
Proteins in the Nutrient-Sensing and DNA Damage Checkpoint Pathways Cooperate to Restrain Mitotic Progression following DNA Damage
Checkpoint pathways regulate genomic integrity in part by blocking anaphase until all chromosomes have been completely replicated, repaired, and correctly aligned on the spindle. In Saccharomyces cerevisiae, DNA damage and mono-oriented or unattached kinetochores trigger checkpoint pathways that bifurcate to regulate both the metaphase to anaphase transition and mitotic exit. The sensor-associated kinase, Mec1, phosphorylates two downstream kinases, Chk1 and Rad53. Activation of Chk1 and Rad53 prevents anaphase and causes inhibition of the mitotic exit network. We have previously shown that the PKA pathway plays a role in blocking securin and Clb2 destruction following DNA damage. Here we show that the Mec1 DNA damage checkpoint regulates phosphorylation of the regulatory (R) subunit of PKA following DNA damage and that the phosphorylated R subunit has a role in restraining mitosis following DNA damage. In addition we found that proteins known to regulate PKA in response to nutrients and stress either by phosphorylation of the R subunit or regulating levels of cAMP are required for the role of PKA in the DNA damage checkpoint. Our data indicate that there is cross-talk between the DNA damage checkpoint and the proteins that integrate nutrient and stress signals to regulate PKA.
Published in the journal:
. PLoS Genet 7(7): e32767. doi:10.1371/journal.pgen.1002176
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002176
Summary
Checkpoint pathways regulate genomic integrity in part by blocking anaphase until all chromosomes have been completely replicated, repaired, and correctly aligned on the spindle. In Saccharomyces cerevisiae, DNA damage and mono-oriented or unattached kinetochores trigger checkpoint pathways that bifurcate to regulate both the metaphase to anaphase transition and mitotic exit. The sensor-associated kinase, Mec1, phosphorylates two downstream kinases, Chk1 and Rad53. Activation of Chk1 and Rad53 prevents anaphase and causes inhibition of the mitotic exit network. We have previously shown that the PKA pathway plays a role in blocking securin and Clb2 destruction following DNA damage. Here we show that the Mec1 DNA damage checkpoint regulates phosphorylation of the regulatory (R) subunit of PKA following DNA damage and that the phosphorylated R subunit has a role in restraining mitosis following DNA damage. In addition we found that proteins known to regulate PKA in response to nutrients and stress either by phosphorylation of the R subunit or regulating levels of cAMP are required for the role of PKA in the DNA damage checkpoint. Our data indicate that there is cross-talk between the DNA damage checkpoint and the proteins that integrate nutrient and stress signals to regulate PKA.
Introduction
Progression through mitosis in yeast requires the ordered destruction of two inhibitors: securin or Pds1 and the mitotic or B type cyclin, Clb2. The inhibitors are ubiquitinated and targeted for destruction by the anaphase promoting complex or cyclosome (APC/C) in conjunction with the specificity factors Cdc20 and Hct1/Cdh1 [1]–[3]. Destruction of Pds1 allows the separase Esp1 to cleave the cohesins to allow anaphase [4], [5]. Exit from mitosis and the establishment of pre-replication complexes requires the inactivation of the mitotic cyclin dependent kinase (Cdk) complex, achieved in part by the APC/C-mediated proteolysis of the mitotic cyclins, including Clb2, and by increased levels of the Cdk inhibitor Sic1 [6]–[9].
During anaphase the separase Esp1 triggers the release of Cdc14 from the nucleolus by modulating the phosphorylated status of its interacting partner Net1 via regulation of the phosphatase PP2ACdc55 [10]–[14]. When Net1 is phosphorylated Cdc14 is released and can exit the nucleolus to dephosphorylate Cdk1 substrates such as Hct1/Cdh1, Sic1, Pds1. Cdc14 release and dephosphorylation of its targets are important for progression through anaphase and for activation of the mitotic exit network (MEN) [3], [7], [15]–[18].
Accurate transmission of chromosomes to each daughter cell requires that cells block anaphase until all chromosomes have been completely replicated and correctly aligned on the spindle. Similarly, cells that have incurred DNA damage in late S phase or G2 repair the damage before they progress through mitosis. In S. cerevisiae, DNA damage and mono-oriented or unattached kinetochores trigger checkpoint pathways that bifurcate to regulate both the metaphase to anaphase transition and mitotic exit [19]–[23]. In addition, several stress-activated pathways including the cAMP dependent protein kinase (PKA) pathway have been identified in S. cerevisiae that inhibit mitosis by targeting proteins involved in mitotic progression [24]–[28].
The sensor-associated kinase, Mec1 is activated in response to DNA damage and together with a signal amplifier, Rad9, phosphorylates two downstream kinases, Chk1 and Rad53 [29]. Activation of Chk1 prevents anaphase by preventing the destruction of securin [19], [30]; and activation of Rad53 causes inhibition of the mitotic exit network, helps to prevent securin destruction [31], [32], and also prevents segregation of damaged chromosomes by restricting spindle elongation [33].
We showed that PKA had a role in the DNA damage checkpoint. PKA supported mitotic arrest by regulating the phosphorylation of Cdc20 and helped maintain high levels of the mitotic inhibitors, securin (Pds1) and Clb2. In addition, we showed that phosphorylation of Cdc20 was both Mec1 - and PKA-dependent, and that overexpression of PKA catalytic subunits partially rescued the checkpoint defect of mec1-21 cells [34]. In the work described here, we set out to identify the mechanism by which PKA is regulated in response to DNA damage. PKA in its inactive form is a tetramer consisting of two catalytic and two regulatory subunits. In yeast three genes encode the catalytic subunits, TPK1, TPK2 and TPK3 and one gene encodes the R subunit, BCY1 [35]–[37]. R subunit interaction with cAMP causes the tetramer to disassociate rendering the catalytic subunits active. Thus, activation of PKA is regulated by increasing the intracellular levels of cAMP. At least two other signaling pathways in yeast regulate a transient increase in cAMP levels. The Ras pathway, which can be stimulated by intracellular acidification and the glucose signaling pathway (reviewed in [38]). The glucose kinase, Hxk2, is required for the transient increase in cAMP levels in both the Ras and glucose signaling pathways [39].
Regulation of PKA can also occur via post-translational modification, protein interactions and sub-cellular localization of the catalytic or R subunits. These mechanisms of regulation have been more thoroughly studied in metazoan cells in which phosphorylation of the regulatory subunit localizes the complex to a specific sub-cellular compartment via interaction with proteins called A protein Kinase Anchoring Proteins (AKAPs). The AKAPs help mediate the specificity of PKA signaling in the cell [40]. In yeast the R subunit is phosphorylated when cells are deprived of glucose and in response to heat shock or addition of calcium to the media [41], [42]. The Yak1 kinase is required for the phosphorylation of the R subunit when the cells are grown in a non-fermentable carbon source [41], and Mck1, a GSK-3 kinase, is required for the phosphorylation of the R subunit in response to heat shock [42]. This phosphorylation is required for re-distribution of the R subunit from the nucleus to both the nucleus and cytoplasm [42]. In addition, Zds1 and Zds2 have been implicated in the regulation of the localization of the R subunit to the cytoplasm in response to the same stresses that lead to R subunit phosphorylation [41], [42].
Here we show that the checkpoint regulated phosphorylation of the R subunit of PKA has a role in restraining mitosis following DNA damage, suggesting that there is cross-talk between the DNA damage checkpoint and the PKA pathways. In addition we found that proteins that regulate the phosphorylation of the R subunit, and proteins that regulate the levels of intracellular cAMP are required for the role of PKA in the DNA damage checkpoint.
Results
The R subunit is phosphorylated in response to DNA damage
We have previously shown that the PKA pathway plays a role in blocking securin and Clb2 destruction following DNA damage [34]. Our findings also suggested that Mec1 and PKA are in the same signaling pathway and that PKA was likely acting downstream of Mec1 in response to DNA damage.
PKA can be regulated by many mechanisms including regulation of cAMP levels, localization of the PKA holoenzyme or catalytic subunits, interactions with other proteins, and phosphorylation of the catalytic and R subunits [38], [43]. We first set out to determine whether the R subunit was phosphorylated in response to DNA damage, and whether this phosphorylation was dependent on Mec1. The cdc13-1 allele was used to activate the DNA damage checkpoint that blocks mitotic progression [44]. Growth of cdc13-1 cells at a restrictive temperature results in the inactivation of the telomere binding protein, Cdc13, which causes single stranded DNA at the telomeres. This single stranded DNA is recognized as DNA damage in G2/M, and the DNA damage checkpoint is activated, blocking anaphase and mitotic exit [44], [45]. In protein extracts from synchronized cells grown at the restrictive temperature for cdc13-1, a slower migrating form of the R subunit was detected by western analysis at a time when the DNA damage signal was present (Figure 1A). A similar slower migrating form of the R subunit was detected in protein extracts from cells growing in YP ethanol as previously shown [41].

In order to show that the slower migrating form of the R subunit was due to phosphorylation, the R subunit was isolated by immuno-precipitation from extracts prepared from cells with a DNA damage signal. The immunoprecipitated complexes were treated with phosphatase in the presence or absence of phosphatase inhibitors. Treatment with alkaline phosphatase resulted in a loss of the slower migrating form of the R subunit, while the slower migrating form of the R subunit was maintained when the immunoprecipitated-complexes were treated with both phosphatase and phosphatase inhibitors (Figure 1B). These results indicated that the R subunit was modified by phosphorylation following activation of the DNA damage checkpoint.
The R subunit was also phosphorylated in cells grown at 30°C (in the absence of heat shock) and treated with the DNA damaging agent, methylmethane sulfonate (MMS) (Figure 1C). Pre-anaphase arrest due to activation of the spindle checkpoint by treatment with nocodazole (in the absence of a DNA damage signal) did not result in R subunit phosphorylation (Figure 1C). This finding indicated that phosphorylation of the R subunit in cdc13-1 cells was not due to a cell cycle position effect and that activation of the spindle checkpoint does not result in R subunit phosphorylation.
The R subunit was not phosphorylated in cdc13-1 cells lacking the upstream checkpoint kinase, Mec1, (Figure 1D and Figure S4). This result suggested that Mec1 is required for the phosphorylation of the R subunit in response to DNA damage and supports the hypothesis that Mec1 regulates PKA when there is a DNA damage signal.
Mck1 is required for R subunit phosphorylation following DNA damage and has a role in the DNA damage checkpoint
The GSK3 kinase Mck1 was found to be required for the phosphorylation of the R subunit in response to heat shock [42]. We recapitulated these findings in our lab and found that R subunit phosphorylation in response to heat shock was dependent on Mck1 but not on the DNA damage kinase Mec1 (data not shown). Thus, the requirement for Mec1 for R subunit phosphorylation was specific to a DNA damage signal. We next examined whether Mck1 regulated the phosphorylation of the R subunit in the DNA damage checkpoint. R subunit proteins from cdc13-1 or cdc13-1 mck1Δ cells raised to the non-permissive temperature of 32°C for cdc13-1 were analyzed by Western analysis. No phosphorylated R subunit was detected in cdc13-1 cells lacking MCK1 (Figure 2A). Our data indicate that Mck1 responds to different signals to regulate R subunit phosphorylation and that in the case of DNA damage the signal required the sensor-associated kinase Mec1.

We previously showed that PKA and Chk1 work in parallel pathways to inhibit mitotic progression in the DNA damage checkpoint response [34]. To determine whether Mck1 has a role in the DNA damage checkpoint, we examined whether deletion of MCK1 removed the remaining DNA damage-induced mitotic delay in cells lacking CHK1. Because PKA pathways have a role in regulating the G1/S transition, we released the cells into the cdc13-1 induced checkpoint from an hydroxyurea (HU) arrest (which synchronizes the cells in late S-phase) and monitored their progression through mitosis. The number of cells that had failed in the checkpoint-mediated arrest was determined by scoring the number of cells that exhibited a late mitotic phenotype (separated nuclei) in the presence of a DNA damage signal. cdc13-1 cells lacking MCK1 or TPK2 were proficient in the checkpoint arrest and remained arrested throughout the experiment (Figure 2B and data not shown), however, cdc13-1 chk1Δ cells lacking MCK1 or TPK2 exhibited more cells with a late mitotic phenotype at earlier time points than the cdc13-1 chk1Δ cells (Figure 2B). These results indicated that Mck1 had a role in the checkpoint-mediated arrest similar to the role of Tpk2.
To determine whether Mck1 was acting in the same pathway as PKA to restrain mitosis following DNA damage, we tested whether deletion of MCK1 would further enhance the checkpoint defect of cdc13-1 chk1Δ tpk2Δ cells. Cells were released into the cell cycle from an α-factor induced G1 arrest. Only cells lacking TPK2 were used in this experiment to eliminate any differences in cell cycle entry due to role of PKA in the G1/S transition. cdc13-1 chk1Δ cells lacking TPK2 failed in the checkpoint with the same kinetics as cdc13-1 chk1Δ cells lacking both TPK2 and MCK1 (Figure 2C), suggesting that Mck1 and Tpk2 were acting in the same pathway following DNA damage.
Because Mck1 and Mec1 were required for the phosphorylation of the R subunit following DNA damage, and Mck1 had been previously identified as a high-copy suppressor of the lethality associated with deletion of RAD53 [46], we wanted to determine whether Mck1 was acting downstream of Mec1 to restrain mitosis following DNA damage. cdc13-1 or cdc13-1 mec1-21 cells containing an empty vector or MCK1 on a multi-copy vector were released from G1 (α-factor block) at 32°C and the cells were stained with DAPI to determine the number of cells exhibiting late mitotic phenotype at the indicated time points. Overexpression of Mck1 partially alleviated the checkpoint defect of mec1-21 cells following DNA damage (Figure 2D), suggesting that Mck1 was acting downstream of Mec1 to restrain mitosis following DNA damage. Overexpression of MCK1 did not delay entry into S phase (appearance of small budded cells) (Figure 2E, and Figure S6B). Cells with a high copy plasmid encoding MCK1 reached large budded state with an undivided nucleus with the same kinetics as vector controls (Figure 2F and Figure S6C). This was the case whether the cells were released from the G1 block into YPD or minimal media (Figure S6E–S6H).
Mck1 has been shown to play a role in restraining the G2/M cell cycle transition through activation of Swe1, a kinase that negatively regulates Cdk1 [47]. To test whether Mck1 was acting in the Swe1 pathway to restrain entry into mitosis we deleted SWE1 in a chk1Δ mutant and found that SWE1 deletion did not enhance the checkpoint defect of a chk1Δ cell (Figure 2G). These findings suggested that the role of Mck1 following DNA damage was to block mitotic progression, and not to block mitotic entry via Swe1.
Phosphorylation of the R subunit is required for the role of PKA in the DNA damage checkpoint
Two serine rich clusters on the R subunit were shown to be required for the phosphorylation and cytoplasmic localization of the R subunit in response to heat shock and growth in non-fermentable carbon sources [41] (Figure 3A). However, mutation of cluster I or cluster II serines did not compromise R subunit function in response to carbon source or heat shock responses [41]. To determine whether these sites were phosphorylated in response to DNA damage, protein from cells given a DNA damage signal expressing wild type R subunit or the R subunit with serines in cluster I or cluster II [41] mutated to alanines were analyzed by western blot. The wild type R subunit was phosphorylated under these conditions but the phosphorylation was greatly reduced when the serines in cluster I or II were changed to alanine (Figure 3A). These results suggested that the R subunit is phosphorylated on serine residues located in both clusters I and II.

Since Mck1 is both required for the R subunit phosphorylation and has a role in the DNA damage checkpoint similar to PKA, we hypothesized that the role of PKA in the DNA damage checkpoint required phosphorylation of the R subunit on cluster I and cluster II serines. Therefore, phosphorylation defective mutants should have the same phenotype as deletion of a PKA catalytic subunit. cdc13-1 bcy1Δ or cdc13-1 chk1Δ bcy1Δ cells expressing wild type BCY1 or bcy1CI or bcy1CII mutants were released into the cell cycle from an HU-mediated arrest in late S-phase into a DNA damage signal, and the number of cells exhibiting a late mitotic phenotype were counted. Expression of the phosphorylation defective R subunit mutants enhanced the checkpoint defect of a chk1Δ cell to the same extent as deletion of a PKA catalytic subunit (Figure 3B). Furthermore, expression of the phosphorylation defective R subunit mutants did not further enhance the checkpoint defect of cells lacking both CHK1 and MCK1 (Figure 3C). These results suggested that phosphorylation of the serines in cluster I and II play a role in the DNA damage checkpoint and that the role of Mck1 in the DNA damage checkpoint is via regulation of the R subunit by phosphorylation on one or more of the serine residues in clusters I and II.
Re-localization of the R subunit following DNA damage
It was previously reported that R subunit undergoes re-localization to the cytoplasm upon heat shock and that this shift in localization was dependent on serine residues in clusters I and II [42]. Based on our observation that the phosphorylation of cluster I and II serines is important for the mitotic delay following DNA damage, we hypothesized that the DNA damage signal would alter the localization of the R subunit. To address this, the localization of GFP-tagged Bcy1 was analyzed by microscopy in WT and cdc13-1 cells. The number of cells with nuclear accumulation of Bcy1-GFP was scored under asynchronous growth conditions, DNA damage, heat shock or by arresting cells in M phase with nocodazole. Upon induction of DNA damage, we observed a shift in the localization of the R subunit from the nucleus into the cytoplasm (Figure 3D and Figure S2). This re-localization was similar to that observed in cells following heat shock (Figure S2). As previously reported, asynchronous or nocodazole treated WT cultures had a high percentage of cells with nuclear Bcy1. These data suggest that the activation of the DNA damage checkpoint, as elicited by a cdc13-1 mutant, promotes the export of the R subunit from the nucleus into the cytoplasm.
Zds2 plays a role in the DNA damage checkpoint
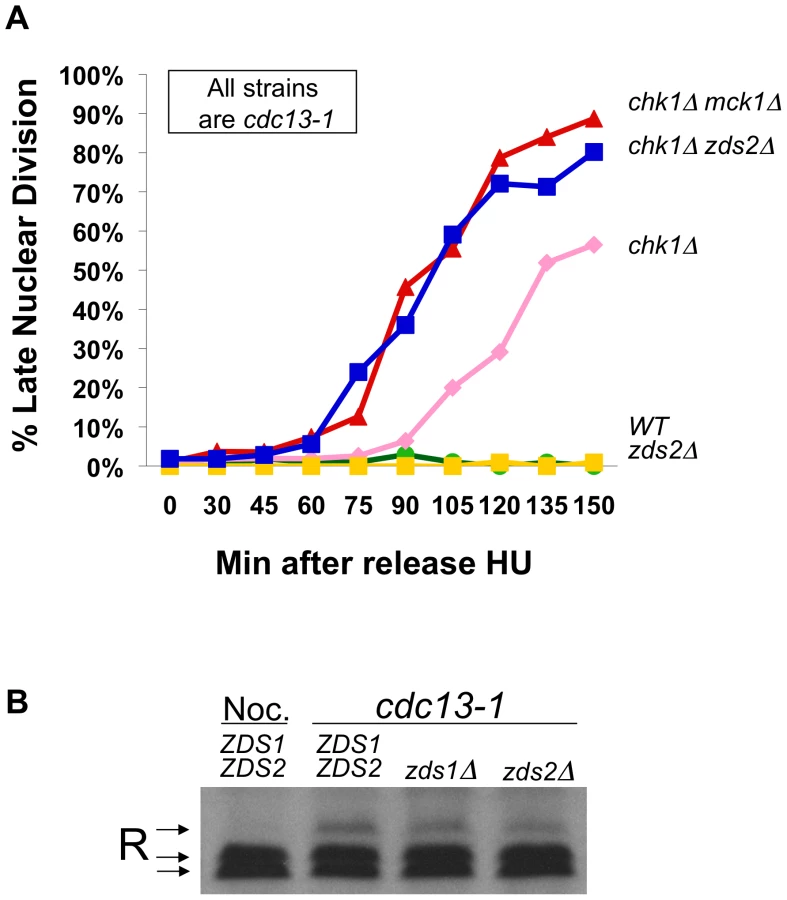
Two proteins, Zds1 and Zds2, have been implicated in regulating the cytoplasmic localization of the R subunit in the same conditions in which the R subunit is phosphorylated. Zds1 was required for the cytoplasmic localization of the R subunit in glucose restricting conditions [41], and Zds1 and its homologue, Zds2, were required for cytoplasmic localization in response to heat shock and addition of extra-cellular calcium [42]. To test whether Zds1 or Zds2 had a role in the DNA damage checkpoint, we analyzed the rate at which cdc13-1, or cdc13-1 chk1Δ cells lacking ZDS1, ZDS2, or TPK2 failed in the checkpoint mediated arrest by scoring the number of cells exhibiting a late mitotic phenotype. cdc13-1, cdc13-1 zds1Δ and cdc13-1 zds2Δ cells were proficient in restraining mitosis, deletion of ZDS1 delayed the rate at which chk1Δ cells failed in the checkpoint mediated arrest (Figure S1A). This result was not surprising as cells lacking Zds1 have a delay in G2 and mitosis under normal growth conditions [47]. However cdc13-1 chk1Δ lacking ZDS2, MCK1 or TPK2 exhibited more cells with a late mitotic phenotype at earlier time points than cdc13-1 chk1Δ (Figure 4A and Figure S1B). The rate at which cdc13-1 chk1Δ lacking ZDS2, MCK1 or TPK2 failed in the checkpoint was identical, suggesting that Zds2 has a role in the checkpoint similar to the role of Tpk2 and Mck1. Neither Zds1 nor Zds2 was required for the phosphorylation of the R subunit following DNA damage (Figure 4B), indicating that Zds2 has a role in the checkpoint independent of the phosphorylation of the R subunit.
cAMP signaling and Hxk2 are both required for the PKA role in the DNA damage checkpoint-induced delay in mitosis
Our genetic experiments support the existence of a signaling complex that includes phosphorylated R subunit, PKA catalytic subunit(s), and possibly Zds2. To examine whether cAMP is required to activate this complex following DNA damage, proteins involved in the regulation of cAMP signaling were deleted or mutated in cdc13-1 chk1Δ cells. In this way we could gain insight as to whether or not cAMP was required for the role of PKA in the DNA damage checkpoint. A cdc35-1 mutation, which causes inactivation of adenylate cyclase when the cells are grown at the restrictive temperature [48], [49], was introduced into cdc13-1 and cdc13-1 chk1Δ cells so that we could determine whether adenylate cyclase had a role in the DNA damage checkpoint. The cells were released from a G1 arrest into the cell cycle. A DNA damage signal was generated and/or adenylate cyclase was inactivated as the cells progressed through S phase by raising the temperature to 35°C. Cells were scored for nuclear division as in previous assays. Due to the timing of Cdc35 inactivation in cdc35-1 cells, only 40% of cdc13-1 cdc35-1 entered S phase as evidenced by budding, consequently only the large-budded fraction was scored for this strain in the later timepoints. All other strains were uniformly large budded at later timepoints. Inactivation of adenylate cyclase enhanced the checkpoint defect of the chk1Δ cell (Figure 5B), suggesting that cAMP is required to activate the PKA complexes that play a role in the DNA damage checkpoint.

Hexokinase 2 (Hxk2) is required for transient activation of cAMP signaling upon addition of glucose to cells growing in a non-fermentable carbon source and in cells with intracellular acidification [39]. We show here that deletion of HXK2 exacerbated the checkpoint defect of cdc13-1 chk1Δ cells (Figure 5A). The cdc13-1 chk1Δ hxk2Δ cells failed in the checkpoint with similar kinetics as a cdc13-1 rad9Δ cell (Figure 5A), indicating that the majority of the checkpoint response was gone in cdc13-1 chk1Δ hxk2Δ cells. Deletion of HXK2 also enhanced the checkpoint defect of chk1Δ cells with similar kinetics as inactivation of adenylate cyclase (Figure 5B) and did not enhance the checkpoint defect of cdc13-1 chk1Δ tpk2Δ cells (Figure 5C), suggesting that Hxk2 is acting in the same pathway as PKA to help restrain mitosis following DNA damage. In addition, Hxk2 was not required for phosphorylation of the R subunit following DNA damage (Figure 5D).
Mck1, Zds1, and Zds2 play roles in the re-localization of the R subunit following DNA damage
Both Zds1 and Zds2 have been shown to be involved in regulating the cytoplasmic localization of the phosphorylated R subunit under conditions in which the phosphorylation of the R subunit was Mck1 dependent [42]. Thus we examined whether Zds1, Zds2 and Hxk2, which did not play a role in R subunit phosphorylation following damage, could regulate PKA by causing the cytoplasmic localization of the R subunit. We also examined whether the cytoplasmic localization following DNA damage was dependent on Mck1, as it was required for the R subunit phosphorylation in this response. Localization of GFP-tagged Bcy1 was analyzed by microscopy in WT BCY1-GFP cells, cdc13-1 BCY1-GFP, cdc13-1 mck1Δ BCY1-GFP, cdc13-1 hxk2Δ BCY1-GFP, cdc13-1 zds1Δ BCY1-GFP and cdc13-1 zds2Δ BCY1-GFP cells released from a G1 block into the cell cycle at the restrictive temperature for cdc13-1. As shown before, G2/M cdc13-1 cells had a significant decrease in nuclear enrichment of Bcy1-GFP signal in the nucleus, indicating cytoplasmic re-localization of the R subunit following a DNA damage signal (Figure 6A, compare nuclear enrichment of Bcy1-GFP in CDC13 vs cdc13-1 cells). We found that the re-localization of R subunit in G2/M cells with a DNA damage signal was completely dependent on Mck1. In fact there was no difference in the cells with damage signal lacking Mck1 (cdc13-1 mck1Δ BCY1-GFP ) and the cells with no damage signal (CDC13 BCY1-GFP). The re-localization of the R subunit was also dependent on Zds1, Zds2 and Hxk2.

These results support a model (Figure 6B) in which phosphorylation and relocalization of the R subunit as well as cAMP are both required to establish the PKA signal following DNA damage in order to help restrain mitotic progression.
Discussion
How a holoenzyme such as PKA can interpret different stimuli to elicit a distinct cellular response is a timely question. PKA specificity of signaling in mammalian cells is regulated at least in part by phosphorylation of the catalytic or regulatory subunits, as well as interactions with specific proteins, and sub-cellular localization. Previous studies in yeast have shown that the R subunit of PKA is phosphorylated in response to various signals; however, the phosphorylation of the R subunit did not have a role in regulating responses previously associated with PKA activation [42]. In budding yeast the PKA pathway antagonizes expression of genes that are required for cells to tolerate stress from starvation, heat shock and oxidative damage [50]. Those signals and DNA damage, which lead to phosphorylation and redistribution of the R subunit, correlate with global down-regulation of PKA [50]. Therefore, phosphorylation of the R subunit could be a mechanism to allow localized activation of the holoenzyme following stress.
We have shown that the catalytic subunits of PKA have a role in restraining mitosis after DNA damage. Our data presented here indicated that phosphorylation of the R subunit in response to DNA damage could contribute to specifically target PKA to substrates involved in restraining mitosis. Previous studies showed that growing cells in non-fermentable carbon sources led to phosphorylation of the R subunit, and that addition of glucose to cells growing in a non-fermentable carbon source caused a transient increase of intracellular cAMP. The PKA activity following glucose addition drives cells into S phase and it had been proposed that PKA activity also helped restrain mitosis if the daughter cell had not reached an appropriate size [27]. Therefore we propose that the checkpoint and nutrient-sensing pathways share a signaling node to restrain mitosis under two different conditions: 1) following nutrient-induced rapid transition through the cell cycle and 2) DNA damage.
Model for activation of PKA by the DNA damage checkpoint
Previous studies showed that growing cells in non-fermentable carbon sources led to phosphorylation of the R subunit [41], and that addition of glucose to cells growing in a non-fermentable carbon source caused a transient increase of intracellular cAMP [51]. From these data we postulated that activation of PKA in response to DNA damage is carried out in two steps: the phosphorylation of a small population of the R subunit, probably to provide the specificity of the signal, and a requirement for cAMP to activate PKA catalytic activity. We showed that in addition to the serine residues in clusters I and II of the R subunit, adenylate cyclase was also required for the DNA damage checkpoint mediated arrest that could be measured in cells lacking CHK1. We have identified four proteins that have a role in regulating PKA activity in the DNA damage checkpoint. These proteins were previously implicated in regulation of PKA by 1) regulating the phosphorylation of the R subunit (Mck1), 2) regulating the cytoplasmic localization of the R subunit (Zds2) [42], and 3) regulation of cAMP levels (Hxk2, Cdc35) [39]. We found that all four proteins had a similar role to that of the PKA catalytic subunits in restraining mitosis suggesting that they function in the same pathway. However only Mck1, but not Zds2 or Hxk2, was required for the phosphorylation of the R subunit.
Mck1 was originally identified and characterized by Shero et al., as a high copy suppressor of a defect in chromosome segregation caused by a single base pair insertion in CDEIII, one of the three conserved DNA binding domains in the centromere of a marker chromosome fragment [52]. Disruption of Mck1 alone did not cause chromosome segregation defects but the mck1Δ cells were benomyl sensitive. Based on this role of Mck1, it will be interesting to determine in the future whether Bcy1 phosphorylation after DNA damage plays a role in preventing chromosome segregation defects. Based on the functions previously attributed to these proteins and the evidence presented here we propose that in response to DNA damage the R subunit is phosphorylated in a checkpoint - and Mck1-dependent fashion. The phosphorylated R subunit, which could still be associated with the catalytic subunits, is further regulated by Zds2, possibly by protein-protein interaction or sub-cellular localization. In the final step of regulation, Hxk2-regulated cAMP levels are required for activation of PKA signaling to help restrain mitosis.
Our findings also implicate a possible role for Ras2 in PKA signaling in the DNA damage checkpoint as the cdc35-1 allele encodes a protein with a single amino acid substitution in the Ras-binding domain [49]. In fact, we recently reported that deletion of the IRA1 and IRA2 genes encoding negative regulators of Ras prevents cellular recovery from a cdc13-1-induced DNA damage induced arrest. The ira1Δ ira2Δ recovery defect required the PKA catalytic subunit Tpk2 and the PKA phosphorylation sites on the anaphase promoting complex specificity factor Cdc20, indicating a link between the recovery defect and PKA regulation of mitosis [53]. Our data support a model in which the R subunit phosphorylation and cAMP signaling are working together to achieve the PKA-mediated restraint of mitosis following DNA damage (Figure 6B).
Model for Zds2 regulation of PKA following DNA damage
Zds2 may also help provide specificity to PKA signaling. We found that deletion of Zds2, but not Zds1, enhanced the checkpoint defect of chk1Δ cells. In fact, deletion of ZDS1 delayed the rate at which chk1Δ cells failed in the checkpoint-mediated arrest (Figure S1A). Cells lacking ZDS1 have a delay in G2 and mitosis under normal growth conditions [47]. Thus, Zds1 may play a role in restraining anaphase that cannot be uncovered due to the fact that lack of Zds1 triggers a G2 delay. Furthermore, it was previously shown that both Zds1 and Zds2 were involved in regulating the cytoplasmic localization of the phosphorylated R subunit under conditions in which the phosphorylation of the R subunit was Mck1 dependent [42]. We show here that the cytoplasmic localization of the R subunit following DNA damage requires both Zds1 and Zds2. Based on these data we cannot rule out a role for Zds1 in restraining mitosis following DNA damage.
The Cdc14 phosphatase plays a key role in reversing Cdk phosphorylation and reducing mitotic cyclin/Cdk activity required for cells to exit mitosis [7]. It was recently shown that Zds1 and Zds2 are required for the timely activation of Cdc14 during mitotic exit. Cdc14 is sequestered in the nucleolus by the inhibitory protein Net1/Cfi1 prior to anaphase. This is achieved by the role of the phosphatase PP2ACdc55 in maintaining Net1/Cfi1 in the de-phosphorylated state. Separase, along with other proteins of the Cdc Fourteen Early Anaphase Release (FEAR) pathway, regulate Cdc14 release during anaphase by downregulation of PP2ACdc55, which results in Net1 phosphorylation. Zds1 and Zds2 interact with PP2ACdc55 and are required for the separase-mediated downregulation of PP2ACdc55 to allow Cdc14 release [54]. One possible role for Zds2 (and Zds1) in restraining mitosis would be that these proteins may act as scaffolds that maintain PP2A in an active state by regulating PKA localization. In this manner PKA could be antagonizing separase in the regulation of PP2A to release Cdc14.
Little is known about how Zds1 and Zds2 regulate R subunit localization, however it is possible that Zds2 is required for the export of the phosphorylated R subunit from the nucleus, but not the PKA catalytic subunit thereby maintaining active catalytic subunits in the nucleus to help restrain mitosis. Alternatively Zds2 could act to retain the holoenzyme in the cytoplasm. Zds1 has been shown to localize to the bud neck and cortex when overexpressed [55], and complexes at the daughter spindle pole body, bud neck and daughter bud cortex have been shown to play roles in the spatio-temporal regulation of mitotic progression including regulators of MEN [56]–[58] and components of PP2ACdc55 [59]. Therefore, Zds2 (and Zds1) could localize the PKA holoenzyme at multiple sites where it could play a role in regulation of mitotic exit: spindle pole body, daughter cortex or at the bud neck.
Models for the role of cAMP during the DNA damage-induced checkpoint delay
In yeast increased cAMP due to intracellular acidification, or addition of glucose to cells growing in a non-fermentable carbon source require Hxk2 [39]. Our data show that either deletion of HXK2 or inactivation of adenylate cyclase enhanced the checkpoint defect of chk1Δ cell suggesting that Hxk2 regulates cAMP levels following DNA damage. Since we do not know whether or not cAMP levels increase in response to DNA damage, there are at least two models by which Hxk2 could be regulating cAMP in the checkpoint response. In the first model we propose that a basal level of cAMP, which is maintained by Hxk2 is sufficient for the activation of PKA in the checkpoint response. Alternatively the checkpoint could lead to increased cAMP levels in the cell by regulating Hxk2 and possibly Ras2 (Figure 6B).
Our data supports a model in which the DNA damage checkpoint can regulate the PKA pathway to induce specific PKA signaling in order to phosphorylate substrates that act to restrain mitosis. We identified three proteins (Mck1, Zds2, Hxk1) that along with adenylate cyclase (Cdc35) and the R subunit (Bcy1) have a novel role in the DNA damage checkpoint via regulation of PKA. Mutation or mis-regulation of PKA subunits has been associated with chromosomal instability in cancer cells [60]. It will be interesting to determine whether PKA also participates in the response to DNA damage in mammals.
Materials and Methods
Strains and plasmids
Strains used in this study are listed in Table 1. Yeast strains were generated using standard genetic techniques. For deletion of BCY1, a DNA fragment was generated by the PCR, which contained the URA3 gene flanked by 50 bp of sequences homologous to the 5′ and 3′-UTRs of BCY1. The DNA fragment was then transformed into Y300, resulting in the replacement of BCY1 with URA3 by homologous recombination. Gene disruption was confirmed by PCR. The BCY1:URA3 was introduced into the strains used in these studies by crossing.

Deletion of ZDS1, MCK1, and HXK2 was carried out by using a construct generated by the PCR which resulted in amplification of the genomic DNA surrounding the gene that had been replaced with KanMX. Genomic DNA from strains from the deletion strain collection (Open Biosystems) were used as template for these reactions. The BCY1-GFP strains were generated by crossing clone YIL033C from the GFP-fusion library (Invitrogen) into Y300 background five times followed by crossing into cdc13-1 strain in Y300 background.
cdc35-1 strains were generated by crossing the cdc35-1 allele from CMY282 [49] into the Y300 strain background before crossing into cdc13-1 strains.
Plasmids used in this study are listed in Table 2. MCK1 including upstream and downstream sequences was amplified using the primers: CTGGATCCTCTTCCCTCTTTCCCAATT, and GCTCTAGATAAACAGCGGATCA AAGG which contained a BamHI and XbaI site respectively. The amplified sequence was ligated into a pRS425 vector that had been cut with BamHI and XbaI.

pJS11 was generated by subcloning BCY1 from pXP1 [61] into pRS425 using BamHI and HindIII. pYCJ1 was generated as described in [41] pYCJ2 was generated using a three step PCR method using pJS11 and the primers:
Reverse: CTGGCTCGAGCTTGAGCTTGAGCTGCTTGAGGTCTGGAAAATGAC
Forward CAAGCTCAAGCTCGAGCCAGAGCGGCTGTTATGTTCAAATCCCCC which generated the serine to alanine mutations described in [41]. The resulting PCR product was used to amplify the region upstream of the mutated sites using pJS11 as the template and the forward primer: CGTCCGACTTTCTTCAGTTC. The PCR product generated in the second PCR was used as the forward primer and the reverse primer: CGTCATACATGAGTCTCTTC were used to amplify the region between BspEI and BsrGI sites containing the nucleotide changes to generate the serine to alanine mutations.
The resulting PCR product replaced the fragment generated by digestion of pJS11 with BspEI and BsrGI.
Growth conditions
Cells were grown in YPD rich medium or SC-Leu medium. When α-factor was added to synchronize cells SC-Leu or YPD at pH 3.9 were used.
Visualization of nuclei
Cells were grown to OD600 = 0.5–0.8 at 22°C. Cells were synchronized in G1 using α-factor (10 µg/ml) or in late S-phase using 200 mM Hydroxyurea (HU) as indicated. Unless indicated otherwise, the temperature was raised to 32°C for 60 min. prior to release. Cells were washed and re-suspended in YPD pH 3.9 at 32°C to release the cells into the cell cycle. To stop the cells from undergoing multiple cell cycles, α-factor was added back to the cells when cells released from a G1 block entered S-phase (re-budded) or immediately upon release for cells released from a late S-phase arrest. The cells were fixed and permeabilized using 70% ethanol. Cells were re-hydrated in PBS and stained with 0.1 mg/ml 4′, 6-diamidino-2-phenylindole dihydrochloride (DAPI, Sigma, St. Louis, MO). Cells were mounted on glass slides coated with 0.1% poly-L-lysine for microscopy.
Western analysis
Protein extracts were prepared by trichloro-acetic acid (TCA) precipitation as previously described [62]. Proteins were separated on 10% acrylamide/0.067% bis-acylamide gels, and transferred to nitrocellulose membranes. Bcy1 was detected by Western analysis using anti-Bcy1 antibody (Santa Cruz Biotechnology, Santa Cruz, CA). The immune complexes were detected by chemiluminescence (NEN).
Phosphatase assay
Protein from cdc13-1 cells grown to OD600 = 0.5 at 22°C and incubated at 32°C for 120 min. was isolated by TCA precipitation. TCA precipitated protein was re-solubilized by boiling in buffer containing 1% SDS and 10 mM Tris pH 8. Yeast lysis buffer with no SDS was used to dilute the solubilized protein solution so that the final SDS concentration was 0.1%. Bcy1 was immuno-precipitaed using anti-Bcy1 antibody (Santa Cruz) and protein-A sepharose beads (Amersham). Bcy1 bound to the beads was treated with alkaline phophatase (Boehringer Mannheim) and/or phosphatase inhibitors (1 mM sodium orthovanadate, and 1 mM sodium fluoride). Bcy1 was released from the beads by boiling in Laemmli sample buffer (4% SDS, 20% glycerol, 10% 2-mercaptoethanol, contaning bromophenol blue and 0.125 M Tris-HCl pH 6.8). Immuno-precipitated proteins were resolved and analyzed as described above.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Cohen-FixOPetersJMKirschnerMWKoshlandD 1996 Anaphase initiation in Saccharomyces cerevisiae is controlled by the APC-dependent degradation of the anaphase inhibitor Pds1p. Genes Dev 10 3081 3093
2. VisintinRPrinzSAmonA 1997 CDC20 and CDH1: a family of substrate-specific activators of APC-dependent proteolysis. Science 278 460 463
3. IrnigerSPiattiSMichaelisCNasmythK 1995 Genes involved in sister chromatid separation are needed for B-type cyclin proteolysis in budding yeast. Cell 81 269 278
4. CioskRZachariaeWMichaelisCShevchenkoAMannM 1998 An ESP1/PDS1 complex regulates loss of sister chromatid cohesion at the metaphase to anaphase transition in yeast. Cell 93 1067 1076
5. UhlmannFLottspeichFNasmythK 1999 Sister-chromatid separation at anaphase onset is promoted by cleavage of the cohesin subunit Scc1. Nature 400 37 42
6. VermaRAnnanRSHuddlestonMJCarrSAReynardG 1997 Phosphorylation of Sic1p by G1 Cdk required for its degradation and entry into S phase. Science 278 455 460
7. VisintinRCraigKHwangESPrinzSTyersM 1998 The phosphatase Cdc14 triggers mitotic exit by reversal of Cdk-dependent phosphorylation. Mol Cell 2 709 718
8. ShouWSeolJHShevchenkoABaskervilleCMoazedD 1999 Exit from mitosis is triggered by Tem1-dependent release of the protein phosphatase Cdc14 from nucleolar RENT complex. Cell 97 233 244
9. JaspersenSLCharlesJFTinker-KulbergRLMorganDO 1998 A late mitotic regulatory network controlling cyclin destruction in Saccharomyces cerevisiae. Mol Biol Cell 9 2803 2817
10. VisintinRHwangESAmonA 1999 Cfi1 prevents premature exit from mitosis by anchoring Cdc14 phosphatase in the nucleolus [see comments]. Nature 398 818 823
11. ShouWAzzamRChenSLHuddlestonMJBaskervilleC 2002 Cdc5 influences phosphorylation of Net1 and disassembly of the RENT complex. BMC Mol Biol 3 3
12. StegmeierFAmonA 2004 Closing mitosis: the functions of the Cdc14 phosphatase and its regulation. Annu Rev Genet 38 203 232
13. QueraltELehaneCNovakBUhlmannF 2006 Downregulation of PP2A(Cdc55) phosphatase by separase initiates mitotic exit in budding yeast. Cell 125 719 732
14. QueraltEUhlmannF 2008 Cdk-counteracting phosphatases unlock mitotic exit. Curr Opin Cell Biol 20 661 668
15. JinFLiuHLiangFRizkallahRHurtMM 2008 Temporal control of the dephosphorylation of Cdk substrates by mitotic exit pathways in budding yeast. Proc Natl Acad Sci U S A 105 16177 16182
16. LimHHGohPYSuranaU 1998 Cdc20 is essential for the cyclosome-mediated proteolysis of both Pds1 and Clb2 during M phase in budding yeast. Curr Biol 8 231 234
17. BaumerMBrausGHIrnigerS 2000 Two different modes of cyclin clb2 proteolysis during mitosis in Saccharomyces cerevisiae. FEBS Lett 468 142 148
18. WaschRCrossFR 2002 APC-dependent proteolysis of the mitotic cyclin Clb2 is essential for mitotic exit. Nature 418 556 562
19. SanchezYBachantJWangHHuFLiuD 1999 Control of the DNA damage checkpoint by chk1 and rad53 protein kinases through distinct mechanisms. Science 286 1166 1171
20. WangYHuFElledgeSJ 2000 The Bfa1/Bub2 GAP complex comprises a universal checkpoint required to prevent mitotic exit. Curr Biol 10 1379 1382
21. HuFWangYLiuDLiYQinJ 2001 Regulation of the Bub2/Bfa1 GAP complex by Cdc5 and cell cycle checkpoints. Cell 107 655 665
22. AlexandruGZachariaeWSchleifferANasmythK 1999 Sister chromatid separation and chromosome re-duplication are regulated by different mechanisms in response to spindle damage. Embo J 18 2707 2721
23. LiR 1999 Bifurcation of the mitotic checkpoint pathway in budding yeast. Proc Natl Acad Sci U S A 96 4989 4994
24. ElledgeSJ 1996 Cell cycle checkpoints: preventing an identity crisis. Science 274 1664 1672
25. ClarkeDJGimenez-AbianJF 2000 Checkpoints controlling mitosis. Bioessays 22 351 363
26. YamadaHKumadaKYanagidaM 1997 Distinct subunit functions and cell cycle regulated phosphorylation of 20S APC/cyclosome required for anaphase in fission yeast. J Cell Sci 110 Pt 15 1793 1804
27. AnghileriPBranduardiPSternieriFMontiPVisintinR 1999 Chromosome separation and exit from mitosis in budding yeast: dependence on growth revealed by cAMP-mediated inhibition. Exp Cell Res 250 510 523
28. HeoSJTatebayashiKIkedaH 1999 The budding yeast cohesin gene SCC1/MCD1/RHC21 genetically interacts with PKA, CDK and APC. Curr Genet 36 329 338
29. NybergKAMichelsonRJPutnamCWWeinertTA 2002 TOWARD MAINTAINING THE GENOME: DNA Damage and Replication Checkpoints. Annu Rev Genet 36 617 656
30. WangHLiuDWangYQinJElledgeSJ 2001 Pds1 phosphorylation in response to DNA damage is essential for its DNA damage checkpoint function. Genes Dev 15 1361 1372
31. HuFWangYLiuDLiYQinJ 2001 Regulation of the Bub2/Bfa1 GAP complex by Cdc5 and cell cycle checkpoints. Cell 107 655 665
32. AgarwalRTangZYuHCohen-FixO 2003 Two distinct pathways for inhibiting pds1 ubiquitination in response to DNA damage. J Biol Chem 278 45027 45033
33. ZhangTNirantarSLimHHSinhaISuranaU 2009 DNA damage checkpoint maintains CDH1 in an active state to inhibit anaphase progression. Dev Cell 17 541 551
34. SearleJSSchollaertKLWilkinsBJSanchezY 2004 The DNA damage checkpoint and PKA pathways converge on APC substrates and Cdc20 to regulate mitotic progression. Nat Cell Biol 6 138 145
35. CannonJFTatchellK 1987 Characterization of Saccharomyces cerevisiae genes encoding subunits of cyclic AMP-dependent protein kinase. Mol Cell Biol 7 2653 2663
36. TodaTCameronSSassPZollerMScottJD 1987 Cloning and characterization of BCY1, a locus encoding a regulatory subunit of the cyclic AMP-dependent protein kinase in Saccharomyces cerevisiae. Mol Cell Biol 7 1371 1377
37. TodaTCameronSSassPZollerMWiglerM 1987 Three different genes in S. cerevisiae encode the catalytic subunits of the cAMP-dependent protein kinase. Cell 50 277 287
38. TheveleinJMde WindeJH 1999 Novel sensing mechanisms and targets for the cAMP-protein kinase A pathway in the yeast Saccharomyces cerevisiae. Mol Microbiol 33 904 918
39. RollandFWankeVCauwenbergLMaPBolesE 2001 The role of hexose transport and phosphorylation in cAMP signalling in the yeast Saccharomyces cerevisiae. FEM Yeast Res 1 33 45
40. ColledgeMScottJD 1999 AKAPs: from structure to function. Trends Cell Biol 9 216 221
41. GriffioenGBranduardiPBallariniAAnghileriPNorbeckJ 2001 Nucleocytoplasmic distribution of budding yeast protein kinase A regulatory subunit Bcy1 requires Zds1 and is regulated by Yak1-dependent phosphorylation of its targeting domain. Mol Cell Biol 21 511 523
42. GriffioenGSwinnenSTheveleinJM 2003 Feedback inhibition of cell wall integrity signaling by Zds1 involves Gsk3 phosphorylation of a cAMP-dependent protein kinase regulatory subunit. J Biol Chem 18 18
43. GriffioenGTheveleinJM 2002 Molecular mechanisms controlling the localisation of protein kinase A. Curr Genet 41 199 207
44. WeinertTAKiserGLHartwellLH 1994 Mitotic checkpoint genes in budding yeast and the dependence of mitosis on DNA replication and repair. Genes Dev 8 652 665
45. GarvikBCarsonMHartwellL 1995 Single-stranded DNA arising at telomeres in cdc13 mutants may constitute a specific signal for the RAD9 checkpoint. Mol Cell Biol 15 6128 6138
46. DesanyBAAlcasabasAABachantJBElledgeSJ 1998 Recovery from DNA replicational stress is the essential function of the S-phase checkpoint pathway. Genes Dev 12 2956 2970
47. MizunumaMHirataDMiyaokaRMiyakawaT 2001 GSK-3 kinase Mck1 and calcineurin coordinately mediate Hsl1 down-regulation by Ca2+ in budding yeast. Embo J 20 1074 1085
48. BouteletFPetitjeanAHilgerF 1985 Yeast cdc35 mutants are defective in adenylate cyclase and are allelic with cyr1 mutants while CAS1, a new gene, is involved in the regulation of adenylate cyclase. Embo J 4 2635 2641
49. DubacqCGueroisRCourbeyretteRKitagawaKMannC 2002 Sgt1p contributes to cyclic AMP pathway activity and physically interacts with the adenylyl cyclase Cyr1p/Cdc35p in budding yeast. Eukaryot Cell 1 568 582
50. GornerWDurchschlagEMartinez-PastorMTEstruchFAmmererG 1998 Nuclear localization of the C2H2 zinc finger protein Msn2p is regulated by stress and protein kinase A activity. Genes Dev 12 586 597
51. TheveleinJM 1994 Signal transduction in yeast. Yeast 10 1753 1790
52. SheroJHHieterP 1991 A suppressor of a centromere DNA mutation encodes a putative protein kinase (MCK1). Genes Dev 5 549 560
53. WoodMDSanchezY 2010 Deregulated Ras signaling compromises DNA damage checkpoint recovery in S. cerevisiae. Cell Cycle 9 3353 3363
54. QueraltEUhlmannF 2008 Separase cooperates with Zds1 and Zds2 to activate Cdc14 phosphatase in early anaphase. J Cell Biol 182 873 883
55. BiEPringleJR 1996 ZDS1 and ZDS2, genes whose products may regulate Cdc42p in Saccharomyces cerevisiae. Mol Cell Biol 16 5264 5275
56. FraschiniRD'AmbrosioCVenturettiMLucchiniGPiattiS 2006 Disappearance of the budding yeast Bub2-Bfa1 complex from the mother-bound spindle pole contributes to mitotic exit. J Cell Biol 172 335 346
57. MaekawaHPriestCLechnerJPereiraGSchiebelE 2007 The yeast centrosome translates the positional information of the anaphase spindle into a cell cycle signal. J Cell Biol 179 423 436
58. PereiraGSchiebelE 2005 Kin4 kinase delays mitotic exit in response to spindle alignment defects. Mol Cell 19 209 221
59. GentryMSHallbergRL 2002 Localization of Saccharomyces cerevisiae protein phosphatase 2A subunits throughout mitotic cell cycle. Mol Biol Cell 13 3477 3492
60. MatyakhinaLLenherrSMStratakisCA 2002 Protein kinase A and chromosomal stability. Ann N Y Acad Sci 968 148 157
61. PanXHeitmanJ 1999 Cyclic AMP-dependent protein kinase regulates pseudohyphal differentiation in Saccharomyces cerevisiae. Mol Cell Biol 19 4874 4887
62. FoianiMMariniFGambaDLucchiniGPlevaniP 1994 The B subunit of the DNA polymerase alpha-primase complex in Saccharomyces cerevisiae executes an essential function at the initial stage of DNA replication. Mol Cell Biol 14 923 933
63. AllenJBZhouZSiedeWFriedbergECElledgeSJ 1994 The SAD1/RAD53 protein kinase controls multiple checkpoints and DNA damage-induced transcription in yeast. Genes Dev 8 2401 2415
64. SanchezYDesanyBAJonesWJLiuQWangB 1996 Regulation of RAD53 by the ATM-like kinases MEC1 and TEL1 in yeast cell cycle checkpoint pathways. Science 271 357 360
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2011 Číslo 7
Nejčtenější v tomto čísle
- A Rice Plastidial Nucleotide Sugar Epimerase Is Involved in Galactolipid Biosynthesis and Improves Photosynthetic Efficiency
- Genome-Wide Association Study Identifies Novel Restless Legs Syndrome Susceptibility Loci on 2p14 and 16q12.1
- Loss of the BMP Antagonist, SMOC-1, Causes Ophthalmo-Acromelic (Waardenburg Anophthalmia) Syndrome in Humans and Mice
- Gene-Based Tests of Association