
Retention and Loss of RNA Interference Pathways in Trypanosomatid Protozoans
RNA interference (RNAi) pathways are widespread in metaozoans but the genes required show variable occurrence or activity in eukaryotic microbes, including many pathogens. While some Leishmania lack RNAi activity and Argonaute or Dicer genes, we show that Leishmania braziliensis and other species within the Leishmania subgenus Viannia elaborate active RNAi machinery. Strong attenuation of expression from a variety of reporter and endogenous genes was seen. As expected, RNAi knockdowns of the sole Argonaute gene implicated this protein in RNAi. The potential for functional genetics was established by testing RNAi knockdown lines lacking the paraflagellar rod, a key component of the parasite flagellum. This sets the stage for the systematic manipulation of gene expression through RNAi in these predominantly diploid asexual organisms, and may also allow selective RNAi-based chemotherapy. Functional evolutionary surveys of RNAi genes established that RNAi activity was lost after the separation of the Leishmania subgenus Viannia from the remaining Leishmania species, a divergence associated with profound changes in the parasite infectious cycle and virulence. The genus Leishmania therefore offers an accessible system for testing hypothesis about forces that may select for the loss of RNAi during evolution, such as invasion by viruses, changes in genome plasticity mediated by transposable elements and gene amplification (including those mediating drug resistance), and/or alterations in parasite virulence.
Published in the journal:
. PLoS Pathog 6(10): e32767. doi:10.1371/journal.ppat.1001161
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1001161
Summary
RNA interference (RNAi) pathways are widespread in metaozoans but the genes required show variable occurrence or activity in eukaryotic microbes, including many pathogens. While some Leishmania lack RNAi activity and Argonaute or Dicer genes, we show that Leishmania braziliensis and other species within the Leishmania subgenus Viannia elaborate active RNAi machinery. Strong attenuation of expression from a variety of reporter and endogenous genes was seen. As expected, RNAi knockdowns of the sole Argonaute gene implicated this protein in RNAi. The potential for functional genetics was established by testing RNAi knockdown lines lacking the paraflagellar rod, a key component of the parasite flagellum. This sets the stage for the systematic manipulation of gene expression through RNAi in these predominantly diploid asexual organisms, and may also allow selective RNAi-based chemotherapy. Functional evolutionary surveys of RNAi genes established that RNAi activity was lost after the separation of the Leishmania subgenus Viannia from the remaining Leishmania species, a divergence associated with profound changes in the parasite infectious cycle and virulence. The genus Leishmania therefore offers an accessible system for testing hypothesis about forces that may select for the loss of RNAi during evolution, such as invasion by viruses, changes in genome plasticity mediated by transposable elements and gene amplification (including those mediating drug resistance), and/or alterations in parasite virulence.
Introduction
In metazoans, RNAi interference and related pathways play many key roles including regulation of mRNA levels and translation, chromatin silencing, programmed DNA rearrangements, genome surveillance, and defense against invading viruses. The phylogenetic distribution of key genes required for RNA interference such as Argonaute and Dicer suggests that this pathway may have been present in the common eukaryote ancestor [1]. However the situation for eukaryotic microbes is complex: some have active RNAi pathways, others lack RNAi genes and activity, and demonstration of RNAi has proven elusive in some species bearing reasonable homologs of canonical genes such as Argonaute [2]–[7].
The trypanosomatid protozoa comprise three major lineages, broadly grouped as the African trypanosomes (Trypanosoma brucei), South American trypanosomes (T. cruzi) and a lineage encompassing a number of genera associated with insects or plants, ultimately leading to the mammalian parasite Leishmania [8]. Functional and genome sequencing data have shown that species within the African trypanosome lineage such as T. brucei contain an active RNAi pathway and genes, including an Argonaute “slicer” (AGO1; [2]) and two Dicers (DCL1 and DCL2; [9], [10]). In contrast, T. cruzi, L. major and L. donovani lack these activities and associated genes [11]–[14]. However the genome of L. braziliensis (subgenus Viannia) contains orthologs of T. brucei AGO1, DCL1 and DCL2 [15], suggesting this group might retain a functional RNAi pathway. Given the uncertainties of extrapolating from RNAi genes to functions noted in other eukaryotic microbes [2]–[4], we sought to establish whether the RNAi machinery functions in L. braziliensis, and explored its utility as a genetic tool. Furthermore, we made evolutionary comparisons to map when the RNAi pathway was lost, and we discuss potential selective forces impacting on the parasite that may have contributed to the demise of RNAi during Leishmania evolution.
Results
siRNA formation in L. braziliensis
Dicer is required to process long dsRNA to small interfering RNAs (siRNAs), which in trypanosomes are 24–26 nt long [16]. A convenient marker of RNAi activity is siRNA formation from endogeneous retroelements [17], and Northern blot analysis of L. braziliensis RNAs revealed the presence of small RNAs of the expected sizes arising from the retroelement SLACS, similar to T. brucei siRNAs (Fig. S1; [16]).
We then developed a green fluorescent protein (GFP)-based RNAi reporter assay for siRNA formation, as well as target mRNA and protein levels. Initially we experienced unexpected difficulty in L. braziliensis transfection, when using episomal constructs previously developed in one of our labs that function effectively in many Leishmania species, and in many laboratories [18]. The basis for this effect is not definitively known, as addressed in the discussion, but we suspect it is due to the tendency of episomal vectors to be transcribed from both strands, which in an RNAi-proficient species would strongly inhibit episomal gene expression [11], [13]. Thus in all studies reported here, transfection was accomplished following integration of DNA constructs into the ribosomal small subunit RNA (SSU) locus, using the appropriately digested DNA from pIR1SAT-based vectors, or derivatives thereof [19]. In trypanosomatids, processing of polycistronic RNA precursors by 5′ trans-splicing and 3′ polyadenylation produces capped mRNAs that can direct protein synthesis [20].
First we generated a GFP ‘stem-loop’ (long hairpin) construct, containing two copies of an AT-rich GFP reporter (GFP65) in an inverted orientation separated by a short loop (Fig. 1A). This GFP stem-loop construct (GFP65-StL) was flanked by Leishmania sequences required for efficient 5′ and 3′ end mRNA formation, and was expressed following integration into the parasite small subunit ribosomal RNA locus (SSU rRNA; Fig. 1A) in L. braziliensis strain M2903.

Northern blot analysis with a GFP65 probe showed that expression of GFP65-StL gave rise to a variety of products (Fig. 1D, lane 2). The largest of these likely correspond to unprocessed transcripts, while the smaller ones likely correspond to degradation products, which could occur irrespective of whether RNAi pathways are active. Importantly, abundant levels of 24–26 nt siRNAs were seen (Figs. 1B and 1E). In contrast, similarly small RNAs were not detected with probes to the SAT drug resistance marker, which is not found in an inverted repeat (data not shown). These data suggested that L. braziliensis expresses a robust Dicer-like activity.
Demonstration of RNAi activity
We used two GFP reporters, one encoded by the AT-rich ORF (GFP65) used in the GFP65-StL construct above, and the second by a GC-rich ORF (GFP+). These genes differ in most 3rd codon positions, but their protein products only differ by a single amino acid. Alignment of these genes showed that the longest tracts of identical nucleotides were less than 14 nt (Fig. S2). GFP65 or GFP+ was then expressed separately following integration into the SSU rRNA locus, in wild-type (WT) L. braziliensis or the GFP65-StL transfectant that produces GFP65 siRNAs.
As expected, expression of GFP65 or GFP+ led to high levels of GFP mRNA and protein in WT lines, as did expression of GFP+ within the GFP65-StL transfectant (Fig. 1D, F, G). In contrast, clonal lines arising from introduction of GFP65 into the GFPST-StL transfectant showed only trace amounts of GFP65 mRNA (Fig. 1D), and the level of GFP protein was below the limit of detection by western blotting (<1% in these studies; Fig. 1G) or flow cytometry (Fig. 1C). These data established that GFP65-derived dsRNA mediated selective ablation of the AT-rich GFP65 but not the GC-rich GFP+.
Similar studies were carried out with a luciferase (LUC) reporter, expressed alone or in combination with a LUC stem-loop construct, revealing strongly-reduced LUC expression (90–300 fold; Fig. S3, and other studies below).
RNAi activity against endogenous L. braziliensis genes
We then tested the activity of the RNAi pathway on several endogenous genes. In transient transfections performed using several protocols and dsRNAs synthesized in vitro against the L. braziliensis α-tubulin, Northern blot analysis showed at best a 63% decrease in α -tubulin mRNA (Fig. 2A). This contrasts with T. brucei where such protocols readily yield >95% reduction in tubulin mRNA expression [21]. This perhaps reflects the lower efficacy of transient transfection attained thus far in Leishmania [11].

Since inducible expression systems were unavailable, we focused on stably expressed ‘stem-loop’ constructs targeting a panel of nonessential genes in L. braziliensis, including ones mediating synthesis of the abundant glycoconjugate lipophosphoglycan (LPG1, LPG2, LPG3; [22]), hypoxanthine-guanine phosphoribosyltransferase (HGPRT), or the genes PFR1 and PFR2, which encode major components of the paraflagellar rod, a component of the trypanosomatid flagellum required for motility [23]. These StL-transfectants showed a variable decrease in mRNA levels when estimated by qPCR, ranging from no effect (LPG1) to more than 10-fold reduction (LPG2, LPG3; Fig. 2B). However, Northern blot analysis showed a nearly complete absence of LPG2 mRNA (Fig. 2C), suggesting that the qPCR values are likely underestimates, possibly due to the presence of RNA degradation intermediates able to act as templates (these are evident in Fig. 2C). Despite the reductions in mRNA levels, LPG levels were at best only 3-fold lower in the LPG2-StL or LPG3-StL transfectants, with considerable clonal variability (Fig. 2E; data for LPG3-StL not shown). This suggests that L. braziliensis requires only low levels of LPG biosynthetic proteins, similar to the relatively small effects of RNAi on trypanosome glycoconjugate biosynthetic genes [24]. Both HGPRT mRNA and protein levels showed 3–4 fold decreases in HGPRT-StL transfectants (Fig. 2B, D).
One of the earliest reports of stable phenotypic modulation by RNAi in trypanosomes involved down regulation of a paraflagellar rod protein [25], [26]. The paraflagellar rod is a complex assembly of proteins required for motility, which in trypanosomatids includes two major proteins, termed PFR1 and PFR2 in Leishmania [23], [27], [28]. Introduction of PFR1-StL or PFR2-StL constructs into L. braziliensis yielded viable transfectants that grew normally, but lacked the paraflagellar rod, as visualized in longitudinal or transverse EM sections, and exhibited motility defects (Fig. 3). These phenotypes closely resemble those seen in L. mexicana PFR1 and PFR2 gene deletion mutants [23].

Multiple attempts to introduce ‘stem-loop’ α - or β-tubulin constructs were unsuccessful, as anticipated for essential genes (not shown). Collectively, the strength of the RNAi effect for these phenotypic reporters suggests that RNAi may function sufficiently well to assess the functions of many genes in L. braziliensis.
RNAi of AGO1 establishes its role in the RNAi pathway in L. braziliensis
In other organisms RNAi is mediated by the combined activity of a number of proteins, ultimately converging on the endonucleolytic ‘slicer’ activity of the Argonaute protein, which is encoded by the single AGO1 gene in trypanosomes and L. braziliensis [15], [17]. To establish a critical role for L. braziliensis AGO1 in RNAi, we employed the seemingly counterintuitive approach of ‘RNAi of RNAi genes’, where introduction of dsRNAs targeting RNAi pathway genes inhibits RNAi activity, albeit not to the same level seen in null RNAi pathway gene knockouts [17], [29]–[31]. To facilitate comparisons of the efficacy of RNAi, we developed a single RNAi ‘self reporter’ construct which simultaneously expressed two mRNAs, one encoding a luciferase ORF (LUC) and a second encoding a luciferase ORF stem-loop (LUC-StL). This minimized experimental variability and the number of transfections required, allowing the assessment of RNAi efficacy by the introduction of a single construct. When introduced into WT L. braziliensis, the ‘LUC RNAi self reporter’ (LUC-SR) showed low levels of luciferase activity, about 4-fold over background and comparable to that obtained with lines expressing LUC and LUC-StL independently after successive transfections (Fig. 4). In contrast, introduction of the LUC reporter alone resulted in activities nearly 1000-fold over background (Fig. 4).

We then introduced a construct expressing an AGO1 stem-loop (AGO1-StL) into the LUC RNAi reporter line (LUC-SR). These transfectants showed an average of 100-fold increased luciferase expression relative to LUCSR transfectants, signifying a considerable reduction in the efficiency of RNAi (Fig. 4). As expected from studies in other organisms cited above, inhibition of RNAi activity was partial, as these values were still about 10-fold less than seen in WT cells transfected with the LUC reporter construct alone (Fig. 4). These data thus implicate AGO1 as an essential component of the RNAi pathway of L. braziliensis.
Mapping of the point in Leishmania evolution at which RNAi activity and RNAi pathway genes were lost
We explored the prevalence of RNAi pathways in other Trypanosomatid species by comparative genomics. PCR assays detected AGO1 and/or DCL1 genes in all isolates of the Leishmania subgenus Viannia tested (L. braziliensis, L. guyanensis, L. panamensis) but not in Leishmania (Sauroleishmania) tarentolae, L. mexicana, L. major or L. donovani (data not shown). Partial genome sequencing of a close non-parasitic ‘outgroup’, Crithidia fasciculata revealed AGO1, DCL1 and DCL2. To confirm the presence or absence of a functional RNAi pathway, we expressed the GFP65-StL RNA in L. tarentolae, L. mexicana, L. panamensis, L. guyanensis and Crithidia fasciculata, and monitored siRNA formation by Northern blotting. Consistent with the observed distribution of RNAi pathway genes, GFP siRNAs were made only in Crithidia, L. guyanensis and L. panamensis (Fig. 5, S4). Transfection with the GFP reporters showed strong reductions in GFP expression in L. panamensis, comparable to that seen with L. major in Fig. 1 (data not shown), and we show in a later section that RNAi is active in L. guyanensis using a luciferase reporter The level of GFP expression in Crithidia with the Leishmania vectors used was too low to utilize for quantification of the strength of RNAi by flow cytometry (data not shown).
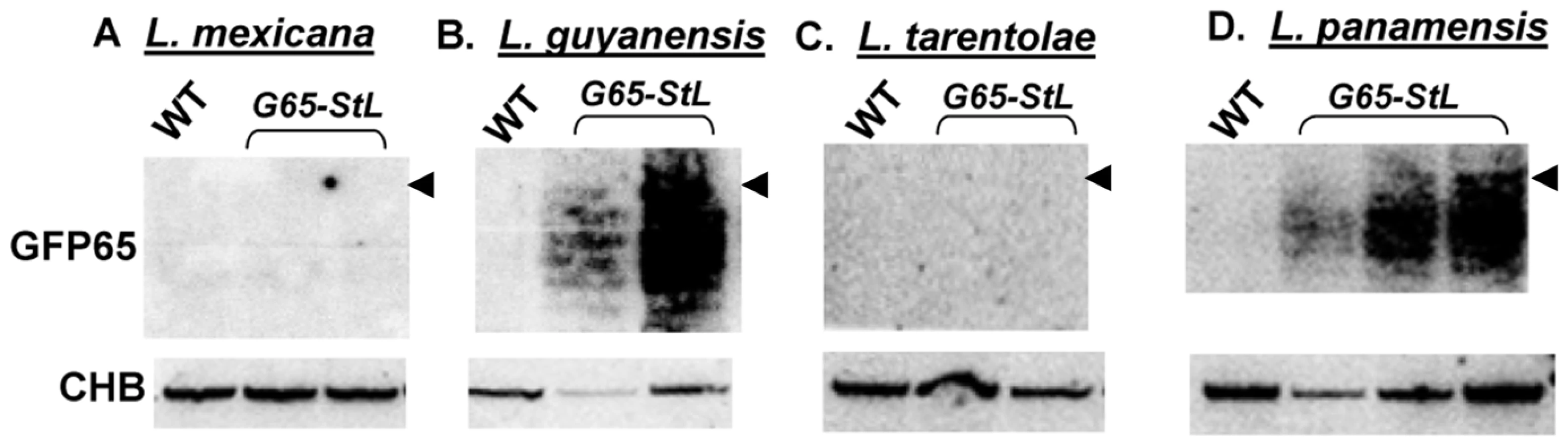
Association of these findings with the trypanosomatid evolutionary tree (Fig. 6A) through evolutionary parsimony identified a single point when the RNAi pathway was lost during evolution, located after the divergence of members of the subgenus Viannia from the remaining species complexes (Fig. 7). Importantly, this corresponds precisely to the point when RNAi genes were lost in evolution, as deduced by comparative genomics and evolutionary parsimony. Inspection of the sequenced Leishmania genomes shows that all RNAi-deficient Leishmania now contain only remnant, highly degenerate pseudogenes (AGO1) or have undergone gene deletion (as revealed by ‘synteny gaps’ for DCL1 and DCL2) for known trypanosomatid RNAi genes. Since species retaining only a partial set of intact RNAi genes have not been reported, from these data we cannot identify which essential RNAi pathway gene was lost first at this distant point in Leishmania evolution. Presumably, once a gene critical for RNAi activity was inactivated, the remaining genes of the pathway become superfluous and fall prey to evolutionary drift, as seen in many other metabolic pathways during evolution.

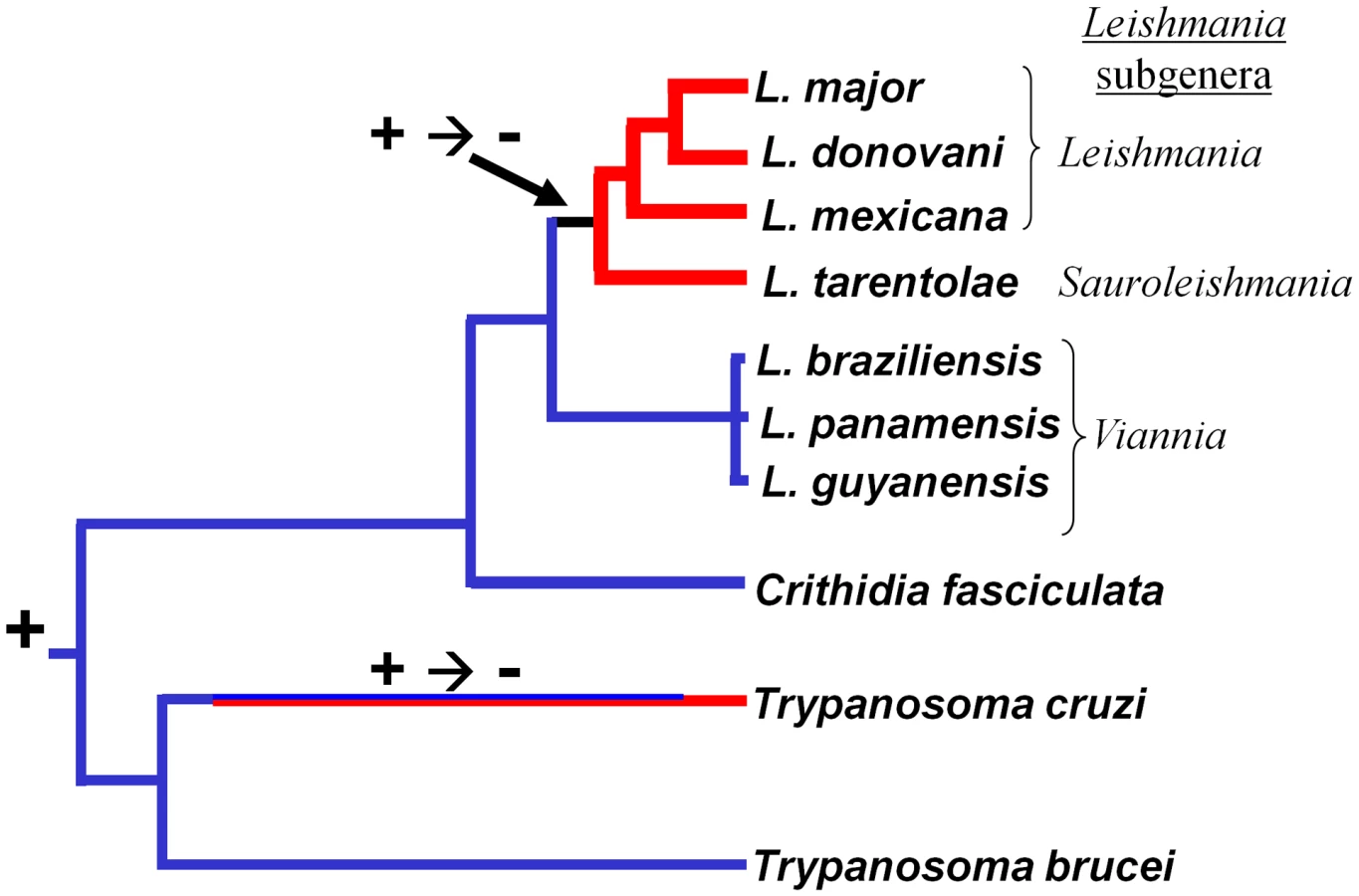
RNAi pathways were probably present in the common eukaryote ancestor [1], and the evolutionary relationships of the available trypanosomatid RNAi pathway proteins closely resemble those of housekeeping protein-based phylogenies (shown for AGO1 and DCL1 in Fig. 6 B–D). While the L. braziliensis AGO1 gene is not syntenic with that of T. brucei [15], [32] the congruency of the RNAi gene and ‘housekeeping’ gene phylogenies renders the possibility of lateral gene transfer and/or independent acquisitions unlikely. Thus, RNAi most likely was lost twice independently in trypanosomatids, once in the lineage leading to T. cruzi, and a second time in the lineage leading to Leishmania, subsequent to the divergence of most Leishmania groups from the non-parasitic species Crithidia fasciculata and the Leishmania subgenus Viannia (Fig. 7).
RNAi activity in virus+ vs. virus-free Leishmania
We and others have speculated that one of the forces contributing to the loss of RNAi in eukaryotic microbes may be invasion or loss of RNA viruses [13], [33]. Significantly, dsRNA viruses termed LRVs are found in many (but not all) strains and/or species from the Leishmania subgenus Viannia, including L. braziliensis [34], [35]. We reasoned that studies of the efficacy of RNAi in extant Leishmania bearing or lack LRVs could provide some insight into their potential roles in evolution.
Using specific PCR primers for LRVs we showed that the L. braziliensis strain M2903 used here lacked LRVs, consistent with previous reports [36], [37]. Unfortunately methods for the introduction and/or cure of LRV from Leishmania are not well developed, precluding tests of isogenic L. braziliensis engineered to harbor the LRV virus. Similarly, just one isogenic virus-free derivative of an LRV-containing Leishmania has been described; L. guyanensis is closely related to L. braziliensis (Fig. 7), and a virus-free derivative arose fortuitously in the course of other studies [38]. The efficiency of RNAi in these lines was evaluated by introduction of the luciferase RNAi reporter (LUC-SR) described earlier, relative to transfectants expressing only LUC. Multiple clonal lines were obtained, and LUC expression was measured in six randomly selected lines (Fig. 8A). Importantly, the level of luciferase expression seen in the lines expressing only LUC were comparable between the closely related Viannia species M2903 L. braziliensis and M4147 L. guyanensis (Fig. 8A). All lines and transfectants were shown to retain or lack the LRV1-4 by RT-PCR tests as expected (Fig. 8B).

While the RNAi pathway was active in the LRV+ L. guyanensis M4147, its efficiency was only about 30-fold (3.8% LUC-SR vs. LUC), compared to the 300-fold reduction seen in the virus free L. braziliensis M2903 (0.34% LUC-SR/LUC; Fig. 8A). The WT LRV+ LgM4147 strain also showed reduced efficiency of RNAi relative to M2903, in studies using successively transfected GFP reporter and GFP-StL constructs (data not shown). Significantly, the LRV-free line LgM4147/pX63HYG showed a similar 30-fold efficiency of RNAi in these studies (3.3% LUC SR/LUC). These data suggest that the reduced RNAi efficiency seen in L. guyanensis M4147 does not require the continued presence of the virus.
Discussion
L. braziliensis has a strongly active RNAi pathway able to reduce target gene expression
Our studies have established that L. braziliensis possesses a functional RNAi pathway, which enables the down-regulation of a variety of reporter and endogenous genes when assayed at the mRNA or protein levels. RNAi of AGO1 was used to confirm a requirement for the sole argonaute gene AGO1 in this process. As seen in many organisms, strong reductions in mRNA expression were seen, often accompanied by phenotypic changes, albeit of variable strength. As anticipated, it was not possible to introduce stem-loop constructs for essential genes such as α - or β-tubulins. Studies of such genes will require the development of inducible expression systems in Leishmania, which while promising have not yet reached the point of utility attained in trypanosomes.
Strong phenotypes were produced by the knockdown of two genes implicated in flagellar motility and paraflagellar rod synthesis (PFR1 and PFR2), closely approximating the phenotypes seen in gene deletion mutants in L. mexicana [23]. In contrast, at best only weak phenotypes were produced by knockdowns of three LPG biosynthetic genes, in keeping with findings in trypanosomes where it has proven difficult to down-regulate expression of genes implicated in glycoconjugate synthesis far enough to attain phenotypic effects. Overall, the results to date suggest that the range in efficacy of RNAi knockdowns, as judged by various phenotypic criteria, is comparable to that seen in trypanosomes and other organisms, and thus is likely to be similarly useful in the systematic analysis of Leishmania gene function in the future.
Factors potentially impacting on the evolutionary loss of RNAi in Leishmania
Given the importance of RNAi pathways in many fundamental aspects of eukaryotic biology, it is unsurprising that it has been lost relatively few times during evolution. While the critical roles of RNAi in metazoan gene regulation would likely select strongly against such attenuation, eukaryotic microbes lacking RNAi have arisen sporadically [1], [2]. This in turn raises the question of under what circumstances RNAi might occur. We consider three working hypotheses for selective pressures that may act independently or in concert to drive this loss in Leishmania.
Viruses
We proposed previously that viral pressure could act as a selective force for the loss of RNAi in Leishmania evolution [11], [13]. In one scenario, invasion by LRVs at some point in Leishmania evolution could lead to an attenuation of the RNAi response, as many RNA viruses are prone to attack by cellular RNAi pathways [39]. Attenuation could be achieved through down regulation of the RNAi pathway by the host cell, or through viral genes targeting key RNAi pathway activities. While some RNA viruses encode inhibitors of RNAi, no studies have been undertaken as yet for Leishmania LRVs. The challenge for this model is to explain what forces would prompt cells to favor RNA virus retention over disruptions arising from perturbation or loss of the RNAi pathway. Interestingly, LRV infection has been proposed to be advantageous to Leishmania, possibly by modulating host immune responses in a way beneficial to parasite survival [40], [41]. In support of this hypothesis, recently we have obtained preliminary in support of the proposal that LRV-containing L. guyanensis show increased survival and pathogenicity (L-FL, KO, S. Hickerson and SMB, unpublished data; N. Fasel, personal communication). Selection for the presence of LRV able to promote parasite survival could thus provide a selective force promoting down-regulation of RNAi activity targeting RNA viruses.
While one cannot perform experimental tests in the ancestral Leishmania, one prediction is that in extant species or strains now harboring Leishmania LRVs, attenuation of the RNAi response may occur. Here we compared the efficacy of RNAi seen in the virus-free L. braziliensis M2903 used in the majority of our studies with a closely related species L. guyanensis that bears the cytosolic dsRNA virus LRV1-4 [35], [36] (Fig. 8). While the RNAi pathway remained highly active in the LRV-infected L. guyanensis, its activity as assayed with LUC or GFP reporters was attenuated ∼10-fold relative to that seen in virus-free L. braziliensis (Fig. 8A). Although tools for the introduction of LRV are not well-developed, one line of L. guyanensis has been described which was cured of LRV [38]. Notably the efficiency of RNAi in the virus free line was similar to that of the LRV1-4 containing line (Fig. 8A), showing that the attenuated RNAi response did not require the continued presence of virus. This implies that attenuation occurred through a down-regulation of the cellular RNAi pathway occurred in the LRV-bearing L. guyanensis. If a similar process occurred in the evolutionary lineage leading to extant RNAi-deficient Leishmania species, it could in turn have facilitated a later transition to a complete loss of RNAi activity. Future development of methods for more readily introducing and curing LRV infections will permit further tests of these hypotheses, as will the advent of RNAi-deficient lines of Leishmania braziliensis and other Viannia species. However, the data already in hand are consistent with the possibility of a biologically relevant interplay between parasite RNAi pathways and viral infection during evolution, as seen in viral infections of metazoans.
Increased genome plasticity
A second selective force arises from consideration of the impact of genome plasticity in Leishmania. The ability of mobile elements to produce mutations and genomic rearrangements are well known, and in trypanosomes and other eukaryotes RNAi pathways may help protect against such events [42]–[44]. Importantly, the RNAi-competent L. braziliensis genome contains several classes of mobile elements, including retrotransposons, while RNAi-deficient L. major and L. infantum appear to lack active transposons [15]. While the forces leading to the loss of mobile elements are unknown, their departure could have freed the parasite from the need to maintain activities including RNAi which act to mitigate their effects.
Gene amplification is another important form of genomic plasticity in Leishmania, often occurring in the form of extra-chromosomal circular DNAs associated with drug resistance [45], [46]. In contrast, extra-chromosomal gene amplifications have not been seen in T. brucei, a difference potentially attributable to its active RNAi pathway [11], [13] since circular amplicons tend to be transcribed from both strands [47]. Consistent with this model, extrachromosomal gene amplifications are uncommon in RNAi-proficient L. braziliensis [48], and we found that transfections with a variety of circular DNAs were generally unsuccessful, causing us to rely exclusively on integrative constructs in this work. This does not imply that episomal circular DNAs will never arise in RNAi-proficient species; but when found, their transcription will be subject to RNAi effects and/or they will contain cis-acting elements that confer a high degree of strand specificity [49]. These requirements might act to constrain the emergence of episomal elements in RNAi-proficient species.
Thus the loss of RNAi could be seen as ‘freeing’ the genome of RNAi-deficient Leishmania from several constraints limiting genome plasticity. In this regards, loss of RNAi may be viewed as ‘mutator’ phenotype, similar to the ‘ARMed’ phenotype described recently in the malaria parasite Plasmodium falciparum or the high mutability phenotypes associated with elevated bacterial virulence in humans [50], [51].
Phenotypic selection
Lastly, loss of RNAi may have been selected directly through effects on Leishmania virulence during evolution. The RNAi machinery affects gene expression at multiple levels, and its loss could lead to profound changes in parasite biology that could alter parasite virulence. Such direct alterations in gene expression may act in concert with the genomic alterations described above. The Leishmania subgenus Viannia is an early diverging clade within the genus [52], and these species exhibit a number of distinct features including the nature of the immune response in the mammalian host, the composition of their surface glycocalyx, and their behavior within the sand fly vector [8], [53]. Any such systematic differences between the RNAi-proficient Viannia subgenus and the RNAi-null Leishmania species groups could potentially reflect changes associated gene expression mediated by the RNAi pathway.
Could RNAi be engineered into RNAi-deficient Leishmania?
Our findings provoke the question of whether the RNAi machinery could be transplanted from L. braziliensis into its close RNAi-deficient relatives. This would be useful given the extensive previous work on species such as L. major and L. donovani, as well as providing a tool for understanding the RNAi machinery. This feat was recently accomplished in Saccharomyces cerevisiae, which required only the introduction of Argonaute and Dicer from the closely related species S. castellii [33]. However, reintroduction of RNAi in L. major or L. donovani may require restoration of a more extensive suite of genes. While only three RNAi genes have been confirmed in trypanosomatids (an Argonaute and two Dicers) [9], [10], [17], preliminary data suggest a requirement for at least two additional genes (E. Ullu and C. Tschudi; unpublished data). Importantly, all 5 genes are absent in the genomes available for RNAi-deficient Leishmania species. In other eukaryotes the RNAi machinery includes as many as 9 proteins or more [15], [31], [54]. Another obstacle may be the tendency of RNAi-deficient species such as L. major to transcribe the antisense chromosomal strand at low levels [55], as well as to synthesize antisense transcripts [56], [57]. This suggests the possibility that introduction of an active RNAi pathway into L. major could be lethal [11], [58]. Thus re-introduction of RNAi into RNAi-deficient Leishmania species will be a challenging task; nonetheless, efforts to introduce this suite of genes from RNAi proficient L. braziliensis are underway.
In summary, we have shown that the RNAi pathway is functional in Leishmania braziliensis. These data provide some optimism for the application of RNAi approaches as a tool for the study of these predominantly asexual organisms, by forward and reverse genetic approaches. While less experimentally developed, L. braziliensis has the potential to emerge as an attractive model, and the advent of RNAi-based tools should provide a further stimulus for this effort. In the long term, delivery of siRNAs targeting essential parasite genes may prove an effective route to chemotherapeutic treatment of RNAi-proficient Leishmania. Lastly, the Leishmania provide an attractive system for testing hypotheses about forces leading to the evolutionary loss of RNAi, including the role of viral pressure, changes in genome plasticity, and virulence. As drug resistance mediated by gene amplification is one manifestation of gene plasticity, these findings have practical implications to parasite chemotherapy.
Materials and Methods
Northern blotting
RNA extraction procedures and Northern analyses were carried out as described [16]. The 5′UTR of L. braziliensis α-tubulin mRNA plus the first 317 nt of the ORF were PCR-amplified from genomic DNA and inserted between the HindIII and XbaI sites of plasmid vector pPD19.36, which contains two opposing T7 RNA Polymerase promoters [59]. The synthesis of dsRNA was according to Ngo et al. [21]. The same DNA was used as a probe in the α-tubulin Northern. PCR products of GFP+ or GFP65 ORFs were used as probes for the GFP Northerns. A portion (nt 3160 to nt 4482) of the L. braziliensis SLACS (LbrM08−V2.0700) was PCR-amplified with primers (LBSLACS1399F: 5′-GCCAGAGAGGTGGTGAGGGTG and LBSLACSORFa-R: 5′-GAGCTCGAGAAAGGTCCACCACCCCGA) from M2903 genomic DNA and TA cloned to generate a sense radiolabeled RNA probe for Northern analysis of small RNAs. For LPG2 (LbrM20_V2.2700) the probe was a PCR fragment (nt 1 to nt 411) amplified with primers SMB3219 and SMB3220 (Table S1).
RNA preparation and quantitative real-time PCR (qRT-PCR)
Leishmania total RNA was isolated using the Trizol reagent (Invitrogen), treated with DNAse and purified using MEGAclear columns (Ambion). Reverse transcription (RT) was performed according to the manufacture instructions using Superscript III First-Strand reverse transcriptase (Invitrogen) in a 20 µl reaction containing 1µg purified RNA. Controls containing the same amount of RNA but lacking reverse transcriptase or template were used to rule out DNA or other contamination. For test RNAs, primers were designed to amplify ∼100 bp amplicons within the target ORF but outside of the stem-fragment, and tested using L. braziliensis gDNA. PCRs were performed using the SYBR Green (Applied Biosystems) and the ABI PRISM 7000 Sequence Detection System instrument (Applied Biosystems). PCR amplifications were performed as follows: 50°C for 2 min and 95°C for 10 sec then followed by 40 cycles of 95°C for 15 sec, 60°C for 1min. The generation of specific PCR products was confirmed by melting curve analysis and agarose gel electrophoresis. Each primer set was individually tested for four StL transfectants (2 for StL-F and 2 for StL-R; except 4 for LPG3-StL-F). All samples were performed in triplicate. Control samples of H2O were included in each experiment. Amplification of SSU rRNA was used as internal control to normalize the parallel reaction of target amplicons.
Leishmania strains
L. braziliensis M2903 (MHOM/BR/75/M2903), L. guyanensis M4147 (MHOM/BR/75/M4147) and L. panamensis WR120 (MHOM/PA/74/WR120) were obtained from Diane McMahon-Pratt (Yale University), L. braziliensis strain M2904 from Angela Cruz (U. Sao Paulo Riberao Preto), L. tarentolae strain TarII was obtained from M. Ouellette and B. Papadopoulou (U. Laval), L. mexicana (MNYZ/BZ/62/M379) from David Russell (Cornell University), and Crithidia fasciculata Cf-C1 from Larry Simpson (UCLA). The LRV-bearing strain of L. guyanensis M4147 (MHOM/BR/75/M4147) and a virus free derivative M4147/pX63-HYG [38] were obtained from Jean L. Patterson (Southwest Foundation for Biomedical Research, San Antonio, Texas). The identities of all Viannia strains used were confirmed by partial and/or complete sequencing of the AGO1 or other genes (not shown).
Viannia species were grown in freshly prepared Schneider's Insect Medium (Sigma-Aldrich Cat. No. S9895) supplemented with 10% heat-inactivated fetal bovine serum, 2 mM L-glutamine, 500 units penicillin/ml and 50 µg/ ml− streptomycin (Gibco Cat No. 5070). Other Leishmania and Crithidia were propagated in M199 medium supplemented with 10% heat-inactivated fetal bovine serum, hemin, adenine, biopterin and biotin [60].
Transient and stable transfection
For each transfection, 10 ml of log phase L. braziliensis were resuspended in 100 µl human T-cell Nucleofector solution (Amaxa Cat No. VPA-1002) mixed with 5 µl of 4 µg/ µl of α-tubulin dsRNA or control dsRNA and subjected to nucleofection with an Amaxa Nucleofector with program U-033 using the kit's cuvette. The transfection mixture was transferred immediately to 10 ml of complete medium and kept in 28°C for 3 hrs. RNA from 9 ml cells was taken for Northern blot analysis with an α-tubulin hybridization probe.
Stable transfections were performed using the high voltage (1400V) procedure described previously [11]. Following electroporation organisms were grown in drug-free media overnight, and then plated on semisolid media [60] to obtain clonal lines. For selections using the SAT marker, parasites were plated on 50–100 µg/ml nourseothricin (clonNAT, Werner BioAgents, Germany), and with the PHLEO marker, parasites were plated on 0.2–2 µg/ml phleomycin (Sigma). After colonies emerged (typically <2 weeks) they were recovered and grown to stationary phase in 1 ml media, and passaged thereafter in 10 and 0.1 µg /ml nourseothricin and phleomycin, respectively. The plating efficiency of untransfected L. braziliensis M2903 ranged from 60–95% and the transfection efficiency from 50–220 colonies / 20 µg DNA.
AGO1 sequencing
The generation of whole genome shotgun sequence data from Crithidia fasciculata strain Cf-C1 by 454 sequencing technology will be described fully elsewhere. Blast searches using L. braziliensis AGO1 were used to identify homologous sequences, which were then assembled manually into several large contigs. PCR primers were designed to amplify missing gaps, and the 5′ end of the mRNA was obtained by RT-PCR using a forward miniexon primer (CFSLB 5′-AAGTATCAGTTTCTGTACTTTATTG) and reverse CfAGO1 specific primer (SMB2895 : 5′-AAGCAGTTCGTCTCCACCGTCACCTG). Then a nested PCR was done with CfSLB and CfAGO1 primers (SMB 2894 : 5′ - GTGATGCCGCCCTCCTCGCGGTCACG). The PCR products were TA cloned and sequenced. The CfAGO1 sequence was deposited in GenBank (EU714010). We noted a polymorphism in the CfAGO1 sequence, introducing a stop codon yielding a truncated protein terminating after amino acid 198. The consequences of this polymorphism (if any) have not been investigated further. The sequence of the L. guyanensis M4147 AGO1 ORF was determined by direct sequencing of the PCR amplicon obtained with primers B2468 (5′-ATGTTGGCGCTAAACGCAGGTTC) and B2469 (5′ - CTACAGGTAGTGCATCGTGGGGC), and deposited in GenBank (accession number FJ234150).
Detection of LRV virus
RT-PCR reactions were performed as described above, with two sets of primers to detect LRV viruses described previously [38] (set 1, primers SMB2472/2473 and set 2, primers SMB3850/3851 (Table S1).
Constructs
The constructs used in this work are derivatives of pIR1SAT (B3541) [11] or pIR1PHLEO (B4054, this work), which have two expression sites (XbaI/SmaI, site a, and BglII, site b). High fidelity thermostable polymerases such as recombinant Pfu DNA polymerase (Stratagene) were used for PCR, and constructs were confirmed by restriction mapping and sequencing of all relevant regions. Unless otherwise indicated, all constructs were digested with SwaI and the linear SSU-targeting fragment purified for subsequent transfection by electroporation.
Reporters
pIR1PHLEO (B4054) was created by replacing the SAT marker of pIR1SAT with the PHLEO marker (M. Cunningham, unpublished data). pIR1PHLEO-GFP+(a) (B5793), pIR1PHLEO-GFP65(a) (B5779) and pIR1-GFP65*(a) (B5959) were constructed by generating ORF cassettes of the respective genes and inserting into the XbaI (a) site. The GFP+ ORF was taken from pXG-GFP+ (B2799), GFP65 from pXG-GFP65 (B2355), and GFP65* was obtained by site-specific mutagenesis of pIR1PHLEO-GFP65 (QuickChange Multi Site-Directed Mutagenesis, Stratagene), changing nt 193 from T to A, resulting in a S65T mutation. A luciferase (LUC) ORF was amplified using pGL3-basic (Promega) as template, with primers adding flanking BglII sites, and a CCACC initiation sequence preceding the initiation codon. The modified LUC ORF was inserted into pGEM-T (Promega) yielding pGEM-Luciferase (B6033); the LUC ORF was then extracted by BglII digestion and inserted into the BglII site of B5959 to create pIR1PHLEO-GFP65*(a)-LUC(b) (B6034).
Stem-Loop (StL) constructs
pIR1SAT-GFP65-StL(b) (B4733) was described previously [11]. For other StL constructs, we assembled a stem-loop consisting of the target gene sequences in inverted orientation, separated by a PEX11-MYC(3) loop/stuffer fragment used previously in pIRGFP Stem-Loop (B4733), and inserted this into either the ‘a’ or ‘b’ expression sites of pIR1SAT. In these constructs the ‘stem’ sequences were organized either in divergent or convergent orientations (DIV or CONV) relative to the target gene sequence, and the stuffer fragment similarly could be in a ‘sense’ or ‘antisense’ orientation relative to PEX11 (F or R). The specific target genes and regions studied included LPG1 (LbrM25_V2.0010, nt 11–592); LPG2 (LbrM20_V2.2700, nt 411–1021); LPG3 (LbrM29_V2.0780, nt 1657–2236); HGPRT (LbrM21_V2.0990, nt 127–626); α-tubulin (LbrM13_V2.0190, nt 736–1309); β-tubulin (LbrM33_V2.0930, nt 470–1004); PFR1 (LbrM31_V2.0160, nt 900–1593); PFR2 (LbrM16_V2.1480, nt 951–1644), AGO1 (LbrM11_V2.0360, nt 247–1070) and LUC (LUC+ from Promega pGL3-Basic, nt 281–788). These steps yielded constructs pIR1SAT-LPG1-StL (b,DIV,R)(B6128), pIR1SAT-LPG1-StL (b,DIV,F) (6132), pIR1SAT-LPG2-StL(b,DIV,R) (B6137), pIR1SAT-LPG2-StL(b,DIV,F) (B6138), pIR1SAT-LPG3-StL(b,DIV,F) (B6140), pIR1SAT-HGPRT-StL(b,DIV,F) (B6136), pIR1SAT-HGPRT-StL(b,DIV,R) (B6135), pIR1SAT-PFR1-StL(b,DIV,F) (B6294), pIR1SAT-PFR2-StL(b,DIV,F) (B6282), pIR1SAT-αTub-StL(b,DIV,F) (B6283), pIR1SAT-βTub-StL(b,DIV,F) (B6295) and pIR1SAT-LUC-StL(b,CONV,F) (B6185), or pIR1SAT-LUC-StL(b,DIV,F) (B6190).
LUC self reporter (LUC SR) and RNAi of AGO1
A single construct enabling tests of RNAi activity was generated by inserting the LUC ORF into the ‘b’ site and a LUC Stem-Loop into the ‘a’ site of a modified pIR vector (pIR2SAT-LUC-StL(a)-LUC(b) (B6386). This construct is referred to as the ‘LUC RNAi self reporter’ or ‘LUC SR’. For RNAi studies of AGO1, an analogous construct was made with a HYG marker (pIR2HYG-LUC-StL(a)-LUC(b), strain B6447). A pIR1SAT-LbrAGO1-StL(b) construct was used for RNAi tests (B6524).
LPG, Western blots and GFP flow cytometry
Western blots were performed as described elsewhere using anti-GFP (Abcam Cat No. 6556, 1∶2500) or anti-L. donovani HGPRT antiserum (1∶5000; J. Boitz and B. Ullman, Oregon Health Sciences University) as the primary antibody, and detected using goat anti-rabbit IgG as the secondary antibody (1∶10000, Jackson ImmunoResearch Laboratories, Inc. catalog number 111-035-003). Parasites expressing GFPs were analyzed using a Becton-Dickenson FACS Calibur, using fluoroscein excitation/emission parameters. LPG was purified and quantitated from L. braziliensis lines grown in logarithmic phase (4–5×106 cells/ml) as described [61]. Purified LPG was subjected to western blotting with antisera CA7AE which recognizes the Gal(β1,4)Man(α1-P) repeat units of the L. braziliensis LPG [62].
Luciferase assay
106 logarithmic phase promastigotes were suspended in 200 µl media containing 30 µg/ ml of luciferin (Biosynth AG) and added to a 96-well plate (Black plate, Corning Incorporated, NY, U.S.A.). After 10 min incubation, the plate was imaged using a Xenogen IVIS photoimager (Caliper LifeSciences), and luciferase activity quantitated as photons/sec (p/s).
Transmission electron microscopy
Promastigotes were fixed in 2% paraformaldehyde/2.5% glutaraldehyde (Polysciences Inc., Warrington, PA) in 100 mM phosphate buffer, pH 7.2 for 1 hr at room temperature. Samples were washed in phosphate buffer and postfixed in 1% osmium tetroxide (Polysciences Inc., Warrington, PA) for 1 hr. Samples were then rinsed extensively in water prior to en bloc staining with 1% aqueous uranyl acetate (Ted Pella Inc., Redding, CA) for 1 hr. Following several rinses in water, samples were dehydrated in a graded series of ethanol solutions and embedded in Eponate 12 resin (Ted Pella Inc.). Sections of 95 nm were cut with a Leica Ultracut UCT ultramicrotome (Leica Microsystems Inc., Bannockburn, IL), stained with uranyl acetate and lead citrate, and viewed on a JEOL 1200 EX transmission electron microscope (JEOL USA Inc., Peabody, MA).
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. CeruttiH
Casas-MollanoJA
2006 On the origin and functions of RNA-mediated silencing: from protists to man. Curr Genet 50 81 99
2. UlluE
TschudiC
ChakrabortyT
2004 RNA interference in protozoan parasites. Cell Microbiol 6 509 519
3. MilitelloKT
RefourP
ComeauxCA
DuraisinghMT
2008 Antisense RNA and RNAi in protozoan parasites: working hard or hardly working? Mol Biochem Parasitol 157 117 126
4. BaumJ
PapenfussAT
MairGR
JanseCJ
VlachouD
2009 Molecular genetics and comparative genomics reveal RNAi is not functional in malaria parasites. Nucleic Acids Res 37 3788 3798
5. BraunL
CannellaD
OrtetP
BarakatM
SautelCF
2010 A complex small RNA repertoire is generated by a plant/fungal-like machinery and effected by a metazoan-like Argonaute in the single-cell human parasite Toxoplasma gondii. PLoS Pathog 6 e1000920
6. LinfordAS
MorenoH
GoodKR
ZhangH
SinghU
2009 Short hairpin RNA-mediated knockdown of protein expression in Entamoeba histolytica. BMC Microbiol 9 38
7. PruccaCG
SlavinI
QuirogaR
EliasEV
RiveroFD
2008 Antigenic variation in Giardia lamblia is regulated by RNA interference. Nature 456 750 754
8. BanulsAL
HideM
PrugnolleF
2007 Leishmania and the leishmaniases: a parasite genetic update and advances in taxonomy, epidemiology and pathogenicity in humans. Adv Parasitol 64 1 109
9. ShiH
TschudiC
UlluE
2006 An unusual Dicer-like1 protein fuels the RNA interference pathway in Trypanosoma brucei. RNA 12 2063 2072
10. PatrickKL
ShiH
KolevNG
ErsfeldK
TschudiC
2009 Distinct and overlapping roles for two Dicer-like proteins in the RNA interference pathways of the ancient eukaryote Trypanosoma brucei. Proc Natl Acad Sci U S A 106 17933 17938
11. RobinsonKA
BeverleySM
2003 Improvements in transfection efficiency and tests of RNA interference (RNAi) approaches in the protozoan parasite Leishmania. Mol Biochem Parasitol 128 217 228
12. DaRochaWD
OtsuK
TeixeiraSM
DonelsonJE
2004 Tests of cytoplasmic RNA interference (RNAi) and construction of a tetracycline-inducible T7 promoter system in Trypanosoma cruzi. Mol Biochem Parasitol 133 175 186
13. BeverleySM
2003 Protozomics: trypanosomatid parasite genetics comes of age. Nat Rev Genet 4 11 19
14. IvensAC
PeacockCS
WortheyEA
MurphyL
AggarwalG
2005 The genome of the kinetoplastid parasite, Leishmania major. Science 309 436 442
15. PeacockCS
SeegerK
HarrisD
MurphyL
RuizJC
2007 Comparative genomic analysis of three Leishmania species that cause diverse human disease. Nat Genet 39 839 847
16. DjikengA
ShiH
TschudiC
UlluE
2001 RNA interference in Trypanosoma brucei: cloning of small interfering RNAs provides evidence for retroposon-derived 24–26-nucleotide RNAs. RNA 7 1522 1530
17. ShiH
DjikengA
TschudiC
UlluE
2004 Argonaute protein in the early divergent eukaryote Trypanosoma brucei: control of small interfering RNA accumulation and retroposon transcript abundance. Mol Cell Biol 24 420 427
18. LeBowitzJH
CoburnCM
McMahon-PrattD
BeverleySM
1990 Development of a stable Leishmania expression vector and application to the study of parasite surface antigen genes. Proc Natl Acad Sci USA 87 9736 9740
19. CapulAA
BarronT
DobsonDE
TurcoSJ
BeverleySM
2007 Two functionally divergent UDP-Gal nucleotide sugar transporters participate in phosphoglycan synthesis in Leishmania major. J Biol Chem 282 14006 14017
20. ClaytonCE
2002 Life without transcriptional control? From fly to man and back again. EMBO J 21 1881 1888
21. NgoH
TschudiC
GullK
UlluE
1998 Double-stranded RNA induces mRNA degradation in Trypanosoma brucei. Proc Natl Acad Sci USA 95 14687 14692
22. TurcoSJ
SpäthGF
BeverleySM
2001 Is lipophosphoglycan a virulence factor? A surprising diversity between Leishmania species. Trends Parasitol 17 223 226
23. MagaJA
SherwinT
FrancisS
GullK
LeBowitzJH
1999 Genetic dissection of the Leishmania paraflagellar rod, a unique flagellar cytoskeleton structure. J Cell Sci 112 Pt 16 2753 2763
24. ChangT
MilneKG
GutherML
SmithTK
FergusonMA
2002 Cloning of Trypanosoma brucei and Leishmania major genes encoding the GlcNAc-phosphatidylinositol de-N-acetylase of glycosylphosphatidylinositol biosynthesis that is essential to the African sleeping sickness parasite. J Biol Chem 277 50176 50182
25. de SouzaW
Souto-PadronT
1980 The paraxial structure of the flagellum of trypanosomatidae. J Parasitol 66 229 236
26. BastinP
SherwinT
GullK
1998 Paraflagellar rod is vital for trypanosome motility. Nature 391 548
27. SchlaeppiK
DeflorinJ
SeebeckT
1989 The major component of the paraflagellar rod of Trypanosoma brucei is a helical protein that is encoded by two identical, tandemly linked genes. J Cell Biol 109 1695 1709
28. BastinP
MatthewsKR
GullK
1996 The paraflagellar rod of kinetoplastida: solved and unsolved questions. Parasitol Today 12 302 307
29. BernsteinE
CaudyAA
HammondSM
HannonGJ
2001 Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature 409 363 366
30. DudleyNR
LabbeJC
GoldsteinB
2002 Using RNA interference to identify genes required for RNA interference. Proc Natl Acad Sci USA 99 4191 4196
31. KimJK
GabelHW
KamathRS
TewariM
PasquinelliA
2005 Functional genomic analysis of RNA interference in C. elegans. Science 308 1164 1167
32. BerrimanM
GhedinE
Hertz-FowlerC
BlandinG
RenauldH
2005 The genome of the African trypanosome Trypanosoma brucei. Science 309 416 422
33. DrinnenbergIA
WeinbergDE
XieKT
MowerJP
WolfeKH
2009 RNAi in budding yeast. Science 326 544 550
34. PattersonJL
1993 The current status of Leishmania RNA virus I. Parasitol Today 9 135 136
35. TarrPI
AlineRFJr
SmileyBL
SchollerJ
KeithlyJ
1988 LR1: a candidate RNA virus of Leishmania. Proc Natl Acad Sci USA 85 9572 9575
36. WidmerG
ComeauAM
FurlongDB
WirthDF
PattersonJL
1989 Characterization of a RNA virus from the parasite Leishmania. Proc Natl Acad Sci USA 86 5979 5982
37. GuilbrideL
MylerPJ
StuartK
1992 Distribution and sequence divergence of LRV1 viruses among different Leishmania species. Mol Biochem Parasitol 54 101 104
38. RoYT
ScheffterSM
PattersonJL
1997 Hygromycin B resistance mediates elimination of Leishmania virus from persistently infected parasites. J Virol 71 8991 8998
39. DingSW
VoinnetO
2007 Antiviral immunity directed by small RNAs. Cell 130 413 426
40. OggMM
CarrionRJr
BotelhoAC
MayrinkW
Correa-OliveiraR
2003 Short report: quantification of leishmaniavirus RNA in clinical samples and its possible role in pathogenesis. Am J Trop Med Hyg 69 309 313
41. GuptaV
DeepA
2007 An insight into the Leishmania RNA virus. Indian J Med Microbiol 25 7 9
42. ShiH
ChamondN
TschudiC
UlluE
2004 Selection and characterization of RNA interference-deficient trypanosomes impaired in target mRNA degradation. Eukaryot Cell 3 1445 1453
43. PatrickKL
LuzPM
RuanJP
ShiH
UlluE
2008 Genomic rearrangements and transcriptional analysis of the spliced leader-associated retrotransposon in RNA interference-deficient Trypanosoma brucei. Mol Microbiol 67 435 447
44. GirardA
HannonGJ
2008 Conserved themes in small-RNA-mediated transposon control. Trends Cell Biol 18 136 148
45. BeverleySM
1991 Gene amplification in Leishmania. Annu Rev Microbiol 45 417 444
46. BorstP
OuelletteM
1995 New mechanisms of drug resistance in parasitic protozoa. Annu Rev Microbiol 49 427 460
47. de LafailleMAC
LabanA
WirthDF
1992 Gene expression in Leishmania: analysis of essential 5′ DNA sequences. Proc Natl Acad Sci USA 89 2703 2707
48. DiasFC
RuizJC
LopesWC
SquinaFM
RenziA
2007 Organization of H locus conserved repeats in Leishmania (Viannia) braziliensis correlates with lack of gene amplification and drug resistance. Parasitol Res 101 667 676
49. PatnaikPK
KulkarniSK
CrossGA
1993 Autonomously replicating single-copy episomes in Trypanosoma brucei show unusual stability. EMBO J 12 2529 2538
50. RathodPK
McErleanT
LeePC
1997 Variations in frequencies of drug resistance in Plasmodium falciparum. Proc Natl Acad Sci USA 94 9389 9393
51. DenamurE
MaticI
2006 Evolution of mutation rates in bacteria. Mol Microbiol 60 820 827
52. StevensJR
NoyesHA
SchofieldCJ
GibsonW
2001 The molecular evolution of Trypanosomatidae. Adv Parasitol 48 1 56
53. BatesPA
2007 Transmission of Leishmania metacyclic promastigotes by phlebotomine sand flies. Int J Parasitol 37 1097 1106
54. DornerS
LumL
KimM
ParoR
BeachyPA
2006 A genomewide screen for components of the RNAi pathway in Drosophila cultured cells. Proc Natl Acad Sci USA 103 11880 11885
55. Martinez-CalvilloS
YanS
NguyenD
FoxM
StuartK
2003 Transcription of Leishmania major Friedlin chromosome 1 initiates in both directions within a single region. Mol Cell 11 1291 1299
56. BelliSI
MonneratS
SchaffC
MasinaS
NollT
2003 Sense and antisense transcripts in the histone H1 (HIS-1) locus of Leishmania major. Int J Parasitol 33 965 975
57. KaplerGM
BeverleySM
1989 Transcriptional mapping of the amplified region encoding the dihydrofolate reductase-thymidylate synthase of Leishmania major reveals a high density of transcripts, including overlapping and antisense RNAs. Mol Cell Biol 9 3959 3972
58. BeverleySM
2003 Genetic and genomic approaches to the analysis of Leishmania virulence.
MarrJM
NilsenT
KomunieckiR
Molecular & Medical Parasitology New York Academic Press
59. TimmonsL
FireA
1998 Specific interference by ingested dsRNA. Nature 395 854
60. KaplerGM
CoburnCM
BeverleySM
1990 Stable transfection of the human parasite Leishmania major delineates a 30-kilobase region sufficient for extrachromosomal replication and expression. Mol Cell Biol 10 1084 1094
61. OrlandiPAJr
TurcoSJ
1987 Structure of the lipid moiety of the Leishmania donovani lipophosphoglycan. J Biol Chem 262 10384 10391
62. SoaresRP
CardosoTL
BarronT
AraujoMS
PimentaPF
2005 Leishmania braziliensis: a novel mechanism in the lipophosphoglycan regulation during metacyclogenesis. Int J Parasitol 35 245 253
63. DjikengA
ShiH
TschudiC
ShenS
UlluE
2003 An siRNA ribonucleoprotein is found associated with polyribosomes in Trypanosoma brucei. RNA 9 802 808
64. TamuraK
DudleyJ
NeiM
KumarS
2007 MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Molecular Biology and Evolution 24 1596 1599
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2010 Číslo 10
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Měli bychom postcovidový syndrom léčit antidepresivy?
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- Retroviral RNA Dimerization and Packaging: The What, How, When, Where, and Why
- Viral Replication Rate Regulates Clinical Outcome and CD8 T Cell Responses during Highly Pathogenic H5N1 Influenza Virus Infection in Mice
- Antimicrobial Peptides: Primeval Molecules or Future Drugs?
- Crystal Structure of DotD: Insights into the Relationship between Type IVB and Type II/III Secretion Systems