
Requirements for Receptor Engagement during Infection by Adenovirus Complexed with Blood Coagulation Factor X
Human adenoviruses from multiple species bind to coagulation factor X (FX), yet the importance of this interaction in adenovirus dissemination is unknown. Upon contact with blood, vectors based on adenovirus serotype 5 (Ad5) binds to FX via the hexon protein with nanomolar affinity, leading to selective uptake of the complex into the liver and spleen. The Ad5:FX complex putatively targets heparan sulfate proteoglycans (HSPGs). The aim of this study was to elucidate the specific requirements for Ad5:FX-mediated cellular uptake in this high-affinity pathway, specifically the HSPG receptor requirements as well as the role of penton base-mediated integrin engagement in subsequent internalisation. Removal of HS sidechains by enzymatic digestion or competition with highly-sulfated heparins/heparan sulfates significantly decreased FX-mediated Ad5 cell binding in vitro and ex vivo. Removal of N-linked and, in particular, O-linked sulfate groups significantly attenuated the inhibitory capabilities of heparin, while the chemical inhibition of endogenous HSPG sulfation dose-dependently reduced FX-mediated Ad5 cellular uptake. Unlike native heparin, modified heparins lacking O - or N-linked sulfate groups were unable to inhibit Ad5 accumulation in the liver 1h after intravascular administration of adenovirus. Similar results were observed in vitro using Ad5 vectors possessing mutations ablating CAR - and/or αv integrin binding, demonstrating that attachment of the Ad5:FX complex to the cell surface involves HSPG sulfation. Interestingly, Ad5 vectors ablated for αv integrin binding showed markedly delayed cell entry, highlighting the need for an efficient post-attachment internalisation signal for optimal Ad5 uptake and transport following surface binding mediated through FX. This study therefore integrates the established model of αv integrin-dependent adenoviral infection with the high-affinity FX-mediated pathway. This has important implications for mechanisms that define organ targeting following contact of human adenoviruses with blood.
Published in the journal:
. PLoS Pathog 6(10): e32767. doi:10.1371/journal.ppat.1001142
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1001142
Summary
Human adenoviruses from multiple species bind to coagulation factor X (FX), yet the importance of this interaction in adenovirus dissemination is unknown. Upon contact with blood, vectors based on adenovirus serotype 5 (Ad5) binds to FX via the hexon protein with nanomolar affinity, leading to selective uptake of the complex into the liver and spleen. The Ad5:FX complex putatively targets heparan sulfate proteoglycans (HSPGs). The aim of this study was to elucidate the specific requirements for Ad5:FX-mediated cellular uptake in this high-affinity pathway, specifically the HSPG receptor requirements as well as the role of penton base-mediated integrin engagement in subsequent internalisation. Removal of HS sidechains by enzymatic digestion or competition with highly-sulfated heparins/heparan sulfates significantly decreased FX-mediated Ad5 cell binding in vitro and ex vivo. Removal of N-linked and, in particular, O-linked sulfate groups significantly attenuated the inhibitory capabilities of heparin, while the chemical inhibition of endogenous HSPG sulfation dose-dependently reduced FX-mediated Ad5 cellular uptake. Unlike native heparin, modified heparins lacking O - or N-linked sulfate groups were unable to inhibit Ad5 accumulation in the liver 1h after intravascular administration of adenovirus. Similar results were observed in vitro using Ad5 vectors possessing mutations ablating CAR - and/or αv integrin binding, demonstrating that attachment of the Ad5:FX complex to the cell surface involves HSPG sulfation. Interestingly, Ad5 vectors ablated for αv integrin binding showed markedly delayed cell entry, highlighting the need for an efficient post-attachment internalisation signal for optimal Ad5 uptake and transport following surface binding mediated through FX. This study therefore integrates the established model of αv integrin-dependent adenoviral infection with the high-affinity FX-mediated pathway. This has important implications for mechanisms that define organ targeting following contact of human adenoviruses with blood.
Introduction
Adenoviruses are non-enveloped, icosahedral double-stranded DNA viruses of 70–90nm diameter. 54 different human serotypes have been identified to date and are classified into species based on their ability to agglutinate human, monkey or rat erythrocytes [1]. Adenoviruses cause a range of illnesses depending on the route of initial infection, largely dictated by inherent adenoviral tropism. These illnesses are usually self-limiting but can become potentially life-threatening in certain circumstances. For example, species C adenoviruses 1, 2 and 5 initially cause respiratory tract infections after inhaled droplet transmission [2] but are associated with fulminant hepatitis in bone marrow transplant patients [3], [4]. Invasive adenovirus infection following liver transplant is relatively common, occurring in approximately 10% of paediatric and 6% of adult liver transplantion recipients (reviewed in [5]) and may be due to latent donor-associated infection of the transplanted organ. Adenovirus has also been detected in peripheral blood from immunocompromised patients [6], [7], [8], [9] a significant proportion of whom then go on to develop potentially fatal disseminated adenoviral disease. Taken together, these studies underline the clinical significance of these common human pathogens. The primary and secondary receptor systems used by adenoviruses for cellular uptake following contact with different environments in vivo are thus of particular interest and importance.
The species C adenovirus Ad5 can efficiently infect a wide variety of cell types. In vitro studies have demonstrated that cell tethering is mediated by a primary interaction of the Ad5 fiber knob domain with the coxsackievirus and adenovirus receptor (CAR; [10], reviewed in [11]), while the subsequent internalisation of Ad5 particles is dependent on binding of αvβ3/αvβ5 integrins to an RGD motif in the adenovirus penton base [12], [13]. Several in vivo studies, however, have shown that direct interaction with CAR is not required for uptake of Ad5 into the liver [14], [15], [16], [17], which is the primary target organ after contact of adenovirus with the bloodstream in rodent models and in non-human primates (reviewed in [18]). Moreover, CAR is now thought to be localised primarily to tight junctions in intact epithelium, rendering it inaccessible to viral particles (reviewed in [19]). Instead, recent studies have demonstrated that uptake of Ad5 into the liver is mediated by a high affinity interaction with blood coagulation factor X (FX), which putatively ‘bridges’ the hexon protein in the adenovirus capsid to heparan sulfate proteoglycans (HSPGs) expressed on the surface of hepatocytes [17], [20], [21]. Ad5 utilises the host FX protein, which circulates at approximately 8–10 µg/ml in the bloodstream, for cell binding since the cell surface interaction of the Ad5: FX complex is mediated through the serine protease domain of FX and not through a direct interaction of the virus with the cell surface [21]. This is of particular significance in the context of disseminated adenoviral disease affecting immunocompromised patients, since typing studies have found a predominance of species C adenoviruses in peripheral blood samples from these individuals [6], [22], [23]. Interestingly, recent surface plasmon resonance (SPR) studies have demonstrated that of 22 Ad species tested, from species A, B, C and D, 14 can bind FX [21] indicating that the interaction of Ad5 with FX may be highly conserved. Only adenoviruses from species D lacked the capacity to bind FX.
HSPGs are widely-expressed molecules composed of a core protein to which one or more heparan sulfate (HS) glycosaminoglycan (GAG) sidechains are covalently linked (reviewed in [24]). Their core protein diversity, structural heterogeneity and high negative charge (imparted by the HS-GAG sidechains, which consist of highly-sulfated disaccharide repeats of N-acetylglucosamine and glucuronic/iduronic acid) ensure that HSPGs play important roles in many biological processes [25]. Furthermore, several viral pathogens including the human immunodeficiency virus-1 (HIV) [26], human papilloma virus (HPV) [27], adeno-associated virus (AAV) [28] and herpes simplex virus (HSV) [29] exploit HSPGs as primary attachment receptors in different tissues and cell types. Although in vitro and in vivo studies suggest that the Ad5:FX complex interacts with membrane HSPGs [17], [30] the specific receptor requirements underlying FX-mediated adenoviral uptake have not been characterised. Interestingly, liver HS have been shown to possess a specialised structure, with much higher levels of N - and O-sulfation than HS from other tissues [31], [32], [33]. Viral interactions with HS sidechains at the cell surface are frequently associated with the presentation of a particular ‘sulfation signature’ [34], [35]. The substantial liver specificity of systemically disseminating Ad5 may therefore be due to the preferential interaction of Ad5:FX complexes with highly-sulfated liver HS.
Here, we investigated the receptor requirements for FX-mediated Ad5 cellular uptake in vitro and in vivo. We first showed in a number of cell lines that the interaction of the Ad5:FX complex with the cell surface was entirely independent of CAR and αv integrins. By analysing Ad5 binding and uptake into enzymatically-pretreated cells we demonstrated that the primary attachment of the Ad5: FX complex was specifically mediated by HS sidechains. Detailed immunocytochemical analysis in vitro revealed delayed FX-mediated cell entry and cytosolic transport to the microtubule organising centre (MTOC) of a fluorescently-labelled Ad5 mutant lacking the penton base RGD motif, showing that rapid and efficient post-attachment kinetics is dependent on engagement of αv integrins. Chemical inhibition or genetic ablation of endogenous HS sulfation completely abrogated FX-mediated Ad attachment and cell uptake, indicating that the Ad5: FX complex interacts with a specific HS sulfation pattern, while heparin-mediated inhibition of adenoviral gene transfer in vitro and Ad attachment to liver slices ex vivo was significantly attenuated in the absence of heparin N - and, in particular, O-linked sulfate groups. In vivo, Ad5 liver accumulation 1 h after intravenous administration to mice was significantly and dose-dependently inhibited by pre-inoculation with unmodified heparin but not by de-sulfated heparins. Immunohistochemical analysis of liver sections from mice intravenously injected with fluorescently-labelled Ad5 revealed localisation of Ad particles in CD31+ hepatic sinusoids and surrounding hepatocytes. We have thus integrated the established model of αv integrin-dependent adenoviral infection with the FX-mediated pathway leading to liver uptake of Ad5.
Results
Heparan sulfate sidechains play an important role in FX-mediated Ad5 cell uptake in vitro
We utilised Ad5CTL (vector based on wild type Ad5 capsid), Ad5KO1 (CAR binding mutated), Ad5PD1 (integrin-binding mutant) or Ad5KP (both mutations) – see Methods for details. To assess the importance of HSPG heparan sulfate (HS) sidechains in Ad5 cell binding and uptake mediated by interaction with human FX we pre-treated human HepG2 hepatoma and SKOV3 ovarian carcinoma cells with heparinase III prior to performing Ad5 cell attachment and gene transfer experiments in the presence or absence of FX. Both HepG2 and SKOV3 cells express HSPGs, however unlike HepG2 cells, SKOV3 cells express very low levels of CAR [36]. Heparinase III is a heparin lyase that specifically cleaves N-acetylated (NAc) and transition domains of HS and can be used in vitro and in vivo to inhibit HS-mediated viral attachment [27], [37]. Cleavage of HS at NAc and transition domains was assayed by FACS. Heparinase III digestion significantly and dose-dependently reduced the percentage of cells positive for antibody 10E4, while significantly increasing the percentage of cells positive for antibody 3G10, confirming the substrate activity of heparinase III treatment (Fig. S1). To verify that heparinase III treatment had no effect on the widely-expressed GAG chondroitin sulfate, which is primarily composed of acetylgalactosamine hexosamine groups, cell surface chondroitin sulfate was quantified by FACS using the CS-56 mouse monoclonal antibody. While treatment of SKOV3 cells with chondroitinase ABC dose-dependently reduced the percentage of CS-56 positive cells, heparinase III treatment had no effect (Fig. S1), indicating that heparinase III treatment has no effect on chondroitin sulfate. Ad5 attachment to cells was substantially increased in the presence of FX in both HepG2 and SKOV3 cells (Fig. 1A and 1B; p<0.01). Similar results were observed when Ad5 mutants ablated for CAR-binding and/or αv integrin binding (Ad5KO1, Ad5PD1 or Ad5KP; Fig. 1A) were used, in agreement with previous studies showing that the FX-mediated increase in virus cell attachment and gene transfer is CAR - and αv integrin - independent [17], [30]. Similar FX-mediated enhancement of virus uptake was also observed in a panel of CARhigh and CARlow cell lines when Ad5CTL and the αv integrin-binding mutant Ad5PD1 were compared (Fig. S2). As expected levels of FX-mediated enhancement in gene transfer were lower in CARhigh cells compared to CARlow cells (Fig. S1) since the CAR and FX pathways are both efficient in vitro pathways in the former. The FX-mediated increase in virus cell attachment was significantly attenuated by heparinase III pretreatment of HepG2 (p<0.01) or SKOV3 (p<0.05) cells (Fig. 1B). FX-mediated gene transfer was also significantly decreased after heparinase III pretreatment (Fig. 1C) of HepG2 and SKOV3 cells (p<0.01). We next carried out virus transport experiments in vitro using an Alexa488-labelled virus in the presence or absence of heparin. Heparin and HS sidechains have very similar structures, although heparin displays higher general sulfation than HS [25]. Heparin is therefore frequently used as a competitive inhibitor for HS binding [38], [39]. In the absence of heparin, Ad5:FX complexes were rapidly and efficiently transported to the MTOC in SKOV3 cells within an hour of adding fluorescently-tagged Ad5CTL:FX complexes to cells, as demonstrated by colocalisation with the MTOC marker pericentrin (Fig. 1D, upper panel). In the presence of heparin, however, FX-mediated attachment of fluorescently-tagged Ad5CTL to the cell surface was completely abrogated (Fig. 1D, lower panel), confirming that HS sidechains mediate Ad5CTL:FX attachment to the cell surface. Similar results were observed in SKOV3 and A549 human lung adenocarcinoma cells (Fig. 1E). Taken together, these results indicate that FX-mediated Ad5CTL cell attachment in vitro is dependent on the presence of HS sidechains.
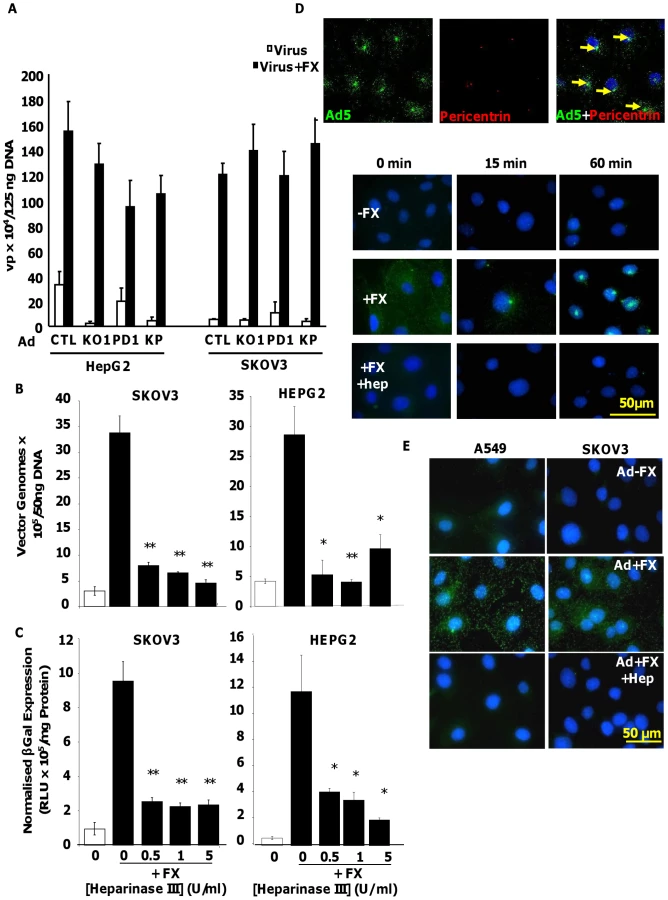
Optimal post-attachment cell entry and cytosolic transport of Ad5 is dependent on an intact penton base RGD motif
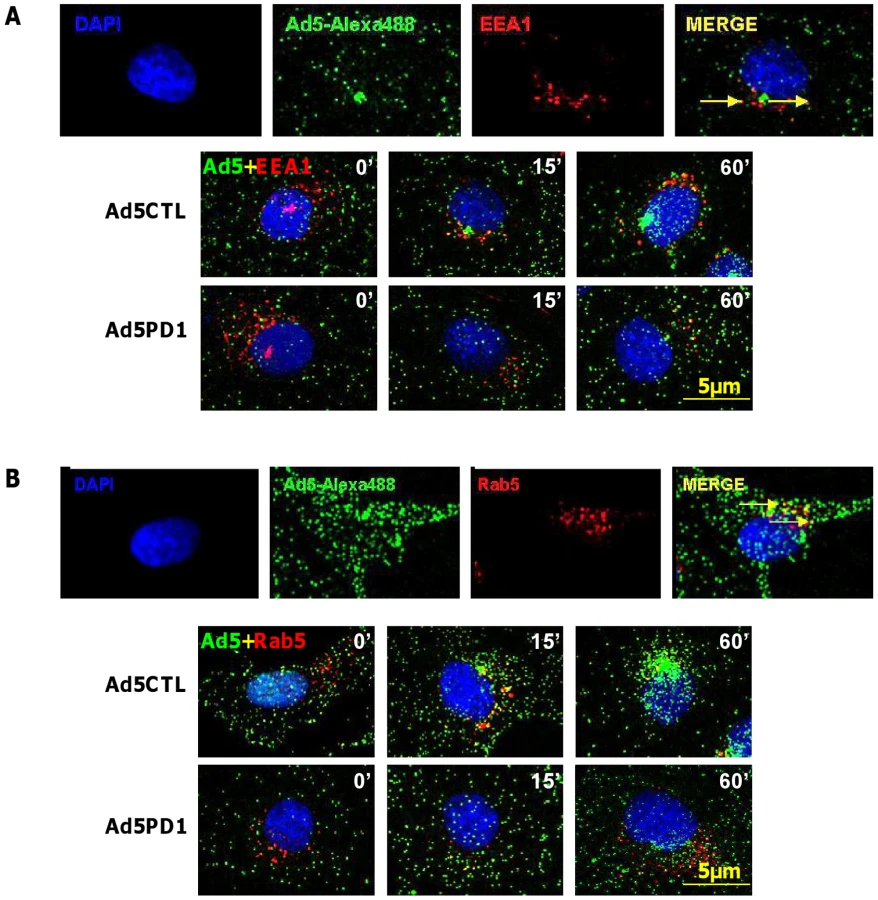
Although in vitro studies have shown that an intact penton base RGD motif is required for efficient endosome escape after CAR-mediated attachment of Ad5 to the cell surface [40] several in vivo studies indicate that ablation of the penton base RGD motif does not significantly affect liver uptake after systemic administration of Ad5 [14], [15], [16], [17], [41]. Having established that FX-mediated attachment of Ad5CTL to the cell surface requires HSPGs, we next assessed the role of αv integrins during FX-mediated Ad5 cell uptake. In vitro cell tracking experiments were carried out in CARlow SKOV3 [36] and CARhigh A549 cells [42] using Ad5 vectors with a mutated penton base RGD motif (Ad5PD1). Both Ad5CTL and Ad5PD1 efficiently bound the cell membrane in the presence of FX (Fig. 2A, B). In SKOV3 cells at 15 and 60 minutes post-internalisation, both Ad5CTL and Ad5PD1 particles had entered endosomal compartments as demonstrated by partial colocalisation with the early endosomal marker EEA1 and Rab5 (Fig. 2A, B). Similar results were observed in A549 cells (data not shown). MTOC colocalisation was quantified by assessing the proportion of cells in a 40× microscope field with colocalisation of fluorescently-labelled Ad5 particles (green) with the MTOC marker pericentrin (red; see upper panel in Fig. 3A, quantification Fig. 3B–C). Cell entry and cytosolic transport kinetics of the CAR binding-ablated vector Ad5KO1 closely resembled Ad5CTL, confirming that FX-mediated cell uptake does not require CAR (Fig. S3). Conversely, only 20–25% MTOC colocalisation was observed for Ad5PD1 in SKOV3 and A549 cells at the same timepoint (Fig. 3B and Fig. 3C, respectively). These data suggest that an integrin-mediated post-internalisation signal is required for optimal transport of Ad5CTL to the MTOC after FX-mediated cell surface attachment of Ad5CTL to HSPGs. To confirm the role of integrins we next performed a short hairpin (sh)RNA approach to knockdown αv integrin expression in SKOV3 cells and assessed the effect of this on intracellular transport of Ad5CTL. Target knockdown was confirmed using TaqMan and flow cytometric analysis compared to mock-transfected and scrambled control shRNA cells (Fig. 4A, B). Knockdown led to a significant reduction in the localisation of Ad5CTL to the peri-nuclear compartment (Fig. 4B–C) thus confirming the importance of integrin engagement for transport via the FX-mediated pathway. We next determined the effect of kinase inhibitors that are known to affect cell entry and intracellular transport of adenovirus [43]. We used H89 dihydrochloride (an inhibitor of PKA), L Y294002 hydrochloride (an inhibitor of PI3K) and SB 203580 hydrochloride (an inhibitor of p38 MAPK). We observed that co-incubation of Ad5CTL-transduced cells in the presence of inhibitors of either PKA, PI3K or p38 MAPK was able to significantly reduce Ad5CTL-mediated transport to the MTOC in the presence of FX (Fig. S4). Since these molecules are linked to activation of cellular integrins during Ad internalisation or intracellular transport this further suggests that integrins are a component of the viral entry cycle in the presence of FX.


Sulfation of HSPGs is critical for cell attachment and uptake of Ad5:FX
Having established that FX-mediated Ad5CTL cell surface attachment required HS sidechains, we next investigated whether HS sidechain sulfation affects FX-mediated Ad5CTL cell uptake. In vitro experiments were therefore carried out in HepG2 and SKOV3 cells pretreated with increasing concentrations of sodium chlorate, a selective inhibitor of sulfation [44]. We confirmed that sodium chlorate treatment inhibited sulfation in a dose-dependent manner (Fig. S5A), reduced binding of Ad5CTL to cells in the presence of FX (Fig. S5B) in the absence of cellular toxicity (Fig. S5C). Gene transfer was used as a marker of successful cellular internalisation, cytosolic transport and nuclear uptake. FX-mediated Ad5CTL gene transfer was inhibited in a dose-dependent manner by pretreatment with sodium chlorate in both HepG2 and SKOV3 cells (Fig. 5A), suggesting that FX-mediated Ad5 uptake in vitro may depend on HS sidechain sulfation. No effect on basal levels of Ad5CTL uptake was observed. To assess the importance of HS sulfation for FX-mediated Ad5CTL transduction, cell attachment and transduction assays were carried out in CHO cell lines deficient in HS biosynthesis enzymes. Although FX induced a 60-fold increase in Ad5 cell attachment and uptake in parental CHO-K1 (CAR-) cells, no FX-mediated increase was observed in CHO-pgsA745 cells, which do not express xylosyltransferase-1 (XT1) and are consequently defective in HS-GAG synthesis [45] (Fig. 5B–C). The FX-mediated increase in Ad5 cell attachment and transduction was also significantly attenuated in N-deacetylase/N-sulfotransferase-1 (NDST1)-deficient CHO-pgsE606 cells, which synthesise HS chains with significantly reduced O - and, in particular, N sulfate groups [46] (Fig. 5B–C). Interestingly, FX was unable to increase Ad5CTL cell attachment and uptake in CHO-pgsF17 cells, which are 2-O-sulfotransferase-deficient and therefore lack 2-O-sulfated residues [47] (Fig. 5B–C). Similar results were obtained in all cell lines using the αv integrin-binding mutant AdPD1 (Fig. 5B and 5C). Taken together, these data suggest that attachment of the Ad5CTL:FX complex to cell surface HSPGs in vitro requires HS sidechain sulfation.

Removal of sulfate groups attenuates the ability of heparin to inhibit Ad5:FX binding and cellular uptake in vitro and ex vivo
To confirm the importance of HS sidechain sulfation for FX-mediated Ad5CTL cell attachment and uptake, in vitro gene transfer and ex vivo attachment experiments were carried out in the presence of heparan sulfates or heparins with biosynthetically-modified sulfation. Bovine intestinal heparin and porcine intestinal heparan sulfate are very highly-sulfated [39]. In contrast, porcine kidney heparan sulfate possesses fewer N - and O - sulfate groups, while de-N-sulfated heparin lacks N-sulfated glucosamine residues [39] and de-O-sulfated heparin lacks O-sulfate groups [39], [48]. Competitive inhibition experiments in vitro and ex vivo were carried out in SKOV3 cells or mouse liver slices, respectively, in the presence or absence of FX.
Dose-dependent inhibition of FX-enhanced Ad5CTL gene transfer into SKOV3 cells was observed in the presence of all heparins and heparan sulfates (Fig. 6A). However the IC50 values for the highly-sulfated bovine intestinal heparin and porcine intestinal heparan sulfate (5.1 µg/ml and 5.2 µg/ml respectively) were lower than the IC50 values for the de-sulfated heparins (39.1 µg/ml and 73.4 µg/ml for de-N-sulfated or de-O-sulfated heparins respectively) or heparan sulfate with reduced sulfation (porcine kidney heparan sulfate, 17.7 µg/ml) (Fig. 6A). Similar results were obtained when CAR - or αv integrin binding-ablated viruses were used (Table S1).
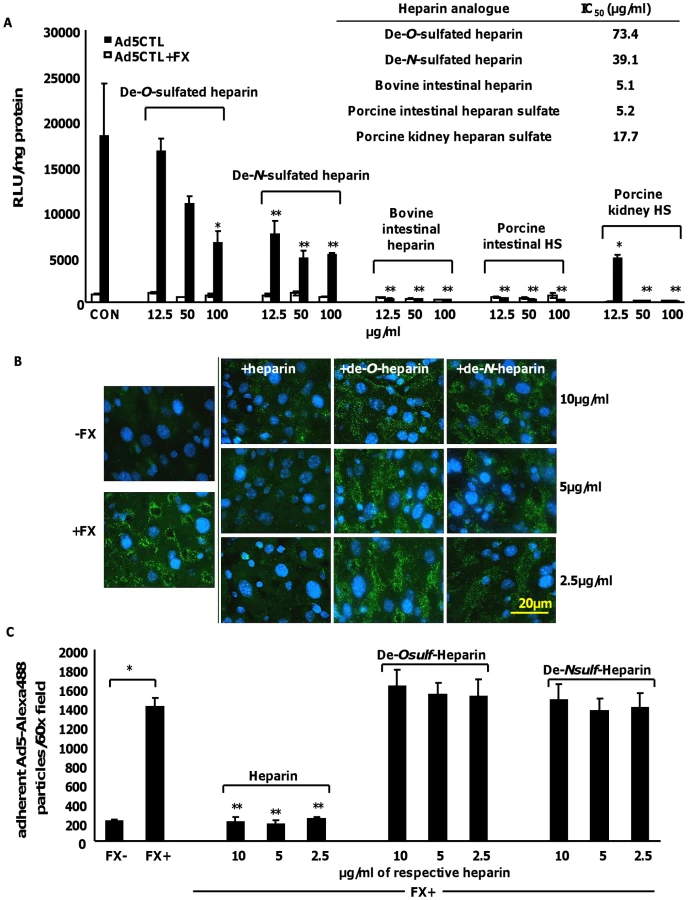
Next, the attachment of fluorescently-labelled Ad5CTL (green) to mouse liver slices ex vivo was analysed. Adherent Ad5CTL particles were quantified by analysing captured images of 60× microscope fields using the ImageJ automated cell counting function. Co-incubation with FX significantly increased the attachment of fluorescently-labelled Ad5CTL to liver sections (Fig. 6B and Fig. 5C). At the concentrations used, heparin significantly inhibited FX-mediated attachment of Ad5CTL to liver sections (p<0.01; Fig. 6B–C). However neither de-O-sulfated nor de-N-sulfated heparin inhibited the attachment of Ad5CTL to liver sections in the presence of FX. These results indicate that binding of the Ad5CTL-FX complex to heparin/HS in vitro requires the presence of highly anionic sulfate groups, supporting the hypothesis that FX-mediated Ad5CTL cell uptake is dependent on HS sidechain sulfation.
Inhibition of Ad liver accumulation in vivo by heparin is sulfation-dependent
To identify whether the sulfation status of hepatocyte HS contributes to the liver uptake of Ad5CTL from the circulation, competitive inhibition experiments were carried out in vivo in the presence of heparins with biosynthetically-modified sulfation. Mice were inoculated with increasing concentrations of heparins prior to intravascular administration of Ad5CTL. Ad5CTL liver uptake at an early timepoint post-inoculation was then quantified by assessing viral genome accumulation 1 h post-administration. Cellular localisation of Ad5CTL in mouse livers was analysed by staining liver sections with CD31 to visualise the vasculature in conjunction with fluorescently labelled Ad5CTL. We have previously shown that the FX-pathway is hepatocyte-specific and Kupffer cell uptake is unaffected by pharmacological modulation or genetic approaches to modify FX binding as Kupffer cell uptake is a scavenging process [49], [50], [51], [52]. We therefore used macrophage-depleted mice to allow selective visualisation of HSPG uptake mechanisms via the FX pathway in vivo at an early time point post injection.
Heparin pre-inoculation at both 20 mg/kg and 50 mg/kg significantly and dose-dependently inhibited Ad5CTL accumulation in the liver 1 h post-inoculation (p<0.01)(Fig. 7A). Although no effect was observed at either dose of de-O-sulfated heparin (20 mg/kg or 50 mg/kg), significantly fewer Ad5CTL genomes were detected in livers of mice pre-treated with 50 mg/kg de-N-sulfated heparin (p<0.05)(Fig. 7A). Immunofluorescence staining for CD31 was performed to facilitate identification of endothelial sinusoids in the liver architecture. Sections from control mice inoculated with fluorescently-labelled Ad5CTL (green) showed accumulation of Ad5CTL particles in liver sinusoids and on the surface of hepatocytes (Fig. 7B, upper panel). Administration of heparin (both 20 mg/kg and 50 mg/kg doses) or high-dose de-N-sulfated heparin clearly reduced accumulation of labelled Ad5 while de-O-sulfated heparin had no effect at either dose (Fig. 7B, lower panel).

Taken together, our data indicate that the hepatic uptake observed after intravenous administration of Ad5CTL is dependent on HS sidechain sulfation, with a particular requirement for O-sulfation. In conjunction with previous studies showing that liver HS is highly enriched in 2-O-sulfated residues [31] our in vitro, ex vivo and in vivo results further suggest that the hepatic tropism of Ad5CTL may be due to preferential binding of the Ad5:FX complex to HS sidechains.
Discussion
Previous studies have shown that the liver uptake of Ad5 after exposure to the circulation is dependent on FX binding directly to the hexon protein in the Ad5 capsid, putatively via a FX-mediated interaction with hepatocyte membrane HSPGs [17], [30]. In the present study we investigated the functional receptor requirements for the Ad5CTL:FX complex using a variety of in vitro, ex vivo and in vivo experimental approaches.
We have demonstrated that FX-mediated Ad5CTL cell attachment in vitro requires the presence of HS sidechains but not CAR or αv integrins. FX-mediated Ad5CTL binding to CARhigh HepG2 and CARlow SKOV3 cells was not affected by fiber knob or penton base mutations ablating CAR - or αv integrin-interacting motifs, respectively, indicating that neither CAR nor αv integrins were required for the FX-mediated primary interaction with the cell surface. Conversely, cleavage of HS sidechains by heparinase III pretreatment significantly inhibited FX-mediated Ad5 attachment to and uptake into HepG2 and SKOV3 cells. Heparin, a highly-sulfated HS analogue, also abrogated FX-mediated adenoviral cell surface binding. Moreover, no FX-mediated enhancement of Ad5CTL cell binding or gene transfer was observed in CHO-pgsA745 cells which do not display HS sidechains. Taken together, these results clearly demonstrate that the primary interaction of the Ad5CTL:FX complex with the cell surface is mediated via HS sidechains. Although our data indicate that CAR - or αv integrins are not required for FX-mediated attachment of Ad5CTL to the cell surface, intracellular transport experiments using a mutant with an ablated penton base RGD motif (Ad5PD1) and a knockdown shRNA approach revealed that efficient and rapid post-internalisation transport of virus particles to the nucleus requires engagement of RGD-interacting integrins. A similar delay in intracellular transport of Ad5PD1 was observed in CARhigh A549 and CARlow SKOV3 cells, suggesting that the altered transport was not affected by differences in CAR expression. Interestingly, a previous study investigating the cytoplasmic transport of Ad5 after CAR-mediated cell surface attachment has demonstrated a similar reliance on RGD-integrin interactions [40]. In conjunction with this study, our results indicate that integrin engagement is required for rapid and efficient intracellular transport of Ad5CTL regardless of the primary attachment receptor used. Furthermore, the potential HS sidechain dependence of FX-mediated cell surface attachment suggests that the Ad5CTL:FX complex may utilise HSPGs as attachment factors in a similar manner to other hepatotropic viruses such as HSV [53] hepatitis [34], [54] and AAV-2 [55], [56].
A central aim of the present study was to establish whether FX-mediated Ad5CTL cell uptake is dependent on the degree or type of HS sidechain sulfation. Blocking HS sulfation by pre-incubating cells with increasing concentrations of sodium chlorate dose-dependently inhibited FX-mediated Ad5CTL gene transfer in HepG2 and SKOV3 cells. Moreover, FX-mediated enhancement of Ad5CTL cell attachment and uptake was significantly attenuated in CHO-pgsE606 cells, which have reduced overall sulfation due to a deficiency in the N-deacetylase/N-sulfotransferase-1(NDST1) gene [46]. In addition, IC50 values for porcine kidney heparan sulfate, which possesses fewer sulfate groups than heparin, were approximately 3-fold higher than IC50 values for highly-sulfated bovine intestinal heparin or porcine intestinal heparan sulfate. These data show that FX-mediated Ad5 cell attachment and uptake is dependent on the degree of HS sidechain sulfation. Interestingly, removal of N - or O - sulfate groups significantly attenuated the inhibitory capabilities of heparin on Ad5CTL uptake in vitro, increasing IC50 values approximately 8 - or 14-fold respectively. Furthermore, unlike native heparin, de-O-sulfated and de-N-sulfated heparins were unable to inhibit FX-mediated attachment of fluorescently-labelled Ad5CTL to liver slices ex vivo. Finally, no FX-mediated enhancement of Ad5CTL cell attachment or uptake was observed in CHO-pgsF17 cells, which lack 2-O-sulfate groups due to a deficiency in the 2-O-sulfotransferase gene [47]. Taken together, our results suggest that while the degree of sulfation modulates the FX-mediated uptake of Ad5CTL in vitro, the Ad5CTL:FX complex may also preferentially interact with specific sulfate moieties.
A number of previous studies have examined the biochemical composition of heparan sulfate from different tissues and have shown that liver heparan sulfate is enriched in sulfated moieties, in particular 2-O sulfate groups [31], [33]. Interestingly, hepatic clearance of intravenously-administered very low density lipoprotein (VLDL) is significantly reduced in mice with liver-specific knockout of the heparan sulfate 2-O sulfotransferase enzyme [57], thereby adding mechanistic insight into previously published work documenting increased levels of systemic VLDL in mice with reduced overall liver HS sulfation [32]. These studies show that the specialised structure of liver HS can contribute to the hepatic accumulation and uptake of circulating particles, and indicate how liver HS sulfation may contribute to the accumulation of systemically-disseminated, FX-interacting adenoviruses such as Ad5. The final aim of this study was therefore to investigate the importance of HS sidechain sulfation in FX-mediated Ad5CTL interactions with liver cells in vivo. While pre-injection of native heparin or high-dose de-N-sulfated heparin significantly attenuated Ad5CTL genome accumulation in the livers of macrophage-depleted mice 1 h after intravenous delivery, administration of de-O-sulfated or low-dose de-N-sulfated heparin had no effect. Immunohistochemical analysis of fluorescently-labelled Ad5CTL in liver sections from these mice clearly showed a significant reduction in Ad5CTL accumulation around CD31+ hepatic sinusoids after pre-injection of high-dose native and de-N-sulfated heparin, but not de-O-sulfated heparin. These results are consistent with our in vitro and ex vivo data, suggesting that HS sidechain sulfation (in particular O-sulfation) may contribute to the accumulation of Ad5CTL in the liver at this timepoint after intravenous administration. Taken together, our data indicate that the FX-mediated interaction of Ad5CTL with HSPGs both in vitro and after intravascular administration in vivo involves the presentation of a ‘sulfation signature’.
The domains of FX responsible for mediating Ad5 transduction of hepatocytes have been demonstrated. The Gla domain of FX docks in the cup at the centre of each hexon trimer and the virus:FX complex is then delivered to the hepatocyte surface via a heparin binding exosite in the FX serine protease domain which tethers to HSPGs at the cell surface [21]. While activated FX (FXa) has previously been shown to bind to the cell surface of hepatocytes and tumour cell lines, this interaction was not observed with FX [58]. The cell surface receptors that mediate FXa interactions with hepatocytes were later shown to be tissue factor pathway inhibitor and nexin-1 and required a functional FXa active site [59]. Previously, it was also shown that a Ca2+-mediated interaction between the Gla domain of FX and phospholipid components of the cell membrane mediated cell surface interactions [60], [61]. A similar phospholipid-mediated interaction between FX and platelets has also been reported [62]. FX has also been previously shown to mediate interactions with the cell membrane of other cell types via other identified receptors. For example, in whole human blood FX has been shown to bind monocytes via the αMβ2 integrin (CD11b/CD18), resulting in its activation via cathepsin G-mediated cleavage, although the domain of FX that binds CD11b was not identified [63].
Previous studies have shown that binding of Ad3 fiber knob to HSPGs in vitro is also dependent on HS sidechain sulfation [64]. A putative HSPG-binding region has been identified in the Ad5 fiber shaft (91KKTK94) [41]. However while reduced hepatic uptake was observed after intravascular administration of a virus harbouring a mutation of the KKTK motif (91KKTK94→GAGA), in vitro assays showed that this virus was deficient in intracellular transport [65]. A recent study has clearly demonstrated that fiber is not involved in binding of the Ad5:FX complex to the cell surface, since a fiberless Ad5 mutant showed no significant reduction in FX-mediated cell surface attachment [21]. This study also showed that the Gla domain of FX binds to hypervariable regions in the Ad5 hexon [21], while positively-charged residues in the FX serine protease domain putatively interact with HSPGs [21], [66], [67], [68]. It is therefore likely that the HS sidechain-dependent interaction of Ad5 with the cell surface is mediated by FX ‘bridging’ to hexon capsid proteins rather than by a direct interaction with fiber.
In the past two decades there has been significant interest in the potential use of sulfated polysaccharides such as heparin and heparan sulfates in antiviral therapy (reviewed in [69]). For example, the polyanionic compound PRO 2000 competitively inhibits attachment of the HIV-1 envelope protein gp120 to HSPGs on CD4+ T cells and is currently under development as a topical antiviral gel to prevent cervical HIV-1 transmission [70], [71]. However undesirable side-effects such as anticoagulation limit the use of certain highly-sulfated, high molecular weight polysaccharides, including heparin. As stated previously, viral interactions with HS sidechains at the cell surface are often associated with the presentation of a particular ‘sulfation signature’. For instance, hepatitis E cell binding is thought to be dependent on 6-O sulfation [34] while the interaction of HSV-1 with the surface of target cells during infection in vivo is mediated by 3-O sulfate moieties [35]. Knowledge of the specific positioning and number of sulfate groups required for optimal virucidal activity has facilitated the development of targeted antiviral polyanions such as carrageenan and cellulose sulfate, which have significantly fewer side-effects [69]. This underlines the therapeutic relevance of fully understanding the sulfation requirements for Ad5:FX attachment to host cell HSPGs. This is of particular importance in the context of disseminated adenoviral disease in immunocompromised patients, as several studies have identified FX-binding species C adenoviruses in peripheral blood samples from these individuals [6], [7], [8], [9]. Further detailed studies on the receptor-mediated interactions of Ad5 in circulation are now required to fully characterise the factors underlying the clinical pathogenicity of this virus.
Materials and Methods
Ethics statement
All animal experiments were approved by the University of Glasgow Animal Procedures and Ethics Committee and performed under UK Home Office licence (PPL 60/3752) in strict accordance with UK Home Office guidelines.
Materials
Purified human blood coagulation factor X (FX) was purchased from Cambridge Biosciences (Cambridge, UK). Heparinase III, chondroitinase ABC, bovine intestinal heparin, porcine intestinal heparan sulfate, porcine kidney heparan sulfate, de-N-sulfated heparin, de-O-sulfated heparin and sodium chlorate were obtained from Sigma (Sigma-Aldrich, Gillingham, UK). Primary antibodies raised against EEA1, Rab5, pericentrin or α-tubulin were obtained from Abcam (Cambridge, UK). Primary antibodies raised against intact heparan sulfate (clone 10E4) or heparinase III-digested heparan sulphate (clone 3G10) were obtained from AMS Biotechnology (Oxford, UK). The primary antibody raised against chondroitin sulfate (clone CS-56) was obtained from Sigma (Sigma-Aldrich, Gillingham, UK). All secondary antibodies were obtained from Invitrogen (Paisley, Scotland, UK). The kinase inhibitors LY 294002 hydrochloride, H 89 hydrochloride and SB 202190 hydrochloride were obtained from Tocris Bioscience (Bristol, UK).
Cell lines, cell culture and virus production
A549 (human lung carcinoma ATCC CCL-185), HT29 (human colorectal adenocarcinoma: ATCC HTB-38), MDA-MB-231 (human breast adenocarcinoma: ATCC HTB-26) NCI-H522 (human lung adenocarcinoma: ATCC CRL-5810), SKOV3 (human ovarian carcinoma: ATCC HTB-77) and SNB19 cells (human glioblastoma: ATCC CRL-2219) were grown in RPMI 1640 medium supplemented with 10% fetal calf serum, 2 mM L-glutamine and 1% penicillin-streptomycin (Invitrogen, Paisley, UK). HepG2 (human hepatocellular carcinoma: ATCC CRL-11997) and 293 (Human Embryonic Kidney: ATCC CRL-1573) cells were grown in Dulbecco's Modified Eagle's Medium (DMEM; Invitrogen, Paisley, UK) supplemented with 10% fetal calf serum, 2 mM L-glutamine and 1% penicillin-streptomycin. CHO-pgsA745 (ATCC: CRL-2242), CHO-pgsE606 (ATCC: CRL-2246) and CHO-pgsF17 cells [47] were grown in Ham's F-12 medium (Invitrogen, Paisley, UK) supplemented with 10% fetal calf serum, 2 mM L-glutamine and 1% penicillin-streptomycin. The E1/E3-deleted Ad5CTL adenovirus encodes a Rous sarcoma virus (RSV) promoter-driven LacZ expression cassette as described previously [66]. Ad5KO1 is based on Ad5CTL and contains a two-amino acid substitution in the fiber knob (S408E, P409A) that ablates CAR binding. Ad5PD1 is also based on Ad5 and contains a substitution of amino acids 337–344 of the penton base gene (HAIRGDTF) with amino acids SRGYPYDVPDYAGTS, ablating RGD-mediated αv integrin-binding. Ad5KP contains both fiber knob (KO1) and penton base (PD1) mutations. Viruses were propagated in 293 cells and purified by CsCl gradient centrifugation. Vector genomes were quantified by SYBR green quantitative polymerase reaction (qPCR) on an Applied Biosystems ABI Prism 7700 sequence detection system using primers directed against the LacZ transgene (forward 5′-ATCTGACCACCAGCGAAATGG-3′ and reverse 5′-CATCAGCAGGTGTATCTGCCG-3′). Viral particles were determined by micro bicinchoninic-acid assay (Perbio Science, Cramlington, UK) using the formula 1 µg protein = 4×109 VP [72].
Virus labelling
Adenoviruses were fluorescently labelled using an Alexa Fluor-488 (green) protein labelling kit according to the manufacturer's instructions (Invitrogen, Paisley, UK). Free label was dialysed from labelled Ad5 using 10,000 molecular weight cut off slide-a-lyzer cassettes (Perbio Science, Cramlington, UK) overnight in 100 mM Tris, 50 mM EDTA. A dye: virus particle ratio of 3∶1 was used for all labelling reactions. Fluorescent dye labelling efficiency was assessed using the ‘proteins and labels’ function on a Nanodrop-1000 spectrophotometer. Infectivity of labelled adenoviruses was verified by in vitro gene transfer assay as described below.
Flow cytometric analysis of heparinase III pretreatment
FACS was performed on SKOV3 cells cultured under the conditions described above. Cells were detached from culture vessels using a 1× citric saline solution and counted using trypan blue exclusion. Cells were then resuspended in serum-free DMEM (SFDMEM) at a concentration of 4×106 cells/ml. Heparinase III or chondroitinase ABC was then added to 50 µl of this cell suspension at the required concentrations (0 U/ml, 0.5 U/ml, 1 U/ml, 5 U/ml) and incubated with cells in a shaking waterbath at 37°C for 1 h. Cells were washed twice in SFDMEM) then incubated with primary antibodies (10E4, 3G10 or CS-56; all mouse monoclonal antibodies) or a matching isotype control in SFDMEM containing 0.1% BSA for 30 min on ice. Cells were then washed twice and incubated with a FITC-labelled secondary antibody in SFDMEM for 30 minutes on ice. Cells were washed twice and cell labelling was then detected on a FACS Canto II flow cytometer (Beckton Dickinson, Oxford, UK) using FACS DIVA software. Viable cells were gated by their FSC/SSC profiles, with a minimum of 5000 gated events analysed per sample. Results are expressed as percentage positively-stained cells per sample, from 3 independent samples analysed in triplicate.
Measurement of sulfated glycosaminoglycan (sGAG) content
Total sulfated glycan content was measured in cell lysates using the Blyscan sulfated GAG assay kit (Biocolour, Newtonabbey, Northern Ireland, UK) according to manufacturer's instructions. Briefly, cultured cells were lysed in RIPA buffer (50 mM Tris, 150 mM NaCl, 0.1% SDS, 0.5% sodium deoxycholate, 1% NP40) then lysates were incubated with a molar excess of the cationic, sulfate-binding dye 1,9 dimethylmethylene blue. Lysates were pelleted and unbound dye was removed. Soluble GAG content was measured by determining the quantity of bound dye by spectrophotometric standard curve analysis at 656 nm. Protein concentrations were measured by Bicinchoninic Acid Assay (Perbio Science, Cramlington, UK) as described above. Data are expressed as µg sGAG/mg protein.
Analysis of Ad5 transport in vitro
Cells were seeded in 4-well chamber slides at 1×105 cells/well 24 h prior to assay. Cells were gently washed with PBS then incubated with 1×104 vp/cell in 300 µl SFDMEM for 1 h on ice. Factor X and bovine intestinal heparin were both used at a concentration of 10 µg/ml. Cells were then gently washed with PBS and incubated at 37°C for 15, 30, 60 or 180 minutes prior to fixation. Localisation of Ad particles at the MTOC was characterised by staining cells using a polyclonal rabbit pericentrin antibody (1∶200 dilution: Abcam, Cambridge, UK) while localisation of Ad particles in early endosomes was characterised by staining cells using a polyclonal rabbit EEA1 antibody (Early Endosome Antigen-1) or a polyclonal rabbit Rab5 antibody at a 1∶200 dilution (Abcam, Cambridge, UK). Cell morphology was assessed using a polyclonal mouse α-tubulin antibody at a 1∶500 dilution (Abcam, Cambridge, UK). Specific binding of primary antibodies was visualised using a goat anti-rat Alexa Fluor 546 (red) secondary antibody in PBS at a dilution of 1∶500. Cells were imaged using a Zeiss confocal imaging system (LSM500). Colocalisation of Ad5 with the MTOC was quantified by visually assessing the percentage of cells with Alexa488-virus and pericentrin co-staining. Data were averaged from 5 40× microscope fields per experimental condition.
Analysis of Ad5 trafficking in vitro in the presence of kinase inhibitors
Cells were seeded in 8-well chamber slides at 5×104 cells/well 24 h prior to assay. Cells were gently washed with PBS then incubated with 100 µM LY 294002 hydrochloride, 40 µM H 89 hydrochloride or 10 µM SB 202190 hydrochloride (Tocris Bioscience, UK) for 30 minutes at 37°C. Cells were gently washed with PBS then incubated with 1×104 vp/cell of Alexa Fluor-488 labelled virus in the presence of 100 µM LY 294002 hydrochloride, 40 µM H 89 hydrochloride or 10 µM SB 202190 hydrochloride in 150 µl SFDMEM for 1 h on ice. Factor X was used at a concentration of 10 µg/ml. Cells were incubated at 37°C for 180 minutes prior to fixation. Localisation of Ad particles at the MTOC was characterised and quantified as previously described.
Analysis of shRNA mediated depletion of cellular αv integrins and effect on FX mediated Ad5 trafficking
To evaluate the effect of depletion of cellular αv integrins on FX mediated Ad5 trafficking, SKOV3 cells (5×104 cells/well in either 24 well plates or 8 well chamber slides) were transfected with shRNA targeting αv integrin, “off-target” control shRNA, or liposomes only (mock transfected), according to manufacturer's instructions. Briefly, 2.5 µl/well of 5 µM shRNA was diluted 50 µl in serum free media before being mixed with 50 µl/well of SFDMEM containing 2 µl Dharmafect transfection reagent. The lipid and shRNA solution were mixed and allowed to stand at room temperature for 30 minutes before the addition of 400 µl/well of complete media. Cells were washed with PBS and 500 µl/well of lipid/shRNA solution was added and allowed to transfect cells for 24 hours prior to analysis of knockdown. We confirmed specific knockdown of αv integrin by detection of αv integrin mRNA by RT-qPCR and by flow cytometry for surface levels of the αv subunit. Total cellular mRNA was harvested using RNeasy mini kit (Qiagen), quantified, and 300 ng of mRNA was converted to cDNA by in vitro reverse transcription. Levels of αv integrin mRNA in 2.5 µl of cDNA were subsequently quantified by TAQman qPCR and normalised to total levels of the housekeeper GAPDH. For analysis of αv integrin knockdown by flow cytometry, cells were detached 48 h post-transfection and incubated with an anti-αv antibody (mAb mouse IgG1 clone L230) for 1 h at 4°C at a final concentration of 10 µg/ml. Cells were washed with SFDMEM, incubated with goat anti-mouse Alexa488-secondary (1∶125 dilution) for a further hour at 4°C, washed again with SFDMEM and resuspended in a final volume of 350µl. Surface levels of the αv integrin subunit were detected using a BD FACS CANTO II flow cytometer, acquiring >10,000 gated events. For cell transport studies, cells were transfected as above in 8-well chamber slides for 24 hours. Cells were subsequently washed and cooled to 4°C, before the addition of fluorescently labelled Ad5 (10,000 vp/cell) in serum free media containing physiological levels of FX. Cells were then placed at 37°C for the stated times, washed, fixed using 4% paraformaldehyde in PBS for 10 minutes before counterstaining with 4′,6-diamidino-2-phenylindole (DAPI) and mounting in Prolong Gold for analysis as previously described.
Analysis of Ad5 attachment ex vivo
Six µm frozen liver sections from macrophage-depleted male MF1 mice were incubated with 1×109 vp of Alexa488-labelled Ad5CTL in SFDMEM in the presence or absence of 10 µg/ml FX and/or increasing concentrations of heparins for 1 h on ice. Sections were then washed twice with PBS and mounted using ProLong Gold antifade reagent with DAPI. Sections were imaged using an Olympus Cell∧M imaging system. To quantify adherent Alexa488-Ad5CTL particles, 40× images captured using an Olympus imaging system and were processed using PaintShop Pro and ImageJ. Viral particles were counted using the semi-automated cell counting tool from ImageJ. An average of 5 captured images were analysed per experimental condition.
In vivo assessment of Ad5 uptake
All animal experiments were approved by the UK Home Office. Male MF1 outbred mice aged between 8–10 weeks (weight approximately 35g) and housed in secure barrier facilities were used for all in vivo experiments. Macrophage depletion was carried out by clodronate liposome pretreatment as described previously [21], [66]. Kupffer cell depletion was confirmed by staining frozen liver sections with a rat anti-mouse F4/80 primary antibody at a 1∶50 dilution (Abcam, Cambridge, UK) and a goat anti-rat Alexa Fluor 546 (red) secondary antibody at a 1∶500 dilution and all sections in macrophage depleted mice showed a complete absence of Kupffer cells to confirm the efficiency of depletion (data not shown). For the analysis of virus genome accumulation in the liver 1 h after intravenous virus administration, 1×1011 vp Ad5CTL in 100 µl PBS was injected into the tail-vein of macrophage-depleted mice 5 minutes after intravenous administration of 20 mg/kg or 50 mg/kg heparins in 100 µl PBS. Mice were sacrificed and perfused with PBS 1h post-inoculation. Livers were harvested and total DNA was purified using the QiaQuick Spin DNA Extraction Kit as described above.
Analysis of Ad5 localisation in vivo
To characterise Ad localisation in vivo, 1×1011 vp Alexa-labelled Ad5CTL in 100 µl PBS was injected into the tail vein 5 minutes after intravenous administration of 20 mg/kg or 5 0mg/kg heparins in 100 µl PBS. Livers were flushed by cardiac PBS perfusion 1 h later to remove non-adherent virus particles and the largest lobe was then embedded and immediately frozen in OCT Tissue-Tek embedding compound. Frozen liver sections (4 µm) were fixed and stained with rat anti-mouse CD31 antibody at a 1∶50 dilution (BD Pharmingen, Oxford, UK) to detect endothelial cells. Specific binding of primary antibodies was visualised using a goat anti-rat Alexa Fluor 546 (red) secondary antibody in PBS at a dilution of 1∶500. Sections were imaged using an Olympus imaging system.
Statistical analysis
Statistical significance was calculated using 2-sample, 2-tailed student's t-tests. P-values of <0.05 or over were considered statistically significant. Results presented are representative data from a minimum of two separate experiments with at least 3 experimental replicates per group. All virus binding and transduction experiments were performed in biological triplicates and on at least three independent occasions. All error bars represent SEM.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. RussellWC
2009 Adenoviruses: update on structure and function. J Gen Virol 90 1 20
2. HongJY
LeeHJ
PiedraPA
ChoiEH
ParkKH
2001 Lower respiratory tract infections due to adenovirus in hospitalized Korean children: epidemiology, clinical features, and prognosis. Clin Infect Dis 32 1423 1429
3. EchavarriaM
2008 Adenoviruses in immunocompromised hosts. Clin Microbiol Rev 21 704 715
4. KojaoghlanianT
FlomenbergP
HorwitzMS
2003 The impact of adenovirus infection on the immunocompromised host. Rev Med Virol 13 155 171
5. Blackwell Munsgardt 2004 Adenovirus. Am J Transplant 4 Suppl 10 101 104
6. LionT
BaumgartingerR
WatzingerF
Matthes-MartinS
SudaM
2003 Molecular monitoring of adenovirus in peripheral blood after allogeneic bone marrow transplantation permits early diagnosis of disseminated disease. Blood 102 1114 1120
7. MunozFM
PiedraPA
DemmlerGJ
1998 Disseminated adenovirus disease in immunocompromised and immunocompetent children. Clin Infect Dis 27 1194 1200
8. Leruez-VilleM
MinardV
LacailleF
BuzynA
AbachinE
2004 Real-time blood plasma polymerase chain reaction for management of disseminated adenovirus infection. Clin Infect Dis 38 45 52
9. TakayamaR
HatakeyamaN
SuzukiN
YamamotoM
HayashiT
2007 Quantification of adenovirus species B and C viremia by real-time PCR in adults and children undergoing stem cell transplantation. J Med Virol 79 278 284
10. BergelsonJM
CunninghamJA
DroguettG
Kurt-JonesEA
KrithivasA
1997 Isolation of a common receptor for Coxsackie B viruses and adenoviruses 2 and 5. Science 275 1320 1323
11. LawLK
DavidsonBL
2005 What does it take to bind CAR? Mol Ther 12 599 609
12. WickhamTJ
MathiasP
ChereshDA
NemerowGR
1993 Integrins alpha v beta 3 and alpha v beta 5 promote adenovirus internalization but not virus attachment. Cell 73 309 319
13. WickhamTJ
FilardoEJ
ChereshDA
NemerowGR
1994 Integrin alpha v beta 5 selectively promotes adenovirus mediated cell membrane permeabilization. J Cell Biol 127 257 264
14. AlemanyR
CurielDT
2001 CAR-binding ablation does not change biodistribution and toxicity of adenoviral vectors. Gene Ther 8 1347 1353
15. MartinK
BrieA
SaulnierP
PerricaudetM
YehP
2003 Simultaneous CAR - and alpha V integrin-binding ablation fails to reduce Ad5 liver tropism. Mol Ther 8 485 494
16. NicolCG
GrahamD
MillerWH
WhiteSJ
SmithTA
2004 Effect of adenovirus serotype 5 fiber and penton modifications on in vivo tropism in rats. Mol Ther 10 344 354
17. ParkerAL
WaddingtonSN
NicolCG
ShayakhmetovDM
BuckleySM
2006 Multiple vitamin K-dependent coagulation zymogens promote adenovirus-mediated gene delivery to hepatocytes. Blood 108 2554 2561
18. Di PaoloNC
ShayakhmetovDM
2009 Adenovirus de-targeting from the liver. Curr Opin Mol Ther 11 523 531
19. CoyneCB
BergelsonJM
2005 CAR: a virus receptor within the tight junction. Adv Drug Deliv Rev 57 869 882
20. KalyuzhniyO
Di PaoloNC
SilvestryM
HofherrSE
BarryMA
2008 Adenovirus serotype 5 hexon is critical for virus infection of hepatocytes in vivo. Proc Natl Acad Sci U S A 105 5483 5488
21. WaddingtonSN
McVeyJH
BhellaD
ParkerAL
BarkerK
2008 Adenovirus serotype 5 hexon mediates liver gene transfer. Cell 132 397 409
22. MadischI
WolfelR
HarsteG
PommerH
HeimA
2006 Molecular identification of adenovirus sequences: a rapid scheme for early typing of human adenoviruses in diagnostic samples of immunocompetent and immunodeficient patients. J Med Virol 78 1210 1217
23. KampmannB
CubittD
WallsT
NaikP
DepalaM
2005 Improved outcome for children with disseminated adenoviral infection following allogeneic stem cell transplantation. Br J Haematol 130 595 603
24. EskoJD
LindahlU
2001 Molecular diversity of heparan sulfate. J Clin Invest 108 169 173
25. BishopJR
SchukszM
EskoJD
2007 Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature 446 1030 1037
26. EndressT
LampeM
BriggsJA
KrausslichHG
BrauchleC
2008 HIV-1-cellular interactions analyzed by single virus tracing. Eur Biophys J 37 1291 1301
27. JohnsonKM
KinesRC
RobertsJN
LowyDR
SchillerJT
2009 Role of heparan sulfate in attachment to and infection of the murine female genital tract by human papillomavirus. J Virol 83 2067 2074
28. SummerfordC
SamulskiRJ
1998 Membrane-associated heparan sulfate proteoglycan is a receptor for adeno-associated virus type 2 virions. J Virol 72 1438 1445
29. ShuklaD
LiuJ
BlaiklockP
ShworakNW
BaiX
1999 A novel role for 3-O-sulfated heparan sulfate in herpes simplex virus 1 entry. Cell 99 13 22
30. ShayakhmetovDM
GaggarA
NiS
LiZY
LieberA
2005 Adenovirus binding to blood factors results in liver cell infection and hepatotoxicity. J Virol 79 7478 7491
31. LyonM
DeakinJA
GallagherJT
1994 Liver heparan sulfate structure. A novel molecular design. J Biol Chem 269 11208 11215
32. MacArthurJM
BishopJR
StanfordKI
WangL
BensadounA
2007 Liver heparan sulfate proteoglycans mediate clearance of triglyceride-rich lipoproteins independently of LDL receptor family members. J Clin Invest 117 153 164
33. VongchanP
WardaM
ToyodaH
ToidaT
MarksRM
2005 Structural characterization of human liver heparan sulfate. Biochim Biophys Acta 1721 1 8
34. KaliaM
ChandraV
RahmanSA
SehgalD
JameelS
2009 Heparan sulfate proteoglycans are required for cellular binding of the hepatitis E virus ORF2 capsid protein and for viral infection. J Virol 83 12714 12724
35. TiwariV
O'DonnellCD
OhMJ
Valyi-NagyT
ShuklaD
2005 A role for 3-O-sulfotransferase isoform-4 in assisting HSV-1 entry and spread. Biochem Biophys Res Commun 338 930 937
36. KimM
ZinnKR
BarnettBG
SumerelLA
KrasnykhV
2002 The therapeutic efficacy of adenoviral vectors for cancer gene therapy is limited by a low level of primary adenovirus receptors on tumour cells. Eur J Cancer 38 1917 1926
37. BoyleMP
EnkeRA
ReynoldsJB
MogayzelPJJr
GugginoWB
2006 Membrane-associated heparan sulfate is not required for rAAV-2 infection of human respiratory epithelia. Virol J 3 29
38. O'DonnellJ
TaylorKA
ChapmanMS
2009 Adeno-associated virus-2 and its primary cellular receptor–Cryo-EM structure of a heparin complex. Virology 385 434 443
39. ZautnerAE
KornerU
HenkeA
BadorffC
SchmidtkeM
2003 Heparan sulfates and coxsackievirus-adenovirus receptor: each one mediates coxsackievirus B3 PD infection. J Virol 77 10071 10077
40. ShayakhmetovDM
EberlyAM
LiZY
LieberA
2005 Deletion of penton RGD motifs affects the efficiency of both the internalization and the endosome escape of viral particles containing adenovirus serotype 5 or 35 fiber knobs. J Virol 79 1053 1061
41. SmithTA
IdamakantiN
RollenceML
Marshall-NeffJ
KimJ
2003 Adenovirus serotype 5 fiber shaft influences in vivo gene transfer in mice. Hum Gene Ther 14 777 787
42. HidakaC
MilanoE
LeopoldPL
BergelsonJM
HackettNR
1999 CAR-dependent and CAR-independent pathways of adenovirus vector-mediated gene transfer and expression in human fibroblasts. J Clin Invest 103 579 587
43. GreberU
PuntenerD
2009 DNA tumour virus entry - from plasma membrane to the nucleus. Semin Cell Dev Biol 20 631 642
44. SafaiyanF
KolsetSO
PrydzK
GottfridssonE
LindahlU
1999 Selective effects of sodium chlorate treatment on the sulfation of heparan sulfate. J Biol Chem 274 36267 36273
45. EskoJD
WeinkeJL
TaylorWH
EkborgG
RodenL
1987 Inhibition of chondroitin and heparan sulfate biosynthesis in Chinese hamster ovary cell mutants defective in galactosyltransferase I. J Biol Chem 262 12189 12195
46. BameKJ
EskoJD
1989 Undersulfated heparan sulfate in a Chinese hamster ovary cell mutant defective in heparan sulfate N-sulfotransferase. J Biol Chem 264 8059 8065
47. BaiX
EskoJD
1996 An animal cell mutant defective in heparan sulfate hexuronic acid 2-O-sulfation. J Biol Chem 271 17711 17717
48. BasuA
KandaT
BeyeneA
SaitoK
MeyerK
2007 Sulfated homologues of heparin inhibit hepatitis C virus entry into mammalian cells. J Virol 81 3933 3941
49. XuZ
TianJ
SmithJS
ByrnesAP
2008 Clearance of adenovirus by Kupffer cells is mediated by scavenger receptors, natural antibodies, and complement. J Virol 82 11705 11713
50. SmithJS
XuZ
TianJ
StevensonSC
ByrnesAP
2008 Interaction of systemically delivered adenovirus vectors with Kupffer cells in mouse liver. Hum Gene Ther 19 547 554
51. ShashkovaEV
DoroninK
SenacJS
BarryMA
2008 Macrophage depletion combined with anticoagulant therapy increases therapeutic window of systemic treatment with oncolytic adenovirus. Cancer Res 68 5896 5904
52. Di PaoloNC
van RooijenN
ShayakhmetovDM
2009 Redundant and synergistic mechanisms control the sequestration of blood-born adenovirus in the liver. Mol Ther 17 675 684
53. ShiehMT
WuDunnD
MontgomeryRI
EskoJD
SpearPG
1992 Cell surface receptors for herpes simplex virus are heparan sulfate proteoglycans. J Cell Biol 116 1273 1281
54. BarthH
SchaferC
AdahMI
ZhangF
LinhardtRJ
2003 Cellular binding of hepatitis C virus envelope glycoprotein E2 requires cell surface heparan sulfate. J Biol Chem 278 41003 41012
55. AsokanA
ConwayJC
PhillipsJL
LiC
HeggeJ
Reengineering a receptor footprint of adeno-associated virus enables selective and systemic gene transfer to muscle. Nat Biotechnol 28 79 82
56. PeraboL
GoldnauD
WhiteK
EndellJ
BoucasJ
2006 Heparan sulfate proteoglycan binding properties of adeno-associated virus retargeting mutants and consequences for their in vivo tropism. J Virol 80 7265 7269
57. StanfordKI
WangL
CastagnolaJ
SongD
BishopJR
Heparan sulfate 2-O-sulfotransferase is required for triglyceride-rich lipoprotein clearance. J Biol Chem 285 286 294
58. SakaiT
KisielW
1990 Binding of human factors X and Xa to HepG2 and J82 human tumor cell lines. Evidence that factor Xa binds to tumor cells independent of factor Va. J Biol Chem 265 9105 9113
59. KazamaY
KomiyamaY
KisielW
1993 Tissue factor pathway inhibitor and protease nexin-1 are major factor Xa binding proteins on the HepG2 cell surface. Blood 81 676 682
60. CraftJ
EppsK
BrancatoS
SerfisA
2003 Incorporation of blood clotting factor X into phospholipid model membranes: fluorescence microscopy imaging and subphase effects surrounding the lipid phase transition region. Journal of Colloid and Interface Science 268 181 187
61. SabharwalAK
PadmanabhanK
TulinskyA
MathurA
GorkaJ
1997 Interaction of calcium with native and decarboxylated human factor X. Effect of proteolysis in the autolysis loop on catalytic efficiency and factor Va binding. J Biol Chem 272 22037 22045
62. ScanduraJM
AhmadSS
WalshPN
1996 A binding site expressed on the surface of activated human platelets is shared by factor X and prothrombin. Biochemistry 35 8890 8902
63. PlesciaJ
AltieriDC
1996 Activation of Mac-1 (CD11b/CD18)-bound factor X by released cathepsin G defines an alternative pathway of leucocyte initiation of coagulation. Biochem J 319 Pt 3 873 879
64. TuveS
WangH
JacobsJD
YumulRC
SmithDF
2008 Role of cellular heparan sulfate proteoglycans in infection of human adenovirus serotype 3 and 35. PLoS Pathog 4 e1000189
65. KritzAB
NicolCG
DishartKL
NelsonR
HolbeckS
2007 Adenovirus 5 fibers mutated at the putative HSPG-binding site show restricted retargeting with targeting peptides in the HI loop. Mol Ther 15 741 749
66. AlbaR
BradshawAC
ParkerAL
BhellaD
WaddingtonSN
2009 Identification of coagulation factor (F)X binding sites on the adenovirus serotype 5 hexon: effect of mutagenesis on FX interactions and gene transfer. Blood 114 965 971
67. RezaieAR
1998 Calcium enhances heparin catalysis of the antithrombin-factor Xa reaction by a template mechanism. Evidence that calcium alleviates Gla domain antagonism of heparin binding to factor Xa. J Biol Chem 273 16824 16827
68. RezaieAR
2000 Identification of basic residues in the heparin-binding exosite of factor Xa critical for heparin and factor Va binding. J Biol Chem 275 3320 3327
69. GhoshT
ChattopadhyayK
MarschallM
KarmakarP
MandalP
2009 Focus on antivirally active sulfated polysaccharides: from structure-activity analysis to clinical evaluation. Glycobiology 19 2 15
70. HuskensD
VermeireK
ProfyAT
ScholsD
2009 The candidate sulfonated microbicide, PRO 2000, has potential multiple mechanisms of action against HIV-1. Antiviral Res 84 38 47
71. FletcherPS
WallaceGS
MesquitaPM
ShattockRJ
2006 Candidate polyanion microbicides inhibit HIV-1 infection and dissemination pathways in human cervical explants. Retrovirology 3 46
72. Von SeggernDJ
KehlerJ
EndoRI
NemerowGR
1998 Complementation of a fibre mutant adenovirus by packaging cell lines stably expressing the adenovirus type 5 fibre protein. J Gen Virol 79 Pt 6 1461 1468
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2010 Číslo 10
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Měli bychom postcovidový syndrom léčit antidepresivy?
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- Retroviral RNA Dimerization and Packaging: The What, How, When, Where, and Why
- Viral Replication Rate Regulates Clinical Outcome and CD8 T Cell Responses during Highly Pathogenic H5N1 Influenza Virus Infection in Mice
- Antimicrobial Peptides: Primeval Molecules or Future Drugs?
- Crystal Structure of DotD: Insights into the Relationship between Type IVB and Type II/III Secretion Systems