
Genome-Wide Scan Identifies , , and as Novel Risk Loci for Systemic Sclerosis
Systemic sclerosis (SSc) is an orphan, complex, inflammatory disease affecting the immune system and connective tissue. SSc stands out as a severely incapacitating and life-threatening inflammatory rheumatic disease, with a largely unknown pathogenesis. We have designed a two-stage genome-wide association study of SSc using case-control samples from France, Italy, Germany, and Northern Europe. The initial genome-wide scan was conducted in a French post quality-control sample of 564 cases and 1,776 controls, using almost 500 K SNPs. Two SNPs from the MHC region, together with the 6 loci outside MHC having at least one SNP with a P<10−5 were selected for follow-up analysis. These markers were genotyped in a post-QC replication sample of 1,682 SSc cases and 3,926 controls. The three top SNPs are in strong linkage disequilibrium and located on 6p21, in the HLA-DQB1 gene: rs9275224, P = 9.18×10−8, OR = 0.69, 95% CI [0.60–0.79]; rs6457617, P = 1.14×10−7 and rs9275245, P = 1.39×10−7. Within the MHC region, the next most associated SNP (rs3130573, P = 1.86×10−5, OR = 1.36 [1.18–1.56]) is located in the PSORS1C1 gene. Outside the MHC region, our GWAS analysis revealed 7 top SNPs (P<10−5) that spanned 6 independent genomic regions. Follow-up of the 17 top SNPs in an independent sample of 1,682 SSc and 3,926 controls showed associations at PSORS1C1 (overall P = 5.70×10−10, OR:1.25), TNIP1 (P = 4.68×10−9, OR:1.31), and RHOB loci (P = 3.17×10−6, OR:1.21). Because of its biological relevance, and previous reports of genetic association at this locus with connective tissue disorders, we investigated TNIP1 expression. A markedly reduced expression of the TNIP1 gene and also its protein product were observed both in lesional skin tissue and in cultured dermal fibroblasts from SSc patients. Furthermore, TNIP1 showed in vitro inhibitory effects on inflammatory cytokine-induced collagen production. The genetic signal of association with TNIP1 variants, together with tissular and cellular investigations, suggests that this pathway has a critical role in regulating autoimmunity and SSc pathogenesis.
Published in the journal:
. PLoS Genet 7(7): e32767. doi:10.1371/journal.pgen.1002091
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002091
Summary
Systemic sclerosis (SSc) is an orphan, complex, inflammatory disease affecting the immune system and connective tissue. SSc stands out as a severely incapacitating and life-threatening inflammatory rheumatic disease, with a largely unknown pathogenesis. We have designed a two-stage genome-wide association study of SSc using case-control samples from France, Italy, Germany, and Northern Europe. The initial genome-wide scan was conducted in a French post quality-control sample of 564 cases and 1,776 controls, using almost 500 K SNPs. Two SNPs from the MHC region, together with the 6 loci outside MHC having at least one SNP with a P<10−5 were selected for follow-up analysis. These markers were genotyped in a post-QC replication sample of 1,682 SSc cases and 3,926 controls. The three top SNPs are in strong linkage disequilibrium and located on 6p21, in the HLA-DQB1 gene: rs9275224, P = 9.18×10−8, OR = 0.69, 95% CI [0.60–0.79]; rs6457617, P = 1.14×10−7 and rs9275245, P = 1.39×10−7. Within the MHC region, the next most associated SNP (rs3130573, P = 1.86×10−5, OR = 1.36 [1.18–1.56]) is located in the PSORS1C1 gene. Outside the MHC region, our GWAS analysis revealed 7 top SNPs (P<10−5) that spanned 6 independent genomic regions. Follow-up of the 17 top SNPs in an independent sample of 1,682 SSc and 3,926 controls showed associations at PSORS1C1 (overall P = 5.70×10−10, OR:1.25), TNIP1 (P = 4.68×10−9, OR:1.31), and RHOB loci (P = 3.17×10−6, OR:1.21). Because of its biological relevance, and previous reports of genetic association at this locus with connective tissue disorders, we investigated TNIP1 expression. A markedly reduced expression of the TNIP1 gene and also its protein product were observed both in lesional skin tissue and in cultured dermal fibroblasts from SSc patients. Furthermore, TNIP1 showed in vitro inhibitory effects on inflammatory cytokine-induced collagen production. The genetic signal of association with TNIP1 variants, together with tissular and cellular investigations, suggests that this pathway has a critical role in regulating autoimmunity and SSc pathogenesis.
Introduction
Systemic sclerosis (MIM181750) is a connective tissue disease characterized by generalized microangiopathy, severe immunologic alterations and massive deposits of matrix components in the connective tissue. Being an orphan disease, SSc presents a major medical challenge and is recognized as the most severe connective tissue disorder with high risk of premature deaths [1]. Epidemiological data on SSc vary in different parts of the world and depend on selection criteria for the study population. Inasmuch, the prevalence of the disease fluctuates across global regions and population-based studies result in higher prevalence than do hospital records-based studies. In North America, the prevalence of SSc has been reported as 0.7–2.8 per 10,000 in a Canadian study, whereas in the U.S. figures of 2.6 per 10,000 versus 7.5 per 10,000 were reported by medical records - versus population-based studies, respectively. In Europe, a prevalence of 1.6 per 10,000 was reported in Denmark, 3.5 per 10,000 in Estonia, 1.58 per 10,000 adults (95% confidence interval, 129–187) in Seine-Saint-Denis in France [2]–[4]. The risk of SSc is increased among first-degree relatives of patients, compared to the general population. In a study of 703 families in the US, including 11 multiplex SSc families, the familial relative risk in first-degree relatives was about 13, with a 1.6% recurrence rate, compared to 0.026% in the general population [5]. The sibling risk ratio was about 15 (ranging from 10 to 27 across cohorts). The only twin study reported to date included 42 twin pairs [6]. The data showed a similar concordance rate in monozygotic twins (4.2%, n = 24) and dizygotic twins (5.6%) (NS) and an overall cross-sectional concordance rate of 4.7%. However, concordance for the presence of antinuclear antibodies was significantly higher in the monozygotic twins (90%) than in the dizygotic twins (40%) suggesting that genetics may be important for the auto-immune part of the disease.
The aetiology of SSc is still unclear but some key steps have been described, in particular early endothelial damage and dysregulation of the immune system with abnormal autoantibody production [7]. At the cellular level, early events include endothelial injury and perivascular inflammation with the release of a large array of inflammatory mediators [8], [9]. In the advanced stage, a progressive activation of fibroblasts in the skin and in internal organs leads to hyperproduction of collagen and irreversible tissue fibrosis [9]. Epidemiological investigations indicate that SSc follows a pattern of multifactorial inheritance [10]. Previous candidate-gene association studies have only identified a handful of SSc risk loci, most contributing to the genetic susceptibility of other autoimmune diseases [9]–[16]. So far, two genome-wide association studies of SSc have been conducted [17], [18]. The studies differ according to the ancestry of the studied population (Korean vs US/European) and the genome-wide association data: map density (∼440 K vs 280 K SNPs) and sample size (∼700 vs ∼7300 subjects). They provided evidence of association with known MHC loci, but only one ‘new’ locus was identified at CD247 in the US/European dataset, variants at CD247 being known to contribute to the susceptibility of systemic lupus erythematosus [18].
The diagnosis of SSc is based on recognized clinical criteria established decades ago however, these do not include specific autoantibodies or recent tools for assessment of the disease [19], [20]. Therefore, phenotypic heterogeneity is a concern for SSc and genetic heterogeneity is also highly probable with regards to data obtained in other connective tissue disorders. Given these considerations, and previous findings in other autoimmune diseases, it is apparent that additional risk variants for SSc remain to be discovered. Therefore, to identify further common variants that contribute to SSc risk in the European population, we conducted a two-stage GWAS, in two case-control samples (total >8,800 subjects).
Results/Discussion
We established a collaborative consortium including groups from 4 European countries (France, Italy, Germany and Eastern-Europe) from which we were able to draw upon a combined sample of over 8,800 subjects (before quality control) and conducted a two-stage genome-wide association study. In stage 1, we genotyped 1,185 samples on Illumina Human610-Quad BeadChip and genotypes obtained using the same chip from 2,003 control subjects were made available to us from the 3C study [21], [22]. After stringent quality control, we finally tested for association in stage-1, 489,814 autosomal SNPs in 2,340 subjects (564 cases and 1,776 controls) (Table 1). We tested for association between each SNP and SSc using the logistic regression association test, assuming additive genetic effects. The quantile-quantile plot and estimation of the genomic inflation factor (λ = 1.035) indicated minimal overall inflation (Figure 1A). The genome-wide logistic association results are presented in Figure 1B. Table S1 provides details for all SNPs with P<10−4, including one SNP exceeding P<10−7, the Bonferroni threshold for genome-wide significance. The three top SNPs were located on 6p21, in the HLA-DQB1 gene: rs9275224, P = 9.18×10−8, OR = 0.69, 95%CI[0.60–0.79]; rs6457617, P = 1.14×10−7 and rs9275245, P = 1.39×10−7 (Figure 1B and Table 2). Several associated SNPs in HLA-DQB1 have already been reported but rs6457617 was also identified as the most associated SNP in the previous US/European GWAS study [18]. Of note, the three SNPs in HLA-DQB1 are in strong LD (r2>0.97). Within the MHC region, the next most associated SNP (rs3130573, P = 1.86×10−5, OR = 1.36[1.18–1.56]) is located in the psoriasis susceptibility 1 candidate 1 (PSORS1C1) gene (Table 2), a candidate gene for psoriasis [23]. Conditional analyses of susceptibility variants within MHC showed that there were two independent association signals at rs6457617 (HLA-DQB1) and at rs3130573 (PSORS1C1). Indeed, the association at PSORS1C1 remained significant (P<2.1×10−5) after controlling for the association at HLA-DQB1 and the association at HLA-DQB1 remained also significant (P<1.5×10−7) after controlling for the association at PSORS1C1 (Table S2).
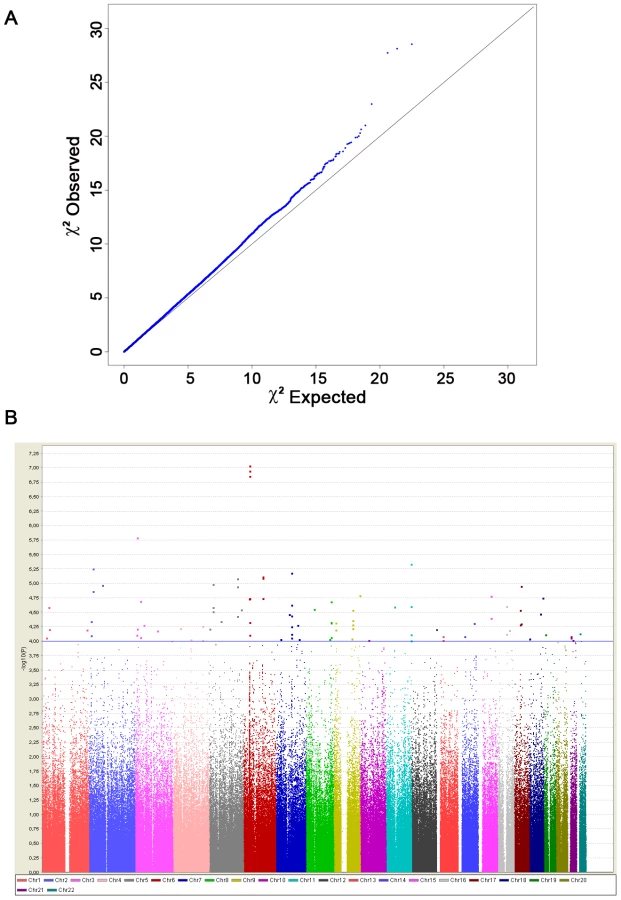


Outside the MHC region, our GWAS analysis revealed 7 top SNPs (P<10−5) that spanned 6 independent genomic regions (Figure 1B and Table S1). Conditional analyses of each of them on HLA-DQB1 showed no significant drop in the association signals (Table S3). The 6 loci having at least one SNP with a P<10−5 were selected for follow-up analysis. Within each locus we selected the SNPs with the strongest (P<10−4) association signals to be genotyped in a post-QC replication sample of 1,682 SSc cases and 3,926 controls (Table 1). To this list we added two top SNPs in HLA-DQB1 and the SNP in PSORS1C1. Finally, we further included 4 SNPs at the two known loci (STAT4 and TNPO3-IRF5) and at the newly identified locus (CD247) by Radstake et al [18]. Out of a total set of 21 SNPs submitted for replication, 20 passed the quality-control analyses.
Stratified association analyses in stage 2 data (Table 2), confirmed the strong association for HLA-DQB1 (rs6457617, P = 1.35×10−28) at 6p21.3 and also with the PSORS1C1 variant (rs3130573, P = 4.98×10−3) at 6p21.1. Of the 6 remaining loci selected in stage 1, only 2 were replicated with nominal P<5% and with same direction of effect. They mapped at 2p24 (rs342070, P = 0.026; rs13021401, P = 0.024) and 5q33 (rs3792783, P = 4.14×10−5; rs2233287, P = 4.38×10−3; rs4958881, P = 2.09×10−3). None of the replicated SNPs showed evidence for heterogeneity of effects among the 4 geographical origins (Breslow-day P>0.10). As expected, ORs estimated in the discovery tended to be higher than those obtained in the replication stage data. Afterwards, association signals from joint analyses of the 2 datasets (Table 2) consistently showed highly significant association for HLA-DQB1 (P = 2.33×10−37), PSORS1C1 (P = 5.70×10−10) and TNIP1 (P = 4.68×10−9), and also showed some evidence of association for RHOB (P = 3.17×10−6). All populations showed same direction of effects (Figure 2). Finally, we also replicated association signals at IRF5 (P = 3.49×10−5; combined-P = 4.13×10−7), at STAT4 (P = 1.9×10−10; combined-P = 2.26×10−13) and at the recently identified new SSc risk locus, CD247 (P = 2.90×10−5; combined-P = 1.30×10−6) (Table 2). In our combined data, the locus-specific PAR estimates were 24% for HLA-DQB1, 4% for TNIP1, 8% for PSORS1C1, 7% for CD247, 8% for STAT4 and 3% with IFR5/TNPO3. The combined PAR estimate was 47.4%.

As secondary analyses, we assessed homogeneity of SNP's effect between sub-categories of SSc (cutaneous sub-types and auto-antibodies). Case-only analyses revealed no significant evidence for heterogeneous ORs between cutaneous sub-types of SSc patients for any of the 5 replicated SNPs at 2p24 or 5q33 loci (Table S4A). Indeed, similar association signals were obtained from case-category association analyses (Table S4B). Altogether, the results did not suggest that the association signals in the newly identified 5q33 locus were driven by a specific sub-type of SSc. Conversely, for HLA-DQB1 and PSORS1C1 we found evidence of heterogeneity in OR estimates in positive vs negative ACA or TOPO auto-antibody SSc patients (Table S4A). Yet, the association signals in each of these sub-types of patients remained strong (Table S4B). These results support the previously reported hypothesis that the magnitude of the HLA-DQB1 effect on SSc susceptibility may depend on auto-antibody status [11]. The GWAS stage had 78% power to detect loci of the effect sizes observed in the discovery sample for TNIP1 variants (OR = 1.50) at a significance of P<10−5. However, it is widely acknowledged that effect sizes of significant GWAS loci are overestimates of true effects and other genes of lower effect sizes are unlikely to reach stringent significant thresholds.
Our GWAS analysis revealed strong association with PSORS1C1, which is ∼1 Mb of HLA-DQB1. Notably, PSORS1C1 is known to be involved in autoimmune response [23]. In the combined data, association with PSORS1C1 was highly significant (P = 5.70×10−10) and remained significant after controlling for the association at HLA-DQB1. Altogether, our results suggest that this region is likely to contain more than one gene playing a role in the pathogenesis of autoimmune disorders [23], [24]. Fine mapping at this locus is warranted to identify causal variants.
The three strongly associated SNPs at the 5q33 locus are located within the TNFAIP3 interacting protein 1 (TNIP1) gene. TNIP1 is a very interesting new candidate gene for SSc. The protein encoded by this gene exerts a negative regulation of NF-kappaB via two sequential activities: deubiquitination of Lys63-based chains and synthesis of Lys48-based chains on the TNF receptor-interacting protein and also inhibition of NF-KappaB processing [25]. TNIP1 interacts with A20 (TNFAIP3) to negatively regulate NF-kappaB. Several recent studies have suggested that the activation of some inflammatory factors may upregulate fibrotic mediators through Toll-like receptors (TLRs), thereby contributing to SSc pathogenesis [8]. It has been shown that TLR engagement leads to A20 induction in macrophages and that TNIP1/A20 is essential for the termination of TLR-induced NF-kappaB activity and proinflammatory cytokine production [26]. Although interactions between TNIP1 and A20 are not well known, A20 also acts as a deubiquitinating enzyme, suggesting a molecular link between deubiquitinating activity and the regulation of TLR signals [26]. Therefore, TNIP1 and A20 may play a critical role in the regulation of downstream TLR signals, and this issue will have to be addressed in SSc. Interestingly, variants at TNIP1 have been shown to be implicated in systemic lupus erythematosus susceptibility [27], [28] and in psoriasis [29]. Furthermore, we have recently reported an association of one TNFAIP3 variant with SSc [30]. In our stage-1 data, evidence of association at TNFAIP3 was nominal (lowest P = 0.047) and no pairwise interaction was found (P>0.06) between TNFAIP3 and TNIP1 variants. Analysis of the LD structure across the TNIP1 gene revealed that the 3 strongly associated SNPs belong to the same LD-block (Figure 3). No residual association signals were observed when rs3792783 and each of the other 2 SNPs were paired in conditional analyses. Therefore, any of them, or other variants yet to be identified, could be the causal variant(s). Interestingly, rs3792783 is located upstream from the transcription start site in exon 2 (Figure 3). It is noteworthy that previously reported lupus TNIP1 variants were located in the same LD-block [27], [28]. Because of the compelling evidence of the potential role of NF-kappaB in autoimmune diseases and our raised new signal association for SSc at TNIP1 (a negative regulator of this pathway) we performed ex vivo and in vitro investigations to assess TNIP1 expression in SSc patients and healthy controls. For SSc patients, the results showed a strikingly reduced expression of TNIP1 in skin tissue (Figure 4A), and of both mRNA (Figure 4B) and protein (Figure 4C) synthesis by cultured dermal fibroblasts. Addressing the question of the potential link between the NF-kappaB pathway and the fibrotic propensity that characterizes SSc, we next assessed the influence of pro-inflammatory cytokines and TNIP1 on the synthesis of extra-cellular matrix by dermal fibroblasts in culture. Using cells from the skin of healthy controls (Figure 5) and SSc patients (Figure 6), we showed that recombinant TNIP1 abrogated collagen synthesis induced by inflammatory cytokines both at the mRNA and protein levels. It must be acknowledged that TNIP1 is described as an intra-cellular protein whereas we used recombinant protein added to cell supernatant in these experiments. The observed effects may be related to different hypotheses. TNIP1 has been described as a nuclear shuttling protein and it could have a chaperon-like activity, highly interacting with other protein that could result in engulfment of TNIP1 through interaction with a cell surface protein. Such intra-cellular effects of extra-cellular proteins has been shown also for the S100 family of proteins that have no leader sequence and for clusterin for which it is postulated that the protein could be taken up by interacting with either a yet unidentified receptor or by a mechanism related to their chaperon-like activity [31]. More work is needed to determine which of these hypotheses has to be retained and to investigate more in depth soluble TNIP1 In this first attempt to explore TNIP1 functional disturbances, we could not investigate a relationship between specific TNIP variants and in vitro or in vivo changes; this will need to be addressed ideally after the identification of the causal variant and using a much larger sample size. Nevertheless, our results raise a potential relationship between inflammation and fibrosis and open a new and highly relevant field of investigation in SSc pathogenesis and in fibrotic disorders.




Our next most associated SNPs at 2p24 are in strong LD (r2 = 0.98) and map ∼30 kb from RHOB. RHOB is the Ras homolog gene family member B that regulates protein signalling and intracellular protein trafficking. RhoB is essential for activity of farnesyltransferase inhibitors and also statins that are two strong potential future drugs in SSc [9], [32]. To our knowledge, association to RHOB has never been reported so far. The signal for association was weaker at this locus and therefore will need to be confirmed in other samples and more investigations are warranted to assess RHOB implication in this disease.
In conclusion, we have conducted a large genome-wide association study of SSc and identified two new SSc-risk loci, PSORS1C1 and TNIP1. We also confirmed the association of SSc with variants at STAT4, IRF5 and CD247, in the European population. We also found compelling evidence of association to a putative new SSc risk locus on 2p24, close to the RHOB gene. None of the newly identified 3 loci have been previously reported associated to SSc. The TNIP1 variants identified do not have precise functional implications; however, their localization within a regulatory region strongly suggests an impact on transcription of the gene. This is supported by our ex vivo and in vitro investigations. Altogether, our results are consistent with a reduced inhibition of NF-kappaB, therefore favoring inflammatory/immune responses and potentially contributing to the overproduction of extra-cellular matrix. This raises a new clue for a link between inflammation and SSc that could also be of importance in other fibrotic disorders.
Material and Methods
Study populations
Stage-1 included 654 SSc patients and 531 controls recruited through the French GENESYS project [11], [13], [30] and 2,003 controls from the French Three-City (3C) cohort [21], [22]. The stage-2 data included an independent collection of 4,492 samples (pre quality controls) from several University Hospitals in France, Italy, Germany and Eastern Europe. It also included 721 Italian controls recruited through nationwide efforts by HYPERGENE consortium and 481 Illumina HumanHap550 for the KORA S4 study [33], recruited in the city of Augsburg, Southern Germany. In both stage 1 and stage 2 samples, SSc patients fulfilled ACR criteria [34] and were classified in cutaneous subsets according to LeRoy's criteria [19]. Table 1 shows the main characteristics of the post-QC SSc patients and controls.
Ethics statement
All participants gave written informed consent, and approval was obtained from the relevant local ethical committees.
Genotyping and quality control analyses
Stage 1
French DNA samples from GENESYS and 3C were genotyped at Integragen and at the Centre National de Génotypage (Evry, France), respectively, with Illumina Human610-Quad BeadChip. Data were subjected to standard quality control procedures using tools implemented in PLINK version 1.07 [35]. Markers were removed if they had a genotype-missing rate >0.03 or a minor allele frequency (MAF)<0.05 or a Hardy-Weinberg P< = 10−5. Samples were removed on low (<98%) call rate, inconsistencies between reported gender and genotype-determined gender and/or genetic relatedness (identity-by-descent estimate >0.12). Applying these QC filters led to the removal of 791 subjects (56 cases, 735 controls). To detect individuals of non-European ancestry, we computed genome-wide average identity-by-state (IBS) distance with PLINK using a thinned map of 55,193 SNPs. To this end, we removed SNPs in extensive regions of LD (Chr.2, Chr.5, Chr.6, Chr.8, Chr.11) [36], and excluded SNPs if any pair within a 1000-SNPs window had r2>0.2. Our data were then merged with genotypes at the same SNPs from 381 unrelated European (CEU), Yoruban (YRI) and Asian (CHB and JPT) samples from the HapMap project. Classical multi-dimensional scaling analysis was applied on the resulting matrix of IBS distances and the first two dimensions were extracted and plotted against each other. The HapMap data were clearly separated into three distinct clusters according to ancestry. Fifty seven of our stage-1 subjects did not cluster within the European group and were excluded from further analyses.
Stage 2
Follow-up analysis was conducted for the set of 17 SNPs that were identified in stage I and for 4 SNPs at STAT4, IRF5/TNPO3 and CD247 loci. De novo genotyping was performed by a competitive allele-specific PCR system (Kbioscience, Hoddeston, UK) [11], [13], [30]. The additional set of Italian and German control samples were previously genotyped using Illumina 1MQuad or Human610Quad bead chip. One SNP (rs9275224 in HLA) failed genotyping. Accuracy of genotyping was assessed using quality control procedures similar to those applied to our stage 1 data; following the quality control analyses, our stage 2 data consisted of 20 SNPs genotyped in a total of 1,682 cases and 3,926 controls (Table 1).
Statistical association analyses
Association analysis of the genotype data was conducted with PLINK (v1.07) software [35]. All reported P values are two sided. In stage 1, we applied logistic regression assuming an additive genetic model. The quantile-quantile plot was used to evaluate overall significance of the genome-wide association results and the potential impact of residual population substructure. A conservative genome-wide significance threshold of 0.05/489,918 = 1.02×10−7 was used.
Stage 2 association and combined analyses were carried out with the Mantel-Haenszel test to control for differences between geographical groups. A Breslow-Day test was performed to assess the heterogeneity of effects in different populations. In the replication analysis, P values<0.05 and direction of effect as observed in the stage-1 data, were considered to indicate statistical significance.
Secondary statistical analyses were conducted to assess independency of multiple association signals within and between loci and homogeneity of effects between subgroups of SSc patients. Case-only association analyses were conducted using the three main clinical variables (Table 1). The LD structure of the identified loci was analyzed using Haploview 4.1 [37] and LD blocks delimited using the D′-based confidence interval method [38]. The locus-specific Population attributable risk (PAR) was calculated for each of the 6 replicated loci (HLA-DQB1, TNIP1, PSORS1C1, STAT4, IFR5/TNPO3 and CD247) according to the following formula: PAR = RAF×(OR-1)/(RAF×(OR-1)+1), where RAF is the frequency of the associated allele in the controls, and OR is the odds ratio associated with the risk allele. The combined PAR was computed as 1−Pj(1−PARj).
Histologic and cytologic investigations
Fibroblast cultures were prepared by outgrowth cultures from lesional skin biopsy specimens of eleven SSc patients and from twelve healthy controls matched for age and sex. The median age of SSc patients was 49 years old (range: 22–67 years) and their median disease duration was 7 years (range: 1–17 years); seven had the limited cutaneous subset and four the diffuse. Immunohistochemistry was performed on paraffin-embedded skin sections from 5 SSc patients and 5 controls using mouse anti-human TNIP1 antibodies (eBioscience, Frankfurt, Germany). Total RNA, issued from cultured dermal fibroblasts, isolation and reverse transcription into complementary DNA were performed as previously described [39]. Gene expression was quantified by SYBR Green real-time PCR, with a specific primer pair available upon request. Protein assessment was performed on western blots, as previously described [40] using mouse anti-human TNIP1 antibodies (eBioscience, CA, USA). In selected experiments, dermal fibroblasts from healthy control subjects and patients with SSc were treated for 24 hours with recombinant TNIP1 (2 µg/ml, Abnova, Tapei City, Taiwan) in the presence or not of the following proinflammatory cytokines: TNFα (20 ng/ml, R&D systems, Abingdon, UK), IL1β (1 µg/ml, Immunotools, Friesoythe, Germany) or IL6 (1 µg/ml, Immunotools). mRNA levels of human α1(I) and α2(I) procollagen were quantified by quantitative real-time PCR, specific primers are available upon request. The collagen content in cell culture supernatants was analyzed with the SirCol collagen assay (Biocolor, Belfast, UK) [41]. Comparisons were performed using Student's T test.
Supporting Information
Zdroje
1. ValentiniGBlackC 2002 Systemic sclerosis. Best Pract Res Clin Rheumatol 16 807 16
2. ThompsonAEPopeJE Increased prevalence of scleroderma in southwestern Ontario: a cluster analysis. J Rheumatol 29 1867 73
3. Le GuernVMahrAMouthonLJeanneretDCarzonM 2004 Prevalence of systemic sclerosis in a French multi-ethnic county. Rheumatology (Oxford) 2004;43 1129 37
4. CzirjakLKissCGLoveiCSütoGVarjúC 2005 Survey of Raynaud's phenomenon and systemic sclerosis based on a representative study of 10,000 south-Transdanubian Hungarian inhabitants. Clin Exp Rheumatol 23 801 8
5. ArnettFCChoMChatterjeeSAguilarMBReveilleJD 2001 Familial occurrence frequencies and relative risks for systemic sclerosis (scleroderma) in three United States cohorts. Arthritis Rheum 44 1359 62
6. Feghali-BostwickCMedsgerTAJrWrightTM 2003 Analysis of systemic sclerosis in twins reveals low concordance for disease and high concordance for the presence of antinuclear antibodies. Arthritis Rheum 48 1956 6
7. GabrielliAAvvedimentoEVKriegT 2009 Scleroderma. N Engl J Med 360 1989 2003
8. LafyatisRYorkM 2009 Innate immunity and inflammation in systemic sclerosis. Curr Opin Rheumatol 21 617 22
9. VargaJAbrahamD 2007 Systemic sclerosis: a prototypic multisystem fibrotic disorder. J Clin Invest 117 557 67
10. AllanoreYDieudePBoileauC 2010 Updating the genetics of Systemic Sclerosis. Curr Opin Rheumatol 22 665 70
11. DieudéPGuedjMWipffJAvouacJFajardyI 2009 Association between the IRF5 rs2004640 functional polymorphism and systemic sclerosis: a new perspective for pulmonary fibrosis. Arthritis Rheum 60 225 33
12. ItoIKawaguchiYKawasakiAHasegawaMOhashiJ 2009 Association of a functional polymorphism in the IRF5 region with systemic sclerosis in a Japanese population. Arthritis Rheum 60 1845 50
13. DieudéPGuedjMWipffJRuizBHachullaE 2009 STAT4 is a genetic risk factor for systemic sclerosis having additive effects with IRF5 on disease susceptibility and related pulmonary fibrosis. Arthritis Rheum 60 2472 9
14. RuedaBBroenJSimeonCHesselstrandRDiazB 2009 The STAT4 gene influences the genetic predisposition to systemic sclerosis phenotype. Hum Mol Genet 18 2071 7
15. GourhPAgarwalSKDivechaDAssassiSPazG 2009 Polymorphisms in TBX21 and STAT4 increase the risk of systemic sclerosis: evidence of possible gene-gene interaction and alterations in Th1/Th2 cytokines. Arthritis Rheum 60 3794 806
16. ArnettFCGourhPSheteSAhnCWHoneyRE 2010 Major histocompatibility complex (MHC) class II alleles, haplotypes, and epitopes which confer susceptibility or protection in the fibrosing autoimmune disease systemic sclerosis: analyses in 1300 Caucasian, African-American and Hispanic cases and 1000 controls. Ann Rheum Dis 69 822 7
17. ZhouXLeeJEArnettFCXiongMParkMY 2009 HLA-DPB1 and DPB2 are genetic loci for systemic sclerosis: a genome-wide association study in Koreans with replication in North Americans. Arthritis Rheum 60 3807 14
18. RadstakeTRGorlovaORuedaBMartinJEAlizadehBZ 2010 Genomewide association study of systemic sclerosis identifies CD247 as a new susceptibility locus. Nat Genet 42 426 9
19. LeRoyECBlackCFleischmajerRJablonskaSKriegT 1988 Scleroderma (systemic sclerosis): classification, subsets and pathogenesis. J Rheumatol 15 202 205
20. WollheimFA 2005 Classification of systemic sclerosis. Visions and reality. Rheumatology 44 1212 6
21. LambertJCHeathSEvenGCampionDSleegersK 2009 Genomewide association study identifies variants at CLU and CR1 associated with Alzheimer's disease. Nat Genet 41 1094 1099
22. C Study Group 2003 Vascular factors and risk of dementia: design of the Three-City Study and baseline characteristics of the study population. Neuroepidemiology 22 316 25
23. FanXYangSHuangWWangZMSunLD 2008 Fine mapping of the psoriasis susceptibility locus PSORS1 supports HLA-C as the susceptibility gene in the Han Chinese population. PLoS Genet 4 e1000038 doi:10.1371/journal.pgen.1000038
24. ReichKHüffmeierUKönigIRLascorzJLohmannJ 2007 TNF polymorphisms in psoriasis: association of psoriatic arthritis with the promoter polymorphism TNF*-857 independent of the PSORS1 risk allele. Arthritis Rheum 56 2056 64
25. WertzIEO'RourkeKMZhouHEbyMAravindL 2004 De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature 430 694 699
26. BooneDLTurerEELeeEGAhmadRCWheelerMT 2004 The ubiquitin-modifying enzyme A20 is required for termination of Toll-like receptor responses. Nat Immunol 5 1052 1060
27. GatevaVSandlingJKHomGTaylorKEChungSA 2009 A large-scale replication study identifies TNIP1, PRDM1, JAZF1, UHRF1BP1 and IL10 as risk loci for systemic lupus erythematosus. 41 228 233
28. HanJWZhengHFCuiYSunLDYeDQ 2009 Genomewide association study in a Chinese Han population identifies nine new susceptibility loci for systemic lupus erythematosus. Nat Genet 41 1234 1237
29. NairRPDuffinKCHelmsCDingJStuartPE 2009 Genomewide scan reveals association of psoriasis with IL-23 and NF-kappaB pathways. Nat Genet 41 199 204
30. DieudéPGuedjMWipffJRuizBRiemekastenG 2010 Association of the TNFAIP3 rs5029939 variant with systemic sclerosis in the European Caucasian population. Ann Rheum Dis 69 1958 64
31. FalgaroneGChiocchiaG 2009 Clusterin: A multifacet protein at the crossroad of inflammation and autoimmunity. Adv Cancer Res 104 139 70
32. GabrielliA 2007 Stimulatory autoantibodies to the PDGF receptor: a link to fibrosis in scleroderma and a pathway for novel therapeutic targets. Autoimmun Rev 7 121 6
33. WichmannHEGiegerCIlligT 2005 KORA-gen-resource for population genetics, controls and a broad spectrum of disease phenotypes. Gesundheitswesen 67, Suppl. 1 S26 S30
34. Anonymous 1980 Subcommittee for Scleroderma Criteria of the American Rheumatism Association Diagnostic and Therapeutic Criteria Committee: Preliminary criteria for the classification of systemic sclerosis (scleroderma). Arthritis Rheum 23 581 90
35. PurcellSNealeBTodd-BrownKThomasLFerreiraMA 2007 PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81 559 575
36. PriceALWealeMEPattersonNMyersSRNeedAC 2008 Long-range LD can confound genome scans in admixed populations. Am J Hum Genet 83 132 135
37. BarrettJCFryBMallerJDalyMJ 2005 Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21 263 265
38. GabrielSBSchaffnerSFNguyenHMooreJMRoyJ 2002 The structure of haplotype blocks in the human genome. Science 296 2225 2229
39. DistlerJHJüngelAHuberLCSchulze-HorselUZwerinaJ 2007 Imatinib mesylate reduces production of extracellular matrix and prevents development of experimental dermal fibrosis. Arthritis Rheum 56 311 322
40. AvouacJWipffJGoldmanORuizBCouraudPO 2008 Angiogenesis in systemic sclerosis: impaired expression of vascular endothelial growth factor receptor 1 in endothelial progenitor-derived cells under hypoxic conditions. Arthritis Rheum 58 3550 3561
41. ReichNMaurerBAkhmetshinaAVenalisPDeesC 2010 The transcription factor Fra-2 regulates the production of extracellular matrix in systemic sclerosis. Arthritis Rheum 62 280 290
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2011 Číslo 7
Nejčtenější v tomto čísle
- A Rice Plastidial Nucleotide Sugar Epimerase Is Involved in Galactolipid Biosynthesis and Improves Photosynthetic Efficiency
- Genome-Wide Association Study Identifies Novel Restless Legs Syndrome Susceptibility Loci on 2p14 and 16q12.1
- Loss of the BMP Antagonist, SMOC-1, Causes Ophthalmo-Acromelic (Waardenburg Anophthalmia) Syndrome in Humans and Mice
- Gene-Based Tests of Association