
Acts as a Tumor Suppressor in a Murine Retinoblastoma Model by Facilitating Tumor Cell Death
CDH11 gene copy number and expression are frequently lost in human retinoblastomas and in retinoblastomas arising in TAg-RB mice. To determine the effect of Cdh11 loss in tumorigenesis, we crossed Cdh11 null mice with TAg-RB mice. Loss of Cdh11 had no gross morphological effect on the developing retina of Cdh11 knockout mice, but led to larger retinal volumes in mice crossed with TAg-RB mice (p = 0.01). Mice null for Cdh11 presented with fewer TAg-positive cells at postnatal day 8 (PND8) (p = 0.01) and had fewer multifocal tumors at PND28 (p = 0.016), compared to mice with normal Cdh11 alleles. However, tumor growth was faster in Cdh11-null mice between PND8 and PND84 (p = 0.003). In tumors of Cdh11-null mice, cell death was decreased 5 - to 10-fold (p<0.03 for all markers), while proliferation in vivo remained unaffected (p = 0.121). Activated caspase-3 was significantly decreased and β-catenin expression increased in Cdh11 knockdown experiments in vitro. These data suggest that Cdh11 displays tumor suppressor properties in vivo and in vitro in murine retinoblastoma through promotion of cell death.
Published in the journal:
. PLoS Genet 6(4): e32767. doi:10.1371/journal.pgen.1000923
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1000923
Summary
CDH11 gene copy number and expression are frequently lost in human retinoblastomas and in retinoblastomas arising in TAg-RB mice. To determine the effect of Cdh11 loss in tumorigenesis, we crossed Cdh11 null mice with TAg-RB mice. Loss of Cdh11 had no gross morphological effect on the developing retina of Cdh11 knockout mice, but led to larger retinal volumes in mice crossed with TAg-RB mice (p = 0.01). Mice null for Cdh11 presented with fewer TAg-positive cells at postnatal day 8 (PND8) (p = 0.01) and had fewer multifocal tumors at PND28 (p = 0.016), compared to mice with normal Cdh11 alleles. However, tumor growth was faster in Cdh11-null mice between PND8 and PND84 (p = 0.003). In tumors of Cdh11-null mice, cell death was decreased 5 - to 10-fold (p<0.03 for all markers), while proliferation in vivo remained unaffected (p = 0.121). Activated caspase-3 was significantly decreased and β-catenin expression increased in Cdh11 knockdown experiments in vitro. These data suggest that Cdh11 displays tumor suppressor properties in vivo and in vitro in murine retinoblastoma through promotion of cell death.
Introduction
Retinoblastoma is initiated by loss of both RB1 alleles, denoted M1 and M2 mutational events [1]. These initiating events are sufficient for the development of the benign tumor, retinoma, but not enough to drive to malignancy; additional mutational events (M3-Mn) are required for development to retinoblastoma [1]–[3]. Early cytogenetic analysis performed on human retinoblastoma samples revealed recurrent chromosomal abnormalities [4]. Five comparative genomic hybridization (CGH) studies and one matrix CGH study confirmed these results and identified common genomic regions of gain and loss in retinoblastoma tumors [5]–[10]. Based on the location of these genomic changes, potential oncogenes (KIF14, E2F3 and DEK) and tumor suppressor genes (p75NTR and CDH11) have been identified and shown to have involvement in retinoblastoma development and progression [2], [11]–[13]. Based on the frequency of and correlation between these mutational events, we proposed a genetic cascade to malignancy. Subsequent to loss of RB1, the most frequent event is gain of 1q (involving KIF14) [11], followed by gain of 6p (involving E2F3 and DEK) [12], [14], and then, loss of 16q (involving CDH11) [13] or gain of MYCN [15].
Understanding the pathway to tumorigenesis is important for the development of new and better therapeutics that can ultimately be used to halt retinoblastoma progression at an early stage. Importantly, delineating the order of mutational events in retinoblastoma, the prototypical model of cancer, is pertinent to the understanding of oncogenesis in general.
In previous work, we narrowed the minimal region of genomic loss on chromosomal arm 16q22.1 to CDH11 [13]. This gene was lost in 58% of 71 retinoblastoma tumors, and its expression showed gradual loss in tumors of the murine retinoblastoma model (TAg-RB) induced by simian virus 40 large T-Antigen (TAg) expression [16], with some advanced tumors (3 of 8) showing loss of Cdh11. Thus, we proposed that Cdh11 acts as a tumor suppressor gene in retinoblastoma.
Gratias et al, 2007, identified a complex pattern of 16q loss of heterozygosity (LOH) in 18 out of 58 retinoblastoma samples. One tumor showed LOH at 16q24, the region where CDH13 is located; however, CDH13 did not show reduced expression in retinoblastoma tumors, confirming our previous findings [13], [17]. Gratias et al, 2007 did not test markers for CDH11 directly, as it was outside their minimal region of loss. They also correlated 16q allelic loss with diffuse intraocular seeding, implicating 16q loss as a late mutational event, in agreement with our proposed sequence of mutational events described in Bowles et al, 2007 [15]. Laurie et al., 2009, recently reported that loss of Cdh11 correlated with optic nerve invasion using an in vivo model of in vitro cell lines derived from an in vivo murine retinoblastoma model [18].
In our present study, we use the TAg-RB retinoblastoma mouse model to study the function of Cdh11 in tumorigenesis. This murine model, unlike any other RB mouse model, displays both molecular and histological features similar to the human disease [2], [11]–[13], [19]. Moreover, it is widely used as a pre-clinical model for testing therapeutics [14], [20]–[25]. Perhaps the strongest resemblance to human tumors is evidenced by its initiation in the inner nuclear layer (INL) of the retina and the presence of Flexner-Wintersteiner rosettes. The latter is an important feature not recapitulated in any of the other mouse models of retinoblastoma [26]–[30].
We now address the roles of Cdh11 in developing retina and retinoblastoma. We report that Cdh11 is developmentally regulated during retinogenesis. We show that Cdh11 loss impacts the number of tumors that develop initially, and that it significantly increases the average tumor volume at PND84 per tumor initiating cell defined at PND8 in animals with mutant Cdh11 alleles with respect to animals with wild type alleles. We also show clear in vivo and in vitro evidence that more cell death occurs in tumors with wild type alleles than with mutant Cdh11 alleles, while cell proliferation remains unchanged regardless of Cdh11 allele status. Taken together, these data provide substantial evidence to suggest that in retinoblastoma Cdh11 acts as a tumor suppressor by facilitating cell death.
Results
Spatio-temporal expression and co-localization of cadherin-11 in the developing retina
To assess the role of Cdh11 in the murine retina, we analyzed the spatio-temporal expression of cadherin-11 by immunostaining. Cadherin-11 was highly expressed by cells that typically differentiate at ED (embryonic day) 18.5 (Figure 1A). At PND3, expression was observed in areas where cells are migrating, and this became more visible in individual cells at PND6. At PND15 (data not shown) and adult (PND60), cadherin-11 was expressed in the inner nuclear layer (INL) by Müller glia cell bodies and processes at the outer border of the outer nuclear (ONL) and inner border of the ganglion cell layer (GCL) (Figure 1B).

To identify retinal cell types that express cadherin-11 in the INL, we performed co-localization studies in adult retina, using retinal specific cell type markers. Cadherin-11 co-expressed with markers of horizontal cells and Müller glia cells and their processes, (Figure 2A and 2B) but not with markers of bipolar or amacrine cells (Figure 2C and 2D).

Retinal development in the absence of Cdh11
To examine the role of Cdh11 in the developing retina, we studied littermates of Cdh11 knockout animals. We analyzed retinas of Cdh11+/+, Cdh11+/-, and Cdh11-/- on a 129/C57Bl-6 mixed background at developmental time points ED18.5, PND3, PND6, PND15 and PND60. To accurately compare the retina of varying genotypes, retinal sections were cut every 5 µm throughout the eyes in the papillary-optic nerve plane.
Hematoxylin and eosin (H&E) analysis of retinal sections at all developmental time points revealed no gross phenotypic differences between the Cdh11 genotypes (Figure 3). Staining of retinal cell type markers was performed to determine if Cdh11 influenced differentiation. There was no obvious change in cell populations that expressed Chx-10 (progenitor cells and bipolar cells), neurofilament (160 kDA for horizontal cells), cellular retinaldehyde-binding protein (CRALBP for Müller glia cells) or syntaxin (HPC-1 for amacrine cells) (Figure S1). The number of S-phase cells also seemed unaffected with loss of Cdh11, determined by immunohistochemical analysis of BrdU positive cells (Figure S1).

It is possible that the lack of gross phenotype in Cdh11-/- retinas is due to functional compensation by cadherins similar to Cdh11. Cdh2, also known as neuronal cadherin (N-cadherin), shares 53% amino acid similarity to Cdh11 and is a mesenchymal cadherin like Cdh11 [31]. However, immunohistochemical analysis showed no change in expression of Cdh2 in the absence of Cdh11 (Figure S1).
Cadherin-11 expression in TAg-RB murine retinoblastoma tumors
To evaluate cadherin-11 expression in developing tumors of the TAg-RB mouse model, we stained for cadherin-11 at PND9, PND28, PND35, PND84 and PND140. At PND9, early initiating tumour cells showed complete overlap of TAg and cadherin-11 staining (Figure 4A). At later time points, cadherin-11 expression was gradually lost from tumors: at PND28, some tumors showed loss and others showed expression (Figure 4B); at PND35, most tumors had lost expression of cadherin-11 (Figure 4C); by PND140, large, late stage tumors showed complete absence of cadherin-11 expression (Figure 4D).

Tumor development in TAg-RB mice
TAg-RB tumor development has been characterized (unpublished data). At PND8, TAg was first expressed by single cells in the INL of the retina (Figure 5A). At PND28, clusters of TAg-positive cells emerged (Figure 6A), consistent with multifocal tumors, each derived from single TAg expressing cells already present at PND8. These small tumor foci showed evidence of Homer Wright rosettes (data not shown). At PND84, tumors resembled human retinoblastoma (Figure 7A).



Loss of Cdh11 reduces the number of cells expressing TAg
To examine the tumor suppressor role of Cdh11 in retinoblastoma development, we crossed Cdh11-/- mice with TAg-RB mice and analyzed genotypes Cdh11+/+;TAg+/-, Cdh11+/-;TAg+/-, and Cdh11-/-;TAg+/-, on a mixed 129/C57Bl-6 background. Gross phenotypes at varying time points were assessed by H&E staining.
At PND8, retinal histology of mice with normal and Cdh11 allelic losses showed no differences in H&E staining (Figure 5A). Immunostaining showed that TAg was expressed by large, spindle shaped single cells in the INL (Figure 5A). Tissue sections taken every 300 µm spanning the entire eye were manually counted for TAg-positive cells and the total number of TAg-positive cells per eye was extrapolated to the entire retina based on the total number of sections that were produced per eye (Figure 5B, for detailed report of this method see [32]). A striking reduction in the number of TAg-positive cells was observed in retinas of mice with mutant Cdh11 alleles compared to mice with normal Cdh11 alleles. Animals of the Cdh11+/+;TAg+/- genotype had a mean of 6,417 TAg-positive cells per entire retina compared to 3,874 and 2,230 in Cdh11+/-;TAg+/- and Cdh11-/-;TAg+/- genotypes respectively, describing a significant allele dosage effect (p = 0.01, n = 5) (Figure 5B). As a control and to normalize the total TAg-positive cells per retina, retinal area was measured for each of the selected sections, using the Image J software and then extrapolated to the entire retina. Total retinal areas at PND8 were found to be similar in all Cdh11 genotypes (p = 0.83, n = 5) (Figure 5B). To quantify tumor-initiating cells with respect to retinal area, we determined the ratio of TAg-positive cells per retinal area, which showed a significant reduction correlated with Cdh11 genomic loss (p = 0.01) (Figure 5C). This effect continued at later stages in development, since at PND28, fewer multifocal tumors developed in mice with Cdh11 loss. These data suggest that in this model, the expression of TAg may be dependent on Cdh11.
Evaluating tumor volume at PND28 and PND84
At PND28, we observed a significant decrease in the number of multifocal tumors with decreasing number of functional copies of Cdh11 as assessed by both H&E and TAg stain (Figure 6A). Tumor volumes as a percent of retina were estimated to be 5.0%, 3.2% and 1.5% in Cdh11+/+;TAg+/-, Cdh11+/-;TAg+/- and Cdh11-/-;TAg+/- genotypes respectively (5 animals analyzed per genotype). These analyses describe a significant decrease in tumor volume as Cdh11 alleles are lost (p = 0.016, Figure 6B).
At PND84, tumor morphology of the varying genotypes did not differ by H&E or TAg staining (Figure 7A). All three genotypes showed tumors highly reminiscent of human retinoblastoma, presenting with large tumors originating from the INL, bulging into adjacent layers, and displaying features of Homer Wright rosettes (Figure 7A).
In stark contrast to earlier timepoints, total tumor volume at PND84 was not significantly different in mice of Cdh11+/+;TAg+/-, Cdh11+/-;TAg+/- and Cdh11-/-;TAg+/- genotypes (p = 0.26; n = 8, 8, and 9 respectively, Figure 7B). However, unlike in the younger mice, total retinal size was significantly larger (p = 0.01) in the Cdh11 null mice compared to Cdh11 normal mice (Figure 7B), suggesting that loss of Cdh11 may affect the overall size of the adult retina in TAg mice. Tumor volume as a percentage of the entire retina was not significantly different between genotypes (p = 0.07, Figure 7C). The similarity of tumor volume at PND84 suggests faster tumor growth may be occurring in mice with mutant Cdh11 alleles, considering that fewer multifocal tumors were initially present at PND28. These data suggest two roles for Cdh11 in retina: 1) Cdh11 displays tumor suppressor abilities in vivo and 2) Cdh11 loss affects retinal development in TAg mice, reflected in increase in overall size of the adult retina. This difference was not observed up to PND60 in Cdh11-/- mice (Figure 3).
Faster tumor growth is observed from PND8 to PND84 in mice with mutated Cdh11 alleles
To establish whether tumors developing in Cdh11 mutant animals grew faster, we studied the rate of tumor growth between PND8 and PND84. This was done by calculating the ratio of tumor volume at PND84 (in pixels) to the mean number of TAg-positive cells (single tumor initiating cells) at PND8. The analysis revealed significant differences between the genotypes (p = 0.003, Figure 8A), indicative of faster growing tumors in mice with mutant Cdh11 alleles. We performed a second comparison to account for the difference in retinal size between genotypes at PND84 by calculating the ratio of percent tumor volume per retina at PND84 to the mean number of TAg-positive cells in the entire PND8 retina per genotype. Even after adjusting for retinal size, the tumor volume per initiating cell in mice with mutant Cdh11 alleles remained significantly greater (p = 0.01, data not shown). In addition, we noticed that while the retinal size at PND8 was similar between genotypes (p = 0.83, Figure 5B), the PND84 retinal size was significantly larger (p = 0.01, Figure 7B), suggesting a role for Cdh11 in the retinal development of TAg-RB mice.
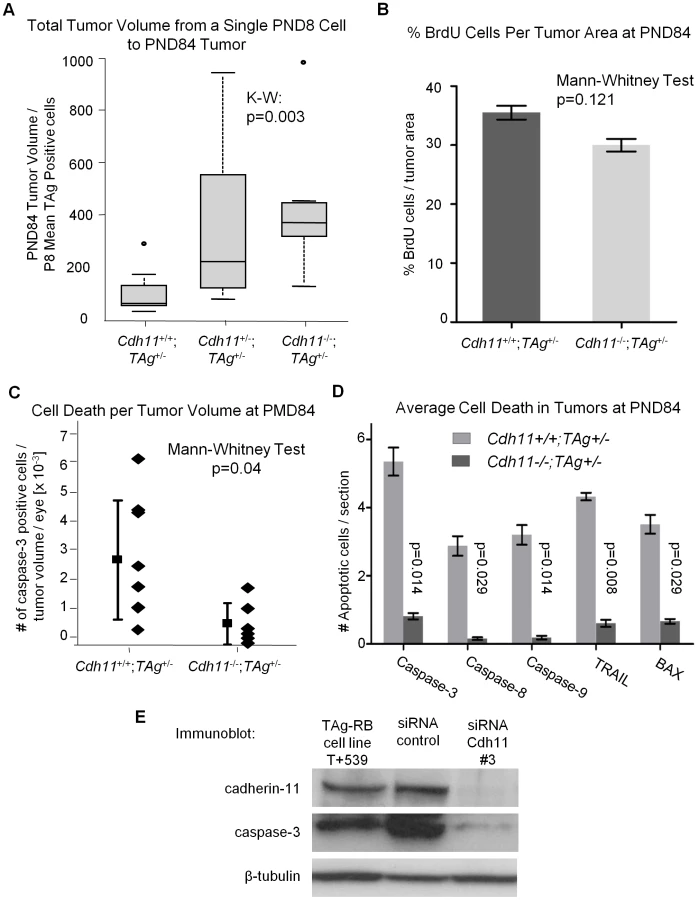
Cdh11 mediates its tumor suppressor function through apoptosis and not proliferation
Since “growth” reflects a positive balance between cell proliferation and cell death we evaluated both cell proliferation and death in tumors at PND84.
At PND84, tumors are well defined and easily quantifiable. We performed PCNA staining (a marker of cells in early G1 and S phase) of PND84 tumors in selected sections of Cdh11+/+;TAg+/- (n = 2) and Cdh11-/-; TAg+/- (n = 2) mice and calculated the percent PCNA positive cells per tumor volume revealing little difference between the two genotypes. To improve the power of this analysis, BrdU incorporation in PND84 tumors was evaluated in an additional larger cohort of animals. Again, we noticed no significant difference between the genotypes (p = 0.121, n = 6 for each genotype, Figure 8B). These data strongly support that Cdh11 is not acting to impede proliferation of tumor cells.
To assess cell death, selected sections of Cdh11+/+;TAg+/- (n = 8) and Cdh11-/-; TAg+/- (n = 6) were manually counted for activated caspase-3 positive cells per tumor area and extrapolated to the entire tumor volume. Non-tumor retina showed no activated caspase-3 positive cells. We found significantly more cell death in tumors of mice with normal Cdh11 alleles than in tumors of mice with mutated Cdh11 alleles (p = 0.04, Figure 8C). Interestingly, β-catenin mRNA was upregulated in the Cdh11-/- TAg+/- mice relative to the Cdh11+/+Tag+/- mice (Figure S2C).
Furthermore, we observed a wide distribution of cell death among Cdh11+/+;TAg+/- mice (mean = 2.90×10−03, standard deviation ±2.08×10−03) compared to mice with mutant Cdh11 alleles (mean = 6.94×10−04, standard deviation ±7.25×10−04, Figure 8C). To further support the role of Cdh11 in apoptosis, we assayed by immunohistochemistry, in an additional cohort of animals, five pro-apoptotic proteins: activated caspases 3, 8, 9, TRAIL and BAX. Depending on the cell death marker, we observed 5 to 10 fold less expression in CDH11 mutant animals than in animals with normal Cdh11 alleles (p<0.03 for all five cell death markers, Figure 8C).
We also assessed cell death in vitro in a primary cell line derived from TAg-RB tumors (T+539). This tumor cell line, when treated with cadherin-11 siRNA, showed significant cadherin-11 knockdown (Figure 8D). Following knockdown, caspase-3 expression was decreased (Figure 8D), providing further evidence that Cdh11 acts to promote apoptosis. In addition, we studied RNA from the T+539 cell line treated either with Cdh11 siRNA or scrambled siRNA by RT-PCR for proliferation markers PCNA and Ki67, and found no difference in expression (Figure S3). These data strongly support the hypothesis that Cdh11 has a pro-apoptotic role in TAg-RB tumors, but does not suggest a role in cell division or proliferation.
Discussion
Cdh11 displays tumor suppressor-like properties in vivo
We have previously described copy number and expression loss of CDH11 in human retinoblastomas, suggesting a tumor suppressor role [13]. We now confirm the tumor suppressor role Cdh11 in retinoblastoma through functional experiments. The 97kD Cdh11 isoform that is retained in the Cdh11 knockout model we studied, has been documented to lack adhesion properties and thus likely represents a non-functional protein [33], [34]. By crossing this CDH11 functional knockout with the TAg-RB mice, we report an unexpected result: Cdh11 allelic loss results in fewer tumor initiating TAg positive cells at PND8 (Figure 5), and consequently fewer multifocal tumors at PND28 (Figure 6) compared to animals with normal Cdh11 alleles. This suggests that TAg transgene expression may be affected by the loss of Cdh11 (Figure 5).
Loss of Cdh11 in Cdh11-/- mice did not affect retinal size up to PND60 (Figure 3). At PND8 retinal volumes were similar in Cdh11+/+;TAg+/-, Cdh11+/+;TAg+/-, and Cdh11+/+;TAg+/- mice, but at PND84, the total retinal size was significantly larger (p = 0.01) in the Cdh11-/-;TAg+/- mice compared to Cdh11+/+;TAg+/- mice (Figure 7B), suggesting that loss of Cdh11, when combined with the expression of TAg, affects the overall size of the adult retina in TAg-RB mice. Our previous studies of the Cdh11-/- retina, quantifying the individual retinal cell types visualized by immunofluorescence with cell-specific antibodies, showed no difference between the Cdh11-/- and wild type retina [35].
At PND84, we show that absolute tumor volume was not statistically different between all three genotypes (total tumor volume alone or as a percentage of the retinal volume). However, since these tumors arise from fewer tumor-initiating cells, we conclude that tumor growth per initiating cell was greater in mice with mutant Cdh11 alleles (Figure 7B and 7C, Figure 8A). We conclude that Cdh11 functions as a tumor suppressor. Since tumor “growth” results from an imbalance between cell death and proliferation, we examined cell proliferation (Figure 8B) and cell death (Figure 8C) in TAg-RB tumors of mice with normal Cdh11 alleles versus mutated Cdh11 alleles. Our data indicate that when Cdh11 is lost, cell death is deficient while proliferation remains unchanged, suggesting that the tumor suppressor function of Cdh11 is mediated through promotion of apoptosis rather than inhibition of cell proliferation. This is further supported by our in vitro data showing significant decrease in caspase-3 and increase in β-catenin expression in Cdh11 knockdown experiments using siRNA (Figure 8D and Figure S2A, S2B), while proliferation markers PCNA and Ki67 remain unchanged (Figure S3).
The spread of tumor volumes across the various time points is narrowed in mice that have lost both Cdh11 alleles. We speculate that tumors in mice with normal Cdh11 alleles could be losing functional Cdh11 at varying timepoints during tumor development, and the wide spread in tumor volume reflects heterogeneity for Cdh11. In contrast, mice with both Cdh11 alleles mutated have more consistent measures of cell death (Figure 8C). This agrees with our previous report where some tumors display loss of Cdh11, while others retain it at later timepoints [13]. In summary, we describe a mechanism by which Cdh11 may be functioning as a tumor suppressor gene in retinoblastoma.
Additional experiments need to be performed to assess the mechanism by which Cdh11 facilitates cell death in these tumors. Our preliminary experiments have shown increased protein and mRNA levels of β-catenin when Cdh11 is knocked down, and increased β-catenin mRNA in PND84 Cdh11-/-TAg+/- mice relative to Cdh11+/+TAg+/- mice (Figure S2A, S2B, S2C). Upon cell-cell contact, cadherin molecules form the adherens junction. The cadherin binds directly to β-catenin, which recruits α-catenin to link the complex to the cytoskeleton. This is necessary to maintain cell-cell adhesion and cellular architecture [36]. These junctions are dynamic and the structure and signaling provided by the complex ultimately determines the cellular phenotype and behavior [37]. β-catenin is additionally a major regulator of the Wnt signaling pathway. The Wnt-signaling pathway is implicated in other cancers [38], [39] and suppresses apoptosis through both β-catenin dependent and independent pathways [40]. Many studies have shown that cadherin protein levels impact canonical Wnt-signaling and β-catenin levels. Gain and loss of function studies support cadherins directly sequestering β-catenin from the nucleus, acting as a sink for the cytosolic pool [41]–[43]. Additionally, downregulation of E-cadherin expression has been paralleled with an upregulation of β-catenin in hepatocellular carcinoma tumors [44]. Next investigations would test the possibility that down regulation of cadherin-11 affects the levels of canonical Wnt signaling in these TAg-RB cell lines, that may lead to the decrease in cell death and faster growing tumors.
Cdh11 supports the tumor initiating cell in the TAg-RB mouse model
Previous studies of cell adhesion molecules in the neural retina have described that expression of cadherin subtypes is restricted to different retinal cell populations. Based on these studies the authors suggested that cadherins play a role in maintaining selective neuronal associations [45], [46]. In order to understand the role of Cdh11 in retinoblastoma progression, we examined its presence during healthy retinal development.
We showed that Cdh11 is developmentally regulated. Expression was restricted to differentiating/migrating retinal cells at E18.5 through to PND6, and to the INL at PND60 (adult) (Figure 1). Cadherin-11 co-expresses with markers of Müller glia cell bodies and processes that span the entire retina (Figure 1B and Figure 2B). Prominent expression of cadherin-11 by retinoblasts at PND3 and PND6 in the developing retina and co-expression with Müller glia and horizontal cell types, suggests roles for cadherin-11 in morphogenesis, such as cell migration, sorting or positioning of these cells (Figure 1A) during retinal development.
The tumor -initiating cell in the TAg-RB mouse model has been identified to belong to a subset of the Müller glia (unpublished data). Our results indicate that when Cdh11 alleles are mutated in TAg-RB mice, fewer cells express TAg and develop into retinoblastoma. It is possible that Cdh11 loss affects the expression of the TAg transgene in this murine model, or that it affects development of the subpopulation of Müller glia that gives rise to the TAg-RB tumours. We were unable to discern the latter, since Cdh11-/- mice do not show a significant change in retinal cell type distribution in the retina, and so few of this retinal subtype express TAg in this model (unpublished data). From these data, we suggest that Cdh11 has an important role in the expression of TAg from the transgene in this murine model.
Summary and significance
We describe the use of the retinoblastoma TAg-RB mouse model to study specific gene function in tumor development. This was achieved by crossing TAg-RB mice to Cdh11-/- mice. We showed that Cdh11 is a suppressor of retinoblastoma progression by using a unique and highly sensitive method to identify and quantify tumor volume. Although fewer multifocal tumors initiate in mice with mutant Cdh11 alleles, suggesting that Cdh11 loss modulates the number of TAg-espressing cells in this murine model, the resulting tumors grow faster, describing a tumor suppressor role for Cdh11 in retinoblastoma progression. Significantly reduced numbers of cells stained for pro-apoptotic proteins in tumors of mice with absent Cdh11 alleles, indicating that promotion of cell death is an important part of the tumor suppressor action of Cdh11.
Materials and Methods
Animals
All animals were maintained and sacrificed using protocols approved by the Animal Care Committee of the Ontario Cancer Institute (OCI) which adhere to the EC Directive 86/609/EEC for animal experiments.
Cdh11-/- mice, background strain 129, were provided by Dr. M. Takeichi [33]. To study the role of Cdh11 in retinal development, one-generation crosses were made between Cdh11-/- 129 and Cdh11+/+ C57-Bl-6 to get a mixed background of 129/C57Bl-6. Littermates, Cdh11+/+, Cdh11+/- and Cdh11-/- on this 129/C57Bl-6, mixed background were sacrificed at developmental time points: embryonic day (ED)18.5, post-natal day (PND)3, PND6, PND15 and PND60. To analyze proliferating cells, pregnant mothers at ED18.5, pups at PND3 and PND6, and adults at PND84 were injected with bromodeoxyuridine (BrdU) reagent (5-bromo-2′-deoxyuridine and 5-fluoro-2′-deoxyuridine, 10∶1, used at 1 ml reagent per 100 g body weight, Cat# 00-0103, Lot# 60203722, Zymed Laboratories) for 2 hours and then sacrificed.
TAg-RB (TAg+/-), background strain C57/Bl-6, mice were provided as a gift from Joan O'Brien [16]. One generation crosses were made between Cdh11-/- and TAg+/- mice to get double heterozygotes, Cdh11+/-; TAg+/-, on a 129/C56/Bl-6 mixed background. Mice were further crossed with Cdh11-/-, Cdh11+/- or Cdh11+/+ of a 129/C56Bl-6 mixed background to get the three genotypes analyzed for this study: Cdh11+/+;TAg+/-, Cdh11+/-;TAg+/- and Cdh11-/-;TAg+/-. These animals were sacrificed at PND8, PND28 and PND84, the latest time point we could study in compliance with our Animal Protocol at the Ontario Cancer Institute.
Genotyping of Cdh11-/- mice and littermates were carried out using PCR conditions: 94°C, 2 min, 1 cycle, [94°C, 30 sec, 50°C, 30 sec, 72°C 30 sec] 30 cycles, 72°C 10 min, and 4°C cool block. Primers used were: forward, 5′ to 3′ (21 bp): ttc agt cgg cag aag cag gac and backward, 5′ to 3′ (19 bp): gtg tat tgg ttg cac cat g, and neo, 5′ to 3′ (23 bp): tct atc gcc ttc ttg acg agt tc. Sizes of expected PCR products were: Cdh11+/+: 240 bp, Cdh11+/-: 480 bp and 240 bp, and Cdh11-/-: 480 bp. Genotyping of TAg+/- mice and their littermates were carried out using similar PCR conditions: 94°C, 2 min, 1cycle, [94°C, 1 min 58°C, 1 min, 72°C 1 min] 30 cycles, 72°C 10 min, 1 cycle and 4°C cool block. Primers used were: forward 5′ to 3′: gac ttt gga ggc ttc tgg gat gca act gag and backward 5′ to 3′: ggc att cca cca ctg ctc cca ttc atc agt. Size of expected PCR product was 420 bp.
Histology and slide selection
Heads and/or eyes were fixed in freshly prepared 4% PFA/PBS for 48 hrs and then stored in 70% Ethanol. Heads were decalcified (8% formic acid following 4% PFA) for approximately 1 week. Both heads and/or eyes were paraffin embedded and 5 µm sectioned.
For Cdh11+/+, Cdh11+/- and Cdh11-/- littermates: Serial sections were made specifically through the papillary-optic nerve plane (approx. 20 sections in total) for consistent comparison between genotypes.
For Cdh11+/+;TAg+/-, Cdh11+/-;TAg+/- and Cdh11-/-;TAg+/- mice: Serial sections were made through the entire eye (approximately 270–420 sections per eye with 5–7 sections made per slide). To estimate tumor volume per eye, we selected one slide every 60th section (approx. one slide every 300 µm) for analysis. A total of about 5–8 slides were analyzed per eye. Only one eye was analyzed per mouse.
Immunohistochemistry
Slides selected for analysis were studied using the immunohistochemical protocol described previously[12]. Briefly, slides were incubated with primary, then biotinylated secondary antibodies, either anti-mouse, anti-rabbit, anti-goat, or anti-sheep, used at a dilution of 1∶200 with 10% DakoCytomation Antibody Diluent in 1% BSA/TBST for 1 hr at room temperature. To visualize TAg, BrdU and Brn3b (ganglion) stained cells, we employed an Immunopure DAB Substrate Kit (Cat# 34065, Pierce). After incubation with primary and biotinylated secondary antibodies, slides were incubated for 1 hr at room temperature in an ABC prepared solution (Vectastain ABC Elite, Vector Laboratories). Stained cells could be visualized after a maximum of half an hour incubation in DAB substrate solution (Pierce) prepared fresh with 10% DAB/Metal Concentrate, 10× (Product# 1856090) made in Stable Peroxide Substrate Buffer, 1× (Product# 1855910). All other proteins were visualized by immunofluorescence; after incubation with primary and secondary antibodies, slides were washed in 1×TBS and then incubated with Streptavidin-Alexa488 or Streptavidin-Alexa594, used at 1∶200, prepared in 1×TBS for 15 min at room temperature. Slides were washed briefly in 1×TBS and incubated in 4′, 6-diamino-2-phenylindole (DAPI) used at 1∶50, followed by wash in 1×TBS and mouniting with DakoCytomation Fluorescent Mounting Medium (S3023). Selected slides were Haematoxylin and eosin (H&E) stained for light microscopy analysis. Table 1 provides a complete list of all antibodies used. Antibodies to recognize specific cell types were: Hes-5 [47], CRALBP [48] and glutamine synthetase [49] (early Müller glia, Müller glia cell bodies and processes), syntaxin [50] (HPC-1 for amacrine cells), neurofilament 160kDa [51] (horizontal cells), Brn3b [52] (ganglion cells) and Chx-10[53] (bipolar cells).

Image analysis and quantification of tumor volume in mouse retina
Of the techniques described to measure tumor volume in murine retinoblastoma, none are useful to quantify small, developing tumors at very early time points [20]–[22],[24],[25]. Therefore, we developed a novel technique to quantitate tumor volume in the eyes of TAg-RB mice by analyzing every 60th section through the entire eye [32]. Tumor development was tracked by staining for TAg. Diamino benzidine (DAB) typically stains TAg cells brown with very little background, however in some cases, background staining is visible in the GCL and retinal pigment epithelium (Figure 4, Figure 5, and Figure 6). Total tumor area per eye was quantified as a percentage of total retinal area (measured in pixels) using Image J software. The selected sections were scanned at the Advanced Optical Microscopy Facility at the Ontario Cancer Institute using an Aperio ScanScope CS. Images were retrieved using ImageScope software and analyzed as a TIFF image using public domain image software: ImageJ: Image Processing and Analysis in Java available from http://rsb.info.nih.gov/ij/. Retinas were manually traced for each eye and area was measured in pixels. For time point, PND8, TAg positive cells in the retina were manually counted under a 40× inverted microscope (Leica DMLB) and for PND28 and PDN84, the traced retinas were converted into an 8-bit format, and using a manually selected threshold tool, the tumor area (DAB stained) within the selected retina was highlighted and measured by the program in pixels. Total retina and tumor areas of all 5–8 analyzed sections per retina in one eye per animal were estimated calculating for percent tumor area [(tumor area in pixels/retina area in pixels) * 100]. For PND8, number of tumor cells per retinal area was used instead. BrdU positive cells were measured in pixels and quantified as an average/tumor area at PND84. Positively stained apoptotic cells were also analyzed at PND84 and manually counted per section obtaining an average number per section.
Statistical analysis
Five animals per genotype were analyzed at PND8 and PND28. Seven animals of Cdh11+/+;TAg+/- genotype, eight animals of Cdh11+/-;TAg+/- genotype, and ten animals of Cdh11-/-;TAg+/- genotype, were analyzed at PND84. The Kruskal-Wallis (K-W) Test was the main statistical method used to investigate differences in tumor and retinal size between the genotypes at various ages. Statistical analyses were performed using SAS version 9.1 (SAS Institute, Cary, NC). All tests are two-sided and p-values equal or less than 0.05 were considered statistically significant.
Cell lines and siRNA knockdown experiments
Cdh11 was knocked down in the TAg-RB derived cell line T+539 using three different stealth siRNAs: MSS202865 (siRNA #1), MSS202866 (siRNA #2) and MSS202867 (siRNA #3) (Invitrogen Cat# 1320003), using GL-2 vector siRNA (Qiagen) as a control. T+539 cells were transfected in triplicate with the siRNA at time of plating, using media without the addition of penicillin and streptomycin. The procedure included transfection of 125 pmol of each siRNA oligo in Lipofectamine 2000 (Invitrogen), in a total of 2 ml plating medium. Cells were incubated for 24 hrs, 48 hrs, 72 hrs, 5 days, 7 days or 10 days. Knockdown was confirmed by immunoblot or RT-PCR for Cdh11 (see below). Ideal inhibition was achieved 7 and 10 days post-transfection.
RNA isolation and RT–PCR
RNA was isolated from the T+539 cell lines using the Trizol method. RNA was isolated from paraffin embedded tissue using modified GTC (guanidine isothyocionate)/ proteinase K protocol. In short tissue was deparafinized through series of incubation in xylene and 100% ethanol followed by incubation in 1M GTC/6 mg/ml proteinase K solution for 6 hrs. GTC/proteinase K was removed by phenol extraction and RNA was precipitated by equal volume of isopropanol.
Primers used for RT-PCR analysis were as follows: mCdh11: forward: 5′ atg agc ctc cca tgt tct tg 3′, and reverse: 5′ggg tga tcg ctc tca cag at 3′; mKi67: forward: 5′ agc ctg tga ggc tga gac at 3′, and reverse: 5′ ttt ctg cca gtg tgc tgt tc 3′; mPCNA: forward: 5′gaa ggc ttc gac aca tac cg 3′ , and reverse: 5′ cag cat ctc caa tgt ggc ta 3′; mTBP: forward: 5′ agc aac tgc agc agc ctc agt aca 3′, and reverse: 5′ tct tcc tga atc cct tta aga tg 3′; mb-catenin: forward: 5′ caa gat gat ggt gtg cca ag 3′, and reverse: 5′ ctg cac aaa caa tgg aat gg 3′.
Protein isolation and immunoblot
Protein isolation and immunoblot analysis were performed as described previously [13]. Dilutions for cadherin-11, caspase-3 and β-catenin antibodies used in immunoblot analysis are included in Table 1.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. GallieBL
CampbellC
DevlinH
DuckettA
SquireJA
1999 Developmental basis of retinal-specific induction of cancer by RB mutation. Cancer Res 59 1731s 1735s
2. DimarasH
CoburnB
PajovicS
GallieBL
2006 Loss of p75 neurotrophin receptor expression accompanies malignant progression to human and murine retinoblastoma. Mol Carcinog 45 333 343
3. DimarasH
KhetanV
HallidayW
OrlicM
PrigodaNL
2008 Loss of RB1 induces non-proliferative retinoma; increasing genomic instability correlates with progression to retinoblastoma. Hum Mol Genet
4. SquireJ
GallieBL
PhillipsRA
1985 A detailed analysis of chromosomal changes in heritable and non-heritable retinoblastoma. Hum Genet 70 291 301
5. ChenD
GallieBL
SquireJA
2001 Minimal regions of chromosomal imbalance in retinoblastoma detected by comparative genomic hybridization. Cancer Genet Cytogenet 129 57 63
6. HerzogS
LohmannDR
BuitingK
SchulerA
HorsthemkeB
2001 Marked differences in unilateral isolated retinoblastomas from young and older children studied by comparative genomic hybridization. Hum Genet 108 98 104
7. LillingtonDM
KingstonJE
CoenPG
PriceE
HungerfordJ
2003 Comparative genomic hybridization of 49 primary retinoblastoma tumors identifies chromosomal regions associated with histopathology, progression, and patient outcome. Genes Chromosomes Cancer 36 121 128
8. MairalA
PinglierE
GilbertE
PeterM
ValidireP
2000 Detection of chromosome imbalances in retinoblastoma by parallel karyotype and CGH analyses. Genes Chromosomes Cancer 28 370 379
9. van der WalJE
HermsenMA
GilleHJ
Schouten-Van MeeterenNY
MollAC
2003 Comparative genomic hybridisation divides retinoblastomas into a high and a low level chromosomal instability group. J Clin Pathol 56 26 30
10. ZielinskiB
GratiasS
ToedtG
MendrzykF
StangeDE
2005 Detection of chromosomal imbalances in retinoblastoma by matrix-based comparative genomic hybridization. Genes Chromosomes Cancer 43 294 301
11. CorsonTW
HuangA
TsaoMS
GallieBL
2005 KIF14 is a candidate oncogene in the 1q minimal region of genomic gain in multiple cancers. Oncogene 24 4741 4753
12. OrlicM
SpencerCE
WangL
GallieBL
2006 Expression analysis of 6p22 genomic gain in retinoblastoma. Genes Chromosomes Cancer 45 72 82
13. MarchongMN
ChenD
CorsonTW
LeeC
HarmandayanM
2004 Minimal 16q genomic loss implicates cadherin-11 in retinoblastoma. Mol Cancer Res 2 495 503
14. GrasemannC
GratiasS
StephanH
SchulerA
SchrammA
2005 Gains and overexpression identify DEK and E2F3 as targets of chromosome 6p gains in retinoblastoma. Oncogene 24 6441 6449
15. BowlesE
CorsonTW
BayaniJ
SquireJA
WongN
2007 Profiling genomic copy number changes in retinoblastoma beyond loss of RB1. Genes Chromosomes Cancer 46 118 129
16. WindleJJ
AlbertDM
O'BrienJM
MarcusDM
DistecheCM
1990 Retinoblastoma in transgenic mice. Nature 343 665 669
17. GratiasS
SchulerA
HitpassLK
StephanH
RiederH
2005 Genomic gains on chromosome 1q in retinoblastoma: consequences on gene expression and association with clinical manifestation. Int J Cancer 116 555 563
18. LaurieN
MohanA
McEvoyJ
ReedD
ZhangJ
2009 Changes in Retinoblastoma Cell Adhesion Associated with Optic Nerve Invasion. Mol Cell Biol
19. DimarasH
GallieBL
2008 The p75 NTR neurotrophin receptor is a tumor suppressor in human and murine retinoblastoma development. Int J Cancer 122 2023 2029
20. AlbertDM
KumarA
StrugnellSA
DarjatmokoSR
LokkenJM
2004 Effectiveness of vitamin D analogues in treating large tumors and during prolonged use in murine retinoblastoma models. Arch Ophthalmol 122 1357 1362
21. DawsonDG
GleiserJ
ZimbricML
DarjatmokoSR
LindstromMJ
2003 Toxicity and dose-response studies of 1-alpha hydroxyvitamin D2 in LH-beta-tag transgenic mice. Ophthalmology 110 835 839
22. Escalona-BenzE
JockovichME
MurrayTG
HaydenB
HernandezE
2005 Combretastatin A-4 prodrug in the treatment of a murine model of retinoblastoma. Invest Ophthalmol Vis Sci 46 8 11
23. MurrayTG
CicciarelliN
O'BrienJM
HernandezE
MuellerRL
1997 Subconjunctival carboplatin therapy and cryotherapy in the treatment of transgenic murine retinoblastoma. Arch Ophthalmol 115 1286 1290
24. TongCT
HowardSA
ShahHR
Van QuillKR
LinET
2005 Effects of celecoxib in human retinoblastoma cell lines and in a transgenic murine model of retinoblastoma. Br J Ophthalmol 89 1217 1220
25. Van QuillKR
DioguardiPK
TongCT
GilbertJA
AabergTMJr
2005 Subconjunctival carboplatin in fibrin sealant in the treatment of transgenic murine retinoblastoma. Ophthalmology 112 1151 1158
26. ChenD
Livne-barI
VanderluitJL
SlackRS
AgochiyaM
2004 Cell-specific effects of RB or RB/p107 loss on retinal development implicate an intrinsically death-resistant cell-of-origin in retinoblastoma. Cancer Cell 5 539 551
27. MacPhersonD
ConkriteK
TamM
MukaiS
MuD
2007 Murine bilateral retinoblastoma exhibiting rapid-onset, metastatic progression and N-myc gene amplification. Embo J 26 784 794
28. MacPhersonD
SageJ
KimT
HoD
McLaughlinME
2004 Cell type-specific effects of Rb deletion in the murine retina. Genes Dev 18 1681 1694
29. Robanus-MaandagE
DekkerM
van der ValkM
CarrozzaML
JeannyJC
1998 p107 is a suppressor of retinoblastoma development in pRb-deficient mice. Genes Dev 12 1599 1609
30. ZhangJ
SchweersB
DyerMA
2004 The first knockout mouse model of retinoblastoma. Cell Cycle 3 952 959
31. HoffmannI
BallingR
1995 Cloning and expression analysis of a novel mesodermally expressed cadherin. Dev Biol 169 337 346
32. DimarasH
MarchongMN
GallieBL
2009 Quantitative analysis of tumor size in a murine model of retinoblastoma. Ophthalmic Genet 30 84 90
33. HorikawaK
RadiceG
TakeichiM
ChisakaO
1999 Adhesive subdivisions intrinsic to the epithelial somites. Dev Biol 215 182 189
34. KawaguchiJ
AzumaY
HoshiK
KiiI
TakeshitaS
2001 Targeted disruption of cadherin-11 leads to a reduction in bone density in calvaria and long bone metaphyses. J Bone Miner Res 16 1265 1271
35. MarchongM
2007 Identification of CDH11 as a Candidate Tumor Suppressor in Retinoblastoma and Characterization of its role in Retina and Retinoblastoma. Toronto University of Toronto
36. AberleH
SchwartzH
KemlerR
1996 Cadherin-catenin complex: protein interactions and their implications for cadherin function. J Cell Biochem 61 514 523
37. WheelockMJ
JohnsonKR
2003 Cadherins as modulators of cellular phenotype. Annu Rev Cell Dev Biol 19 207 235
38. KinzlerKW
VogelsteinB
1996 Lessons from hereditary colorectal cancer. Cell 87 159 170
39. MorinPJ
SparksAB
KorinekV
BarkerN
CleversH
1997 Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science 275 1787 1790
40. AlmeidaM
HanL
BellidoT
ManolagasSC
KousteniS
2005 Wnt proteins prevent apoptosis of both uncommitted osteoblast progenitors and differentiated osteoblasts by beta-catenin-dependent and -independent signaling cascades involving Src/ERK and phosphatidylinositol 3-kinase/AKT. J Biol Chem 280 41342 41351
41. CoxRT
KirkpatrickC
PeiferM
1996 Armadillo is required for adherens junction assembly, cell polarity, and morphogenesis during Drosophila embryogenesis. J Cell Biol 134 133 148
42. FagottoF
FunayamaN
GluckU
GumbinerBM
1996 Binding to cadherins antagonizes the signaling activity of beta-catenin during axis formation in Xenopus. J Cell Biol 132 1105 1114
43. HeasmanJ
CrawfordA
GoldstoneK
Garner-HamrickP
GumbinerB
1994 Overexpression of cadherins and underexpression of beta-catenin inhibit dorsal mesoderm induction in early Xenopus embryos. Cell 79 791 803
44. KanaiY
UshijimaS
HuiAM
OchiaiA
TsudaH
1997 The E-cadherin gene is silenced by CpG methylation in human hepatocellular carcinomas. Int J Cancer 71 355 359
45. Faulkner-JonesBE
GodinhoLN
ReeseBE
PasquiniGF
RuefliA
1999 Cloning and expression of mouse Cadherin-7, a type-II cadherin isolated from the developing eye. Mol Cell Neurosci 14 1 16
46. HonjoM
TaniharaH
SuzukiS
TanakaT
HondaY
2000 Differential expression of cadherin adhesion receptors in neural retina of the postnatal mouse. Invest Ophthalmol Vis Sci 41 546 551
47. HojoM
OhtsukaT
HashimotoN
GradwohlG
GuillemotF
2000 Glial cell fate specification modulated by the bHLH gene Hes5 in mouse retina. Development 127 2515 2522
48. EisenfeldAJ
Bunt-MilamAH
SaariJC
1985 Localization of retinoid-binding proteins in developing rat retina. Exp Eye Res 41 299 304
49. LinserP
MosconaAA
1981 Carbonic anhydrase C in the neural retina: transition from generalized to glia-specific cell localization during embryonic development. Proc Natl Acad Sci 78 7190 7194
50. InoueA
ObataK
AkagawaK
1992 Cloning and sequence analysis of cDNA for a neuronal cell membrane antigen, HPC-1. J Biol Chem 267 10613 10619
51. VaughanDK
LasaterEM
1990 Glial and neuronal markers in bass retinal horizontal and Muller cells. Brain Res 537 131 140
52. FedtsovaNG
TurnerEE
1995 Brn-3.0 expression identifies early post-mitotic CNS neurons and sensory neural precursors. Mech Dev 53 291 304
53. LiuIS
ChenJD
PloderL
VidgenD
van der KooyD
1994 Developmental expression of a novel murine homeobox gene (Chx10): evidence for roles in determination of the neuroretina and inner nuclear layer. Neuron 13 377 393
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2010 Číslo 4
Nejčtenější v tomto čísle
- Whole-Genome SNP Association in the Horse: Identification of a Deletion in Myosin Va Responsible for Lavender Foal Syndrome
- Admixture Mapping Scans Identify a Locus Affecting Retinal Vascular Caliber in Hypertensive African Americans: the Atherosclerosis Risk in Communities (ARIC) Study
- Genetic Tests for Ecological and Allopatric Speciation in Anoles on an Island Archipelago
- Human Telomeres Are Hypersensitive to UV-Induced DNA Damage and Refractory to Repair