditions and ubiquitination assay
The cells were lysed in lysis buffer containing 1% SDS and denatured by heating for 5 minutes. The supernatants were diluted with regular lysis buffer until the concentration of SDS was decreased to 0.1%. The diluted supernatants were subjected for immunoprecipitation as described [7], [25], [27], and the immunoprecipitates and whole cell lysates were analyzed by immunoblots with the indicated antibodies.
In vitro ubiquitination assay
The tested proteins were expressed with a TNT Quick-coupled Transcription/Translation Systems kit (Promega) following instructions of the manufacturer. Ubiquitination was analyzed with an ubiquitination kit (Enzo Life Science) following protocols recommended by the manufacturer.
Fluorescent confocal microscopy
The transfected cells were incubated with the ER-Tracker Blue/White or Mito-Tracker Red (Invitrogen) following protocols recommended by the manufacturer. The cells were then fixed with 4% paraformaldehyde for 10 minutes and observed with an Olympus confocal microscope under a ×60 oil objective.
Subcellular fractionation
The cells were washed with PBS and lysed by douncing 30 times in 2 mL of homogenization buffer (10 mM Tris-HCl pH 7.4, 2 mM MgCl2, 10 mM KCl and 250 mM sucrose) on wet ice. The homogenate was centrifuged at 500×g for 10 minutes twice. The supernatant (S5) was centrifuged at 5,000×g for 30 minutes to precipitate crude mitochondria (P5K). The supernatant (S5K) was further centrifuged at 50,000×g for 60 minutes to generate S50K and P50K.
RNAi experiments
Double-stranded oligonucleotides corresponding to the target sequences were cloned into the pSuper.Retro RNAi plasmids (oligoengine Inc.). The following sequences were targeted for human RNF26 cDNA: #1 : 5′-GAGCAAGAGGAGCGGAAGA-3′; #2 : 5′-GAGAGGATGTCATGCGGCT-3′. The following sequences were targeted for murine Rnf26 cDNA: #1 : 5′-GAGCGGAAGAAGTGTGTTA-3′; #2 : 5′-GATCAACAGTCTAGTCAAC-3′.
Quantitative real-time PCR
Total RNA was isolated from cells using TRIzol reagent (Takara) and subjected to real-time PCR analysis to measure expression of mRNA. Gene-specific primer sequences were as described [27] or as follow: Human RNF26 (Forward: 5′-CAGGACCATCAGAGTGACACCT-3′; Reverse: 5′-GCAACACTGTCTTGCTCTGGTC-3′). Murine Rnf26 (Forward: 5′-TGGCTGCTTTCCTCGCTCACAT-3′; Reverse: 5′-GCAACACCAATCCAGTGAGATGG-3′). Human IRF3 (Forward: 5′-TCTGCCCTCAACCGCAAAGAAG-3′; Reverse: 5′-TACTGCCTCCACCATTGGTGTC-3′). Murine Irf3 (Forward: 5′-CGGAAAGAAGTGTTGCGGTTAGC-3′; Reverse: 5′-CAGGCTGCTTTTGCCATTGGTG-3′).
ELISA
The supernatants of cell culture medium were analyzed with a human IFN-β (PBL), human TNFα or human IL-6 (Boster) ELISA kit following protocols recommended by the manufacturers.
Flag-tagged ubiquitin stable THP-1 cells
The 293 cells were transfected with two packaging plasmids (pGAG-Pol and pVSV-G) together with pMSCV-GFP-Flag-Ub-K11O retroviral plasmids by calcium phosphate precipitation. Twenty-four hours after transfection, cells were incubated with new medium without antibiotics for another twenty-four hours. The recombinant virus-containing medium was filtered with 0.22 µm filter (Millex) and then added into cultured THP-1 cells in the presence of polybrene (4 µg/mL). The infected cells were cultured for at least seven days and sorted by a flow cell sorter before additional experiments were performed.
HA-tagged ubiquitin stable 293 cells
The 293 cells were transfected with pRK7-Neo-HA-Ub-K11O plasmid. Twenty-four hours after transfection, cells were selected with G418 (0.8 µg/mL). Single cell colonies were picked and identified by immunoblot analysis.
RNAi-transduced stable 293, THP-1 and Raw264.7 cells
The 293 cells were transfected with two packaging plasmids (pGAG-Pol and pVSV-G) together with a control, RNF26-RNAi or Rnf26-RNAi retroviral plasmids respectively by calcium phosphate precipitation. Twenty-four hours after transfection, cells were incubated with new medium without antibiotics for another twenty-four hours. The recombinant virus-containing medium was filtered with 0.22 µm filter (Millex) and then added into cultured 293, Raw264.7 or THP-1 cells in the presence of polybrene (4 µg/mL). The infected cells were selected with puromycin (1 µg/mL for 293 and Raw264.7 cells or 0.5 µg/mL for THP-1 cells) for at least seven days before additional experiments were performed.
Accession numbers
UniProtKB/Swiss-Prot accession numbers (parentheses) are indicated for proteins mentioned in text: RNF26 (Q9BY78), MITA (Q86WV6), RNF5 (Q99942), VISA (Q7Z434).
Supporting Information
Zdroje
1. KawasakiT, KawaiT, AkiraS (2011) Recognition of nucleic acids by pattern-recognition receptors and its relevance in autoimmunity. Immunol Rev 243 : 61–73.
2. TakeuchiO, AkiraS (2010) Pattern recognition receptors and inflammation. Cell 140 : 805–820.
3. XuLG, WangYY, HanKJ, LiLY, ZhaiZ, et al. (2005) VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol Cell 19 : 727–740.
4. SethRB, SunL, EaCK, ChenZJ (2005) Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 122 : 669–682.
5. MeylanE, CurranJ, HofmannK, MoradpourD, BinderM, et al. (2005) Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 437 : 1167–1172.
6. KawaiT, TakahashiK, SatoS, CobanC, KumarH, et al. (2005) IPS-1, an adaptor triggering RIG-I - and Mda5-mediated type I interferon induction. Nat Immunol 6 : 981–988.
7. ZhongB, YangY, LiS, WangYY, LiY, et al. (2008) The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity 29 : 538–550.
8. IshikawaH, BarberGN (2008) STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455 : 674–678.
9. JinL, WatermanPM, JonscherKR, ShortCM, ReisdorphNA, et al. (2008) MPYS, a novel membrane tetraspanner, is associated with major histocompatibility complex class II and mediates transduction of apoptotic signals. Mol Cell Biol 28 : 5014–5026.
10. SunW, LiY, ChenL, ChenH, YouF, et al. (2009) ERIS, an endoplasmic reticulum IFN stimulator, activates innate immune signaling through dimerization. Proc Natl Acad Sci U S A 106 : 8653–8658.
11. TakaokaA, WangZ, ChoiMK, YanaiH, NegishiH, et al. (2007) DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature 448 : 501–505.
12. UnterholznerL, KeatingSE, BaranM, HoranKA, JensenSB, et al. (2010) IFI16 is an innate immune sensor for intracellular DNA. Nat Immunol 11 : 997–1004.
13. ZhangZ, YuanB, BaoM, LuN, KimT, et al. (2011) The helicase DDX41 senses intracellular DNA mediated by the adaptor STING in dendritic cells. Nat Immunol 12 : 959–965.
14. KondoT, KobayashiJ, SaitohT, MaruyamaK, IshiiKJ, et al. (2013) DNA damage sensor MRE11 recognizes cytosolic double-stranded DNA and induces type I interferon by regulating STING trafficking. Proc Natl Acad Sci U S A 110 : 2969–2974.
15. SunL, WuJ, DuF, ChenX, ChenZJ (2013) Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339 : 786–791.
16. PaludanSR, BowieAG (2013) Immune sensing of DNA. Immunity 38 : 870–880.
17. IshikawaH, MaZ, BarberGN (2009) STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 461 : 788–792.
18. SaitohT, FujitaN, HayashiT, TakaharaK, SatohT, et al. (2009) Atg9a controls dsDNA-driven dynamic translocation of STING and the innate immune response. Proc Natl Acad Sci U S A 106 : 20842–20846.
19. ChenH, SunH, YouF, SunW, ZhouX, et al. (2011) Activation of STAT6 by STING is critical for antiviral innate immunity. Cell 147 : 436–446.
20. TanakaY, ChenZJ (2012) STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Sci Signal 5: ra20.
21. KonnoH, KonnoK, BarberGN (2013) Cyclic dinucleotides trigger ULK1 (ATG1) phosphorylation of STING to prevent sustained innate immune signaling. Cell 155 : 688–698.
22. JinL, HillKK, FilakH, MoganJ, KnowlesH, et al. (2011) MPYS is required for IFN response factor 3 activation and type I IFN production in the response of cultured phagocytes to bacterial second messengers cyclic-di-AMP and cyclic-di-GMP. J Immunol 187 : 2595–2601.
23. WuJ, SunL, ChenX, DuF, ShiH, et al. (2013) Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 339 : 826–830.
24. CavlarT, AblasserA, HornungV (2012) Induction of type I IFNs by intracellular DNA-sensing pathways. Immunol Cell Biol 90 : 474–482.
25. ZhongB, ZhangL, LeiC, LiY, MaoAP, et al. (2009) The ubiquitin ligase RNF5 regulates antiviral responses by mediating degradation of the adaptor protein MITA. Immunity 30 : 397–407.
26. TsuchidaT, ZouJ, SaitohT, KumarH, AbeT, et al. (2010) The ubiquitin ligase TRIM56 regulates innate immune responses to intracellular double-stranded DNA. Immunity 33 : 765–776.
27. ZhangJ, HuMM, WangYY, ShuHB (2012) TRIM32 protein modulates type I interferon induction and cellular antiviral response by targeting MITA/STING protein for K63-linked ubiquitination. J Biol Chem 287 : 28646–28655.
28. MizushimaN, SugitaH, YoshimoriT, OhsumiY (1998) A new protein conjugation system in human. The counterpart of the yeast Apg12p conjugation system essential for autophagy. J Biol Chem 273 : 33889–33892.
29. BremmA, FreundSM, KomanderD (2010) Lys11-linked ubiquitin chains adopt compact conformations and are preferentially hydrolyzed by the deubiquitinase Cezanne. Nat Struct Mol Biol 17 : 939–947.
30. RapeM (2010) Assembly of k11-linked ubiquitin chains by the anaphase-promoting complex. Subcell Biochem 54 : 107–115.
31. WuT, MerblY, HuoY, GallopJL, TzurA, et al. (2010) UBE2S drives elongation of K11-linked ubiquitin chains by the anaphase-promoting complex. Proc Natl Acad Sci U S A 107 : 1355–1360.
32. WickliffeKE, WilliamsonA, MeyerHJ, KellyA, RapeM (2011) K11-linked ubiquitin chains as novel regulators of cell division. Trends Cell Biol 21 : 656–663.
33. BudhavarapuVN, WhiteED, MahanicCS, ChenL, LinFT, et al. (2012) Regulation of E2F1 by APC/C Cdh1 via K11 linkage-specific ubiquitin chain formation. Cell Cycle 11 : 2030–2038.
34. HuMM, YangQ, ZhangJ, LiuSM, ZhangY, et al. (2014) TRIM38 inhibits TNFalpha - and IL-1beta-triggered NF-kappaB activation by mediating lysosome-dependent degradation of TAB2/3. Proc Natl Acad Sci U S A 111 : 1509–1514.
35. LiY, ChenR, ZhouQ, XuZ, LiC, et al. (2012) LSm14A is a processing body-associated sensor of viral nucleic acids that initiates cellular antiviral response in the early phase of viral infection. Proc Natl Acad Sci U S A 109 : 11770–11775.
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2014 Číslo 9
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Měli bychom postcovidový syndrom léčit antidepresivy?
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- The Secreted Peptide PIP1 Amplifies Immunity through Receptor-Like Kinase 7
- Symbionts Commonly Provide Broad Spectrum Resistance to Viruses in Insects: A Comparative Analysis of Strains
- MIF Contributes to Associated Immunopathogenicity Development
- The Ins and Outs of Rust Haustoria