
Transmission Properties of Human PrP 102L Prions Challenge the Relevance of Mouse Models of GSS
Inherited prion disease (IPD) is caused by pathogenic mutations in the human prion protein (PrP) gene leading to the formation of lethal prions in the brain. To-date the properties of prions causing IPD and their similarities to prions causing other forms of human prion disease remain ill-defined. In the present study we have investigated the properties of prions seen in patients with Gerstmann-Sträussler-Scheinker (GSS) disease associated with the substitution of leucine for proline at amino acid position 102 (GSS P102L). We examined the ability of these prions to infect transgenic mice expressing human mutant 102L PrP, human wild-type PrP or wild-type mice. We found that GSS-102L prions have properties distinct from other types of human prions by showing that they can only infect transgenic mice expressing human PrP carrying the same mutation. Mice expressing wild-type human PrP or wild-type mouse PrP were entirely resistant to infection with GSS-102L prions. We conclude that accurate modeling of inherited prion disease requires the expression of authentic mutant human PrP in transgenic models, as other approaches may generate results that do not mirror the human disease.
Published in the journal:
. PLoS Pathog 11(7): e32767. doi:10.1371/journal.ppat.1004953
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004953
Summary
Inherited prion disease (IPD) is caused by pathogenic mutations in the human prion protein (PrP) gene leading to the formation of lethal prions in the brain. To-date the properties of prions causing IPD and their similarities to prions causing other forms of human prion disease remain ill-defined. In the present study we have investigated the properties of prions seen in patients with Gerstmann-Sträussler-Scheinker (GSS) disease associated with the substitution of leucine for proline at amino acid position 102 (GSS P102L). We examined the ability of these prions to infect transgenic mice expressing human mutant 102L PrP, human wild-type PrP or wild-type mice. We found that GSS-102L prions have properties distinct from other types of human prions by showing that they can only infect transgenic mice expressing human PrP carrying the same mutation. Mice expressing wild-type human PrP or wild-type mouse PrP were entirely resistant to infection with GSS-102L prions. We conclude that accurate modeling of inherited prion disease requires the expression of authentic mutant human PrP in transgenic models, as other approaches may generate results that do not mirror the human disease.
Introduction
Prion diseases are a closely related group of neurodegenerative conditions which affect both humans and animals [1,2]. They are both experimentally and, in some cases, naturally transmissible within and between mammalian species. Cross-species transmission is generally much less efficient than within-species transmissions, being limited by a ‘species’ or transmission barrier [2,3]. Prion diseases in humans include Creutzfeldt-Jakob disease (CJD), Gerstmann-Sträussler-Scheinker disease (GSS), fatal familial insomnia (FFI), kuru and variant CJD (vCJD) [1,4,5].
According to the widely accepted ‘protein-only’ hypothesis [6], the central feature of prion disease is the conversion of host-encoded cellular prion protein (PrPC) to alternative isoforms designated PrPSc [1,2,7]. It is proposed that PrPSc is the infectious agent acting to replicate itself with high fidelity by recruiting endogenous PrPC and that the difference between these isoforms lies purely in the monomer conformation and its state of aggregation [1,2,8] although it is now clear that infectivity can also be associated with protease-sensitive disease-related PrP assemblies distinct from classical PrPSc [9–11] and that infectious and neurotoxic PrP species can be uncoupled [12,13]. Inherited prion disease (IPD) is caused by autosomal-dominant mutations in the human PrP gene (PRNP) and constitute about 15% of all human prion disease [4,14]. Over 40 mutations have been identified, but the precise biochemical mechanisms that lead to disease remain unknown. Within the framework of the protein-only hypothesis, pathogenic mutations in PrP are thought to predispose the mutant proteins to adopt disease-causing conformations and assembly states [2–4].
A proline to leucine substitution at codon 102 (P102L) of human PrP is the most common mutation associated with the GSS phenotype and was first reported in 1989 [15]. Many other kindreds have now been documented worldwide [16], including the original Austrian family reported by Gerstmann, Sträussler, and Scheinker in 1936 [17,18]. Progressive ataxia is the dominant clinical feature, with dementia and pyramidal features occurring later in a disease course typically much longer than that of classical CJD. However, marked variability at both the clinical and neuropathological levels is apparent, with some patients developing a classical CJD-like phenotype with early and rapidly progressive dementia [18–30]. A significant part of this phenotypic variability appears to be contributed by variable propagation of distinct disease-related PrP species generated from either PrP 102L [21,22] or wild type PrP [24,27]. Two distinct abnormal conformers of PrP 102L that generate proteinase K (PK)-resistant fragments of either ~21–30 kDa or ~8 kDa [21,22,24,27,31] have distinct prion transmission properties in 101LL PrP gene knock-in mice [25], while the potential transmissibility or neurotoxicity of abnormal conformers of wild type PrP (that generate PK-resistant fragments of 21–30 kDa [24,27]) remains unknown. Such heterogeneity in disease-related PrP isoforms present in IPD P102L patient brain severely complicates interpretation of transmissions in both conventional and transgenic mice. The conformational selection hypothesis [2,32] predicts that heterogeneous prions formed from PrP in distinct conformations would be differentially selected by hosts expressing different PrP primary sequences. In this regard expression of the homotypic human mutant protein in the host may be critical to accurately model the disease, as only the human mutant protein may be conformationally susceptible to the prion strain involved [2,3,33]. Much of the transgenic modelling of inherited prion disease has however focused on superimposing human PrP mutations onto rodent PrP in order to establish whether infectious prions can be generated de novo. An extremely important consideration in such studies is whether superimposition of pathogenic human PrP mutation into mouse PrP will have the same structural consequences [2,3,33,34]. The possibility of propagating novel prion strains that do not recapitulate the molecular and neuropathological phenotype of the original human disease appears probable [2,3,33] and indeed has been documented with variant CJD transmissions [35].
Recently we established that IPD P102L patient brain isolates could transmit disease with 100% clinical attack rates and short incubation periods to transgenic mice expressing human PrP 102L on a mouse PrP null background (designated 102LL Tg27 mice) [33]. In these transmissions we observed the propagation of the abnormal conformer of PrP 102L that generates protease-resistant fragments of ~21–30 kDa [33]. We also demonstrated that such mice were susceptible to infection with classical CJD prions leading to the generation of prions with altered PrPSc glycoform ratios [33]. The availability of these prions from 102LL Tg27 mice, in which disease-related PrP is entirely composed of PrP 102L (as opposed to the heterogeneous PrP in primary human GSS brain inoculum), now permits direct testing of their host range and in particular the ability of these prions to propagate using wild type human PrP or mouse PrP as substrate. Our findings show that human PrP 102L can support the propagation of distinct prion strains and that human PrP 102L prions have transmission properties strikingly different from those generated in transmission models in which the 102L mutation was superimposed onto mouse PrP.
Results
Efficient transmission of GSS-102L prions on further passage in 102LL Tg27 mice
Prions originating from the primary transmission of three different IPD P102L patient brains to 102LL Tg27 mice [33] (hereafter designated GSS-102L prions) transmitted clinical prion disease with 100% attack rates and short mean incubation periods (~165 days) when passaged in further 102LL Tg27 mice (Table 1).
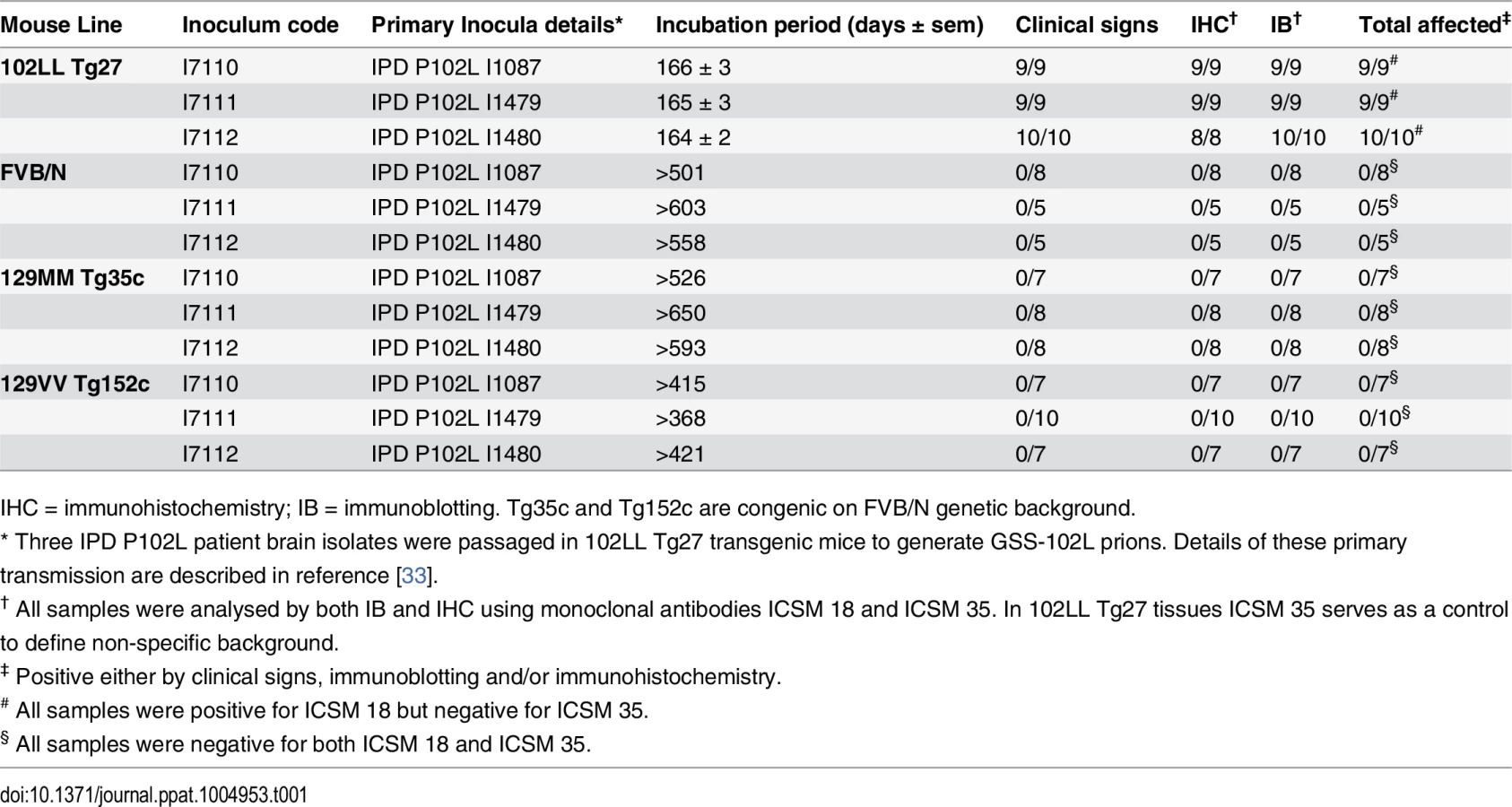
Brain samples of all mice in these transmissions were positive for PK-resistant PrP 102L by immunoblotting using ICSM 18 (Fig 1A) (Table 1).

Both the PK-resistant PrP fragment size (~21–30 kDa) and predominance of the di-glycosylated PrP glycoform mirrored that seen in the 102LL Tg27 mouse brain inoculum (Fig 1A). Immunohistochemistry with ICSM 18 showed extensive abnormal PrP deposition throughout the brain (thalamus shown in Fig 1C) accompanied by prominent astrocytosis and spongiosis. Collectively, these findings establish that IPD P102L prions propagate with high efficiency when serially passaged in 102LL Tg27 mice.
GSS-102L prions fail to transmit prion infection to wild type mice or transgenic mice expressing wild type human PrP
Much of the previous modeling of IPD P102L has involved superimposing the mutation onto the wild type mouse PrP sequence (reviewed in ref [3]). However, it is unclear if challenge of mouse PrP 101L with human P102L prions would lead to the generation of authentic human prion strains or conversely would lead to the generation of experimental prion strains with different transmission characteristics. Notably, after human P102L prions were passaged once in 101LL PrP gene knock-in mice the resultant prions were shown to readily infect wild type mice [36]. We therefore inoculated the GSS-102L prion isolates that transmitted efficiently on passage in 102LL Tg27 mice to wild type mice and also to transgenic mice expressing wild type human PrP on a mouse PrP null background. Strikingly, in complete contrast to findings with the mouse 101LL PrP gene knock-in model we found that GSS-102L prions failed to produce clinical prion disease or any evidence of sub-clinical prion infection when inoculated into wild type mice (Table 1). Even more remarkably the same GSS-102L prions produced no clinical prion disease or evidence of sub-clinical prion infection when inoculated into transgenic mice expressing wild type human PrP (Table 1). In all of these negative transmissions examination of brain showed no detectable PrPSc by high sensitivity immunoblotting (Fig 1A and 1B, lanes 4 and 5) or abnormal PrP deposition by immunohistochemistry (Fig 1E–1H).
In addition, no evidence for elevated levels of spongiosis or gliosis in comparison to the brain of uninoculated age-matched control mice was observed. Collectively these data establish that GSS-102L prions which replicate with high efficiency in a host expressing human PrP 102L are unable to propagate using wild type human PrP or wild type mouse PrP as substrate.
CJD-102L prions are distinct from GSS 102L prions
In comparison to IPD P102L prions, transmission of classical CJD prions to 102LL Tg27 mice appears to be limited by a transmission barrier [33]. In primary transmissions, although nearly all CJD prion-challenged 102LL Tg27 mice showed evidence for prion infection, only a proportion of mice developed clinical prion disease and then only after prolonged incubation periods [33]. In addition, a change in propagated PrPSc type was observed (which itself is indicative of a transmission barrier [2]) with PrPSc glycoform ratios switching from those present in the CJD inocula to ones that more closely resemble those seen in the brain of IPD P102L patients and IPD P102L prion-challenged Tg27 mice [33]. From these primary transmissions, we were unable to distinguish whether the 102L mutation in the host PrP had directly dictated the strain characteristics of the propagated prions (to essentially become congruent with GSS-102L prions) or whether CJD-like prion strain properties were retained. To investigate this, we passaged prions from CJD-challenged 102LL Tg27 mice (hereafter designated CJD-102L prions) in further 102LL Tg27 mice, in transgenic mice expressing wild type human PrP and in wild type mice (Table 2).

In 102LL Tg27 mice we observed that the barrier to development of clinical prion disease seen at primary transmission of classical CJD prions was not abrogated at secondary passage (Table 2). Although nearly all CJD-102L prion-inoculated mice developed prion infection, as evidenced by detection of PrPSc (Fig 2) and abnormal PrP deposition throughout the brain (Fig 3), clinical prion disease was again only observed in a proportion of inoculated recipients and then only at prolonged incubation periods (Table 2). PrPSc typing showed that the altered PrPSc glycoform ratio of CJD-102L prions generated after primary transmission of classical CJD prions to 102LL Tg27 mice was not maintained after further passage in the same mice. The PrPSc type now appeared to more closely resemble the original classical CJD inoculum with a predominance of mono-glycosylated PrP and was readily distinguishable from the di-glycoslyated PrP dominant glycoform pattern seen after secondary passage of GSS 102L prions in 102LL Tg27 mice (Fig 2A, 2C and 2E) (Table 3). From these transmissions we concluded that GSS-102L prions and CJD-102L prions have incongruent transmission properties after further passage in 102LL Tg27 mice.



Importantly, the disparate nature of CJD-102L prions and GSS-102L prions became obvious after examining the transmission properties of CJD-102L prions in transgenic mice expressing wild type human PrP. In complete contrast to GSS-102L prions, all CJD-102L prion isolates transmitted clinical prion disease to mice expressing wild type human PrP in a fashion analogous to the original CJD inoculum (Table 2). In these transmissions PrPSc was readily detected in brain by immunoblotting (Fig 2) and abnormal PrP deposition was observed throughout the brain by immunohistochemistry (Fig 3).
Humanised transgenic mice expressing human PrP 129 valine on a Prnp null background are highly susceptible to sporadic CJD prions regardless of the PrPSc type or codon 129 genotype of the inoculum [37–43]. These transmissions are typically characterised by 100% attack rates of prion infection producing uniform clinical prion disease after similar short incubation periods of around 200 days. The absence of a transmission barrier to sporadic CJD prions is not however uniformly observed in transgenic mice expressing human PrP 129 methionine on a Prnp null background. Here mismatch at residue 129 between the inoculum and host can significantly affect transmission [41,44–46] as evidenced by more prolonged and variable incubation periods and reduced attack rates [41,43,44]. Remarkably, we observed that CJD-102L prions behaved in a closely similar fashion that corresponded with the codon 129 status of the original CJD inoculum (Table 2). This was striking because all of the CJD-102L prion isolates have PrP with residue 129 methionine. Consistent with the CJD-like transmission properties of CJD-102L prions in transgenic mice expressing wild type human PrP, PrPSc typing of the recipient mouse brain showed that the di-glycosylated dominant PrPSc glycoform ratio of CJD-102L prions in the inoculum had switched to a mono-glycosylated PrPSc dominant pattern which more closely resemble CJD prions (Table 3; Fig 2B, 2D and 2F, lanes 5). Collectively, these data show that CJD-102L prions are distinct from GSS-102L prions and retain the transmission properties of the original CJD prion strains. Notwithstanding these observations, all the CJD-102L prion isolates were obtained after a single passage of classical CJD prions in 102LL Tg27 mice and it remains to be seen whether serial passage on the mutated sequence would lead to similar conservation of CJD phenotype.
Consistent with the finding that classical CJD prions transmit prion infection only occasionally to wild type mice with long and variable incubation periods [37,39,40,42,47] we found that CJD-102L prions were also unable to propagate efficiently in wild type mice (Table 2). We found that only one out of eighteen CJD-102L prion-inoculated wild type mice became infected (Table 2) with all other mice showing no evidence of subclinical prion infection by either PrP immunoblotting or immunohistochemistry.
Discussion
Co-propagation of distinct disease-related PrP conformers in IPD brain, combined with differences in their neuropathological targeting, abundance and potential neurotoxicity, provides a general molecular mechanism underlying phenotypic heterogeneity in patients with the same PRNP mutation. Previously we and others have reported the propagation of distinct isoforms of protease-resistant PrP with divergent properties in IPD P102L patient brain and such molecular heterogeneity severely hampers interpretation of primary transmissions to both conventional and transgenic mice (for review see [3]).
In the present study we have investigated the properties of prions generated in transgenic mice expressing human PrP 102L following the intracerebral inoculation of IPD P102L or classical CJD brain isolates. The resultant prion isolates from these transgenic mouse brain were designated GSS-102L or CJD-102L prions, respectively, and because they are associated exclusively with disease-related conformers of human PrP 102L this enables unequivocal examination of the effects that this point mutation has on prion transmission barriers. Our findings show that GSS-102L and CJD-102L prions are distinct from one another with divergent prion strain transmission properties following further passage in transgenic mice expressing either human PrP 102L or wild type human PrP (Fig 4). Thus human PrP 102L is capable of supporting the propagation of distinct lethal prion strains and these data establish that the point mutation does not restrict PrP 102L to a single dominant pathogenic assembly state when templated by an exogenous prion strain.

Importantly our model has enabled us to isolate and investigate the transmission properties of prions originating from IPD P102L patient brain following amplification exclusively on human 102L PrP. Our data show that 102L PrP prions from IPD P102L patient brain that generate PK-resistant PrP fragments of ~21–30 kDa have prion strain transmission properties distinct from all other prion strains propagated in acquired or sporadic human prion disease. The most outstanding feature of this prion strain is its inability to propagate in transgenic mice expressing wild type PrP. Significantly, the inability of GSS-102L prions to also propagate in wild type mice clearly shows that this prion strain is distinct from prions generated in IPD P102L prion-challenged 101LL PrP gene knock-in mice [36]. The remarkable ease of transmission of 101L-passaged IPD P102L prions to wild type mice [36] contrasts strikingly with our data and suggests that a novel prion strain was propagated by the mutant mouse PrP rather than faithful replication of the authentic human PrP 102L prion strain. We therefore recommend that future transgenic modeling of inherited prion disease should focus exclusively on using models that express the homotypic mutant human PrP primary sequence.
We and others have reported that variable involvement of disease-related conformers of wild type human PrP may contribute to phenotypic heterogeneity in IPD P102L [24,27]. However the mechanism by which abnormal wild type PrP is deposited in P102L patient brain remains ill-defined. Wild type PrP may be recruited by a seeded reaction with 102L PrPSc or may accumulate independently as a consequence of pathological changes associated with disease progression. Notably, the glycoform ratios of proteinase K-resistant fragments of 102L PrP and wild type PrP from P102L patient brain are distinct from each other [24] [27] suggesting that the 102L point mutation powerfully dictates thermodynamic preferences for disease-related PrP assembly states that cannot be adopted by wild type PrP and that a significant transmission barrier may be associated with conversion of wild type PrP by a 102L PrPSc seed. This idea is supported by the observation that PK-resistant wild-type PrP in P102L patient brain does not appear to exceed approximately 10% of total PK-resistant PrP [24,27]. Here we show that GSS-102L prions that propagate efficiently in further 102LL Tg27 transgenic mice fail to produce prion infection in transgenic mice expressing wild type human PrP. Based upon the strength of this transmission barrier we conclude that seeded conversion of wild type PrP by abnormal conformers of 102L PrP that generate proteolytic fragments of ~ 21–30 kDa may, at best, be highly inefficient. From these data it is tempting to speculate that abnormal conformers of 102L PrP that generate protease-resistant fragments of 8 kDa might instead be responsible for variable recruitment of wild type PrP in IPD P102L patient brain. However other explanations may be equally possible. In particular, our transmission experiments do not mirror the situation in IPD P102L patient brain where both PrP 102L and wild type PrP are co-expressed. Thus in IPD P102L patient brain, wild type PrP will be exposed throughout the disease time course to all propagating 102L PrPSc species (rather than only at inoculation) and such prolonged exposure in vivo may be required for the generation of misfolded isoforms of wild type PrP. Alternatively because prion strains appear to comprise a quasispecies maintained under host selection pressure (rather than constituting a single molecular clone) [2,48–50] minor subtypes of 102L PrPSc may be populated differently in individual P102L patients leading to variable degrees of recruitment of wild type PrP. Notwithstanding such possibilities, at present we cannot conclusively resolve whether wild type PrP in IPD P102L patient brain misfolds through a directly seeded conversion reaction with an abnormal 102L PrP template or as a consequence of other pathological changes in the brain. In this regard, transmission experiments in heterozygous transgenic mice expressing both 102L PrP and wild type PrP would not be able to differentiate between these possibilities. Although the mechanism that leads to the accumulation of abnormal wild type PrP continues to remain ill-defined, this remains a potentially important contributor to phenotypic variation, not only in IPD P102L, but also in IPD associated with other PRNP mutations [51–56].
Methods
Ethics statement
Storage and biochemical analyses of post-mortem human brain samples and transmission studies to mice were performed with written informed consent from patients with capacity to give consent. Where patients were unable to give informed consent, assent was obtained from their relatives in accordance with UK legislation and Codes of Practice. Samples were stored and used in accordance with the Human Tissue Authority Codes of Practice and in line with the requirements of the Human Tissue Authority licence held by UCL Institute of Neurology. This study was performed with approval from the National Hospital for Neurology and Neurosurgery and the UCL Institute of Neurology Joint Research Ethics Committee (now National Research Ethics Service Committee, London—Queen Square)—REC references: 03/N036, 03/N038 and 03/N133. Work with mice was performed under approval and licence granted by the UK Home Office (Animals (Scientific Procedures) Act 1986; Project Licence number 70/6454) and conformed to University College London institutional and ARRIVE guidelines (www.nc3rs.org.uk/ARRIVE/).
Transgenic mice
Transgenic mice homozygous for a human PrP102L,129M transgene array and murine PrP null alleles (Prnpo/o) designated Tg(HuPrP102L 129M+/+ Prnpo/o)-27 mice (102LL Tg27) [33] have been described previously and were used without modification. Transgenic mice homozygous for a wild type human PrP129M transgene array and murine PrP null alleles (Prnpo/o) designated Tg(HuPrP129M+/+ Prnpo/o)-35 congenic (129MM Tg35c) were derived by subjecting previously described 129MM Tg35 mice [35,44,57] to commercially available speed congenic backcrossing on FVB/N genetic background (Charles River UK). Similarly, transgenic mice homozygous for a wild type human PrP129V transgene array and murine PrP null alleles (Prnpo/o) designated Tg(HuPrP129V+/+ Prnpo/o)-152 congenic (129VV Tg152c) were derived by subjecting previously described 129VV Tg152 mice [37,39,42] to the speed congenic scheme (Charles River UK). Inbred FVB/NHsd mice were supplied by Harlan UK Ltd.
Transmission studies
Strict bio-safety protocols were followed. Inocula were prepared, using disposable equipment for each inoculum, in a microbiological containment level 3 laboratory and inoculations performed within a class 1 microbiological safety cabinet. Ten mice per group from three transgenic lines, 102LL Tg27, 129MM Tg35c, 129VV Tg152c and FVB/N wild type mice were inoculated with a panel of prion isolates, all previously passaged in 102LL Tg27 transgenic mice and therefore adapted to human 102L PrP. The primary inocula comprised human brain homogenates from three IPD P102L patients, one sporadic CJD patient and three iatrogenic CJD patients. Diagnosis of all cases had been neuropathologically confirmed. The genotype of each mouse was confirmed by PCR of DNA prior to inclusion and all mice were uniquely identified by sub-cutaneous transponders. Disposable cages were used and all cage lids and water bottles were also uniquely identified by transponder and remained with each cage of mice throughout the incubation period. Care of the mice was according to institutional and ARRIVE guidelines. Mice were anaesthetised with a mixture of halothane and O2, and intra-cerebrally inoculated into the right parietal lobe with 30 μl of 1% (w/v) brain homogenate prepared in Dulbecco’s phosphate buffered saline lacking Ca2+ or Mg2+ ions (D-PBS). All mice were thereafter examined daily for clinical signs of prion disease. Mice were killed if they exhibited any signs of distress or once a diagnosis of prion disease was established. At post-mortem brains from inoculated mice were removed, divided sagittally with half frozen and half fixed in 10% buffered formol saline.
Antibodies
Anti-PrP monoclonal antibodies ICSM 18 and ICSM 35 were supplied by D-Gen Ltd, London, UK. ICSM antibodies were raised in Prnpo/o mice against α or β PrP as described elsewhere [58]. ICSM 18 is an IgG1 with an epitope spanning residues 142–153 of human PrP [58]. ICSM 35 is an IgG2b with an epitope spanning residues 93–105 of human PrP [58,59]. ICSM 18 recognizes both human PrP 102L and wild type human PrP whereas ICSM 35 recognizes wild type human PrP but not human PrP 102L [24].
Immunoblotting
Brain homogenates (10% (w/v)) were prepared in D-PBS and aliquots analysed in duplicate with or without proteinase K digestion (50 μg/ml final protease concentration, 1h, 37°C) by electrophoresis and immunoblotting as described previously [60,61]. Duplicate blots were blocked in PBS containing 0.05% v/v Tween-20 (PBST) and 5% w/v non-fat milk powder and probed with ICSM 18 or ICSM 35 anti-PrP monoclonal antibodies (0.2 μ g/ml final concentration in PBST) in conjunction with anti-mouse IgG-alkaline phosphatase conjugated secondary antibody and chemiluminescent substrate CDP-Star (Tropix Inc, Bedford, MA, USA) and visualized on Biomax MR film (Kodak) as described [60,61]. For analysis of PrP glycoforms, blots were developed in chemifluorescent substrate (AttoPhos; Promega) and visualized on a Storm 840 phosphoimager (Amersham) using ImageQuaNT software (Amersham) [31,61].
Immunohistochemistry
Fixed brain was immersed in 98% formic acid for 1 h and paraffin wax embedded. Serial sections of 4 μm nominal thickness were pre-treated with Tris-Citrate EDTA buffer for antigen retrieval [61]. PrP deposition was visualized using ICSM 35 or ICSM 18 as the primary antibody, using an automated immunostaining system (www.ventana.com). Visualization was accomplished with diaminobenzidine staining. Bright field photographs were taken on an ImageView digital camera (www.soft-imaging.de) and composed with Adobe Photoshop.
Zdroje
1. Prusiner SB (1998) Prions. Proc Natl Acad Sci USA 95 : 13363–13383. 9811807
2. Collinge J, Clarke A (2007) A general model of prion strains and their pathogenicity. Science 318 : 930–936. 17991853
3. Wadsworth JD, Asante EA, Collinge J (2010) Contribution of transgenic models to understanding human prion disease. Neuropathol Appl Neurobiol 36 : 576–597. doi: 10.1111/j.1365-2990.2010.01129.x 20880036
4. Collinge J (2005) Molecular neurology of prion disease. J Neurol Neurosurg Psychiatry 76 : 906–919. 15965195
5. Wadsworth JD, Collinge J (2011) Molecular pathology of human prion disease. Acta Neuropathol 121 : 69–77. doi: 10.1007/s00401-010-0735-5 20694796
6. Griffith JS (1967) Self replication and scrapie. Nature 215 : 1043–1044. 4964084
7. Prusiner SB (1982) Novel proteinaceous infectious particles cause scrapie. Science 216 : 136–144. 6801762
8. Caughey B, Baron GS (2006) Prions and their partners in crime. Nature 443 : 803–810. 17051207
9. Safar J, Wille H, Itri V, Groth D, Serban H et al. (1998) Eight prion strains have PrPSc molecules with different conformations. Nat Med 4 : 1157–1165. 9771749
10. Safar JG, Geschwind MD, Deering C, Didorenko S, Sattavat M et al. (2005) Diagnosis of human prion disease. Proc Natl Acad Sci USA 102 : 3501–3506. 15741275
11. Cronier S, Gros N, Tattum MH, Jackson GS, Clarke AR et al. (2008) Detection and characterization of proteinase K-sensitive disease-related prion protein with thermolysin. Biochem J 416 : 297–305. doi: 10.1042/BJ20081235 18684106
12. Sandberg MK, Al Doujaily H, Sharps B, Clarke AR, Collinge J (2011) Prion propagation and toxicity in vivo occur in two distinct mechanistic phases. Nature 470 : 540–542. doi: 10.1038/nature09768 21350487
13. Sandberg MK, Al Doujaily H, Sharps B, De Oliveira MW, Schmidt C et al. (2014) Prion neuropathology follows the accumulation of alternate prion protein isoforms after infective titre has peaked. Nat Commun 5 : 4347. doi: 10.1038/ncomms5347 25005024
14. Beck JA, Poulter M, Campbell TA, Adamson G, Uphill JB et al. (2010) PRNP allelic series from 19 years of prion protein gene sequencing at the MRC Prion Unit. Hum Mutat 31: E1551–E1563. doi: 10.1002/humu.21281 20583301
15. Hsiao K, Baker HF, Crow TJ, Poulter M, Owen F et al. (1989) Linkage of a prion protein missense variant to Gerstmann-Straussler syndrome. Nature 338 : 342–345. 2564168
16. Kovacs GG, Trabattoni G, Hainfellner JA, Ironside JW, Knight RS et al. (2002) Mutations of the prion protein gene phenotypic spectrum. J Neurol 249 : 1567–1582. 12420099
17. Kretzschmar HA, Honold G, Seitelberger F, Feucht M, Wessely P et al. (1991) Prion protein mutation in family first reported by Gerstmann, Straussler, and Scheinker. Lancet 337 : 1160. 1674033
18. Hainfellner JA, Brantner-Inthaler S, Cervenáková L, Brown P, Kitamoto T et al. (1995) The original Gerstmann-Straussler-Scheinker family of Austria: Divergent clinicopathological phenotypes but constant PrP genotype. Brain Pathol 5 : 201–211. 8520719
19. Kretzschmar HA, Kufer P, Riethmuller G, DeArmond SJ, Prusiner SB et al. (1992) Prion protein mutation at codon 102 in an Italian family with Gerstmann-Straussler-Scheinker syndrome. Neurology 42 : 809–810. 1348851
20. Barbanti P, Fabbrini G, Salvatore M, Petraroli R, Cardone F et al. (1996) Polymorphism at codon 129 or codon 219 of PRNP and clinical heterogeneity in a previously unreported family with Gerstmann-Straussler-Scheinker disease (PrP-P102L mutation). Neurology 47 : 734–741. 8797472
21. Piccardo P, Dlouhy SR, Lievens PMJ, Young K, Bird TD et al. (1998) Phenotypic variability of Gerstmann-Straussler-Scheinker disease is associated with prion protein heterogeneity. J Neuropathol Exp Neurol 57 : 979–988. 9786248
22. Parchi P, Chen SG, Brown P, Zou W, Capellari S et al. (1998) Different patterns of truncated prion protein fragments correlate with distinct phenotypes in P102L Gerstmann-Sträussler-Scheinker disease. Proc Natl Acad Sci USA 95 : 8322–8327. 9653185
23. Majtenyi C, Brown P, Cervenakova L, Goldfarb LG, Tateishi J (2000) A three-sister sibship of Gerstmann-Straussler-Scheinker disease with a CJD phenotype. Neurology 54 : 2133–2137. 10851377
24. Wadsworth JD, Joiner S, Linehan J, Cooper S, Powell C et al. (2006) Phenotypic heterogeneity in inherited prion disease (P102L) is associated with differential propagation of protease-resistant wild-type and mutant prion protein. Brain 129 : 1557–1569. 16597650
25. Piccardo P, Manson JC, King D, Ghetti B, Barron RM (2007) Accumulation of prion protein in the brain that is not associated with transmissible disease. Proc Natl Acad Sci USA 104 : 4712–4717. 17360589
26. Webb TE, Poulter M, Beck J, Uphill J, Adamson G et al. (2008) Phenotypic heterogeneity and genetic modification of P102L inherited prion disease in an international series. Brain 131 : 2632–2646. doi: 10.1093/brain/awn202 18757886
27. Monaco S, Fiorini M, Farinazzo A, Ferrari S, Gelati M et al. (2012) Allelic origin of protease-sensitive and protease-resistant prion protein isoforms in Gerstmann-Straussler-Scheinker disease with the P102L mutation. PLoS ONE 7: e32382. doi: 10.1371/journal.pone.0032382 22384235
28. Popova SN, Tarvainen I, Capellari S, Parchi P, Hannikainen P et al. (2012) Divergent clinical and neuropathological phenotype in a Gerstmann-Straussler-Scheinker P102L family. Acta Neurol Scand 126 : 315–323. doi: 10.1111/j.1600-0404.2011.01628.x 22211828
29. Rusina R, Fiala J, Holada K, Matejckova M, Novakova J et al. (2012) Gerstmann-Straussler-Scheinker syndrome with the P102L pathogenic mutation presenting as familial Creutzfeldt-Jakob disease: a case report and review of the literature. Neurocase 19 : 41–53. doi: 10.1080/13554794.2011.654215 22494260
30. Riudavets MA, Sraka MA, Schultz M, Rojas E, Martinetto H et al. (2013) Gerstmann-Straussler-Scheinker syndrome with variable phenotype in a new kindred with PRNP-P102L mutation. Brain Pathol 24 : 142–147. doi: 10.1111/bpa.12083 23944754
31. Hill AF, Joiner S, Beck J, Campbell TA, Dickinson A et al. (2006) Distinct glycoform ratios of protease resistant prion protein associated with PRNP point mutations. Brain 129 : 676–685. 16415305
32. Collinge J (1999) Variant Creutzfeldt-Jakob disease. Lancet 354 : 317–323. 10440324
33. Asante EA, Gowland I, Grimshaw A, Linehan JM, Smidak M et al. (2009) Absence of spontaneous disease and comparative prion susceptibility of transgenic mice expressing mutant human prion proteins. J Gen Virol 90 : 546–558. doi: 10.1099/vir.0.007930-0 19218199
34. Hart T, Hosszu LL, Trevitt CR, Jackson GS, Waltho JP et al. (2009) Folding kinetics of the human prion protein probed by temperature jump. Proc Natl Acad Sci USA 106 : 5651–5656. doi: 10.1073/pnas.0811457106 19321423
35. Wadsworth JD, Asante EA, Desbruslais M, Linehan J, Joiner S et al. (2004) Human prion protein with valine 129 prevents expression of variant CJD phenotype. Science 306 : 1793–1796. 15539564
36. Manson JC, Jamieson E, Baybutt H, Tuzi NL, Barron R et al. (1999) A single amino acid alteration (101L) introduced into murine PrP dramatically alters incubation time of transmissible spongiform encephalopathy. EMBO J 18 : 6855–6864. 10581259
37. Collinge J, Palmer MS, Sidle KCL, Hill AF, Gowland I et al. (1995) Unaltered susceptibility to BSE in transgenic mice expressing human prion protein. Nature 378 : 779–783. 8524411
38. Collinge J, Sidle KC, Meads J, Ironside J, Hill AF (1996) Molecular analysis of prion strain variation and the aetiology of 'new variant' CJD. Nature 383 : 685–690. 8878476
39. Hill AF, Desbruslais M, Joiner S, Sidle KCL, Gowland I et al. (1997) The same prion strain causes vCJD and BSE. Nature 389 : 448–450. 9333232
40. Telling GC, Scott M, Mastrianni J, Gabizon R, Torchia M et al. (1995) Prion propagation in mice expressing human and chimeric PrP transgenes implicates the interaction of cellular PrP with another protein. Cell 83 : 79–90. 7553876
41. Korth C, Kaneko K, Groth D, Heye N, Telling G et al. (2003) Abbreviated incubation times for human prions in mice expressing a chimeric mouse-human prion protein transgene. Proc Natl Acad Sci USA 100 : 4784–4789. 12684540
42. Wadsworth JD, Joiner S, Linehan JM, Desbruslais M, Fox K et al. (2008) Kuru prions and sporadic Creutzfeldt-Jakob disease prions have equivalent transmission properties in transgenic and wild-type mice. Proc Natl Acad Sci USA 105 : 3885–3890. doi: 10.1073/pnas.0800190105 18316717
43. Kobayashi A, Asano M, Mohri S, Kitamoto T (2007) Cross-sequence transmission of sporadic Creutzfeldt-Jakob disease creates a new prion strain. J Biol Chem 282 : 30022–30028. 17709374
44. Asante EA, Linehan J, Desbruslais M, Joiner S, Gowland I et al. (2002) BSE prions propagate as either variant CJD-like or sporadic CJD-like prion strains in transgenic mice expressing human prion protein. EMBO J 21 : 6358–6366. 12456643
45. Beringue V, Le Dur A, Tixador P, Reine F, Lepourry L et al. (2008) Prominent and persistent extraneural infection in human PrP transgenic mice infected with variant CJD. PLoS ONE 3: e1419. doi: 10.1371/journal.pone.0001419 18183299
46. Kong Q, Zheng M, Casalone C, Qing L, Huang S et al. (2008) Evaluation of the human transmission risk of an atypical bovine spongiform encephalopathy prion strain. J Virol 82 : 3697–3701. doi: 10.1128/JVI.02561-07 18234793
47. Telling GC, Scott M, Hsiao KK, Foster D, Yang S-L et al. (1994) Transmission of Creutzfeldt-Jakob disease from humans to transgenic mice expressing chimeric human-mouse prion protein. Proc Natl Acad Sci USA 91 : 9936–9940. 7937921
48. Li J, Browning S, Mahal SP, Oelschlegel AM, Weissmann C (2010) Darwinian evolution of prions in cell culture. Science 327 : 869–872. doi: 10.1126/science.1183218 20044542
49. Collinge J (2010) Medicine. Prion strain mutation and selection. Science 328 : 1111–1112. doi: 10.1126/science.1190815 20508117
50. Browning S, Baker CA, Smith E, Mahal SP, Herva ME et al. (2011) Abrogation of complex glycosylation by Swainsonine results in strain - and cell-specific inhibition of prion replication. J Biol Chem 286 : 40962–73. doi: 10.1074/jbc.M111.283978 21930694
51. Gabizon R, Telling G, Meiner Z, Halimi M, Kahana I et al. (1996) Insoluble wild-type and protease-resistant mutant prion protein in brains of patients with inherited prion disease. Nat Med 2 : 59–64. 8564843
52. Chen SG, Parchi P, Brown P, Capellari S, Zou WQ et al. (1997) Allelic origin of the abnormal prion protein isoform in familial prion diseases. Nat Med 3 : 1009–1015. 9288728
53. Silvestrini MC, Cardone F, Maras B, Pucci P, Barra D et al. (1997) Identification of the prion protein allotypes which accumulate in the brain of sporadic and familial Creutzfeldt-Jakob disease patients. Nat Med 3 : 521–525. 9142120
54. Capellari S, Cardone F, Notari S, Schinina ME, Maras B et al. (2005) Creutzfeldt-Jakob disease associated with the R208H mutation in the prion protein gene. Neurology 64 : 905–907. 15753435
55. Xiao X, Cali I, Dong Z, Puoti G, Yuan J et al. (2013) Protease-sensitive prions with 144-bp insertion mutations. Aging (Albany NY) 5 : 155–173. 23515139
56. Cardone F, Principe S, Schinina ME, Maras B, Capellari S et al. (2014) Mutant PrP(CJD) prevails over wild-type PrP(CJD) in the brain of V210I and R208H genetic Creutzfeldt-Jakob disease patients. Biochem Biophys Res Commun 454 : 289–294. doi: 10.1016/j.bbrc.2014.10.051 25450391
57. Asante EA, Li YG, Gowland I, Jefferys JG, Collinge J (2004) Pathogenic human prion protein rescues PrP null phenotype in transgenic mice. Neurosci Lett 360 : 33–36. 15082172
58. Khalili-Shirazi A, Quaratino S, Londei M, Summers L, Tayebi M et al. (2005) Protein conformation significantly influences immune responses to prion protein. J Immunol 174 : 3256–3263. 15749856
59. Khalili-Shirazi A, Summers L, Linehan J, Mallinson G, Anstee D et al. (2005) PrP glycoforms are associated in a strain-specific ratio in native PrPSc. J Gen Virol 86 : 2635–2644. 16099923
60. Wadsworth JD, Joiner S, Hill AF, Campbell TA, Desbruslais M et al. (2001) Tissue distribution of protease resistant prion protein in variant CJD using a highly sensitive immuno-blotting assay. Lancet 358 : 171–180. 11476832
61. Wadsworth JD, Powell C, Beck JA, Joiner S, Linehan JM et al. (2008) Molecular diagnosis of human prion disease. Methods Mol Biol 459 : 197–227. doi: 10.1007/978-1-59745-234-2_14 18576157
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2015 Číslo 7
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Měli bychom postcovidový syndrom léčit antidepresivy?
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- RNA Virus Reassortment: An Evolutionary Mechanism for Host Jumps and Immune Evasion
- Activation of TLR2 and TLR6 by Dengue NS1 Protein and Its Implications in the Immunopathogenesis of Dengue Virus Infection
- N-acetylglucosamine Regulates Virulence Properties in Microbial Pathogens
- Characterization of a Prefusion-Specific Antibody That Recognizes a Quaternary, Cleavage-Dependent Epitope on the RSV Fusion Glycoprotein