
GITR Intrinsically Sustains Early Type 1 and Late Follicular Helper CD4 T Cell Accumulation to Control a Chronic Viral Infection
The natural rodent pathogen LCMV clone 13 causes a persistent viral infection in mice and has successfully predicted several immunological factors that are relevant to human chronic viral infection such as HIV. LCMV clone 13 infection is ultimately controlled by cell-mediated and humoral immune responses by day 60–90 post-infection in CD4 T cell-sufficient mice. While it has been known for several years that CD4 T cell help is critical for control of LCMV clone 13, research to date has been largely limited to the regulatory factors that contribute to late CD4 T cell dysfunction, with little knowledge of the role of T cell co-stimulatory factors in sustaining CD4 T cells to help cell-mediated and humoral immune responses. Using GITR-deficient mice, we show that the co-stimulatory molecule GITR plays a critical cell-intrinsic role in early CD4 T cell accumulation to support cytotoxic T cell responses and late LCMV-specific IgG production. The early effects of Th1 on CTL responses are IL-2-dependent. Mice lacking GITR have up to 35-fold higher viral burden relative to GITR-sufficient controls. Taken together, we demonstrate a critical cell-intrinsic role for GITR is sustaining CD4 T cell responses to control chronic LCMV infection. Thus, GITR on CD4 T cells may critically contribute to the initial viral set-point in infections such as HIV.
Published in the journal:
. PLoS Pathog 11(1): e32767. doi:10.1371/journal.ppat.1004517
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004517
Summary
The natural rodent pathogen LCMV clone 13 causes a persistent viral infection in mice and has successfully predicted several immunological factors that are relevant to human chronic viral infection such as HIV. LCMV clone 13 infection is ultimately controlled by cell-mediated and humoral immune responses by day 60–90 post-infection in CD4 T cell-sufficient mice. While it has been known for several years that CD4 T cell help is critical for control of LCMV clone 13, research to date has been largely limited to the regulatory factors that contribute to late CD4 T cell dysfunction, with little knowledge of the role of T cell co-stimulatory factors in sustaining CD4 T cells to help cell-mediated and humoral immune responses. Using GITR-deficient mice, we show that the co-stimulatory molecule GITR plays a critical cell-intrinsic role in early CD4 T cell accumulation to support cytotoxic T cell responses and late LCMV-specific IgG production. The early effects of Th1 on CTL responses are IL-2-dependent. Mice lacking GITR have up to 35-fold higher viral burden relative to GITR-sufficient controls. Taken together, we demonstrate a critical cell-intrinsic role for GITR is sustaining CD4 T cell responses to control chronic LCMV infection. Thus, GITR on CD4 T cells may critically contribute to the initial viral set-point in infections such as HIV.
Introduction
During chronic viral infections, exemplified by the clone 13 variant of lymphocytic choriomeningitis virus (LCMV cl 13), persistent antigen presentation results in the functional exhaustion of the T cell response, characterized by persistent upregulation of inhibitory molecules and a progressive loss of T cell effector functions [1]. Ultimately, LCMV cl 13 is cleared by 60–90 days p.i. due to both T - and B-cell responses. While there have been a number of studies on the role of T cell inhibitory receptors and anti-inflammatory cytokines during chronic infection [2–7], rather less has been done to study the role of co-stimulatory receptors in this context. Co-stimulatory TNFR family members are of particular interest in this regard because they are often induced upon antigen receptor signaling, leading to their co-expression with inhibitory receptors during a persistent infection [8–10]. CD4 T cell help is critical for the control of chronic infections. The removal of CD4 T cells from mice prior to infection with LCMV cl 13 [11–13], or the loss of CD4 T cells during progressive HIV infection [14] leads to increased viral burden, immune dysregulation, and functional T cell exhaustion. While CD4 cells are clearly implicated in the control of chronic viral infections, the co-stimulatory signals that contribute to CD4 T cell help remain poorly defined. Evidence to date suggests that there is significant heterogeneity in the potency and mechanisms of T cell modulation by members of the TNFR superfamily during chronic viral infection [8, 9, 15–18].
The Glucocorticoid-induced TNFR related protein (GITR) and its ligand (GITRL) are induced upon activation of a number of immune cell types [19]. GITR is expressed at low levels on resting T cells, but its expression rapidly increases upon activation. Although constitutively expressed on Foxp3+ regulatory T cells (Treg), GITR is dispensable for Treg function but can play a role in the accumulation of both regulatory and effector T cells in vivo [20, 21]. In many experimental systems, it has been difficult to determine the key cell types through which GITR mediates its effects. Controversy over the relative role of GITR on effector versus regulatory T cells persists, though these effects may be context-dependent [19, 22, 23]. GITR-deficient mice are viable, reproduce normally, and have no obvious immunological defects in the naïve state [24]. Studies using GITR-deficient mice or an agonist anti-GITR antibody have shown an immune stimulatory role for GITR in the context of viral infections, but the mechanisms underlying these phenomena are not entirely clear [19].
Here we address the role of GITR during chronic viral infection using GITR-deficient mice infected with LCMV cl 13 [25]. We find that GITR-/- mice have 2–3-fold fewer LCMV-specific CD8 T cells post-priming and throughout the infection, with concomitant increases in viral load. A previous study showed that GITR on TCR transgenic CD8 T cells contributes to the control of influenza infection [26]. Surprisingly however, during LCMV cl 13 infection, the effect of GITR was largely CD4 T cell-intrinsic and independent of Tregs. GITR-deficiency impaired the accumulation of IFNγ+IL-2+ T helper 1 (Th1) cells as well as follicular helper T cells (Tfh) and the production of LCMV-specific IgG. Mixed bone marrow chimeras and adoptive transfer studies of LCMV-specific CD4 and CD8 T cells demonstrate that the effects of GITR are CD4 T cell-intrinsic. Neutralization of IL-2 in GITR+/+ mice reduced the immune response to the level observed in GITR-/- mice, whereas IL-2 neutralization in GITR-/- mice had no effect, revealing a central role for this cytokine in GITR-dependent control of LCMV cl 13. Taken together, our studies provide evidence for a critical role for GITR in the post-priming accumulation of IL-2+ Th1 cells to help cell-mediated immunity as well as Tfh accumulation to sustain the humoral arm of the immune response to control a persistent viral infection.
Results
GITR is required for CD8 T cell accumulation and function post-priming and control of chronic LCMV infection
To evaluate the role of GITR in a chronic infection, we infected wild-type mice (GITR+/+) or mice with ablated Tnfrsf18 (GITR-/-) [24] with LCMV cl 13. At both acute and chronic time points (days eight and 45 p.i.), GITR-/- mice had significantly increased viral load (Fig. 1A). These effects were particularly striking in the kidney, where GITR-/- mice had 35-fold higher viral load than GITR+/+ mice at day eight p.i. GITR-/- mice had substantially fewer LCMV-specific CD8 T cells in peripheral blood (Fig. 1B, for gating strategy see S1 Fig.). Following initial infection and T cell priming at day five p.i., there was no difference in the number of splenic LCMV-specific CD8 T cells between GITR+/+ and GITR-/- mice (Fig. 1C). However, between days five and eight p.i., a significant impairment in the frequency and total number of splenic LCMV-specific CD8 T cells in GITR-/- mice materializes, and this effect becomes even more striking at day 45 p.i. (Fig. 1, D–F).
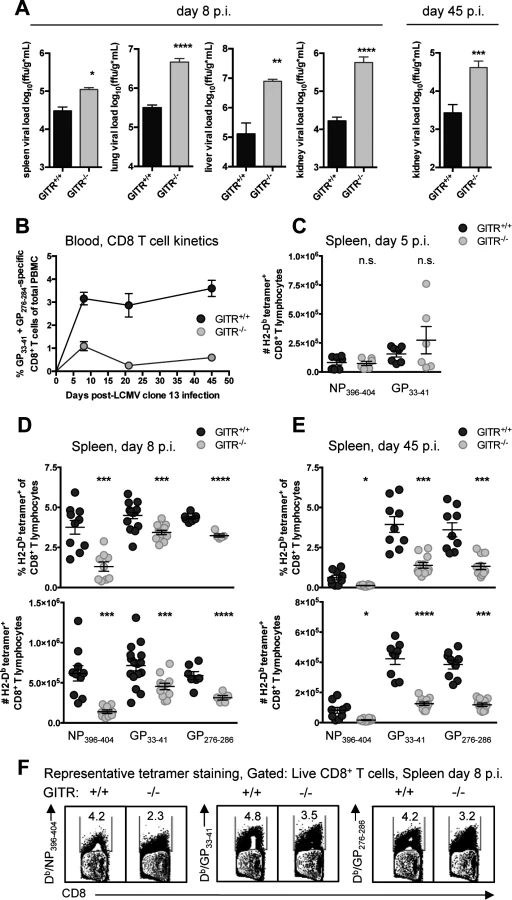
Co-signaling molecules PD-1 and Tim-3 impair CD8 T cell responses to LCMV cl 13 [4, 5]. The majority of the LCMV-specific CD8 T cells in GITR+/+ and GITR-/- mice were PD-1+ Tim-3+ after LCMV cl 13 infection, but CD8 T cells from GITR-/- mice expressed significantly higher levels of PD-1 and Tim-3 per cell at days eight and 45 p.i. (Fig. 2, A and B). CD8 T cells from GITR+/+ or GITR-/- mice produced similar amounts of IFNγ following peptide restimulation at day eight p.i., but by day 45 p.i., the GITR-/- mice had fewer IFNγ+ CD8 T cells with lower IFNγ production per cell (Fig. 2, C and D). Moreover, at all time points analyzed, fewer of the IFNγ-producing CD8 T cells from GITR-/- mice exhibited a multifunctional phenotype as defined by TNF and surface CD107a expression (a surrogate marker for cellular cytotoxic activity) (Fig. 2, E and F). These findings reveal a key role for GITR in the CD8 T cell response and control of chronic LCMV infection. We also examined the role of GITR with the acutely infecting Armstrong variant of LCMV and observed approximately two - to three-fold more GP33–41-specific CD8 T cells in GITR+/+ compared to GITR-/- at day eight p.i. (p<0.01 in two of the three experiments, total of ten mice per group), however both GITR+/+ and GITR-/- mice cleared LCMV Armstrong infection by day eight p.i.

GITR potentiates the CD8 T cell response through CD8 T cell-extrinsic effects
Previous work has shown an intrinsic role for GITR in sustaining the survival of TCR transgenic CD8 T cells during acute influenza virus infection [26]. To determine if CD8 T cell-intrinsic effects were also responsible for defects in LCMV cl 13 control in GITR-/- mice, we crossed GITR-/- with P14 mice, which have a transgenic TCR specific for the Db-restricted LCMV GP33–41 epitope [27]. We transferred 103 purified CD8 T cells from CD45.1 GITR+/+ or GITR-/- P14 littermates into CD45.2 congenic mice one day prior to LCMV cl 13 infection (Fig. 3A). P14 T cells lacking GITR showed no impairment in expansion, and in fact showed a slight increase in frequency (Fig. 3B). There was also no difference in IFNγ production or degranulation, PD-1 or Tim-3 expression between the GITR-sufficient or GITR-deficient P14 T cells (Fig. 3, C–E). Moreover, the absence of GITR only on the transferred P14 T cells had no effect on viral control at day eight p.i. (Fig. 3F). Thus, the effect of GITR in potentiating the CD8 T cell response to LCMV cl 13 is CD8 T cell-extrinsic.

GITR critically regulates early Th1 responses to LCMV
To evaluate the importance of CD4 T cells in this model, we depleted CD4 cells from GITR+/+ and GITR-/- mice prior to LCMV cl 13 infection (Fig. 4A). CD4-depleted GITR+/+ mice had fewer and less functional LCMV-specific CD8 T cells, and 100-fold higher viral load relative to non-CD4-depleted GITR+/+ mice, consistent with previous reports of CD4 depletion increasing the severity of LCMV cl 13 infection [11–13]. However, the qualitative and quantitative differences between GITR+/+ and GITR-/- CD8 T cell responses and viral control at day eight p.i. were largely lost in CD4-depleted mice (Fig. 4, B–E), suggesting that CD4 T cells are necessary for the effect of GITR on the CD8 T cell response to LCMV cl 13 as well as viral control.

We next analyzed the effect of GITR on CD4 T cell responses. GITR-/- mice had similar total numbers of CD4 T cells in the spleen following LCMV infection at day eight p.i. However, fewer of the GITR-/- CD4 T cells expressed the signature Th1 transcription factor T-bet (Fig. 4F, for gating strategy see S2 Fig.) and there was a striking three-fold reduction in the percent of CD4 T cells that were IFNγ+ or IFNγ+IL-2+ following GP61–80-restimulation at day eight p.i., a defect that was maintained out to day 45 p.i. (Fig. 4, G and H). In contrast, GITR+/+ and GITR-/- littermates had similar proportions and absolute numbers of Foxp3+ Tregs (Fig. 4F). Moreover, depletion of Tregs did not recapitulate the effects of total CD4 T cell depletion on the CD8 T cell response (S3 Fig.).
IL-2 is necessary for the effect of GITR on CD8 T cell responses
IL-2 is important for CD8 T cell accumulation and function in the context of chronic LCMV infection [28–30]. Therefore, we evaluated whether the effects of GITR on the T cell response were dependent on IL-2. Of note, we did not detect any IL-2+ CD8 T cells after ex vivo peptide stimulation at day eight p.i., consistent with previous reports [31]. As the CD8 T cell deficit in GITR-/- mice is not realized until after day five p.i., we administered 0.5mg of blocking anti-IL-2 to mice on days four and six p.i., and evaluated T cell responses at day eight p.i. IL-2 blockade reduced the LCMV-specific Th1 and CD8 T cell responses in GITR+/+ mice to the level observed in GITR-/- mice, whereas IL-2 blockade had no impact on the T cell responses in GITR-/- mice (Fig. 4, I and J). Thus, GITR is critical for Th1 responses and IL-2-dependent help for CD8 T cells.
GITR enhances follicular helper CD4 T cell responses to LCMV cl 13 and the production of LCMV-specific IgG
GITR impacts viral load both early and late during LCMV cl 13 infection (Fig. 1A). As Tfh have emerged as an important CD4 T cell subset that promotes late B cell responses to LCMV cl 13 infection [32, 33], we examined the effects of GITR on Tfh. At day eight p.i., we noted elevated levels of GITR expression on Tfh compared to non-Tfh CD4+ SMARTA cells (SMARTA mice express a transgenic TCR specific for the I-Ab-restricted LCMV GP61–80) (Fig. 5A) [34]. Evaluation of the Tfh response in peripheral blood of GITR+/+ and GITR-/- mice between day seven and 45 p.i. indicated a significantly lower frequency of Tfh in GITR-/- mice compared to GITR+/+ mice at late (days 21 and 45 p.i.) but not early times during the response to LCMV cl 13 (Fig. 5B). T follicular regulatory cells (Tfr) are Foxp3+ T cells that localize to germinal centers to regulate the response [35, 36]. The spleens of GITR+/+ and GITR-/- mice had similar proportions of Tfh and Tfr at day eight p.i., but at day 45 p.i. GITR-/- mice exhibited significantly fewer Tfh and there was a significant increase in the proportion of Tfr in GITR-/- compared to GITR+/+ mice (Fig. 5, C and D). Moreover, GITR-/- mice had significant impairment in the LCMV-specific IgG response at day 21–45 p.i. (Fig. 5E).

GITR acts intrinsically on CD4 T cells
Although CD4 T cells are required for the effect of GITR on the CD8 T cell response to LCMV, it was conceivable that GITR on another cell population could indirectly affect CD4 T cell help [19]. To evaluate a cell-intrinsic effect of GITR, we used mixed bone marrow chimeras, in which GITR+/+ and GITR-/- cells compete in the same mouse. If GITR intrinsically affects a specific cell type, then the GITR-/- cells will be at a competitive disadvantage, whereas if the effect of GITR is extrinsic then GITR+/+ and GITR-/- cells would be equally affected by the lack of GITR in another cell compartment and their ratio would be constant. To this end, we reconstituted lethally irradiated C57BL/6 mice with 1 : 1 mixture of CD45.2 GITR+/+:CD45.1 GITR-/- bone marrow cells (Fig. 6A). Although we used CD45.2 recipients, we have found that in separate control experiments using identical radiation schedules, >95% of lymphocytes were of donor origin at three months post-reconstitution. Consistent with the minimal contribution from residual T cells following lethal irradiation, we verified that the CD45.2:CD45.1 ratio in peripheral blood was 1 : 1, with similar proportions of CD4, CD8 T cells and Foxp3+ Tregs prior to infection (Fig. 6B).

At day eight p.i., there was a slight but significant effect of GITR on total CD4 cells, with fewer CD4 T cells in the GITR-/- compartment (Fig. 6C). Among the CD4 cells, there was a two-fold reduction in GITR-/- Tregs and a three-fold reduction in GITR-/- Tfh (Fig. 6, C and D). Additionally, there was a three-fold reduction in IFNγ-producing GITR-/- Th1 cells after LCMV GP61–80 restimulation, of which fewer co-produced IL-2 (Fig. 6, E and F). Taken together, these data demonstrate a key CD4 T cell-intrinsic role of GITR during LCMV cl 13 infection, with the most dramatic effects on the Th1 and Tfh responses.
At day eight p.i., there was no difference in the frequency of GITR+/+ and GITR-/- NP394–404-specific CD8 T cells, whereas the frequency of GITR-/- GP33–41-specific T cells was significantly reduced compared to the GITR+/+ population (Fig. 6G), however in contrast to the complete knockout mouse (Fig. 2), the levels of PD-1 and Tim-3 were similar between the GITR+/+ and GITR-/- compartments (Fig. 6H). The frequency of CD8 T cells with the ability to produce cytokines and degranulate after GP33–41 peptide restimulation was also decreased in GITR-/- compared to GITR+/+ (Fig. 6, I–L). These data show that under competitive conditions, GITR intrinsically affects GP33–41 but not NP396–404-specific accumulation, whereas effects of GITR on T cell inhibitory receptor levels are cell-extrinsic, and likely due to increased viral load in the complete GITR-deficient mouse.
GITR affects the accumulation but not the initial rate of division or the differentiation of LCMV-specific CD4 T cells
To distinguish between the effects of GITR on CD4 T cell accumulation versus differentiation, we transferred a 1 : 1 mixture of GITR+/+:GITR-/- TCR transgenic SMARTA cells one day prior to LCMV cl 13 infection (Fig. 7A). We used the congenic marker CD45.1 and GITR expression to identify GITR+/+ and GITR-/- SMARTA cells within the same mouse (Fig. 7B). Differences in GITR+/+ and GITR-/- SMARTA accumulation showed a trend starting at day five p.i., with significant differences by day eight p.i., with a 3.2 : 1 ratio (Fig. 7, C and D). The ratio of GITR+/+:GITR-/- CD44hi, Tfh, and Th1 SMARTA cells was uniformly 3 : 1, indicating that GITR is not skewing the differentiation of particular CD4 effector T cell sub-populations, rather it contributes to the accumulation of all CD4 T cells (Fig. 7E). While there was a lower frequency of GITR - CD4 T cells producing IFNγ after GP61–80 peptide restimulation, GITR+ and GITR - IFNγ+ SMARTA produced a similar amount of IFNγ per cell. There was also a consistent trend toward fewer GITR - IL-2+ IFNγ+ SMARTA cells relative to the GITR+ SMARTA (Fig. 7, F and G).

To determine whether GITR co-stimulation enhanced early division or post-priming accumulation of CD4 T cells, we transferred a mixture of 5×105 each of GITR+/+ and GITR-/- CFSE-labeled CD45.1 SMARTA cells into naive C57BL/6 (CD45.2) mice. CFSE dilution at days three and five p.i. was comparable between GITR- SMARTA and GITR+ SMARTA (Fig. 7H), suggesting that GITR does not influence initial division, but rather affects post-priming accumulation of CD4 T cells.
GITR induces NF-κB and mTORC1 activation in LCMV-specific CD4 T cells
The accumulation of T cells during their clonal expansion is well known to require survival signals, such as those mediated by NF-κB, as well as upregulation of protein translation and glycolysis, as mediated by mTOR [37–40]. Therefore, to investigate a possible mechanism underlying the enhanced accumulation of GITR+ SMARTA, a 1 : 1 mixture of GITR+/+ and GITR-/- CD45.1 SMARTA were transferred into C57BL/6 (CD45.2) mice and phsopho(p)-p65 NF-κB and (p)S6, a downstream mTOR target were evaluated in SMARTA cells at day three p.i. without further stimulation. Consistent with GITR-dependent NF-κB activation in CD8 T cells [26, 41–43], (p)p65 NF-κB was increased in GITR+/+ compared to GITR-/- SMARTA cells at day three p.i. and (p)S6 was also increased, consistent with GITR-dependent mTORC1 activation (Fig. 7I).
mTORC1 can be activated by Akt, a kinase that critically regulates T cell metabolism and expansion [37, 38]. Akt activation has been noted downstream of other TNFRs, such as OX40 [37, 44]. Therefore, we evaluated Akt activation following GITR cross-linking. GITR+/+ SMARTA splenocytes were activated in vitro with LCMV GP61–80 and IL-2 to induce GITR expression, then serum starved for 12 hours, followed by treatment with agonistic anti-GITR (DTA-1) or Rat IgG control. DTA-1 enhanced Akt activation as demonstrated by increased (p)Akt Thr308 at 15 and 30 minutes post-stimulation (Fig. 7J). Taken together, these studies show that GITR intrinsically affects post-priming accumulation but not early division of CD4 T cells with effects on NF-κB and the Akt-mTORC1-S6 signaling axis detected at day three p.i.
Discussion
The mechanism by which CD4 T cell responses contribute to the control of persistent infection, or how co-stimulation regulates CD4 T cell responses to infection remains incompletely defined [45]. Here we show that GITR directly augments the CD4 Th1 response to LCMV cl 13, which in turn impacts IL-2-dependent CD8 T cell expansion and viral control. The critical role of GITR on the CD4 T cells for control of LCMV cl 13 is demonstrated by the loss of GITR-mediated protection when CD4 T cells are depleted. Signaling downstream of GITR could be detected as early as day three p.i., consistent with an effect of GITR on CD4 T cell accumulation starting around day five and delayed effects on CD8 T cells appearing between days five and eight p.i. These findings are also consistent with strong induction of GITRL by day two p.i. with LCMV cl 13 [46]. These results demonstrate an important role for GITR early in the immune response that impacts post-priming accumulation of CD4 T cells to allow help for CD8 T cells. CD4 T cell-intrinsic GITR also reciprocally affects the Tfh and Tfr responses and, consequently, LCMV-specific IgG production. As Tfh and antibody responses are thought to contribute to the late control of persistent LCMV infection [32], this effect of GITR on Tfh and Tfr likely impacts the late control of the infection rather than the early control, when Tfh and anti-LCMV IgG levels are comparable between GITR+/+ and GITR-/- mice.
Previous work demonstrated a CD8 T cell-intrinsic role for GITR on survival of adoptively transferred CD8 T cells during acute respiratory infection of mice with influenza A virus, with no impact on cell division [26]. Here we saw similar effects in the competitive bone marrow chimera experiment (Fig. 6). However, the CD4-depletion studies and the P14 GITR+/+ and GITR-/- adoptive transfer studies support a CD8 T cell-extrinsic effect of GITR. LCMV cl 13 infection results in systemic prolonged inflammation, perhaps providing additional signals to compensate for the lack of GITR on CD8 T cells during the early stages of LCMV cl 13 infection. Nonetheless, the effects of GITR on CD8 T cells can be revealed under competitive conditions, but are clearly not sufficient to compensate for the requirement for GITR on the CD4 T cells to allow help for CD8 T cell and antibody responses to chronic LCMV infection.
LCMV-specific CD4 T cells can rescue exhausted CD8 T cell responses to LCMV cl 13, although the precise mechanism of this CD4 T cell helper function remains open to speculation [47]. CD25-deficient CD8 T cells have defective persistence during chronic LCMV infection [30], and IL-2 can be used therapeutically to augment the CD8 T cell response to Armstrong (acute) and cl 13 (chronic) LCMV infection [28, 29]. Here we show that IL-2 blockade in GITR+/+ mice resulted in an immune response indistinguishable from that in GITR-/- mice, in which IL-2 blockade had no effect. These findings imply that the effect of GITR on CD4 help for CD8 T cells is IL-2 dependent. Although we have not directly shown that the CD4 T cells are the source of IL-2, taken together with the CD4 depletion studies, the IL-2 blocking data are consistent with a lack of early IL-2+ Th1 accumulation as the underlying cause of the defective CD8 T cell accumulation and viral control in GITR-/- mice at day eight p.i.
GITR-deficiency did not result in a change in the proportion or number of Tregs. However, under conditions of competition, there was a two-fold reduction in the number of GITR-/- Tregs, consistent with previous findings that GITR may have a role in peripheral maintenance of Tregs [20]. When CD4 T cells were depleted, the dominant effect was a net increase in viral load, consistent with a loss of T cell help rather than the removal of a regulatory CD4 T cell population in GITR-/- mice. Nonetheless, it is possible that GITR on effector cells could render them resistant to Tregs [20]. Therefore we used DEREG mice to further assess the role of Tregs in our model (S3 Fig.). Diphtheria toxin (DT) was used to deplete Tregs from depletion of regulatory T cells (DEREG) mice, which express the human DT receptor under the Foxp3 promoter. DT was highly toxic in the LCMV cl 13 model, even in the non-DEREG mice. Viral load in this model was three to four orders of magnitude higher than in non-DT treated GITR+/+ mice, making the results difficult to interpret. Moreover, four of 26 LCMV cl 13 infected GITR+/+ mice treated with DT died, whereas all GITR-/- mice survived, likely due to reduced levels of inflammatory cytokines. Although the data from these experiments are difficult to interpret given the high level of toxicity, it is clear that Th1 as well as CD8 T cell differences between GITR+/+ and GITR-/- mice are largely retained even when >90% of Tregs were depleted. These data argue against a major role for GITR on the CD4 or CD8 effector T cells in preventing the action of Tregs in this model.
Late in LCMV cl 13 infection Tfh accumulate and provide help for antibody responses [32]. GITR-/- mice have impaired Tfh responses with a slight concomitant increase in the Tfr population [35, 36] and a modest decrease in LCMV-specific antibodies. Competitive bone marrow chimeras revealed that GITR also has cell-intrinsic effects on Tfh. GITRL is largely absent on total B cells, whereas a low level of GITRL can be detected on GL-7+ Fas+ germinal center B cells at days eight and 21 p.i. [46]. It is unlikely that the defect in LCMV-specific IgG production is due to direct effects of GITR on the B cell response, as GITR is largely dispensable for B cell development and activation [48].
Competitive adoptive transfer experiments showed that the absence of GITR on CD4 T cells resulted in a three-fold defect in total LCMV-specific CD4 T cells, Th1, and Tfh sub-populations equivalently, indicating that the Th1 deficit in GITR-/- mice is not due to altered CD4 T cell differentiation. We demonstrated that differences in GITR+/+ and GITR-/- CD4 numbers are not due to differences in early CD4 T cell proliferation, but instead due to defects in post-priming accumulation of CD4 T cells. GITR induces classical NF-κB to induce the pro-survival molecule Bcl-xL in CD8 T cells [26]. T cell accumulation also requires upregulation of glycolytic and biosynthetic pathways, such as induced by the Akt-mTORC1 pathway [38]. Here we show that GITR can directly activate Akt and induce phosphorylation of the downstream mTORC1 target S6, as well as activate the classical NF-κB pathway in CD4 T cells as early as day three p.i. Collectively, these signals allow the accumulation of IL-2+ Th1 cells, thereby allowing them to help the early CD8 T cell response, as well as to sustain the CD4 response as it progresses towards a Tfh fate.
In sum, we report here a critical CD4 T cell-intrinsic role for GITR in control of chronic LCMV infection. We showed that GITR co-stimulation activates the Akt-mTORC1-S6 signaling axis as well as classical NF-κB signaling to collectively promote CD4 T cell expansion. GITR-deficient mice have defective post-priming accumulation of IL-2+ Th1 cells, which precedes an impaired CD8 T cell response and loss of viral control. The effects of GITR on both Th1 and CD8 T cells are IL-2-dependent. We also reveal a novel role for GITR in sustaining the Tfh response and LCMV-specific IgG production. These findings define a critical role for CD4 T cell-intrinsic GITR signaling in early accumulation of CD4 T cells, help for CD8 T cells and viral control.
Materials and Methods
Reagents and antibodies
Biotinylated H-2Db/GP33–41: KAVYNFATM, H-2Db/GP276–286: SGVENPGGYCL, and H-2Db/NP396–404: FQPQNGQFI monomers were obtained from the National Institute for Allergy and Infectious Disease tetramer facility (Emory University, Atlanta, GA), and tetramerized with streptavidin APC (Molecular Probes). Anti-CD107a (clone 15A7), anti-CD8 (clone 53–6.7), anti-CXCR5 (clone 2G8), and anti-TNF (clone MP6-XT22) were purchased from BD Biosciences. Anti-phospho-Akt Thr308 (C31E5E) and anti-phospho-S6 ribosomal protein Ser235/236 (clone D57.2.2E) were purchased from Cell Signaling Technologies. Lectin PNA from Arachis hypogaea (Cat. #L21409) and viability stain (Cat. #L34955) were purchased from Molecular Probes. Anti-CD4 (clone RM4–5) and CD45.1 (clone A20) were purchased from BioLegend. FITC IgG isotype control, anti-Fas (clone 15A7), anti-CD25 (clone PC61.5), anti-LAP (clone TW7–16B4), anti-PD-1 (clone J43), anti-Tim-3 (clone RMT3–23), anti-ICOS (clone 7E.17G9), anti-CD3 (clone 1245–2C11), anti-CD45.1 (clone A20), anti-CD45.2 (clone 104), anti-CTLA-4 (clone UC10–4B9), anti-B220 (clone RA3–6B2), anti-CD16/32 (clone 93), anti-Foxp3 (clone FJK-16s), anti T-bet (clone eBio4B10), anti-IL-2 (clone JES6–5H4), anti-IFNγ (clone XMG1.2), phospho-NF-κB p65 Ser529 (clone MCFA30), Streptavidin-PE and -APC, and fixable viability dye eFluor 506 (Cat. #65–0866–14) were purchased from eBioscience. Anti-IL-2 (clone S4B6–1) was purchased from Bio X Cell (West Lebanon, NH). Un-nicked Diphtheria Toxin was purchased from List Biological Laboratories, Inc. (Campbell, CA) and was used to deplete Foxp3+ Tregs from DEREG mice [49] by administering 1μg i.p. DT per two days from day -2 to day six p.i.
Mice and LCMV
LCMV Armstrong and cl 13 (obtained from P. Ohashi, University of Toronto and M. Oldstone, Scripps Research institute) were propagated on BHK and L929 cells, respectively (kindly provided by Pamela Ohashi, Princess Margaret Cancer Centre, Toronto, Ontario). GITR+/+ and GITR-/- mice were infected intravenously with 2 × 106 ffu of LCMV Armstrong or LCMV clone 13 where indicated. GITR-/- mice were kindly provided by Dr. C. Riccardi (University of Perugia) and Dr. P. Pandolfi (Harvard Medical School). GITR-/- mice were analyzed by SNP analysis across 1500 SNPs differing in the 129 and B6 genome and found to be a minimum of ninety two per cent C57BL/6 (TCAG, University of Toronto). Effects of GITR on CD4 and CD8 T cell effector responses were similar when compared to commercial C57BL/6 mice or when we used F1 or F2 littermate controls. However, the proportion of Tregs in the mice after infection differed in commercial mice versus littermates, therefore all Treg data are reported from experiments with littermate controls. DEREG [49], now available from Jackson Laboratories, P14 [27] and SMARTA [34] mice were obtained from Dr. P. Ohashi (Princess Margaret Hospital, University of Toronto) and additionally crossed to GITR-/- CD45.1 mice and compared with their F2 littermates. All animals were housed under specific pathogen free conditions at the Terrence Donnelly Centre for Cellular and Biomolecular Research (University of Toronto). Where indicated, CD4 T cells were depleted by administering 0.5mg of purified anti-CD4 (clone GK1.5) two days before LCMV cl 13 infection, or IL-2 was blocked by administering 0.5mg anti-IL-2 (clone S4B6–1) given at days four and six p.i. All animal procedures were approved by the animal care committee of the University of Toronto (Protocol approval number: 20009988) in accordance with the Canadian Council on Animal care, a National Regulatory body through which the University of Toronto holds a certificate of good animal care (http://www.ccac.ca/en_/assessment/certification).
Intracellular cytokine staining
Splenocytes were cultured in complete medium with Brefeldin A and monensin (BD, Cat. #554715) with 1μg/mL Db-restricted peptides: NP396–404, GP33–41, or GP276–286 (Anaspec, Fremont, CA) for five hours, or 4μg/mL I-Ab-restricted GP61–80 with 20U/mL rmIL-2 (eBioscience) for five hours. Cells were then surface stained, fixed and permeabilized (BD), and stained for intracellular cytokine production.
LCMV focus forming assay
Organs were harvested and immediately placed on dry ice. Organs were later thawed, homogenized, and supernatant dilutions (range: 100–105) were used to infect an MC57 cell monolayer under a 2% methylcellulose-MEM overlay. 48 hours later, monolayers were fixed with 4% PFA, permeabilized with 1% Triton X-100, and stained with Rat anti-LCMV mAb (clone VL-4). Following secondary Goat anti-Rat-HRP, a colorimetric reaction with o-Phenylenediamine (Sigma-Aldrich) was used to quantify LCMV-infected foci.
LCMV-specific IgG quantification
LCMV-specific IgG ELISAs were performed as previously described [50], with modification to the purification of LCMV. LCMV was purified on a CsCl gradient of 1.45g/L and 1.2g/L in 10mM Tris (pH 8.1). Plates were coated with 10ng of inactivated purified LCMV cl 13 and blocked with Superblock in TBST (Thermo Fisher Scientific), followed by one-hour incubation with dilutions of plasma from infected animals. 1 : 2,000 HRP-tagged anti-mouse secondary antibody was then added and incubated for one hour. 3,3’,5, 5’-tetramethylbenzidine (Sigma Aldrich) was used for colorimetric reaction and absorbance was measured at 456nm.
Adoptive transfer-based assays and bone marrow chimeras
Purification of CD8 T cells for P14 adoptive transfers or CD4 T cells for SMARTA adoptive transfers were achieved with negative selection kits according to manufacturer’s protocols (StemCell Technologies, Vancouver, BC, Cat. 19753A and 19752, respectively). For P14 and SMARTA transfers, cells were transferred intraperitoneally one day prior to LCMV infection, and where indicated, were labeled with 10μM Carboxyfluorescein Diacetate Succinimidyl Ester (CFSE). GITR+/+: GITR-/- mixed bone marrow chimeras were generated by intravenously reconstituting lethally irradiated C57BL/6 mice with a 1 : 1 mixture of GITR-/- CD45.1: GITR+/+ CD45.2 bone marrow cells for a total of 5 × 106 cells. Following irradiation and reconstitution, mice were given 2mg/mL neomycin sulfate (Bio-Shop, Burlington, ON) for two consecutive weeks, after which mice were rested for an additional 90 days before use.
Phosphoflow assays
For in vitro studies, SMARTA splenocytes were stimulated with 4μg/mL I-Ab-restricted GP61–80 with 40U/mL recombinant murine IL-2 in complete RPMI containing 10% FCS for two days. Cells were rested overnight in serum-free Aim V medium with 1X β-Mercaptoethanol (Invitrogen). Live SMARTA cells were stimulated with 10μg/mL anti-GITR (DTA-1, hybridoma provided by S. Sakaguchi, Osaka University) or Rat IgG isotype control (Jackson) each followed by 5μg/mL Goat anti-Rat (Jackson). Samples were prepared for flow cytometry using BD Phosflow Buffer I (BD, Cat. #557870). Similar protocols were used for direct ex vivo staining of freshly isolated splenocytes at day three p.i.
Data analysis and statistics
Samples were acquired with FACS Canto, LSR II, or LSR Fortessa (BD Biosciences) with FACSDiva software. Flow cytometry data were analyzed with FlowJo v9 (Tree Star, Inc., Ashland, OR). All statistical analyses were performed using GraphPad Prism v6 (La Jolla, CA). Unpaired Student’s t-tests were used to compare two groups, with P values indicated on figures: * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Ng CT, Snell LM, Brooks DG, Oldstone MB (2013) Networking at the level of host immunity: immune cell interactions during persistent viral infections. Cell Host Microbe 13 : 652–664. doi: 10.1016/j.chom.2013.05.014 23768490
2. Wherry EJ (2011) T cell exhaustion. Nature Immunology 131 : 492–499. doi: 10.1038/ni.2035
3. Virgin HW, Wherry EJ, Ahmed R (2009) Redefining chronic viral infection. Cell 138 : 30–50. doi: 10.1016/j.cell.2009.06.036
4. Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, et al. (2006) Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 439 : 682–687. doi: 10.1038/nature04444 16382236
5. Jin HT, Anderson AC, Tan WG, West EE, Ha SJ, et al. (2010) Cooperation of Tim-3 and PD-1 in CD8 T-cell exhaustion during chronic viral infection. Proc Natl Acad Sci U S A 107 : 14733–14738. doi: 10.1073/pnas.1009731107 20679213
6. Jones RB, Ndhlovu LC, Barbour JD, Sheth PM, Jha AR, et al. (2008) Tim-3 expression defines a novel population of dysfunctional T cells with highly elevated frequencies in progressive HIV-1 infection. J Exp Med 205 : 2763–2779. doi: 10.1084/jem.20081398 19001139
7. Brooks DG, Trifilo MJ, Edelmann KH, Teyton L, McGavern DB, et al. (2006) Interleukin-10 determines viral clearance or persistence in vivo. Nat Med 12 : 1301–1309. doi: 10.1038/nm1492 17041596
8. Wang C, McPherson AJ, Jones RB, Kawamura KS, Lin GH, et al. (2012) Loss of the signaling adaptor TRAF1 causes CD8+ T cell dysregulation during human and murine chronic infection. J Exp Med 209 : 77–91. doi: 10.1084/jem.20110675 22184633
9. Boettler T, Moeckel F, Cheng Y, Heeg M, Salek-Ardakani S, et al. (2012) OX40 facilitates control of a persistent virus infection. PLoS Pathog 8: e1002913. doi: 10.1371/journal.ppat.1002913 22969431
10. Wherry EJ, Ha SJ, Kaech SM, Haining WN, Sarkar S, et al. (2007) Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity 27 : 670–684. doi: 10.1016/j.immuni.2007.09.006 17950003
11. Matloubian M, Concepcion RJ, Ahmed R (1994) CD4+ T cells are required to sustain CD8+ cytotoxic T-cell responses during chronic viral infection. J Virol 68 : 8056–8063. 7966595
12. Zajac AJ, Blattman JN, Murali-Krishna K, Sourdive DJ, Suresh M, et al. (1998) Viral immune evasion due to persistence of activated T cells without effector function. J Exp Med 188 : 2205–2213. doi: 10.1084/jem.188.12.2205 9858507
13. Battegay M, Moskophidis D, Rahemtulla A, Hengartner H, Mak TW, et al. (1994) Enhanced establishment of a virus carrier state in adult CD4+ T-cell-deficient mice. J Virol 68 : 4700–4704. 7911534
14. Moir S, Chun TW, Fauci AS (2011) Pathogenic mechanisms of HIV disease. Annu Rev Pathol 6 : 223–248. doi: 10.1146/annurev-pathol-011110-130254 21034222
15. Arens R, Tesselaar K, Baars PA, van Schijndel GM, Hendriks J, et al. (2001) Constitutive CD27/CD70 interaction induces expansion of effector-type T cells and results in IFNgamma-mediated B cell depletion. Immunity 15 : 801–812. doi: 10.1016/S1074-7613(01)00236-9 11728341
16. De Milito A, Morch C, Sonnerborg A, Chiodi F (2001) Loss of memory (CD27) B lymphocytes in HIV-1 infection. AIDS 15 : 957–964. doi: 10.1097/00002030-200105250-00003 11399977
17. Matter M, Odermatt B, Yagita H, Nuoffer JM, Ochsenbein AF (2006) Elimination of chronic viral infection by blocking CD27 signaling. J Exp Med 203 : 2145–2155. doi: 10.1084/jem.20060651 16923852
18. Tesselaar K, Arens R, van Schijndel GM, Baars PA, van der Valk MA, et al. (2003) Lethal T cell immunodeficiency induced by chronic costimulation via CD27-CD70 interactions. Nat Immunol 4 : 49–54. doi: 10.1038/ni869 12469117
19. Clouthier DL, Watts TH (2014) Cell-specific and context-dependent effects of GITR in cancer, autoimmunity, and infection. Cytokine Growth Factor Rev 25 : 91–106. doi: 10.1016/j.cytogfr.2013.12.003 24484736
20. Stephens GL, McHugh RS, Whitters MJ, Young DA, Luxenberg D, et al. (2004) Engagement of glucocorticoid-induced TNFR family-related receptor on effector T cells by its ligand mediates resistance to suppression by CD4+CD25+ T cells. J Immunol 173 : 5008–5020. doi: 10.4049/jimmunol.173.8.5008 15470044
21. van Olffen RW, Koning N, van Gisbergen KP, Wensveen FM, Hoek RM, et al. (2009) GITR triggering induces expansion of both effector and regulatory CD4+ T cells in vivo. J Immunol 182 : 7490–7500. doi: 10.4049/jimmunol.0802751 19494272
22. Shevach EM, Stephens GL (2006) The GITR-GITRL interaction: co-stimulation or contrasuppression of regulatory activity? Nat Rev Immunol 6 : 613–618. doi: 10.1038/nri1867 16868552
23. Ephrem A, Epstein AL, Stephens GL, Thornton AM, Glass D, et al. (2013) Modulation of Treg cells/T effector function by GITR signaling is context-dependent. Eur J Immunol 43 : 2421–2429. doi: 10.1002/eji.201343451 23722868
24. Ronchetti S, Nocentini G, Riccardi C, Pandolfi PP (2002) Role of GITR in activation response of T lymphocytes. Blood 100 : 350–352. doi: 10.1182/blood-2001-12-0276 12070049
25. Ahmed R, Oldstone MB (1988) Organ-specific selection of viral variants during chronic infection. J Exp Med 167 : 1719–1724. doi: 10.1084/jem.167.5.1719 3367096
26. Snell LM, McPherson AJ, Lin GH, Sakaguchi S, Pandolfi PP, et al. (2010) CD8 T cell-intrinsic GITR is required for T cell clonal expansion and mouse survival following severe influenza infection. J Immunol 185 : 7223–7234. doi: 10.4049/jimmunol.1001912 21076066
27. Pircher H, Burki K, Lang R, Hengartner H, Zinkernagel RM (1989) Tolerance induction in double specific T-cell receptor transgenic mice varies with antigen. Nature 342 : 559–561. doi: 10.1038/342559a0 2573841
28. Blattman JN, Grayson JM, Wherry EJ, Kaech SM, Smith KA, et al. (2003) Therapeutic use of IL-2 to enhance antiviral T-cell responses in vivo. Nat Med 9 : 540–547. doi: 10.1038/nm866 12692546
29. West EE, Jin HT, Rasheed AU, Penaloza-Macmaster P, Ha SJ, et al. (2013) PD-L1 blockade synergizes with IL-2 therapy in reinvigorating exhausted T cells. J Clin Invest 123 : 2604–2615. doi: 10.1172/JCI67008 23676462
30. Bachmann MF, Wolint P, Walton S, Schwarz K, Oxenius A (2007) Differential role of IL-2R signaling for CD8+ T cell responses in acute and chronic viral infections. Eur J Immunol 37 : 1502–1512. doi: 10.1002/eji.200637023 17492805
31. Wherry EJ, Blattman JN, Murali-Krishna K, van der Most R, Ahmed R (2003) Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J Virol 77 : 4911–4927. doi: 10.1128/JVI.77.8.4911-4927.2003 12663797
32. Fahey LM, Wilson EB, Elsaesser H, Fistonich CD, McGavern DB, et al. (2011) Viral persistence redirects CD4 T cell differentiation toward T follicular helper cells. J Exp Med 208 : 987–999. doi: 10.1084/jem.20101773 21536743
33. Crotty S (2011) Follicular helper CD4 T cells (TFH). Annu Rev Immunol 29 : 621–663. doi: 10.1146/annurev-immunol-031210-101400 21314428
34. Oxenius A, Bachmann MF, Zinkernagel RM, Hengartner H (1998) Virus-specific MHC-class II-restricted TCR-transgenic mice: effects on humoral and cellular immune responses after viral infection. Eur J Immunol 28 : 390–400. doi: 10.1002/(SICI)1521-4141(199801)28 : 01%3C390::AID-IMMU390%3E3.3.CO;2-F 9485218
35. Linterman MA, Pierson W, Lee SK, Kallies A, Kawamoto S, et al. (2011) Foxp3+ follicular regulatory T cells control the germinal center response. Nat Med 17 : 975–982. doi: 10.1038/nm.2425 21785433
36. Chung Y, Tanaka S, Chu F, Nurieva RI, Martinez GJ, et al. (2011) Follicular regulatory T cells expressing Foxp3 and Bcl-6 suppress germinal center reactions. Nat Med 17 : 983–988. doi: 10.1038/nm.2426 21785430
37. So T, Croft M (2013) Regulation of PI-3-Kinase and Akt Signaling in T Lymphocytes and Other Cells by TNFR Family Molecules. Front Immunol 4 : 139. doi: 10.3389/fimmu.2013.00139 23760533
38. Han JM, Patterson SJ, Levings MK (2012) The Role of the PI3K Signaling Pathway in CD4(+) T Cell Differentiation and Function. Front Immunol 3 : 245. doi: 10.3389/fimmu.2012.00245 22905034
39. Chen C, Edelstein LC, Gelinas C (2000) The Rel/NF-kappaB family directly activates expression of the apoptosis inhibitor Bcl-x(L). Mol Cell Biol 20 : 2687–2695. doi: 10.1128/MCB.20.8.2687-2695.2000 10733571
40. Chi H (2012) Regulation and function of mTOR signalling in T cell fate decisions. Nat Rev Immunol 12 : 325–338. doi: 10.1038/nri3198 22517423
41. Esparza EM, Arch RH (2005) Glucocorticoid-induced TNF receptor functions as a costimulatory receptor that promotes survival in early phases of T cell activation. J Immunol 174 : 7869–7874. doi: 10.4049/jimmunol.174.12.7869 15944292
42. Kanamaru F, Youngnak P, Hashiguchi M, Nishioka T, Takahashi T, et al. (2004) Costimulation via glucocorticoid-induced TNF receptor in both conventional and CD25+ regulatory CD4+ T cells. J Immunol 172 : 7306–7314. doi: 10.4049/jimmunol.172.12.7306 15187106
43. Ronchetti S, Nocentini G, Bianchini R, Krausz LT, Migliorati G, et al. (2007) Glucocorticoid-induced TNFR-related protein lowers the threshold of CD28 costimulation in CD8+ T cells. J Immunol 179 : 5916–5926. doi: 10.4049/jimmunol.179.9.5916 17947665
44. So T, Choi H, Croft M (2011) OX40 complexes with phosphoinositide 3-kinase and protein kinase B (PKB) to augment TCR-dependent PKB signaling. J Immunol 186 : 3547–3555. doi: 10.4049/jimmunol.1003156 21289304
45. Swain SL, McKinstry KK, Strutt TM (2012) Expanding roles for CD4(+) T cells in immunity to viruses. Nat Rev Immunol 12 : 136–148. doi: 10.1038/nri3152 22266691
46. Clouthier DL, Zhou AC, Watts TH (2014) Anti-GITR agonist therapy intrinsically enhances CD8 T cell responses to chronic LCMV thereby circumventing LCMV-induced downregulation of co-stimulatory GITRL on APC. J Immunol 193. doi: 10.4049/jimmunol.1401002 25281716
47. Aubert RD, Kamphorst AO, Sarkar S, Vezys V, Ha SJ, et al. (2011) Antigen-specific CD4 T-cell help rescues exhausted CD8 T cells during chronic viral infection. Proc Natl Acad Sci U S A 108 : 21182–21187. doi: 10.1073/pnas.1118450109 22160724
48. Teodorovic LS, Riccardi C, Torres RM, Pelanda R (2012) Murine B cell development and antibody responses to model antigens are not impaired in the absence of the TNF receptor GITR. PLoS One 7: e31632. doi: 10.1371/journal.pone.0031632 22328941
49. Lahl K, Loddenkemper C, Drouin C, Freyer J, Arnason J, et al. (2007) Selective depletion of Foxp3+ regulatory T cells induces a scurfy-like disease. J Exp Med 204 : 57–63. doi: 10.1084/jem.20061852 17200412
50. von Herrath M, Whitton JL (2001) Animal models using lymphocytic choriomeningitis virus. Curr Protoc Immunol Chapter 19: Unit 19 10. doi: 10.1002/0471142735.im1910s36
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2015 Číslo 1
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Měli bychom postcovidový syndrom léčit antidepresivy?
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- Infections in Humans and Animals: Pathophysiology, Detection, and Treatment
- : Trypanosomatids Adapted to Plant Environments
- Environmental Drivers of the Spatiotemporal Dynamics of Respiratory Syncytial Virus in the United States
- Dengue Virus RNA Structure Specialization Facilitates Host Adaptation