
Cytokinesis-Based Constraints on Polarized Cell Growth in Fission Yeast
The rod-shaped fission yeast Schizosaccharomyces pombe, which undergoes cycles of monopolar-to-bipolar tip growth, is an attractive organism for studying cell-cycle regulation of polarity establishment. While previous research has described factors mediating this process from interphase cell tips, we found that division site signaling also impacts the re-establishment of bipolar cell growth in the ensuing cell cycle. Complete loss or targeted disruption of the non-essential cytokinesis protein Fic1 at the division site, but not at interphase cell tips, resulted in many cells failing to grow at new ends created by cell division. This appeared due to faulty disassembly and abnormal persistence of the cell division machinery at new ends of fic1Δ cells. Moreover, additional mutants defective in the final stages of cytokinesis exhibited analogous growth polarity defects, supporting that robust completion of cell division contributes to new end-growth competency. To test this model, we genetically manipulated S. pombe cells to undergo new end take-off immediately after cell division. Intriguingly, such cells elongated constitutively at new ends unless cytokinesis was perturbed. Thus, cell division imposes constraints that partially override positive controls on growth. We posit that such constraints facilitate invasive fungal growth, as cytokinesis mutants displaying bipolar growth defects formed numerous pseudohyphae. Collectively, these data highlight a role for previous cell cycles in defining a cell's capacity to polarize at specific sites, and they additionally provide insight into how a unicellular yeast can transition into a quasi-multicellular state.
Published in the journal:
. PLoS Genet 8(10): e32767. doi:10.1371/journal.pgen.1003004
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1003004
Summary
The rod-shaped fission yeast Schizosaccharomyces pombe, which undergoes cycles of monopolar-to-bipolar tip growth, is an attractive organism for studying cell-cycle regulation of polarity establishment. While previous research has described factors mediating this process from interphase cell tips, we found that division site signaling also impacts the re-establishment of bipolar cell growth in the ensuing cell cycle. Complete loss or targeted disruption of the non-essential cytokinesis protein Fic1 at the division site, but not at interphase cell tips, resulted in many cells failing to grow at new ends created by cell division. This appeared due to faulty disassembly and abnormal persistence of the cell division machinery at new ends of fic1Δ cells. Moreover, additional mutants defective in the final stages of cytokinesis exhibited analogous growth polarity defects, supporting that robust completion of cell division contributes to new end-growth competency. To test this model, we genetically manipulated S. pombe cells to undergo new end take-off immediately after cell division. Intriguingly, such cells elongated constitutively at new ends unless cytokinesis was perturbed. Thus, cell division imposes constraints that partially override positive controls on growth. We posit that such constraints facilitate invasive fungal growth, as cytokinesis mutants displaying bipolar growth defects formed numerous pseudohyphae. Collectively, these data highlight a role for previous cell cycles in defining a cell's capacity to polarize at specific sites, and they additionally provide insight into how a unicellular yeast can transition into a quasi-multicellular state.
Introduction
Many cells polarize in response to intrinsic and extrinsic signals. As cell polarization is generally multifaceted, cells must integrate both negative and positive cues for successful cellular morphogenesis. In various organisms, the cell cycle provides a platform on which these cues are organized (for reviews, see [1], [2]), thereby ensuring distinct polarization events occur at the appropriate location, time, and context.
The fission yeast Schizosaccharomyces pombe represents a genetically tractable organism for studying cell cycle regulation of growth polarity (for reviews, see [3], [4]). Wild-type S. pombe extend solely at their two cell tips, lengthening their rod-shaped bodies while retaining fairly constant widths. After cell division, S. pombe grow only at old ends, so-called because they served as ends of the dividing mother cell. Then, at a point in G2 known as new end take off (NETO), new ends, which arise from cell division, also initiate growth [5]. NETO is not required for cell viability, and myriad mutants defective in this process have been identified [3], [4]. Yet, beyond requirements for S-phase completion and a minimal interphase cell size [5], additional cell cycle controls on NETO have not been identified.
As in other cell polarization events, cytoskeletal rearrangements accompany growth transitions in S. pombe. Prior to NETO, microtubule plus end-associated proteins Tea1 and Tea4 ride growing microtubule ends to both cell tip cortices [6]–[9], where they anchor based on their association with membrane proteins [10], [11]. Upon NETO, Tea4 recruits formin For3, which had before only been tethered to old ends, into a complex with itself and Tea1 at new ends [8]. As over-expression of a Tea1-For3 fusion can drive NETO prematurely [8], this association likely brings For3 into the proximity of formin activators at new ends, stimulating For3 catalysis of F-actin cables that will deliver growth cargo to this tip. Not surprisingly, loss of Tea1, Tea4, and/or For3 impairs fission yeast polarization and elongation [8], [9], [12], [13]. Actin patches, which guide endocytic vesicle internalization and constitute a second F-actin structure, also re-polarize to both cell tips upon NETO [14]. Disruption of proteins comprising these structures similarly jeopardizes growth polarity establishment [15]–[17]. Thus, alteration in protein composition at cell tips is coupled tightly to cytoskeletal rearrangements.
In addition to promoting cell tip growth, several tip-localized cell polarity factors, including Tea1 and Tea4, direct the cell division plane away from cell ends and towards the cell middle for cytokinesis [18], the process by which daughter cells undergo physical separation following nuclear division. However, whether the process of cytokinesis reciprocally modulates cell polarity is unclear. Some observations hint that the cell division machinery may play a role in directing cell polarity. As was previously noted, new ends formed by cell division initiate growth well after old ends. In mutants in which cells remain physically connected at division sites for multiple cell divisions, internal cells can grow, though this occurs sub-apically adjacent to septa [19], [20]. Moreover, many polarity factors localize to the cell division site [4], [21]–[23]; nonetheless, only cell tip-localized populations of these polarity proteins have been demonstrated to contribute to growth polarity in S. pombe.
As in most eukaryotes, cytokinesis occurs in S. pombe through the assembly and constriction of an actomyosin-based cytokinetic ring (CR) [24]. In addition to actin and myosin, several accessory proteins regulate the dynamics and organization of this structure. For one, Cdc15, which contains an N-terminal F-BAR domain and a C-terminal SH3 domain characteristic of the pombe Cdc15 homology protein family [25], has been posited to link CR proteins to the cortical membrane at the division site [26]. Cdc15-binding proteins at the CR include formin, myosin, and the C2 domain protein Fic1 [27], [28]. Fic1 localizes to both interphase cell tips and the cell division site [28], though its specific functions at these sites have not been described. Fic1's budding yeast ortholog, Inn1, contributes to cytokinesis by linking the CR to the ingressing membrane and by participating in septum formation [29], [30]. Septa form in both budding and fission yeasts as cell wall is deposited behind the constricting CR [31]. A conserved signaling network, known as the septation initiation network (SIN) in S. pombe, triggers septum deposition during cytokinesis [32]. Together with the CR, septa provide mechanical force for membrane closure at the cell division site [33]. Subsequent septum degradation allows for abscission [34], [35]. Clearly, various remodeling events must occur at the cell division site for cytokinesis to complete efficiently. Whether such remodeling events also influence daughter cell behavior has never been examined.
While wild-type S. pombe classically grow in a single-celled form, multiple fission yeasts, including S. pombe, possess the ability to assume an invasive, hyphal-like state [20], [36]. The ability of pathogenic fungi to undergo such a morphogenetic switch contributes significantly to fungal infections [37]. Though non-pathogenic, S. pombe, similar to the budding yeast Saccharomyces cerevisiae [38], can transition into invasive growth as a foraging response to low nutrients [36]. Invasive S. pombe form structures that technically qualify as pseudohyphae, for, unlike as in hyphal growth, cytokinetic constriction occurs [39], [40]. Pseudohyphae likely maintain their hyphal-like structure due to cellular adherence and preferential growth at old ends [39], [40]. Intriguingly, it has been postulated that single-celled fission yeast evolved from multicellular, filamentous fungi, with transcriptional networks that ensure efficient cell separation playing predominant roles in the evolution of a single-celled state [41]. Though S. pombe pseudohyphae do not commonly exhibit aborted cytokineses or multicellularity, it is an attractive hypothesis that inefficient, but not entirely defective, cytokinesis might somehow mark new ends to impair their growth and promote the dimorphic switch in S. pombe.
In this manuscript, we define a novel cell cycle control on S. pombe growth polarity, namely that the process of cytokinesis imposes limitations on new end growth competency. Here, we focus on Fic1, which we show to be involved in the re-establishment of polarized cell growth at new ends following cell division. Specifically, we demonstrate that Fic1 polarity function requires its localization to the CR but not to interphase cell tips, and that its protein-protein interactions at the CR, including that with Cdc15, promote bipolar cell growth in the ensuing cell cycle. We present evidence that loss of Fic1 impairs disassembly of the cell division apparatus, with parts of this machinery persisting at new ends following CR constriction. Additional mutants defective in late cytokinesis also exhibit impaired new end growth. Importantly, premature activation of NETO signaling does not fully rescue bipolar growth in cells with late cytokinesis defects, suggesting that cytokinesis-based constraints on S. pombe growth polarity play a central role in defining new end growth competency. We propose that such constraints can provide a mechanistic understanding of how S. pombe and possibly other fungi transition into invasive hyphal-like growth.
Results
The S. pombe Cytokinesis Factor Fic1 Promotes the Establishment of Bipolar Cell Growth
Recently, our laboratory identified Fic1, which was implicated in cytokinesis based on its protein and genetic interactions and its localization to the CR [28]. In addition to defects in cytokinesis, deletion of S. pombe fic1+, which is a non-essential gene, resulted in an abnormally high percentage of cells that grew only from one end (i.e., monopolar cells) (Figure 1A–1C). Tip growth was judged using calcofluor staining, as birth scars formed at previous division sites do not stain well with calcofluor and growth can be assessed using the position of these scars relative to cell tips (Figure S1A) [5]. The growth defects observed upon fic1+ disruption suggested that Fic1 not only participates in cytokinesis but also in the establishment of bipolar cell growth.

Although the upstream NETO factors Tea1 and Tea4 localized normally to both cell tips in fic1Δ cells (Figure S1B–S1C), other cell tip proteins implicated in growth polarity regulation exhibited unusual localization patterns in this mutant. Unlike wild-type cells with mostly bipolar actin patch distribution (Figure 1D–1E), a variety of mutants defective in bipolar cell growth exhibit monopolar actin patches [8], [21]–[23]. As in such mutants, the actin patch marker Crn1-GFP [42] accumulated preferentially at one cell end in a high percentage of fic1Δ cells (Figure 1D–1E). Signaling through Rho GTPases controls actin patch organization in S. pombe [13], [43], and the Rho1 activator Rgf1 [21], which was GFP-tagged and imaged with the spindle pole body marker Sid4-RFP [44], likewise predominated on one end of many fic1Δ cells (Figure 1F–1G). Not surprisingly, in both wild-type and fic1Δ cells, Rgf1-GFP and Crn1-RFP concentrated at the same ends (Figure S1D), which were confirmed by calcofluor staining to be the growing ends of fic1Δ cells (Figure S1E). Consistent with Fic1 affecting both actin and Rho networks, deletion of fic1+ was synthetically sick with deletion of genes encoding factors involved in F-actin nucleation (WASp Wsp1) and Rho GTPase regulation (RhoGEF Rgf1 and RhoGAP Rga1) (Figure S1F). Thus, we conclude that the absence of Fic1 upsets patterning of some but not all polarity factors.
To discern whether new and/or old ends were defective in resuming growth following cell division in fic1Δ cells, we performed time-lapse DIC imaging to trace birth scars in live cells. As expected, nearly all wild-type cells underwent NETO prior to subsequent septation (Figure 2A and 2C). However, following roughly two-thirds of fic1Δ cell divisions, either one or both daughter cells failed to initiate new end growth prior to the next septation (Figure 2B–2C). The most predominant growth pattern in fic1Δ cells was that in which one daughter cell underwent NETO while the other did not (Figure 2B–2C), with nearly 70% of those daughter cells that did not exhibit NETO being the younger daughter cell. Unlike tea1Δ and tea4Δ cells, in which one daughter cell commonly fails at its new end and the other daughter cell fails at its old end (Figure 2D) [8], [22], [23], fic1Δ cells were specifically defective in the re-establishment of growth at new ends following cell division (Figure 2B–2C). Intriguingly, tea1Δ fic1Δ double mutants grew mainly in a tea1Δ pattern, though nearly one-fifth of cell divisions produced a T-shaped daughter cell (Figure 2D–2E). Consistent with this, roughly 10% of tea1Δ fic1Δ cells were T-shaped at 25°C, while T-shaped tea1Δ cells were almost never observed at this temperature (Figure 2F). T-shapes always arose in cells that the tea1Δ growth pattern dictated should grow at their new ends (Figure 2D–2E) but that actually grew at neither (Figure 2E and 2G), suggesting these cells polarize at sites other than their tips because growth is inhibited at both ends. These data confirmed that the polarity defect caused by loss of Fic1 stochastically impacts new end growth in a variety of genetic backgrounds. Importantly, fic1Δ new ends that failed to extend in one cell cycle initiated growth as an old end in the next cell cycle, suggesting the defect in growth polarity caused by loss of Fic1 was not permanent. Consistent with a delay but not a block in growth, new ends that initiated growth prior to the next septation did so much later on average in fic1Δ cells than in wild-type cells (120 min versus 75 min) (Figure 2H).

To test whether fic1Δ's polarity defect was independent of S phase completion, we arrested fic1Δ cells in late G2 using cdc25-22, a temperature-sensitive allele of the phosphatase that activates cyclin-dependent kinase at the G2-M transition. As was previously observed [5], otherwise wild-type cells blocked in G2 almost always underwent NETO (Figure 2I–2J). However, roughly half of fic1Δ cells remained monopolar (Figure 2I–2J), indicating that the fic1Δ polarity defect occurs irrespective of S phase completion. To test whether fic1Δ cells were too small to initiate NETO, we measured cell lengths at division. Though slightly shorter on average than wild-type cells (13.3 µm versus 15.3 µm), all fic1Δ cells were longer at division than the minimum length required for NETO (∼9 µm) (Figure 2K) [5]. Therefore, it is unlikely that the fic1Δ growth polarity defect is caused by reduced cell length. These data underscore that loss of Fic1 disrupts the establishment and timing of NETO independently of previously described cell cycle controls.
Fic1 Protein–Protein Interactions at the CR Support Subsequent Polarized Cell Growth at New Ends
Though many cell polarity factors localize to the cell division site in addition to interphase cell tips, only the actions of these proteins at interphase cell tips have been demonstrated to be relevant to polarity regulation. As was observed previously [28], cytoplasmic Fic1-GFP localizes to cell tips during interphase and later to the CR during cell division (Figure 3A). We also detected another pool of Fic1-GFP lining the division site as the CR constricted (Figure 3A). Given this localization pattern and the specific new end growth defect of fic1Δ cells, we asked whether Fic1 affected the timing of NETO via its functions at the cell division site.

Like S. cerevisiae Inn1 [30], Fic1 is comprised of an N-terminal C2 domain and a C-terminal stretch of PxxP motifs (Figure 3B). As was found for Inn1 [29], [30], the C terminus of Fic1 (“Fic1C”, amino acids 127–272), expressed from its endogenous locus and GFP-tagged, was sufficient for CR localization, as judged by co-localization with the CR protein Cdc15-mCherry (Figure 3C). In contrast, a GFP-tagged N-terminal C2 domain fragment (“Fic1N”, amino acids 1–126) was never observed at the CR (Figure 3C) though it was produced in vivo (Figure S2). Importantly, medial-localizing Fic1C, unlike Fic1N, supported proper growth polarity establishment (Figure 3D–3F), and, in contrast to full-length Fic1-GFP, Fic1C-GFP was not detected at tips of interphase cells (Figure 3G). We thus conclude that Fic1, unlike other characterized growth polarity factors, does not exert its polarity function at cell tips during interphase, but instead does so at the cell division site during cytokinesis.
Because Fic1's C terminus was necessary and sufficient for proper growth polarity, we examined whether protein-protein interactions at the CR mediated by Fic1's C-terminal PxxP motifs, which bind SH3 domains, govern Fic1's polarity function. Fic1 was originally identified based on its interaction with Cdc15's SH3 domain [28]. As would be expected if association of Cdc15 with Fic1's C terminus is important in establishing the timing of NETO, calcofluor-stained cdc15ΔSH3 cells, which are viable but lack Fic1-Cdc15 interaction [28], exhibited growth polarity defects (Figure 4A–4C and Figure S3A–S3B).

To address the consequence of specifically disrupting Fic1-Cdc15 interaction, we determined which of Fic1's C-terminal PxxP motifs interact(s) with Cdc15's SH3 domain. Previous yeast-two hybrid data indicated Fic1 amino acids 190–269 mediate direct association with Cdc15's SH3 domain [28]. This region contains four of the eleven PxxP motifs within Fic1's C terminus (Figure S3C). To identify which are relevant for Cdc15 interaction, yeast two-hybrid assays using single and combinations of proline to alanine mutations were performed (Figure S3D). Mutation of PxxPs 10 and 11 in combination, or P257 of PxxP 11 alone, abolished the two-hybrid interaction (Figure S3D), and the P257A mutation eliminated co-immunoprecipitation of Fic1-FLAG3 with Cdc15 in vivo (Figure 4D). Supporting the idea that the Fic1-Cdc15 interaction is most relevant during cell division, Fic1-GFP did not accumulate preferentially in Cdc15-mCherry puncta at interphase cell tips (Figure S3E) and co-immunoprecipitation of Fic1-FLAG3 with Cdc15 was considerably stronger in mitosis than in interphase (Figure 4D). This is similar to other Cdc15 protein-protein interactions, which become enriched upon Cdc15 dephosphorylation at mitosis [26]. fic1-P257A cells exhibited monopolar growth defects similar to fic1Δ and cdc15ΔSH3 cells (Figure 4A–4C), confirming that binding of Fic1's C terminus to Cdc15 is critical for Fic1's polarity function. Even so, Fic1-P257A-GFP still localized to the CR (Figure S3F), indicating that medial localization of Fic1 during cytokinesis is necessary but not sufficient for re-establishment of proper growth polarity following cell division.
To corroborate that PxxP-mediated protein-protein interactions at the cytokinetic ring play a predominant role in Fic1's polarity function, we tested whether other interactors participate in S. pombe polarity regulation. The SH3 protein Imp2 has previously been shown to function redundantly with Cdc15 and bind Fic1 during cytokinesis [28]. Consistent with additional Fic1 interactions guiding growth polarity, loss of Imp2 also severely compromised bipolar cell growth (Figure 4A–4C and Figure S3A–S3B). In S. cerevisiae, the Fic1 ortholog Inn1 binds to another SH3 protein, Cyk3 [29], [45], and complexing of these two proteins with the Cdc15 homolog Hof1 has been suggested to direct septum formation and cell separation [29]. We found that S. pombe Cyk3 co-immunoprecipitated with Fic1 in mitosis (Figure 4E), and we also detected direct interaction between S. pombe Cyk3's SH3 domain and Fic1 via yeast two-hybrid (Figure S3G). Accordingly, these interactions appear to be conserved. As was also described in a recent study [46], we found that Cyk3-GFP localized to the CR and division site during cytokinesis, and it was retained at new ends immediately following cell division (Figure 4F). Consistent with these proteins performing a common function, loss of Cyk3 resulted in growth polarity defects similar to those seen upon loss of Fic1 or its interaction with Cdc15 or Imp2 (Figure 4A–4C). Thus, Fic1 collaborates with associated proteins at the CR to execute its growth polarity function, and we postulate that its C terminus acts as an adaptor molecule for SH3 proteins to ensure integration of distinct processes during cytokinesis. Of note, Fic1-P257A-GFP still localized to the CR in imp2Δ cyk3Δ cells (Figure S3H), indicating other CR proteins besides Cdc15, Imp2, and Cyk3 bind Fic1 and likely participate in polarity-relevant events at the division site.
Loss of Fic1 Impedes CR Disassembly and Leads to Persistence of Factors at the Division Site
To discern how loss of Fic1 scaffold function during cytokinesis impacts subsequent new end growth, we next defined what aspects of cytokinesis are perturbed in fic1Δ cells. Previous data demonstrated that fic1Δ was synthetically lethal with sid2-250 [28], a temperature-sensitive allele of the SIN kinase Sid2. Consistent with Fic1 and associated factors working in parallel to the SIN, we found that fic1Δ and cyk3Δ suppressed the hyperactive SIN allele cdc16-116 (Figure S4A), and that fic1Δ and cyk3Δ were synthetically sick or lethal with a variety of SIN alleles conferring loss of SIN function (Figure S4A–S4B). These genetic data implied that Fic1 most likely functions during late stages of cytokinesis. In line with this idea, the percentage of fic1Δ cells that had undergone ingression but were still joined at their division sites was more than four times that of wild-type cells (Figure 5A–5B). When cells were arrested in G2 using the cdc25-22 allele, this difference increased, with the percentage of joined cells roughly 15 times greater in the absence of Fic1 (Figure 5A–5B). Similar to S. cerevisiae inn1Δ cells [29] and S. pombe cdc15ΔSH3 cells [28], many G2-arrested fic1Δ daughter cells that were still joined at division sites exhibited membranous bridges (Figure S4C). These findings verified that the completion of cell division is perturbed in the absence of Fic1.

Consistent with early cytokinesis events proceeding appropriately without Fic1, time-lapse imaging of myosin regulatory light chain Rlc1-GFP [47], [48] along with spindle pole body marker Sid4-GFP revealed that the CR formed and constricted normally in fic1Δ cells (Figure 5C–5D). However, at the termination of CR constriction, parts of the CR persisted at the division plane (Figure 5E–5G and Figure S4D). During cytokinesis, the septum closes behind the constricting CR, and septum closure can be visualized using the β-glucan synthase GFP-Cps1 [49], [50]. As cytokinesis progresses, two GFP-Cps1 dots marking the leading edge of the septum can be seen getting progressively closer in the division plane, and these dots eventually join into one just as the CR completes constriction (Figure 5F). We found that Rlc1-mCherry3 remained at the division site following septum closure on average longer in fic1Δ cells compared to wild-type cells (22 min versus 8 min) (Figure 5E–5F). Consistent with these remnants representing the CR as a whole and not just Rlc1, phalloidin staining revealed atypical actin-rich masses, in addition to normal actin patches, flanking septa in fic1Δ cells (Figure 5G). By expressing LifeAct-GFP, we verified that these abnormal actin masses co-localized to a high degree with Rlc1-mCherry3 in a fic1Δ genetic background (Figure S4D). Thus, we conclude that the CR does not disassemble properly at the conclusion of cell division in fic1Δ cells.
In addition to CR-associated factors, glucanase Eng1-GFP [35] persisted at ingressed division sites significantly longer in fic1Δ cells compared to wild-type cells (on average, 51 min versus 21 min) (Figure S4E–S4F). Because glucanases execute septum degradation [34], [35], these data suggest that cell wall turnover is inefficient at fic1Δ septa. We thus conclude that loss of Fic1 jeopardized the completion of cell division, stalling remodeling of new ends in the next cell cycle.
Mutants with Late Cytokinesis Defects Likewise Exhibit New End Growth Polarity Errors
Because faulty cytokinesis led to persistence of parts of the cell division machinery at fic1Δ division planes, we speculated that these remnants might deter subsequent polarized growth at new ends. If this were the case, one would expect other mutants with late cytokinesis defects to also show erroneous new end growth. Previous data had indicated that Fic1-associated Imp2 contributes to CR disassembly, with imp2Δ cells exhibiting abnormal actin structures flanking previous division sites [51]. Though we had shown that imp2Δ cells are defective in bipolar cell growth (Figure 4A–4C), we wanted to confirm that their growth defect was specific to new ends. Using time-lapse DIC imaging, we found that roughly 75% of imp2Δ cell divisions produced at least one daughter cell that failed at new end growth (Figure 6A). Interestingly, both imp2Δ daughter cells failed at new end growth in the majority of cases (Figure 6A–6B). Therefore, proper disassembly of CR components correlates with new end competency for polarized growth.

In addition to showing CR disassembly defects, fic1Δ cells also exhibited delays in septum remodeling at the division site. We therefore tested if disruption of septum degradation could likewise impact polarized growth. Loss of Eng1 or its cooperating glucanase, Agn1 [34], resulted in high percentages of monopolar growth (Figure 6C–6D and Figure S5A). Moreover, the growth defect of eng1Δ daughter cells was specific to new ends (Figure 6A–6B), and, similar to fic1Δ cells, eng1Δ daughter cells that initiated NETO prior to the next septation did so on average later than wild-type cells (129 min versus 75 min) (Figure S5B). Anillin-like Mid2 and the septin ring, of which Spn1 and Spn4 form the core [52], target these glucanases into a ring structure around septa [53]. Loss of any of these proteins likewise impaired bipolar cell growth (Figure 6C–6D and Figure S5A). In addition, though the majority of spn1Δ daughter cells failed at new end growth (Figure 6A–6B), those that initiated NETO prior to the next septation took longer on average to do so than wild-type cells (95 min versus 75 min) (Figure S5B). We therefore conclude that defective completion of cell wall remodeling at the division site, in addition to improper disassembly of CR components, compromises NETO efficiency.
The SIN coordinates many aspects of CR and septum regulation during late cytokinesis. Not only does SIN signaling oversee maintenance of a mature, homogenous CR [54], it mediates Cps1 targeting and accumulation at the division site [49], [50]. Loss of SIN signaling during cytokinesis can thus lead to CR fragmentation [54] and abortive septation [49]. Given these phenotypes and the synthetic genetic interactions between fic1Δ and SIN mutants (Figure S4A–S4B), we examined the relevance of the SIN to new end growth control. Temperature-sensitive alleles of genes encoding the SIN kinases Cdc7 and Sid2 caused mild but statistically significant growth polarity defects at semi-restrictive temperature (Figure 6C–6D and Figure S5A). A temperature-sensitive allele of the gene encoding Cps1, which functions downstream of the SIN, caused dramatic defects in establishing bipolar cell growth even at permissive temperature (Figure 6C–6D and Figure S5A). Additionally, a high proportion of cps1-191 cells failed specifically at new end growth (Figure 6A–6B), and those that were able to trigger NETO prior to subsequent septation did so on average later than wild-type cells (107 min versus 75 min) (Figure S5B). Not surprisingly, we were able to detect incomplete ingression of cps1-191 cells shifted to the restrictive temperature during cytokinesis (Figure S5C), again suggesting that these mutants experience remodeling errors at the division site.
Currently, the mechanism of membrane remodeling and scission at the S. pombe division site is unclear. In a variety of other organisms, endosomal sorting complex required for transport (ESCRT)-III factors contribute to this process [55]. ESCRT-III components have not been implicated in S. pombe cytokinesis regulation, though ESCRT-III-associated AMSH (S. pombe Sst2) localizes to the division site [56]. We found that deletions of genes encoding ESCRT-III components Vps2 and Vps24 or ESCRT-III-associated Sst2 were synthetically sick with a variety of loss-of-function cytokinesis alleles, including imp2Δ and cps1-191 (Figure S5D). Interestingly, loss of Vps2, Vps24, or Sst2 resulted in monopolar percentages significantly greater than observed for wild-type cells (Figure 6C–6D and Figure S5A), and nearly half of vps24Δ cell divisions resulted in one or both daughter cells that failed at new end growth prior to the next septation (Figure 6A–6B). Though these phenotypes were less penetrant than in other mutants, we speculate that ESCRT-III function guides membrane remodeling at the conclusion of S. pombe cell division to impact new end polarized growth.
Of note, deletion of rlc1+ or paxillin pxl1+, which function primarily in early actomyosin function at the CR [47], [48], [57], [58], did not alter growth polarity percentages as significantly as other mutations or deletions (Figure 6C–6D and Figure S5A). Indeed, less than half of non-septated rlc1Δ and pxl1Δ cells were monopolar (Figure 6C), and the monopolar septated percentages of these genotypes were more similar to wild-type percentages than were those of the other mutants examined (Figure 6D). We therefore conclude that early steps in cytokinesis do not impact subsequent polarized cell growth as much as the terminal steps in cell division.
Ineffective Cytokinesis Partially Impedes New End-Tip Growth Even upon Constitutive NETO Signaling
If faithful remodeling of the division site is important for growth competency of new ends, then one would expect that prematurely triggering NETO signaling just after cell division should not fully rescue the growth polarity defects of late cytokinesis mutants. To test this, we constructed a mutant that would undergo constitutive NETO. As over-expression of a fusion protein linking cell tip-associated Tea1 with formin For3 induces NETO in G1 [8], we integrated a Tea1-For3 fusion (Figure 7A) into the endogenous tea1+ locus and deleted the single copy of the for3+ gene. We confirmed that the Tea1-For3 fusion protein was produced in vivo (Figure 7B) and verified that this fusion was sufficient to induce NETO in a cdc10-V50 G1 arrest (Figure S6A–S6B). As previously reported [7], double deletion of tea1+ and for3+ resulted in general cell rounding (Figure 7C). However, expression of the Tea1-For3 fusion protein in the absence of Tea1 and For3 individually caused cells to regain their rod-shaped appearance (Figure 7C). Intriguingly, a high percentage of tea1-for3 cells were either septated or exhibited cytokinesis defects (Figure 7C–7D), and tea1-for3 cells were significantly longer at division than wild-type cells (on average, 18.3 µm versus 15.3 µm) (Figure 7C and 7E). Thus, though the endogenous Tea1-For3 fusion protein functioned in prematurely triggering NETO, it also affected cell division.
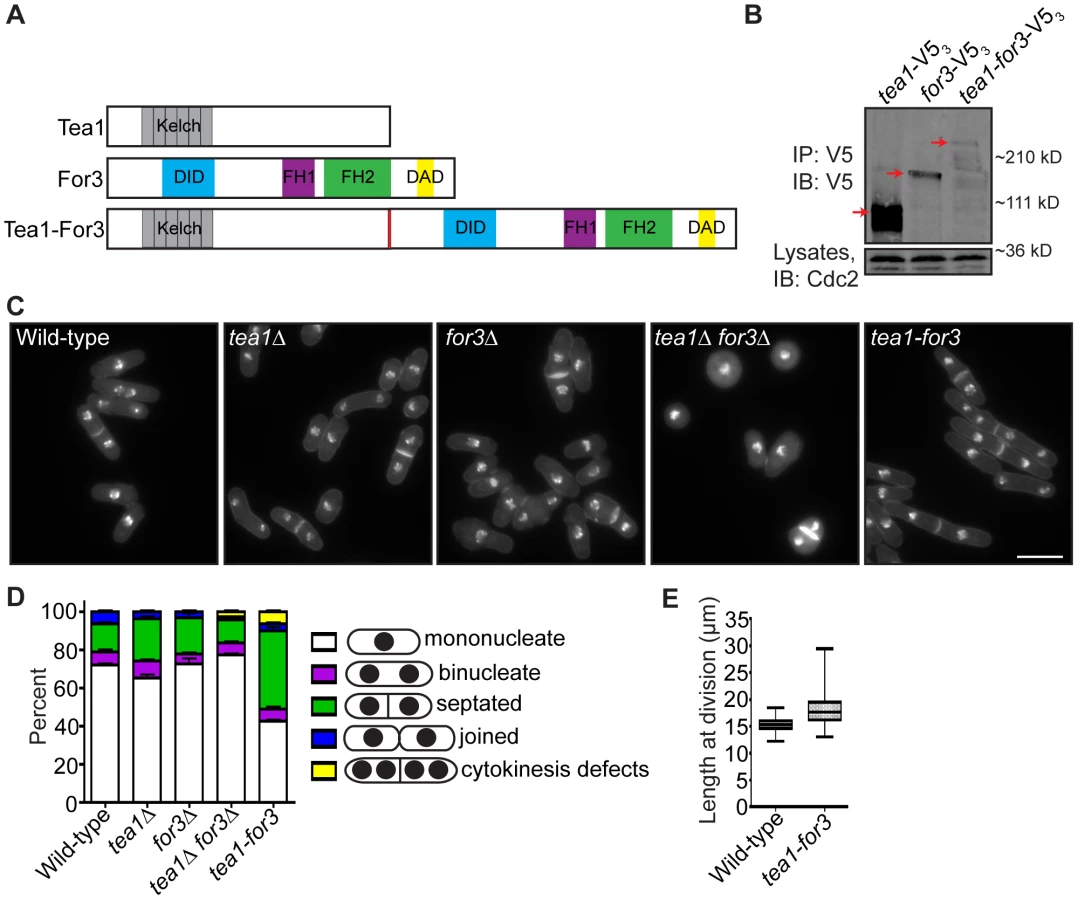
To analyze tea1-for3 cells in real-time, we performed time-lapse DIC imaging. As expected, most tea1-for3 cells underwent NETO before the next cell division (Figure 8A), with nearly 75% of new ends initiating growth within 50 minutes of septum splitting (Figure 8B). Nonetheless, some tea1-for3 outliers took much longer to extend at tips created by cell division (Figure 8B). After grouping the times needed for tip growth to occur at previous division sites relative to the amount of time needed for the mother cell to complete cytokinesis, we found that newly-formed tips that took longer to initiate growth had been formed by more inefficient cytokinesis (Figure 8C–8D). As distal tip growth continued in cells undergoing division (Figure 8D) and appeared unimpeded by additional factors, these findings suggested that faulty cytokinesis imposes constraints at previous division sites that counteract positive polarizing cues. We corroborated this model by expressing the Tea1-For3 fusion in fic1Δ cells. Although tea1-for3 cells were mostly bipolar, tea1-for3 fic1Δ cells showed a high percentage of monopolar growth (Figure 8E–8G). These findings confirmed that efficient completion of cytokinesis is critical for new end growth, even when signaling networks responsible for NETO are prematurely activated.

Errors in Growth Polarity Caused by Faulty Cytokinesis Translate into Heightened Fungal Invasiveness
S. pombe undergoing a dimorphic switch from single-celled to invasive form grow primarily in a monopolar fashion at old ends [39], [40]. Moreover, it has been postulated that cytokinesis errors might contribute to a hyphal-like transition in S. pombe [41]. We therefore considered that cytokinesis-based constraints on S. pombe growth polarity might facilitate invasive growth transitions. Using techniques similar to those described previously [40], [59], we tested whether various cytokinesis mutants displaying defective bipolar growth could form pseudohyphae into 2% agar. Cells lacking Fic1 or its interacting partners Cyk3 or Imp2 were significantly more invasive than wild-type cells (Figure 9A–9B). Like other invasive S. pombe mutants [39], [40], these mutants formed pseudohyphae composed of single cells oriented in filament-like projections (Figure 9C and Figure S7A). In addition to these strains, we found other cytokinesis mutants exhibiting high degrees of monopolar growth (spn1Δ, cdc7-24, and vps24Δ) to also be highly invasive and to form pseudohyphal projections into 2% agar (Figure 9A–9B and Figure S7A). Of note, the vps24Δ strain showed drastically more invasive growth than the others, though the reasons for this are currently unclear. rlc1Δ and pxl1Δ, which possess cytokinesis defects that do not considerably impact polarized cell growth (Figure 6C–6D and Figure S5A), invaded less efficiently on 2% agar than cytokinesis mutants exhibiting NETO defects (Figure 9A–9B). This supports the notion that defective cytokinesis promotes the dimorphic switch most robustly when it results in faulty NETO. As has previously been observed, tea1Δ also invaded well on 2% agar (Figure S7B–S7D). Thus, though cytokinesis-based constraints on growth polarity support enhanced S. pombe invasiveness, other polarity defects, which are not entirely specific to new ends, can do so as well.

Consistent with bipolar growth defects accompanying pseudohyphal growth, tea1-for3 cells, which experience constitutive NETO induction, almost never extended pseudohyphae into 2% agar (Figure 9D–9E). Because cytokinesis-based constraints on growth polarity partially override tip-based NETO signaling, we reasoned that tea1-for3 cells should become more invasive upon loss of Fic1. Indeed, on 2% agar tea1-for3 fic1Δ cells formed pseudohyphae (Figure S7E), which were more numerous than those observed for wild-type and tea1-for3 strains (Figure 9D–9E). Thus, perturbations in cytokinesis cause growth polarity errors that facilitate pseudohyphal growth even upon constant NETO signaling.
Lastly, we asked whether loss of polarity-relevant cytokinesis factors could partially rescue invasiveness of an asp1Δ strain, which is unable to undergo the dimorphic switch due to an inability to sense external cues [40]. Previously, it was demonstrated that asp1Δ cells form a biofilm-like colony on 0.3% agar (Figure 9F) [40]. Growth on 0.3% agar is more sensitive for assaying invasiveness of strains that invade less efficiently, as wild-type colonies form protrusions on 0.3% agar but extend relatively few pseudohyphal projections into 2% agar (Figure 9A–9B and 9F) [40]. We therefore assessed the effect of cytokinesis defects on asp1Δ invasiveness by testing whether asp1Δ strains that also lacked relevant cytokinesis factors still formed biofilms on 0.3% agar. Intriguingly, double deletion strains of asp1Δ with fic1Δ, spn1Δ, or vps24Δ did not form biofilms on 0.3% agar but instead made projections into and on the surface of the agar (Figure 9F). In contrast, double deletion strains of asp1D with either rlc1Δ or pxl1Δ still formed biofilms on 0.3% agar (Figure 9F). We thus conclude that cytokinesis-based constraints on polarized cell growth in S. pombe can foster invasiveness even in the absence of typical nutritional signals.
Discussion
In this study, we have shown how cytokinesis and cell polarity crosstalk to regulate fission yeast morphogenesis. Our data support a model (Figure 10) in which Fic1 acts as an adaptor at the CR, where it guides proper completion of cytokinesis and thereby affects division site remodeling. Loss of Fic1, its interactions, or parallel pathways results in delayed growth at new ends, even upon constitutive activation of NETO signaling. Impaired bipolar cell growth resulting from defective cytokinesis in turn enhances S. pombe invasiveness.

Cytokinesis-Based Regulation of Cell Polarity
The majority of S. pombe monopolar mutants previously analyzed fail at old end growth [4]. However, the cytokinesis mutants studied here were predominantly new end growth defective. As in other organisms [60], numerous S. pombe proteins known to affect growth polarity localize to the division site; this has fostered speculation that signaling at both cell tips and the division site might impact growth zones [23]. However, whether or not the cytokinesis functions of these proteins can specifically impact cell polarity has received little attention, especially in S. pombe. Our data provide evidence directly linking division site organization to S. pombe growth polarity. Because many factors involved in completing cell division likely also impact subsequent polarized growth, we believe our data could explain the involvement of diverse proteins in this process.
In other organisms, cytokinesis proteins appoint local regions of the cell cortex for growth following cell division [61]–[63]. For example, several budding yeast proteins, which remain at the cell cortex following cell division, have been reported to convey a cortical “tag” that marks the position of the next bud site [61], [62]. Similarly, during Drosophila melanogaster neurogenesis, cytokinetic furrow components mark the site from which the first dendrite will sprout [63]. In these cases, cytokinesis factors confer a positive polarizing cue adjacent to previous division sites, which contrasts with our findings in S. pombe where the cell division machinery impedes polarization and growth at new ends created by cell division.
The fact that S. pombe grow at old but not new ends after cell division is somewhat counterintuitive [4], especially because the cell growth machinery concentrates at the division site. Upon the completion of cytokinesis, the S. pombe growth machinery mysteriously shuttles to old ends rather than remaining at new ends. Why does the growth machinery relocate from the division site to old ends? One explanation is that new end cortices must be re-structured to become competent for tip growth. Indeed, specific lipid and cell wall variants contribute to S. pombe cytokinesis [64], [65], and local rearrangements of these may be required for growth activation. Moreover, the persistence of CR factors at new ends might create physical barriers to cytoskeletal elements, such as actin cables, required for tip growth. An inhibitory role for cell division in polarization is supported by studies of mutants that undergo multiple rounds of cytokinesis without physically separating, because internal cells in these structures do not grow into septa but branch adjacently. In a single-celled context, we speculate that when late cytokinetic events are perturbed, inherent delays in cortical re-structuring are exacerbated, causing growth polarity defects at new ends. In cases in which one daughter cell fails at NETO while the other is successful, we suspect these arise due to unequal partitioning of cytokinesis remnants and/or differences in cell cycle stages or life histories of daughter cells.
Consistent with furrow remodeling affecting polarization in other organisms, initial cellular protrusions of dividing mammalian cells orient away from the midbody linking daughter cells during abscission [66]. Only after the completion of cell division does polarization also occur near the latent division site [66]. Moreover, forced entry of HeLa cells with monopolar spindles into cytokinesis results in anucleate daughter cells that, similar to their nucleated counterparts, exhibit membrane protrusions only distal to cleavage furrows [67]. Thus, similar to our model in S. pombe, some factor at the division site cortex, and not a cell's cytosolic constituents, requires remodeling for post-cytokinetic polarization. Recent evidence indicates that mechanosensory pathways can direct cell polarization away from points of tension [68]. As modeling predicts that cortical tension peaks at the division plane during cytokinesis [69], it will likewise be important to assess the relevance of mechanical cues to cytokinesis-based polarization events.
Interplay within NETO and Relevance of New End-Growth Control
Previous work has implied that association of microtubule-associated protein Tea1 with formin For3 at new ends is sufficient for NETO [8]. In our study, we expressed an endogenous Tea1-For3 fusion that could induce premature NETO. However, when cytokinesis was perturbed in tea1-for3 mutants, NETO was delayed. We posit that local cortical abnormalities in cell wall, membrane, or associated factors can partially override typical growth cues in S. pombe, as has been observed in some plants [70]. Upon defective cytokinesis, such abnormalities at the division site may physically inhibit cell growth at new ends. These defects can lead to the formation of T-shaped cells when old end growth is also blocked, as in tea1Δ fic1Δ mutants. Our data underscore robust completion of cytokinesis as a major determinant of S. pombe NETO.
Is it beneficial for a cell to halt new end growth until well after cytokinesis completion? As mentioned previously, human cells undergoing division initially move away from each other, creating a pulling force that could contribute to abscission [66]. Highly adherent mammalian cells can actually complete cytokinesis, with some defects, in the absence of cortical myosin from the cleavage furrow [71]. Constriction-independent cytokinesis was first observed in the amoeba Dictyostelium discoideum [72], which accomplishes this task by likewise polarizing and growing distally to the division site [73]. One could imagine that in cases where S. pombe cell separation is delayed, tip growth at old ends might contribute similar forces to aid in abscission. Premature new end growth signaling might unbalance these forces, leading to exacerbated cytokinesis delays as in some tea1-for3 cells. Premature new end growth might also interfere with remodeling during cytokinesis and thereby result in cell division defects. These findings highlight interdependence between the cell polarization and division machineries in S. pombe.
Fic1 Scaffold Function during Cytokinesis
Our data indicate that Fic1's C terminus, and not its C2 domain, represents its major cytokinetic functional domain, contrasting with data reported for S. cerevisiae Inn1 [29], [30]. Why is Fic1's C2 domain dispensable for Fic1's cytokinesis, and thus polarity, functions? Sequence alignment indicates that there is in fact very low sequence identity between Fic1 and Inn1 C2 domains [30]. If Fic1's C2 domain is unable to perform functions or mediate interactions that Inn1's C2 domain can, it seems reasonable that Fic1-interacting proteins may be able to compensate. Consistent with this idea, over-expression of S. cerevisiae Cyk3 suppresses cytokinesis defects of inn1Δ mutants, suggesting Cyk3 function overlaps with Inn1 [29]. These data support that Fic1's C terminus is an efficient signaling platform, which scaffolds SH3 domain proteins through its PxxP motifs to ensure coherent integration of late cytokinesis signals.
What is the specific function of the Fic1 scaffold during cytokinesis? Our data indicate that loss of Fic1 leads to faulty CR disassembly and prolonged persistence of factors at the division site. CR disassembly defects were also observed in inn1Δ S. cerevisiae mutants, leading to speculation that Inn1 might stabilize the constricting CR by physically linking it to the ingressing membrane [30]. Subsequent findings countered that Inn1's C2 domain cannot bind phospholipids, and it was postulated instead that Inn1 cooperates with Cyk3 to coordinate cell wall deposition [29]. As in fic1Δ cells, septation and CR disassembly defects commonly accompany one another [51], [74]. Because these processes are inextricably linked, it is currently difficult to tease apart which defect precedes the other in fic1Δ cells. Moreover, completion of cytokinesis also requires lipid rearrangements in both animal cells and S. pombe [64], [75], and membrane bridges were observed in fic1Δ cells. We thus envision that Fic1's C terminus links signaling pathways that guide completion of multiple tasks during late cytokinesis and thereby affect new end remodeling. Of note, we believe that defects in early cytokinesis do not significantly alter bipolar growth establishment, as later defects more directly impinge on division site remodeling and have less time to be remedied before the next cell division. Our data furthermore support the notion that CR constriction and disassembly occur independently [74], as CR constriction but not disassembly proceeded appropriately in fic1Δ cells.
Defective Cytokinesis in Invasive Fungal Growth
As fungal hyphae consist of long chains of cells, the transition into hyphal growth requires strict inhibition of cytokinesis. In some yeasts, Cdc14 phosphatase activates the Ace2 transcriptional program [76], which triggers expression of cell separation enzymes [77]. Upon the hyphal transition in Candida albicans, this signaling cascade is disrupted [78], and other transcription factors suppress expression of Ace2 targets [79]. Therefore, cytokinetic inhibition in hyphae is believed to operate largely on a transcriptional level, and reactivation of the Ace2 transcriptional program is thought to be responsible for the evolution of single-celled yeast growth [41].
In this study, we showed that fission yeast cells that undergo defective, yet not wholly abortive, cytokinesis exhibit enhanced invasive capacity. We believe cytokinesis-based constraints on growth polarity assist the transition into pseudohyphal growth because they force S. pombe to orient outwards and grow predominantly from old ends, a pattern commonly observed in S. pombe pseudohyphal growth [39], [40]. However, as demonstrated by tea1Δ cells, other changes in polarity can also enhance S. pombe invasiveness. Though not specifically defective at new end growth, tea1Δ cells grow predominantly in the direction of the mother cell, and these alterations in polarity might likewise favor growth orientations that are more conducive than bipolar growth to the invasive process.
We believe our data suggest that manipulation of cytokinesis proteins, and not necessarily signaling cascades that feed into downstream transcriptional pathways, can directly modulate the dimorphic switch. We thus speculate that the cytokinetic machinery might represent a direct target of the pseudohyphal developmental program. Intriguingly, loss of cytokinesis proteins that affect NETO rescued invasiveness of an asp1Δ mutant, which lacks the ability to detect nutritional cues [40] deemed important for the S. pombe dimorphic switch [36]. Because various environmental cues also regulate hyphal morphogenesis in pathogenic fungi [80], it will be important to assess the relative significance of cytokinesis-based controls on polarized growth for invasiveness in these species.
Materials and Methods
Strains and General Yeast Methods
The S. pombe strains used in this study (Table S1) were grown in either yeast extract (YE) or Edinburgh minimal media with relevant supplements. fic1+, fic1N, fic1C, fic1-P257A, crn1+, tea4+, rlc1+, tea1+, for3+, and tea1-for3 were tagged endogenously at the 3′ end with GFP:kanR, FLAG3:kanR, mCherry3:kanR, RFP:hygR, V5:kanR, or V5:hygR cassettes as previously described [81]. A lithium acetate method [82] was used in S. pombe tagging transformations, and integration of tags was verified using whole-cell PCR and/or fluorescence microscopy. Introduction of tagged loci into other strains was accomplished using standard S. pombe mating, sporulation, and tetrad dissection techniques. For blocking of cdc25-22 strains in G2, cells were grown at 25°C and then shifted to 36°C for 3 h. For blocking of nda3-KM311 strains in prometaphase, cells were grown at 32°C and then shifted to 18°C for 6.5 h. For blocking of cdc10-V50 strains in G1, cells were grown at 25°C and then shifted to 36°C for 4 h. For blocking of cps1-191 cells in a cytokinesis arrest, cells were grown at 25°C and then shifted to 36°C for 3 h.
Mutants and truncations of fic1 were expressed from the endogenous fic1+ locus. To make these strains, a pIRT2 vector was originally constructed in which fic1+ gDNA with 5′ and 3′ flanks was inserted between BamHI and PstI sites of pIRT2. Mutations were then introduced via site-directed mutagenesis. The fic1(aa1-126) construct was made by inserting a stop codon after residue 126. The fic1(aa127-272) construct was created by inserting XhoI sites before both the start codon and residue 127, digesting with XhoI to release the internal fragment, re-ligating the plasmid, and adding a start codon after the remaining XhoI site. fic1Δ was then covered by these pIRT2-fic1 constructs, and stable integrants resistant to 5-FOA were isolated and confirmed by whole-cell PCR and western blotting.
To make the tea1-for3 fusion, a pIRT2 vector was originally constructed in which tea1+ gDNA with 5′ and 3′ flanks was inserted between SacI and SphI sites of pIRT2. Site-directed mutagenesis was performed to replace the tea1+ stop codon with a SmaI/SalI/PstI multiple cloning site. for3+ gDNA was amplified with a small N-terminal linker sequence and inserted between SmaI and PstI in this multiple cloning site (linker residues are Pro-Gly-Ade-Gly-Ade-Gly-Ade accounting for restriction site and added residues). tea1Δ was then covered by this pIRT2-tea1-for3 construct, and stable integrants resistant to 5-FOA were isolated and confirmed by whole-cell PCR and western blotting. An integrant was subsequently mated with for3Δ, such that we could isolate tea1-for3 strains in which tea1+ and for3+ were lacking.
Expression of acyl-GFP [83] was controlled by the thiamine-repressible nmt1 promoter of pREP3 [84], [85]. Expression of LifeAct-GFP [86] was controlled by the thiamine-repressible nmt81 promoter of pREP81 [87]. Expression from these nmt promoters was kept off by addition of 5 µg/mL thiamine to the medium, and expression was induced by washing and culturing in medium lacking thiamine for at least 24 h.
Spot assays to analyze genetic interactions were performed as previously described [88], except that all were done on YE agar. Synthetic interactions were judged based on differences in growth between double mutants and relevant single mutants.
Yeast Two-Hybrid
Yeast two-hybrid analysis was performed as previously described [89], except that the bait and prey plasmids were either empty or encoded Cdc15 SH3 (aa843–927) [28], Cyk3 SH3 (aa1–59), wild-type [28] or mutant Fic1(aa190–269) fragments, or full-length Fic1.
Protein Methods
Cells were lysed by bead disruption in NP40 lysis buffer in either native or denaturing conditions as previously described [90], except with the addition of 0.5 mM diisopropyl fluorophosphate (Sigma-Aldrich). Proteins were immunoprecipitated by anti-FLAG (Sigma-Aldrich), anti-Cdc15 [28], or anti-V5 (Invitrogen) antibodies. Immunoblot analysis of cell lysates and immunoprecipitates was performed using anti-FLAG, anti-Cdc15, anti-V5, anti-GFP (Roche), or anti-Cdc2 (Sigma-Aldrich) antibodies as previously described [88].
Microscopy
Live-cell bright field images as well as all still images of cells expressing proteins endogenously-tagged with GFP, RFP, or mCherry were acquired on a spinning disc confocal microscope (Ultraview LCI; PerkinElmer) equipped with a 100X NA 1.40 PlanApo oil immersion objective, a 488-nm argon ion laser (GFP), and a 594-nm helium neon laser (RFP, mCherry). Images were taken via a charge-coupled device camera (Orca-ER; Hamamatsu Phototonics) and processed using Metamorph 7.1 software (MDS Analytical Technologies; Molecular Devices). Bright field images were used in determining cell lengths at division. Time-lapse GFP images of cyk3-GFP sid4-GFP cells secured on agar pads, which were sealed by Valap (a Vaseline, lanolin, and paraffin mixture), were acquired every 3 min using this system. Z-sections were acquired for all fluorescence images and combined into maximum projections. Cells were grown to log phase at 25°C before such imaging.
Images of yeast cells and pseudohyphae on YE agar plates were acquired by focusing a camera (PowerShot SD750; Canon) through a microscope (Universal; Carl Zeiss) equipped with a 20X NA 0.32 objective.
All other microscopy was performed using a personal DeltaVision microscope system (Applied Precision). This system includes an Olympus IX71 microscope, 60X NA 1.42 PlanApo and 100X NA 1.40 UPlanSApo objectives, fixed - and live-cell filter wheels, a Photometrics CoolSnap HQ2 camera, and softWoRx imaging software. The microscopy performed using this system was as follows:
-
For calcofluor staining, cells were washed in PBS and then resuspended in PBS containing 5 µg/mL calcofluor. After incubation on ice for 30 min, cells were washed three times in PBS and images were acquired using the personal DeltaVision system. Using the proximity of birth scars to cell ends, growth/morphology was scored as one of the following: monopolar (i.e., growth on one end), bipolar (i.e., growth on both ends), monopolar and septated, bipolar and septated, or multiseptated. For cells just completing division, daughter cells were scored as monopolar as long as ingression of the mother cell had progressed to such a degree that birth scars could be easily identified at new ends. All cells stained with calcofluor were grown to log phase at 25°C, except for the following: (1) cdc25-22 mutants, which were grown overnight at 25°C and then shifted to 36°C for 3 h before staining; (2) cdc10-V50 mutants, which were grown overnight at 25°C and then shifted to 36°C for 4 h before staining; and (3) SIN temperature-sensitive mutants and a wild-type control, which were grown overnight at 29°C before staining.
-
For live-cell DIC movies, movies of rlc1-GFP sid4-GFP cells, movies of GFP-cps1 rlc1-mCherry3 cells, and movies of eng1-GFP cells, cells were secured within the ONIX microfluidics perfusion system (CellASIC). Cells were loaded into Y04C plates for 5 sec at 8 psi, and YE liquid media flowed into the chamber at 5 psi during imaging. For the DIC movies, single z-planes were acquired every 20 min. Growth patterns were scored according to the tip growth that had occurred prior to the next septation. Relative cell ages were scored where applicable based on the number of birth scars seen on each daughter cell. Times until NETO were only determined for cells that initiated NETO prior to the next septation, except for tea1-for3 cells, for which tip growth at new ends was monitored until growth at these sites occurred even if this carried into the next cell cycle. During time-lapse DIC imaging, daughter cells were judged as monopolar after septum splitting when birth scars could be identified, and timing for NETO was started at this point. The point of new end growth was noted when evident elongation occurred relative to birth scars formed by cytokinesis. For the fluorescence movies, multiple z-planes were acquired at different intervals, and z-planes were subsequently combined into maximum projections following deconvolution. Cells were grown to log phase at 25°C before acquisition of these movies.
-
For images of nuclei and cell walls in fixed cells, cells were fixed in 70% ethanol for at least 30 min and stained with DAPI and methyl blue. For counting of cells joined at their division sites, cells were also sonicated at 3.5 W following fixation to break weak associations. Single z-planes were acquired. Non-temperature-sensitive cells were grown to log phase at either 25°C or 32°C before imaging, and cdc25-22 mutants were grown overnight at 25°C and then shifted to 36°C for 3 h before fixation.
-
For acyl-GFP images, cells were either fixed or imaged live. cdc25-22 fic1Δ cells were grown overnight at 25°C in the presence of thiamine, grown an additional 21 h in the absence of thiamine to induce acyl-GFP expression, shifted to 36°C for 3 h before 70% ethanol fixation, washed three times in PBS, and then sonicated at 3.5 W to break weak associations. GFP z-planes were acquired, deconvolved, and combined into maximum projections. For acyl-GFP images of cps1-191 cells at restrictive temperature, cells were grown overnight at 25°C in the presence of thiamine, grown an additional 21 h in the absence of thiamine to induce acyl-GFP expression, and shifted to 36°C for 3 h before imaging.
-
For LifeAct-GFP images, cells were grown overnight at 25°C in the presence of thiamine and then grown an additional 30 h to induce expression of LifeAct-GFP. Single z-planes were acquired and deconvolved.
-
For phalloidin staining (adapted from [42]), cells were fixed in formaldehyde (Polysciences Inc.) for 5 min, and fixation was stopped by addition of PBS. Cells were washed three times in PBS and incubated with 0.1% NP40 for 1 min to permeablize cells. Cells were pelleted and washed three more times in PBS. Then, Alexa-Fluor 488 phalloidin (Molecular Probes) was added. Samples were placed on a nutator for 1 h and subsequently imaged. Images were processed using deconvolution.
Invasive Growth Assays
To assay pseudohyphal invasion into 2% agar, 5 µl containing a total of 105 cells were spotted on 2% YE agar and incubated at 29°C for 20 days. Colonies were subsequently placed under a steady stream of water and surface growth was wiped off using a paper towel. These methods were established in previous studies [40], [59].
To assay whether specific mutants rescued invasiveness of an asp1Δ strain on 0.3% agar [40], 1 µl containing 106 cells was spotted on 0.3% YE agar as well as onto 2% agar as a control. Plates were incubated at 29°C for 12 days, at which point colony growth and/or biofilm formation were visualized.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. HowellAS, LewDJ (2012) Morphogenesis and the cell cycle. Genetics 190 : 51–77.
2. ClarkAG, PaluchE (2011) Mechanics and regulation of cell shape during the cell cycle. Results Probl Cell Differ 53 : 31–73.
3. MartinSG, ChangF (2005) New end take off: regulating cell polarity during the fission yeast cell cycle. Cell Cycle 4 : 1046–1049.
4. HuismanSM, BrunnerD (2011) Cell polarity in fission yeast: a matter of confining, positioning, and switching growth zones. Semin Cell Dev Biol 22 : 799–805.
5. MitchisonJM, NurseP (1985) Growth in cell length in the fission yeast Schizosaccharomyces pombe. J Cell Sci 75 : 357–376.
6. BehrensR, NurseP (2002) Roles of fission yeast tea1p in the localization of polarity factors and in organizing the microtubular cytoskeleton. J Cell Biol 157 : 783–793.
7. FeierbachB, VerdeF, ChangF (2004) Regulation of a formin complex by the microtubule plus end protein tea1p. J Cell Biol 165 : 697–707.
8. MartinSG, McDonaldWH, YatesJR3rd, ChangF (2005) Tea4p links microtubule plus ends with the formin for3p in the establishment of cell polarity. Dev Cell 8 : 479–491.
9. MataJ, NurseP (1997) tea1 and the microtubular cytoskeleton are important for generating global spatial order within the fission yeast cell. Cell 89 : 939–949.
10. BichoCC, KellyDA, SnaithHA, GoryachevAB, SawinKE (2010) A catalytic role for Mod5 in the formation of the Tea1 cell polarity landmark. Curr Biol 20 : 1752–1757.
11. SnaithHA, SawinKE (2003) Fission yeast mod5p regulates polarized growth through anchoring of tea1p at cell tips. Nature 423 : 647–651.
12. FeierbachB, ChangF (2001) Roles of the fission yeast formin for3p in cell polarity, actin cable formation and symmetric cell division. Curr Biol 11 : 1656–1665.
13. NakanoK, ImaiJ, AraiR, TohEA, MatsuiY, et al. (2002) The small GTPase Rho3 and the diaphanous/formin For3 function in polarized cell growth in fission yeast. J Cell Sci 115 : 4629–4639.
14. GachetY, HyamsJS (2005) Endocytosis in fission yeast is spatially associated with the actin cytoskeleton during polarised cell growth and cytokinesis. J Cell Sci 118 : 4231–4242.
15. IwakiT, TanakaN, TakagiH, Giga-HamaY, TakegawaK (2004) Characterization of end4+, a gene required for endocytosis in Schizosaccharomyces pombe. Yeast 21 : 867–881.
16. CabreraR, SuoJ, YoungE, ChangEC (2011) Schizosaccharomyces pombe Arc3 is a conserved subunit of the Arp2/3 complex required for polarity, actin organization, and endocytosis. Yeast 28 : 495–503.
17. CastagnettiS, BehrensR, NurseP (2005) End4/Sla2 is involved in establishment of a new growth zone in Schizosaccharomyces pombe. J Cell Sci 118 : 1843–1850.
18. HuangY, ChewTG, GeW, BalasubramanianMK (2007) Polarity determinants Tea1p, Tea4p, and Pom1p inhibit division-septum assembly at cell ends in fission yeast. Dev Cell 12 : 987–996.
19. SipiczkiM, GrallertB, MiklosI (1993) Mycelial and syncytial growth in Schizosaccharomyces pombe induced by novel septation mutations. J Cell Sci 104 (Pt 2) 485–493.
20. SipiczkiM, TakeoK, YamaguchiM, YoshidaS, MiklosI (1998) Environmentally controlled dimorphic cycle in a fission yeast. Microbiology 144 (Pt 5) 1319–1330.
21. GarciaP, TajaduraV, GarciaI, SanchezY (2006) Rgf1p is a specific Rho1-GEF that coordinates cell polarization with cell wall biogenesis in fission yeast. Mol Biol Cell 17 : 1620–1631.
22. TatebeH, ShimadaK, UzawaS, MorigasakiS, ShiozakiK (2005) Wsh3/Tea4 is a novel cell-end factor essential for bipolar distribution of Tea1 and protects cell polarity under environmental stress in S. pombe. Curr Biol 15 : 1006–1015.
23. GlynnJM, LustigRJ, BerlinA, ChangF (2001) Role of bud6p and tea1p in the interaction between actin and microtubules for the establishment of cell polarity in fission yeast. Curr Biol 11 : 836–845.
24. PollardTD, WuJQ (2010) Understanding cytokinesis: lessons from fission yeast. Nat Rev Mol Cell Biol 11 : 149–155.
25. LippincottJ, LiR (2000) Involvement of PCH family proteins in cytokinesis and actin distribution. Microsc Res Tech 49 : 168–172.
26. Roberts-GalbraithRH, OhiMD, BallifBA, ChenJS, McLeodI, et al. (2010) Dephosphorylation of F-BAR protein Cdc15 modulates its conformation and stimulates its scaffolding activity at the cell division site. Mol Cell 39 : 86–99.
27. CarnahanRH, GouldKL (2003) The PCH family protein, Cdc15p, recruits two F-actin nucleation pathways to coordinate cytokinetic actin ring formation in Schizosaccharomyces pombe. J Cell Biol 162 : 851–862.
28. Roberts-GalbraithRH, ChenJS, WangJ, GouldKL (2009) The SH3 domains of two PCH family members cooperate in assembly of the Schizosaccharomyces pombe contractile ring. J Cell Biol 184 : 113–127.
29. NishihamaR, SchreiterJH, OnishiM, VallenEA, HannaJ, et al. (2009) Role of Inn1 and its interactions with Hof1 and Cyk3 in promoting cleavage furrow and septum formation in S. cerevisiae. J Cell Biol 185 : 995–1012.
30. Sanchez-DiazA, MarchesiV, MurrayS, JonesR, PereiraG, et al. (2008) Inn1 couples contraction of the actomyosin ring to membrane ingression during cytokinesis in budding yeast. Nat Cell Biol 10 : 395–406.
31. GuertinDA, TrautmannS, McCollumD (2002) Cytokinesis in eukaryotes. Microbiol Mol Biol Rev 66 : 155–178.
32. KrappA, SimanisV (2008) An overview of the fission yeast septation initiation network (SIN). Biochem Soc Trans 36 : 411–415.
33. JohnsonBF, YooBY, CallejaGB, KozelaCP (2005) Second thoughts on septation by the fission yeast, Schizosaccharomyces pombe: pull vs. push mechanisms with an appendix–dimensional modelling of the flat and variable septa. Antonie Van Leeuwenhoek 88 : 1–12.
34. DekkerN, SpeijerD, GrunCH, van den BergM, de HaanA, et al. (2004) Role of the alpha-glucanase Agn1p in fission-yeast cell separation. Mol Biol Cell 15 : 3903–3914.
35. Martin-CuadradoAB, DuenasE, SipiczkiM, Vazquez de AldanaCR, del ReyF (2003) The endo-beta-1,3-glucanase eng1p is required for dissolution of the primary septum during cell separation in Schizosaccharomyces pombe. J Cell Sci 116 : 1689–1698.
36. Amoah-BuahinE, BoneN, ArmstrongJ (2005) Hyphal Growth in the Fission Yeast Schizosaccharomyces pombe. Eukaryot Cell 4 : 1287–1297.
37. BrandA (2012) Hyphal growth in human fungal pathogens and its role in virulence. Int J Microbiol 2012 : 517529.
38. GagianoM, BauerFF, PretoriusIS (2002) The sensing of nutritional status and the relationship to filamentous growth in Saccharomyces cerevisiae. FEMS Yeast Res 2 : 433–470.
39. DodgsonJ, BrownW, RosaCA, ArmstrongJ (2010) Reorganization of the growth pattern of Schizosaccharomyces pombe in invasive filament formation. Eukaryot Cell 9 : 1788–1797.
40. PohlmannJ, FleigU (2010) Asp1, a conserved 1/3 inositol polyphosphate kinase, regulates the dimorphic switch in Schizosaccharomyces pombe. Mol Cell Biol 30 : 4535–4547.
41. BahlerJ (2005) A transcriptional pathway for cell separation in fission yeast. Cell Cycle 4 : 39–41.
42. PelhamRJJr, ChangF (2001) Role of actin polymerization and actin cables in actin-patch movement in Schizosaccharomyces pombe. Nat Cell Biol 3 : 235–244.
43. ArellanoM, DuranA, PerezP (1997) Localisation of the Schizosaccharomyces pombe rho1p GTPase and its involvement in the organisation of the actin cytoskeleton. J Cell Sci 110 (Pt 20) 2547–2555.
44. ChangL, GouldKL (2000) Sid4p is required to localize components of the septation initiation pathway to the spindle pole body in fission yeast. Proc Natl Acad Sci U S A 97 : 5249–5254.
45. JendretzkiA, CiklicI, RodicioR, SchmitzHP, HeinischJJ (2009) Cyk3 acts in actomyosin ring independent cytokinesis by recruiting Inn1 to the yeast bud neck. Mol Genet Genomics 282 : 437–451.
46. PollardLW, OnishiM, PringleJR, LordM (2012) Fission yeast Cyk3p is a transglutaminase-like protein that participates in cytokinesis and cell morphogenesis. Mol Biol Cell
47. Le GoffX, MotegiF, SalimovaE, MabuchiI, SimanisV (2000) The S. pombe rlc1 gene encodes a putative myosin regulatory light chain that binds the type II myosins myo3p and myo2p. J Cell Sci 113 Pt 23 : 4157–4163.
48. NaqviNI, WongKC, TangX, BalasubramanianMK (2000) Type II myosin regulatory light chain relieves auto-inhibition of myosin-heavy-chain function. Nat Cell Biol 2 : 855–858.
49. CortesJC, IshiguroJ, DuranA, RibasJC (2002) Localization of the (1,3)beta-D-glucan synthase catalytic subunit homologue Bgs1p/Cps1p from fission yeast suggests that it is involved in septation, polarized growth, mating, spore wall formation and spore germination. J Cell Sci 115 : 4081–4096.
50. LiuJ, TangX, WangH, OliferenkoS, BalasubramanianMK (2002) The localization of the integral membrane protein Cps1p to the cell division site is dependent on the actomyosin ring and the septation-inducing network in Schizosaccharomyces pombe. Mol Biol Cell 13 : 989–1000.
51. DemeterJ, SazerS (1998) imp2, a new component of the actin ring in the fission yeast Schizosaccharomyces pombe. J Cell Biol 143 : 415–427.
52. AnH, MorrellJL, JenningsJL, LinkAJ, GouldKL (2004) Requirements of fission yeast septins for complex formation, localization, and function. Mol Biol Cell 15 : 5551–5564.
53. Martin-CuadradoAB, MorrellJL, KonomiM, AnH, PetitC, et al. (2005) Role of septins and the exocyst complex in the function of hydrolytic enzymes responsible for fission yeast cell separation. Mol Biol Cell 16 : 4867–4881.
54. HachetO, SimanisV (2008) Mid1p/anillin and the septation initiation network orchestrate contractile ring assembly for cytokinesis. Genes Dev 22 : 3205–3216.
55. PeelS, MacheboeufP, MartinelliN, WeissenhornW (2011) Divergent pathways lead to ESCRT-III-catalyzed membrane fission. Trends Biochem Sci 36 : 199–210.
56. KourantiI, McLeanJR, FeoktistovaA, LiangP, JohnsonAE, et al. (2010) A global census of fission yeast deubiquitinating enzyme localization and interaction networks reveals distinct compartmentalization profiles and overlapping functions in endocytosis and polarity. PLoS Biol 8: e1000471 doi:10.1371/journal.pbio.1000471
57. GeW, BalasubramanianMK (2008) Pxl1p, a paxillin-related protein, stabilizes the actomyosin ring during cytokinesis in fission yeast. Mol Biol Cell 19 : 1680–1692.
58. PinarM, CollPM, RinconSA, PerezP (2008) Schizosaccharomyces pombe Pxl1 is a paxillin homologue that modulates Rho1 activity and participates in cytokinesis. Mol Biol Cell 19 : 1727–1738.
59. PrevorovskyM, StanurovaJ, PutaF, FolkP (2009) High environmental iron concentrations stimulate adhesion and invasive growth of Schizosaccharomyces pombe. FEMS Microbiol Lett 293 : 130–134.
60. St JohnstonD, AhringerJ (2010) Cell polarity in eggs and epithelia: parallels and diversity. Cell 141 : 757–774.
61. FlescherEG, MaddenK, SnyderM (1993) Components required for cytokinesis are important for bud site selection in yeast. J Cell Biol 122 : 373–386.
62. SnyderM, GehrungS, PageBD (1991) Studies concerning the temporal and genetic control of cell polarity in Saccharomyces cerevisiae. J Cell Biol 114 : 515–532.
63. PollaroloG, SchulzJG, MunckS, DottiCG (2011) Cytokinesis remnants define first neuronal asymmetry in vivo. Nat Neurosci 14 : 1525–1533.
64. LuoJ, MatsuoY, GulisG, HinzH, Patton-VogtJ, et al. (2009) Phosphatidylethanolamine is required for normal cell morphology and cytokinesis in the fission yeast Schizosaccharomyces pombe. Eukaryot Cell 8 : 790–799.
65. StreiblovaE, MalekI, BeranK (1966) Structural changes in the cell wall of Schizosaccharomyces pombe during cell division. J Bacteriol 91 : 428–435.
66. MullinsJM, BieseleJJ (1973) Cytokinetic activities in a human cell line: the midbody and intercellular bridge. Tissue Cell 5 : 47–61.
67. HuCK, CoughlinM, FieldCM, MitchisonTJ (2008) Cell polarization during monopolar cytokinesis. J Cell Biol 181 : 195–202.
68. WeberGF, BjerkeMA, DeSimoneDW (2012) A mechanoresponsive cadherin-keratin complex directs polarized protrusive behavior and collective cell migration. Dev Cell 22 : 104–115.
69. WhiteJG, BorisyGG (1983) On the mechanisms of cytokinesis in animal cells. J Theor Biol 101 : 289–316.
70. PanterisE, GalatisB (2005) The morphogenesis of lobed plant cells in the mesophyll and epidermis: organization and distinct roles of cortical microtubules and actin filaments. New Phytol 167 : 721–732.
71. O'ConnellCB, WheatleySP, AhmedS, WangYL (1999) The small GTP-binding protein rho regulates cortical activities in cultured cells during division. J Cell Biol 144 : 305–313.
72. NeujahrR, HeizerC, GerischG (1997) Myosin II-independent processes in mitotic cells of Dictyostelium discoideum: redistribution of the nuclei, re-arrangement of the actin system and formation of the cleavage furrow. J Cell Sci 110 (Pt 2) 123–137.
73. KingJS, VeltmanDM, GeorgiouM, BaumB, InsallRH (2010) SCAR/WAVE is activated at mitosis and drives myosin-independent cytokinesis. J Cell Sci 123 : 2246–2255.
74. TullyGH, NishihamaR, PringleJR, MorganDO (2009) The anaphase-promoting complex promotes actomyosin-ring disassembly during cytokinesis in yeast. Mol Biol Cell 20 : 1201–1212.
75. EmotoK, UmedaM (2000) An essential role for a membrane lipid in cytokinesis. Regulation of contractile ring disassembly by redistribution of phosphatidylethanolamine. J Cell Biol 149 : 1215–1224.
76. BraceJ, HsuJ, WeissEL (2011) Mitotic exit control of the Saccharomyces cerevisiae Ndr/LATS kinase Cbk1 regulates daughter cell separation after cytokinesis. Mol Cell Biol 31 : 721–735.
77. Colman-LernerA, ChinTE, BrentR (2001) Yeast Cbk1 and Mob2 activate daughter-specific genetic programs to induce asymmetric cell fates. Cell 107 : 739–750.
78. Gonzalez-NovoA, Correa-BordesJ, LabradorL, SanchezM, Vazquez de AldanaCR, et al. (2008) Sep7 is essential to modify septin ring dynamics and inhibit cell separation during Candida albicans hyphal growth. Mol Biol Cell 19 : 1509–1518.
79. WangA, RanigaPP, LaneS, LuY, LiuH (2009) Hyphal chain formation in Candida albicans: Cdc28-Hgc1 phosphorylation of Efg1 represses cell separation genes. Mol Cell Biol 29 : 4406–4416.
80. ErnstJF (2000) Regulation of dimorphism in Candida albicans. Contrib Microbiol 5 : 98–111.
81. BahlerJ, WuJQ, LongtineMS, ShahNG, McKenzieA3rd, et al. (1998) Heterologous modules for efficient and versatile PCR-based gene targeting in Schizosaccharomyces pombe. Yeast 14 : 943–951.
82. KeeneyJB, BoekeJD (1994) Efficient targeted integration at leu1-32 and ura4-294 in Schizosaccharomyces pombe. Genetics 136 : 849–856.
83. TakedaT, KawateT, ChangF (2004) Organization of a sterol-rich membrane domain by cdc15p during cytokinesis in fission yeast. Nat Cell Biol 6 : 1142–1144.
84. MaundrellK (1990) nmt1 of fission yeast. A highly transcribed gene completely repressed by thiamine. J Biol Chem 265 : 10857–10864.
85. MaundrellK (1993) Thiamine-repressible expression vectors pREP and pRIP for fission yeast. Gene 123 : 127–130.
86. RiedlJ, CrevennaAH, KessenbrockK, YuJH, NeukirchenD, et al. (2008) Lifeact: a versatile marker to visualize F-actin. Nat Methods 5 : 605–607.
87. BasiG, SchmidE, MaundrellK (1993) TATA box mutations in the Schizosaccharomyces pombe nmt1 promoter affect transcription efficiency but not the transcription start point or thiamine repressibility. Gene 123 : 131–136.
88. BohnertKA, ChenJS, CliffordDM, Vander KooiCW, GouldKL (2009) A link between aurora kinase and Clp1/Cdc14 regulation uncovered by the identification of a fission yeast borealin-like protein. Mol Biol Cell 20 : 3646–3659.
89. RosenbergJA, TomlinGC, McDonaldWH, SnydsmanBE, MullerEG, et al. (2006) Ppc89 links multiple proteins, including the septation initiation network, to the core of the fission yeast spindle-pole body. Mol Biol Cell 17 : 3793–3805.
90. GouldKL, MorenoS, OwenDJ, SazerS, NurseP (1991) Phosphorylation at Thr167 is required for Schizosaccharomyces pombe p34cdc2 function. EMBO J 10 : 3297–3309.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2012 Číslo 10
Nejčtenější v tomto čísle
- A Mutation in the Gene Causes Alternative Splicing Defects and Deafness in the Bronx Waltzer Mouse
- Mutations in (Hhat) Perturb Hedgehog Signaling, Resulting in Severe Acrania-Holoprosencephaly-Agnathia Craniofacial Defects
- Classical Genetics Meets Next-Generation Sequencing: Uncovering a Genome-Wide Recombination Map in
- Regulation of ATG4B Stability by RNF5 Limits Basal Levels of Autophagy and Influences Susceptibility to Bacterial Infection