
Innate Killing of by Macrophages of the Splenic Marginal Zone Requires IRF-7
Highly phagocytic macrophages line the marginal zone (MZ) of the spleen and the lymph node subcapsular sinus. Although these macrophages have been attributed with a variety of functions, including the uptake and clearance of blood and lymph-borne pathogens, little is known about the effector mechanisms they employ after pathogen uptake. Here, we have combined gene expression profiling and RNAi using a stromal macrophage cell line with in situ analysis of the leishmanicidal activity of marginal zone macrophages (MZM) and marginal metallophilic macrophages (MMM) in wild type and gene targeted mice. Our data demonstrate a critical role for interferon regulatory factor-7 (IRF-7) in regulating the killing of intracellular Leishmania donovani by these specialised splenic macrophage sub-populations. This study, therefore, identifies a new role for IRF-7 as a regulator of innate microbicidal activity against this, and perhaps other, non-viral intracellular pathogens. This study also highlights the importance of selecting appropriate macrophage populations when studying pathogen interactions with this functionally diverse lineage of cells.
Published in the journal:
. PLoS Pathog 6(3): e32767. doi:10.1371/journal.ppat.1000813
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1000813
Summary
Highly phagocytic macrophages line the marginal zone (MZ) of the spleen and the lymph node subcapsular sinus. Although these macrophages have been attributed with a variety of functions, including the uptake and clearance of blood and lymph-borne pathogens, little is known about the effector mechanisms they employ after pathogen uptake. Here, we have combined gene expression profiling and RNAi using a stromal macrophage cell line with in situ analysis of the leishmanicidal activity of marginal zone macrophages (MZM) and marginal metallophilic macrophages (MMM) in wild type and gene targeted mice. Our data demonstrate a critical role for interferon regulatory factor-7 (IRF-7) in regulating the killing of intracellular Leishmania donovani by these specialised splenic macrophage sub-populations. This study, therefore, identifies a new role for IRF-7 as a regulator of innate microbicidal activity against this, and perhaps other, non-viral intracellular pathogens. This study also highlights the importance of selecting appropriate macrophage populations when studying pathogen interactions with this functionally diverse lineage of cells.
Introduction
Mononuclear phagocytes are widely distributed in all tissues, and provide a broad range of homeostatic and immune functions during development and throughout adult life. Nevertheless, the heterogeneity of mature tissue macrophages represents one of the most striking, yet under-studied, features of mononuclear cell differentiation. Expression of a range of transcription factors and cellular receptors has helped define membership of the mononuclear phagocyte system [1],[2],[3], and whereas some tissue macrophages have a capacity for local self-renewal, others are derived from blood-borne monocytes undergoing tissue-specific differentiation [reviewed in [4]]. In lymphoid tissues, macrophage heterogeneity is most strikingly evident. For example in mice, readily distinguishable populations of macrophages are found in the splenic MZ, red pulp, B cell follicles and white pulp [5]. Moreover, within the MZ, SIGNR-1+MARCO+ MZM occupy the outer rim adjacent to the red pulp, and CD169hi MMM border the lymphocyte-rich white pulp [5],[6],[7],[8],[9],[10]. Mice lacking various transcription factors (e.g. relβ and NFκB2), TNF superfamily cytokines (e.g. LTα), and chemokines (e.g. CCL19/21ser) exhibit steady-state defects in MZ macrophage differentiation and/or positioning [11],[12], illustrating the complexity behind this micro-anatomical organisation.
MZM and MMM are well-placed in the marginal sinus to encounter blood borne antigens and pathogens. MZM are avidly phagocytic, are well-characterised as facilitating clearance of Streptococcus pneumoniae from the blood stream [13],[14],[15] and are involved in the initiation of Type I T-independent immune responses [16]. MMM, whilst also phagocytic [17] are better recognised for playing a role in antigen transport into B cell follicles [4] and are also known to be robust producers of IFN-α following infection with viral pathogens [18]. MMM, and to a lesser extent MZM, also express CD169, a sialoadhesin first described on bone marrow ‘stromal’ macrophages involved in the support of erythropoiesis [19] and these cells may provide this or other stromal functions in the spleen. Similarly, in lymph nodes, subcapsular sinus macrophages mark a point of entry for viruses and bacteria entering via the afferent lymphatics and are intimately involved in the initiation of antibody responses [20],[21],[22]. Thus, various specialised macrophages populations are ideally situated within secondary lymphoid tissues to first encounter pathogens that enter the blood and lymphatics.
Nevertheless, despite their importance in providing a ‘gatekeeper’ function, little is known about how these tissue resident macrophages subsequently deal with the pathogens they encounter. Direct study of MZM and MMM has been problematic and only rarely reported [6], largely because of the inherent technical difficulties in isolating these scarce and fragile cells for functional analysis in vitro. Hence, our understanding of the effector functions of MZM and MMM is largely extrapolated from that of other diverse macrophage populations, such as those derived from the peritoneal cavity or grown from bone marrow precursors under the influence of CSF-1. In this study, therefore, we set out to directly identify the mechanism by which splenic MZM and MMM are able to kill intracellular Leishmania parasites. We first identified a stromal macrophage cell line (14M1.4; [23]) that displayed similar innate capacity to kill L. donovani as observed in MZM and MMM. By gene expression profiling, we showed that Leishmania infection stimulated expression of the transcription factor Irf-7, as well as redistribution of cellular IRF-7 from MyD88+ endosomes to the Leishmania-containing phagosome. Initial control of intracellular Leishmania by 14M1.4 cells involved NO and was minimally affected by silencing of Irf-7. However, subsequent leishmanicidal activity strictly required Irf-7 and was NO-independent. Furthermore, Irf-7 loss-of-function could not be overcome by exogenous IFN-α, suggesting a downstream effector mechanism uncoupled from Irf-7-dependent amplification of IFN-α. By immunohistochemistry of spleens from infected mice, we showed that MZM and MMM also responded to infection with heightened expression and phagosomal recruitment of IRF-7. Finally, we showed that MZM and, to a lesser extent, MMM from Irf-7-deficient mice were unable to kill intracellular Leishmania. By focusing on these tissue-resident stromal macrophages, we have thus identified a new role for IRF-7 as a regulator of macrophage anti-leishmanial activity, and demonstrated that this transcription factor plays a role in innate effector responses to a broader range of intra-phagosomal pathogens than previously recognised.
Results
IFN-γ-independent killing of Leishmania by stromal macrophages
Previous studies in BALB/c mice showed that after i.v. injection, L. donovani amastigotes are rapidly and selectively taken up by MZM and MMM, and that over the following 24h, parasite load in the MZ was significantly reduced [17]. We first confirmed this selectivity of uptake in the MZ of C57BL/6 mice, observing greater than 90% of amastigotes in the MZ (within cells staining either for CD169 or SIGNR1) and few parasites in either the deep white pulp or in the red pulp (Figure 1A). To confirm that C57BL/6 mice also expressed rapid leishmanicidal activity and to determine whether this might be mediated by NK cell-derived IFN-γ [24], we treated mice with either control IgG or anti-IFN-γ. Both control and anti-IFN-γ-treated mice reduced parasite load to a similar extent, indicating that this response was largely IFN-γ independent (Figure 1B). We then screened a number of macrophage cell lines (RAW264.7, J774.1, 14M1.4) and primary macrophage populations (bone marrow-derived, peritoneal) for their ability to kill L. donovani amastigotes in the absence of IFN-γ activation. Of the cells tested, 14M1.4 cells were striking in their ability to clear amastigotes over a 24–48h period. Infected 14M1.4 cells reduced parasite load by ∼50% from 6h–12h after infection and sustained this activity over the subsequent 36h, resulting in almost complete clearance of amastigotes by 48h (Figure 1C). In contrast, although RAW264.7 reduced the number of intracellular amastigotes by ∼30–40% from 12h–24h, no further reduction in parasite load was detected after this time. Differences in amastigote numbers were not readily attributable to differential rates of cell division between 14M1.4 cells and other cells tested, with 14M1.4 and RAW264.7 cells showing similar levels of CFSE dilution over 48h (Supplementary Figure S1A). Likewise, equivalent levels of CFSE dilution were observed in intracellular amastigotes isolated from both cell populations, indicating that parasite growth rate was similar in these two host cells (Supplementary Figure S1B). Thus, these data collectively demonstrated that 14M1.4 cells were intrinsically very efficient at killing intracellular amastigotes.

Gene expression profiling of stromal macrophages
To understand further the mechanism(s) by which 14M1.4 cells killed intracellular amastigotes, we used global gene expression profiling to examine the transcriptional response of 14M1.4 cells to L. donovani infection. 977 and 851 genes were found to be differentially expressed in infected vs. control 14M1.4 cells at 12h or 48h p.i. respectively, using a cut-off of a 2-fold change in gene expression (Supplementary Tables S1 and S2). These differentially expressed genes were analysed by generating gene interaction networks using Ingenuity Pathways Analysis software. 10 networks of high interconnectivity (≥20 differentially regulated genes) were constructed for genes differentially regulated at 12h p.i. and 12 hubs at 48h p.i (Supplementary Figure S2 and S3). In contrast, only 136 and 161 genes were differentially regulated in RAW264.7 cells, at 12h and 48h p.i. respectively (Supplementary Tables S3 and S4), and only one hub with high interconnectivity could be identified at each time point (Supplementary Figure S2 and S3). In 14M1.4 cells, the highest scoring networks reflected genes involved in interferon signalling (notably Irf-7, Irf-1, Stat-2), cytokines (notably Il-6) and cardinal interferon response genes (including mx-1, Rsad and various MHC and MHC related genes: Figure 2A and B). At 48h, 31% of genes (9/29) listed in the Ingenuity Pathways ‘Interferon Signalling’ pathway were represented in the differentially induced gene set, all being upregulated. Within the ‘Antigen Presentation Pathway’, 25% (10/39) were either up - or down-regulated, with a marked differential response between MHCI-and MHCII-associated genes (Figure 2B). A range of genes within these networks and associated with pathogen recognition and handling were also down-regulated including Irf-8, recently associated with the innate control of intra-phagosomal pathogens [25], Cx3cr1, a chemokine receptor involved in the NO-dependent bactericidal activity of macrophages [26], Clec7a (Dectin-1) and Clec4a (Mincle), both associated with fungal recognition [27],[28] and Msr1, the macrophage scavenger receptor and its cytoplasmic partner Hook3 [29],[30]. Sc5d, associated with cholesterol synthesis [31] was also down-regulated, in keeping with data indicating that Leishmania infection depletes this membrane lipid [32],[33], as was Emr1 (F4/80; [34]). TGFβ and a network of interconnected genes were also specifically down-regulated at 48h in 14M1.4 cells (Supplementary Figure S3).

We next performed clustering using the CLICK algorithm contained in EXPANDER [35] which identified 550 genes in a single cluster (homogeneity value: 0.895) that were increased in their expression level at both 12h and 48h p.i. (Figure 2C). GO functional classes representing ‘immune response’ (GO:0006955; 5.45%) and ‘defense response’ (GO: 0006952; 0.54%) were significantly enriched in this cluster. We next used PRIMA software [36] to identify transcription factor sequences enriched amongst these 550 induced genes. By this analysis (Figure 2D), we identified Irf-7 and Irf-1 and ISRE, as potential regulators of the response of 14M1.4 cells to L. donovani. Similar analysis of genes down regulated in 14M1.4 cells at both time points did not reveal any significantly enriched transcription factors (data not shown).
Based on the high levels of expression in 14M1.4 cells, absence of expression of an interferon response signature in RAW264.7 cells, and the known role of Irf-7 as a central regulator of the Type I interferon response [37],[38], we focused further attention on Irf-7. Confirmation of the induction of Irf-7 following Leishmania infection was obtained by quantitative RT-PCR (Figure 2E), by intracellular flow cytometry (Figure 2F) and by confocal microscopy (Figure 2G). Amastigotes induced a dose - and time-dependent accumulation of Irf-7 mRNA and IRF-7 protein in 14M1.4 cells that was similar in kinetics but of somewhat reduced magnitude than poly(I:C) a known Toll-like receptor (TLR)-3-dependent inducer of Irf-7. In contrast, RAW264.7 cells and bone marrow-derived macrophages induced minimal levels of Irf-7 mRNA accumulation or IRF-7 protein expression (Supplementary Figure S4). Together, these data indicated that 14M1.4 stromal macrophages, but not RAW 264.7 macrophages responded to L. donovani infection by strong induction of IRF-7.
IRF-7 is recruited to the Leishmania phagosome
In IRF-7/MyD88 co-transfected RAW264.7 and HEK 293 cells and in plasmacytoid DC, IRF-7 and MyD88 co-localise in endosomes [39]. To establish whether such co-localisation also occurred in un-manipulated 14M.1.4 cells and to gain further insight into IRF-7 activation in these cells, we examined the distribution of MyD88 and IRF-7 by confocal microscopy (Figure 3). In resting 14M1.4 cells, IRF7 and MyD88 were largely co-localised in small cytoplasmic vesicles, with little evidence of nuclear IRF-7 (Figure 3A; Video S1, shown as a snapshot in Figure 3E). Following poly(I:C) activation, nuclear translocation of IRF-7 occurred, with residual cytoplasmic IRF-7 remaining associated with MyD88 containing vesicles (Figure 3B). In sharp contrast, although 14M1.4 cells infected for 48h with L. donovani also had nuclear IRF-7, cytoplasmic IRF-7 was now primarily associated with phagosomes containing Leishmania, and rarely with MyD88+ vesicles (Figure 3C). By 48h p.i., almost all L. donovani phagosomes were IRF-7+ (Figure 3D). IRF-7 was almost undetectable in RAW264.7 macrophages, with or without infection, but where low levels of IRF-7 could be seen in rare cells, an association with the L. donovani phagosome was not apparent (Supplementary Figure S4). Of interest, IRF-7 in infected 14M1.4 cells was almost always localised to one pole of the phagosome (Figure 3C insert; Video S2, shown as a snapshot in Figure 3F). The recruitment and polarisation of IRF-7 in the Leishmania phagosome was specific, based on three sets of observations. First, MyD88 was not similarly detected at this location (Figure 3C and F). Second, in 14M1.4 macrophages treated with poly(I:C) and then exposed to latex beads for 48h, no phagosomal recruitment of IRF-7 was observed in spite of high levels of induction of IRF-7 (Figure 3G). Third, TLR3 accumulating on latex and Leishmania phagosomes did not show the polar distribution seen with IRF-7 on Leishmania phagosomes (Figure 3G and H). With TLR3 used to demarcate the phagosome membrane, IRF-7 appeared to have an intra-phagosomal localisation (Figure 3H), an observation that was supported by using LAMP1 as an alternative phagosome membrane marker (Figure 3I and Video S3). Although some co-localisation with LAMP1 was observed, a significant amount of the observed IRF-7 was clearly intra-phagosomal, either on, within or tightly apposed to the amastigote itself. In summary, these data demonstrate that dissociation of endosomal MyD88 and IRF-7 is followed by nuclear and phagosomal recruitment of IRF-7.
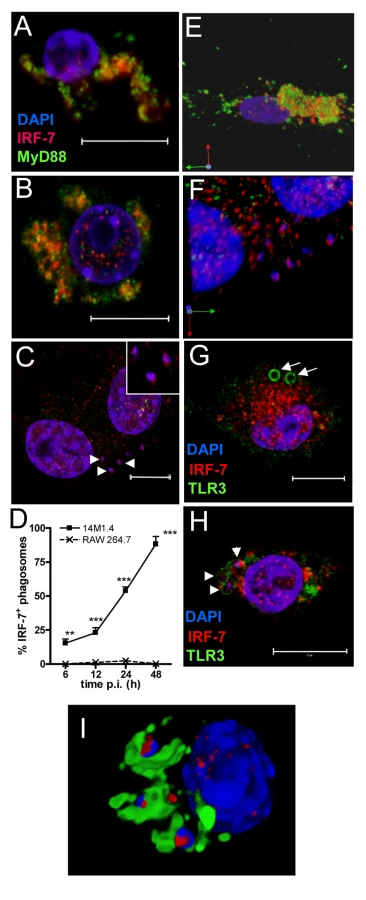
To determine whether the IRF-7 response in 14M1.4 cells actually reflected the response of MZM and MMM to Leishmania infection in situ, we prepared tissue sections from control and L. donovani-infected mice, and analysed IRF-7 expression (Figure 4). IRF-7 expression was minimal in MMM (Figure 4A) and MZM (Figure 4B) of naïve mice, but readily observed at 5h (data not shown) and 24h in MMM (Figure 4C) and MZM (Figure 4D) of infected C57BL/6 mice. At 5h p.i., ∼90% and at 24h p.i. ∼65% of parasite-containing phagosomes had clearly associated IRF-7 staining (Figure 4E), and although it was difficult to always assess the subcellular distribution of IRF-7, a polar distribution, similar to that seen in 14M1.4 cells in vitro, was often observed (Video S4, shown as a snapshot in Figure 4F). Collectively, these data support the contention that the early leishmanicidal activity of splenic MZ macrophage populations is also accompanied by IRF-7 induction and recruitment of IRF-7 to the amastigote-containing phagosome.

IRF-7 is required for leishmanicidal activity
To directly determine whether IRF-7 induction was involved in regulating the leishmanicidal activity of 14M1.4 cells, we performed functional gene knock down of IRF-7 using RNAi. Knock-down clones were screened by RT-PCR (Figure 5A) and immunofluorescence (Figure 5B–E) in the presence or absence of poly(I:C), and two independent clones with strong inhibition of Irf-7 mRNA accumulation were selected for further study (KD#1 and KD#2). Analysis of the fate of L. donovani in Irf-7 knock-down compared to control cells indicated that control of parasite burden was biphasic with respect to the requirement for Irf-7. Amastigote numbers were reduced to a similar extent over the first 12h of infection in all cells tested, suggesting an early phase of leishmanicidal activity that was Irf-7-independent. In contrast, sustained leishmanicidal activity, measured over 12–48h, was completely abrogated by knock-down of Irf-7 (Figure 5F). Thus, the leishmanicidal response of 14M1.4 cells consisted of an immediate Irf-7-independent component and a late-acting Irf-7-dependent component ultimately responsible for amastigote clearance.

Involvement of NO and IFN-α in Irf-7-dependent killing
IRF-7 plays a central role in the amplification of the Type I IFN response [37] and NO has been reported to have a major role as a leishmanicidal effector in macrophages exposed to either Type I IFNs [40] or IFN-γ [41]. We therefore determined whether NO and IFN-α were involved in the killing of Leishmania by 14M1.4 cells. Although only minimal amounts of NO could be detected in the supernatants from infected 14M1.4 cells (Supplementary Figure S5), treatment of 14M1.4 cells with the Nos2 inhibitor aminoguanidine (AMG) blocked their ability to rapidly kill L. donovani as measured over the first 12h p.i. (Figure 5G). Likewise, AMG also blocked the early leishmanicidal activity of Irf-7-deficient 14M1.4 cells (Figure 5H). In contrast, AMG had a minimal effect on the late phase of leishmanicidal activity in 14M1.4 cells (with parasite burden continuing to decrease steadily from 12–48h) or in Irf-7-deficient 14M1.4 cells (with amastigote numbers remaining at similar levels or increasing slightly from 12–48h; Figure 5G, H). Thus, whereas NO played a role in immediate parasite killing, it had limited involvement in the subsequent Irf-7-dependent phase of leishmanicidal activity.
Next, we exposed wild type and knock down 14M1.4 cells to exogenous IFN-α. At early time points (<12h), exogenous IFN-α significantly enhanced the rate at which wild type, but not Irf-7-deficient, 14M1.4 cells killed L. donovani (Figure 5G, H), suggesting that the early augmentation of effector function mediated by exogenous IFN-α required the presence of Irf-7. Importantly, however, at later time points (12–48h) exogenous IFN-α was unable to overcome the loss-of-function associated with Irf-7-deficiency (Figure 5H). Thus, leishmanicidal activity in 14M1.4 macrophages involves i) an early NO-dependent and Irf-7-independent component, which can nevertheless be augmented by IFN-α in an Irf-7-dependent manner and ii) a late-acting NO-independent, Irf-7-dependent component which can not be compensated for by exogenous IFN-α.
Killing of Leishmania in the MZ requires IRF-7
The existence of an Irf-7-dependent leishmanicidal pathway in 14M1.4 cells led us to investigate whether innate killing of Leishmania in the marginal zone was similarly controlled by IRF-7. In contrast to what has been observed with other interferon regulatory factor-deficient mice, no obvious structural differences were observed in the organisation and cellular content of the marginal zone in Irf-7−/− mice before or after infection (Figure 6A–F). However, in contrast to B6 mice, B6.Irf-7−/− mice failed to eliminate intracellular amastigotes over the first 24h of infection (Figure 6G). To determine whether Irf-7-dependent killing was a property of both MMM and MZM, we separately scored the change in amastigote number within these two populations (Figure 6H). In B6 mice, MMM and MZM both significantly reduced their parasite load from 5h to 24h (by 62% and 37% respectively; p<0.001). In Irf-7−/− mice, although MMM were still able to reduce amastigote load by ∼33% (P<0.001), killing activity was clearly reduced in comparison to MMM in wild type mice (p<0.001). For MZM, the impact of Irf-7-deficiency was more striking, with Irf-7−/− MZM failing to display any killing activity. Thus, IRF-7 played an essential role in regulating the innate ability of splenic tissue macrophages to kill L. donovani in vivo. In the course of these studies, we also noted that in B6.Irf-7−/− , but not in B6 mice, amastigotes could be found more readily within red pulp macrophages (Figure 6E and F), suggesting that the barrier function of the marginal zone that prevents early amastigote dissemination may also be impaired by Irf-7 deficiency.

Discussion
MZM and MMM have previously been shown to be involved in the uptake of various blood borne pathogens, and in this way provide an important barrier to systemic infection [7],[10],[13],[14],[15],[17],[18]. Although macrophages in the marginal zone have been shown to be capable of IFN-α production [9],[18], their broader effector function has been largely inferred from the in vitro study of other macrophage populations. Here, using a comparative approach involving direct in vitro and in vivo analysis of microbicidal activity, we provide evidence that IRF-7 plays a central role in regulating the ability of MZM and MMM to control the intra-phagosomal pathogen, L. donovani. Although IRF-7 has long been acknowledged to orchestrate anti-viral immune responses, a role in macrophage effector responses to non-viral pathogens has not been previously described.
IRF-7 has been shown to co-localise with MyD88 in endosomes of IRF-7 and MyD88 transfected cell lines [39], an observation we confirmed here using non-transfected 14M1.4 cells. Our studies extend these observations by showing that upon Leishmania infection, the spatiotemporal link between MyD88 and IRF-7 was lost. Whereas MyD88 remained associated with small cytoplasmic vesicles, IRF-7 was selectively recruited to Leishmania-containing phagosomes, both in vitro in 14M1.4 cells and in vivo in infected MZM and MMM. This recruitment of IRF-7 was specific and not observed with latex bead phagosomes, even in cells also containing Leishmania or in cells highly activated for IRF-7 expression using poly(I:C). It remains to be formally demonstrated whether this change in IRF-7 localisation reflects i) dissociation of IRF-7 from existing complexes with MyD88 and subsequent re-location to the phagosome, or ii) whether such IRF-7 undergoes nuclear translocation, with only de novo synthesised IRF-7 being recruited to phagosomes. Based on the short half life of IRF-7 [42] and the kinetics of mRNA accumulation described here, we currently favour the latter possibility. Interestingly, the absence of detectable MyD88 on the Leishmania phagosome, along with the absence of phagosomal TLR 9 expression (Phillips et al, unpublished observations) suggests that MyD88-dependent phagosomal signaling may not be a significant feature of Leishmania infection in these cells, at least at the time points studied. Similarly, it remains to be determined whether phagosomal IRF-7 plays any functional role in the IRF-7-dependent leishmanicidal activity observed here. On the one hand, phagosomal recruitment of IRF-7 might lead to IRF-7 forming part of a signalosome complex at the phagosome membrane, as has been described for phagosome-recruited AP-1 [43]. On the other hand, our confocal analysis suggests that the bulk of phagosomal IRF7 is intra-phagosomal (as defined by LAMP1) and likely to be associated with the amastigote itself. This observation is strikingly similar to that made by Antoine and colleagues who demonstrated acquisition of phagosomal MHCII (and not LAMP1) by L. amazonensis amastigotes [44] and suggests that the amastigote might act as a ‘sink’ that accumulates phagosome-associated IRF-7. These two possibilities are not mutually exclusive, and we do indeed see low levels of IRF-7 co-localized with LAMP1 apparently in the phagosome membrane. Nevertheless, given that the amastigotes of L. donovani also have tight attachment sites to the phagosomal membrane [45]; and Phillips and Kaye, unpublished), future studies employing immunogold EM, cell fractionation and biochemical approaches will be required to determine the exact localization of IRF-7 in the L. donovani phagosome and whether such IRF-7 plays a functional role.
The mechanism of killing we observe in 14M1.4 cells, in MZM and, to a lesser extent, MMM appears distinct from that previously reported for Leishmania studied in other macrophage populations. Innate IFN-γ-independent control over L. donovani in hepatic and splenic macrophages is a function often attributed to Slc11a1 [46]. However, this phagosomal iron transporter exerts leishmanistatic control over amastigote growth and, in any case, 14M1.4 cells carry the mutant allele of Slc11a1. Similarly, expression profiling suggests that a number of other recently identified anti-microbial pathways are down-regulated in 14M1.4 cells after Leishmania infection. Although a role for NO in IFN-γ-independent killing of Leishmania has also been established [41], our data indicate that the major phase of Irf-7-dependent killing occurs in a largely NO-independent manner. That previous studies have failed to identify this pathway may reflect the stromal origin of 14M1.4 cells and their closer relationship to resident tissue macrophages such as MZM and MMM. For example, RAW264.7 cells, a commonly used cell line in the study of Leishmania-macrophage interactions, responds minimally to infection with IRF-7 induction, and hence this pathway would not be evident in studies using this host-pathogen combination. Indeed, our data emphasize the importance of studying appropriate tissue macrophages when aiming to identify physiologically relevant effector mechanisms. Importantly, our data do not argue against a role for IFN-α in the regulation of Irf-7 [42]. IFN-α could be produced either through an autocrine pathway (as with 14M1.4 cells in vitro) or through a combination of autocrine and paracrine pathways, as is more likely to affect MZM and MMM in vivo. Indeed, plasmacytoid DC (pDC), a major source of IFN-α are readily activated after Leishmania infection [[47] and Sanos and Kaye, unpublished). Rather, our finding that exogenous IFN-α could not compensate for Irf-7 loss-of-function provides evidence for an alternative downstream effector mechanism that cannot be amplified by IFN-α alone. Further studies to identify potential phagosomal effectors regulated by Irf-7 are currently in progress.
Our studies were not designed to directly address the question of recognition of L. donovani during the process of early phagocytic uptake. Nevertheless, recent work has suggested that TLR9 is involved in the recognition of L. infantum and L. major promastigote-derived genomic DNA by CD11c+ cDC and by pDC [48]. In these published studies, however, no attempt was made to determine the immediate consequences of this recognition for the subsequent control of Leishmania survival. Using RAW264.7 macrophages, Descoteaux and colleagues suggested a role for both TLR2 and TLR3 in the phagocytic uptake of L. donovani promastigotes and a role for TLR3 in the leishmanicidal activity of the IFN-gamma-primed macrophages. However, as reported here, RAW264.7 cells do not recapitulate the IRF-7 response observed in 14M1.4 cells, MZM and MMM and these cells show only limited leishmanicidal activity in the absence of IFN-γ activation. Nevertheless, TLR-3 was induced in L. donovani-infected, as well as poly (I:C) treated 14M1.4 cells, but unlike IRF-7, TLR-3 appeared to be promiscuously recruited to phagosome membranes, at least after IRF-7 activation.
CD8+ T cell responses are of notable importance for protection against L. donovani, and vaccines which target CD8+ T cells are particularly effective against this infection in experimental models [49],[50]. Although the role of tissue resident macrophages as antigen presenting cells has recently returned to the fore, with a variety of studies demonstrating antigen capture and presentation to B cells [13], the importance of these cells for the induction of T cell immunity remains controversial [10]. Early studies suggested that innate Type I IFN responses were associated with an increased capacity for cross-priming [51],[52] and might therefore favour the induction of CD8+ T cell responses to intracellular pathogens. However, whilst a recent systems biology analysis of the response to Yellow Fever vaccine has confirmed the induction of a Type I IFN gene signature centred on IRF-7, this signature was not directly predictive of the magnitude of the CD8+ T cell response [53]. Previous studies of MHC gene expression in Leishmania-infected macrophages have reported inhibition of basal and IFN-γ-induced MHCI and MHCII gene expression in BALB/c derived peritoneal macrophages [54]. However, in the presence of the strong Irf-7-mediated response associated with infection of 14M1.4 cells, we observed a reciprocal regulation of genes related to MHCI - and MHCII-restricted antigen presentation, with elevated levels of transcripts for MHCI, TAP1 and tapasin. A similar role for IRF-7 in regulating cross-presentation has also recently been confirmed following adenoviral gene delivery of Irf-7 into peritoneal macrophages [55]. Together, these data suggest that further examination of the role of stromal macrophages, as represented by 14M1.4 cells and macrophages of the splenic marginal zone, in CD8+ T cell priming is now warranted.
In conclusion, we have shown that IRF-7 is indispensable for effective control of L. donovani amastigotes in stromal macrophages in vitro and in vivo. Our results suggest that IRF-7 may play a similarly important role in the containment of other intracellular pathogens.
Materials and Methods
Animals and parasites
C57BL/6 mice (CRUK, Margate, UK) were used at 6–10 weeks of age and housed under specific-pathogen free conditions. B6.Irf-7−/− mice were obtained from the RIKEN BioResource Center (Ibaraki, Japan) with kind permission of T. Taniguchi, University of Tokoyo, Japan. Amastigotes of the Ethiopian LV9 strain of L. donovani were maintained by passage in B6.RAG1−/− mice. Mice were infected with 2–5×107 amastigotes intravenously in 100µl RPMI 1640 (Gibco, Paisley, UK) and killed by cervical dislocation as required.
Ethics statement
All experiments were approved by the University of York Animal Procedures and Ethics Committee and performed under UK Home Office license (‘Immunity and Immunopathology of Leishmaniasis’ Ref # PPL 60/3708).
In vitro infection of cell lines
RAW 264.7 macrophages (American Type Culture Collection, Rockville, MD) and 14M1.4 stromal macrophages ([23]; a gift from D. Zipori, Weizmann Institute of Science, Rehovot, Israel) were maintained in complete Dulbecco's modified Eagle's medium (DMEM; Gibco) containing 5% FCS supplemented with 1 mM sodium pyruvate, 2mM l-glutamine, 0.05 mM 2-mercaptoethanol, 100U/ml penicillin and 100µg/ml streptomycin (all Gibco, Paisley, UK). 14M1.4 cells express low levels of SIGNR1 but undetectable levels of MARCO and CD169 (data not shown). 14M1.4 cells deficient in Irf-7 were generated by transfection with lentiviral particles encoding shRNA for Irf-7 mRNA (SHVRS-NM_016850, Sigma-Aldrich, St. Louis, MO). Transfected cells were positively selected in complete media containing 5µg/ml puromycin (Sigma-Aldrich, St Louis, MO) and clones were generated by limited dilution. Irf-7 mRNA knock-down was screened by RT-PCR and immuno-staining. Before use in infection experiments, cells were collected by scraping with cell scraper (BD Falcon) and 5×105 cells in complete DMEM were added to 24 well plates (Corning Inc. Corning, NY) containing sterilised 13mm diameter coverslips (VWR International, Leics, UK). After 24h, amastigotes were added at a multiplicity of infection (MOI) of 10∶1 (in 300µl complete DMEM). After 1h at 37°C, non-adherent cells and free amastigotes were removed and cells were cultured for various times post infection (p.i.). In some instances, 2µm latex beads (Bangs Laboratories, Fishers, IN) were used in addition to or instead of amastigote infections. Cell lines were treated for 2h with IFN-α (1000U/ml; PBL Interferon Source, Piscataway, NJ) at the time of infection. To quantify infection level, cells were fixed and permeabilised using 4% paraformaldehyde + 0.1% Triton-X 100 (BDH) and coverslips were mounted in Vectashield with DAPI (Vector laboratories, Burlingame, CA). The proportion of cells infected and the number of amastigotes per 100 host cells was calculated from triplicate cultures per treatment group.
Microarray analysis
106 control or infected 14M1.4 and RAW264.7 cells were incubated for 12 and 48 hours in T25 tissue culture flasks. Samples were prepared in triplicate on three separate occasions, with an average infection level of 38±5% for 14M1.4 cells and 36±8% for RAW264.7 cells. RNA was extracted from the cell lines using Trizol according to the manufacturer's protocol (Sigma-Aldrich). One of the triplicate samples from each of the three experiments were used for microarray analysis. RNA concentration and integrity was established using 210 Bioanalyser (Agilent Technologies, Palo Alto, CA). Extracted RNA was reversed transcribed to cDNA using the Affymetrix GeneChip one-cycle target labelling kit (Affymetrix, Santa Clara, CA) according to the manufacturer's recommended protocols and hybridised to GeneChip® Mouse Genome 430 2.0 Genome Arrays. Raw data processing was performed by using the Affymetrix GCOS 1.2 software. After hybridization and scanning, probe cell intensities were calculated and summarized for the respective probe sets by means of the MAS5 algorithm. MAS5 normalised data were collected and analyzed by using the ArrayAssist Expression software, Version 5.5 (Stratagene). Differentially expressed genes were identified by using a two-class t test where significance level was set at p > 0.05. Genes that were >2.0 fold up - or down-regulated between groups were selected. Data from these studies have been deposited in the EBI ArrayExpress data base (Accession #: E-MEXP-2554). Gene lists were susbsequently analysed using Ingenuity Pathway Analysis (Ingenuity Systems, Redwood City, CA) and Expander (Shamir et al., 2005) software.
Cell/parasite proliferation assay
Proliferation of macrophages and L. donovani amastigotes was assessed by analysis of the loss of CFSE (Invitrogen, Paisley, UK) fluorescence. Macrophages were incubated with 10µM CFSE in PBS for 10 minutes at 37°C, washed repeatedly, and then plated as above. At the appropriate times, macrophages were harvested and analysed for CFSE fluorescence. To assess amastigote proliferation, amastigotes labelled with CFSE as above prior to use for infection. At appropriate times p.i., macrophages were lysed with 1.25mg/ml saponin (BDH) in PBS. Released amastigotes were isolated by centrifugation and labelled with 10µM cell tracker blue (Invitrogen, Paisely, UK) for 20 minutes at 37°C, followed by specific anti-L. donovani antibody (1∶50 dilution of heat inactivated serum from an infected hamster) and then goat anti-hamster alexa fluor 647 antibody (10µg/ml; Invitrogen, Paisley, UK). CFSE dilution of macrophages and/or viable parasites was determined by flow cytometry using a cyan flow cytometer (Dako). 20,000–50,000 events were analysed per sample at each time point (n = 3).
Real-time reverse transcription-PCR (RT-PCR)
Macrophage RNA was isolated using an RNeasy kit according to the manufacturer's instructions (Qiagen, UK). RNA was reverse transcribed to cDNA using the first-strand cDNA synthesis kit according to manufacturer's instructions (Invitrogen, Paisley, UK). Primers for the specific amplification of hypoxanthine phosphoribosyltransferase (HPRT) have been previously described (45). Pre-designed primers were used for the specific amplification of Irf-7 and Mx1 (PPM04696E and PPM05520A, respectively: Superarray, Frederick, MD). PCR conditions for all primers were 95°C (15s), 62°C (30s), and 60°C (30s) for 40 cycles. Real-time PCR was performed with the SYBR green PCR kit in an ABI Prism 7000 sequence detection system (Applied Biosystems) according to manufacturer's instructions. Relative expression of target genes was normalized to HPRT and was quantified by the formula 2-ΔΔCT where ΔΔCT = (CT Target − CT HPRT)Treated − (CT Target − CT HPRT)Control. Values are expressed as the mean of triplicates ± SE. Broken line indicates the expression of the untreated control to which all samples were calibrated.
IRF-7 flow cytometry
For IRF-7 protein expression, cells were fixed in 4% PFA and permeabilised on ice for 30 min with 0.1% Triton-X 100 buffer. Cells were incubated with a polyclonal rabbit anti-IRF-7 antibody (Zymed, San Francisco, CA) or control (rabbit IgG Calprologics Inc. Harwick, MA) used at 5µg/ml. Primary antibody was detected using a goat anti-rabbit alexa fluor 546 secondary antibody (Invitrogen, Paisley, UK). Flow cytometric analysis was performed with a CyAn flow cytometer and Summit v4.3 software (Dako).
Fluorescence microscopy
Macrophages were stained for MyD88 and IRF-7 after fixation and permeabilised as above, but at room temperature. Cells were blocked in 2% goat serum (Vector laboratories) in 0.5% BSA (Fluka, UK) in PBS for 2h at room temperature or at 4°C overnight. Polyclonal rabbit anti-IRF-7 or control polyclonal sera was used at 5µg/ml. Monoclonal rat anti-MyD88 (R&D Systems, UK) or rat IgG2a (Invitrogen, Paisley, UK) was used at 5µg/ml. Primary antibodies were incubated with cells for 1h at RT. Primary antibodies were detected using the following secondary antibodies (10µg/ml; 1h at RT) ; goat anti-rabbit alexa fluor 546 (Invitrogen, Paisley, UK), and goat anti-rat alexa fluor 488 (Invitrogen, Paisley, UK). Coverslips were mounted in ProLong gold (Invitrogen, Paisley, UK) mounting medium. Immunofluorescence staining of 8µm cryosections of spleen was performed under same conditions except fixation/permeabilisation was carried out using ice cold acetone for 10 min. Sections were blocked as described above and antibodies were used as follows (5µg/ml); rat anti-CD169 FITC conjugated (Serotec, UK), rat IgG2a FITC isotype (Invitrogen), rat anti-SIGNR1 biotinylated (Bachem, UK), rat IgM biotinylated (Invitrogen). Streptavidin alexa fluor 633 (Invitrogen) was used to detect biotinylated primary antibodies and IRF-7/amastigote staining was carried out as above. Specificity of IRF-7 staining was determined by absence of staining in sections taken from naïve and infected B6.Irf-7−/− mice. Similarly, no specific staining around L. donovani amastigotes was observed following infection of 14M1.4 knock-down clones or following in vivo infection of B6.Irf7−/− mice (Supplementary Figure S6), confirming that the polyclonal anti-IRF-7 antibodies did not react to Leishmania components. Sections counterstained with 1µM DAPI (Invitrogen) in PBS for 10 minutes and mounted. Slides were analysed on a 4 laser (7-line) Zeiss Meta invert motorised microscope (Carl Zeiss, Germany) and images analysed in LSM software (Carl Zeiss, Germany). Total numbers of parasites within the MZ (and their distribution within macrophage subsets) were determined from >5 sections per mouse, scoring all MZ profiles observed in each section. Data were pooled across 3–5 mice per time point and each experiment was repeated independently at least twice.
Statistical analysis
Statistical analysis was performed using two-tailed student t test or Mann Whitney U test (as appropriate), with p <0.05 considered significant.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. HumeDA
RossIL
HimesSR
SasmonoRT
WellsCA
2002 The mononuclear phagocyte system revisited. J Leukoc Biol 72 621 627
2. RavasiT
WellsCA
HumeDA
2007 Systems biology of transcription control in macrophages. Bioessays 29 1215 1226
3. WellsCA
RavasiT
FaulknerGJ
CarninciP
OkazakiY
2003 Genetic control of the innate immune response. BMC Immunol 4 5
4. GordonS
TaylorPR
2005 Monocyte and macrophage heterogeneity. Nat Rev Immunol 5 953 964
5. MebiusRE
KraalG
2005 Structure and function of the spleen. Nat Rev Immunol 5 606 616
6. AtoM
NakanoH
KakiuchiT
KayePM
2004 Localization of marginal zone macrophages is regulated by C-C chemokine ligands 21/19. J Immunol 173 4815 4820
7. GeijtenbeekTB
GrootPC
NolteMA
van VlietSJ
Gangaram-PandayST
2002 Marginal zone macrophages express a murine homologue of DC-SIGN that captures blood-borne antigens in vivo. Blood 100 2908 2916
8. KraalG
JanseM
1986 Marginal metallophilic cells of the mouse spleen identified by a monoclonal antibody. Immunology 58 665 669
9. KraalG
MebiusR
2006 New insights into the cell biology of the marginal zone of the spleen. Int Rev Cytol 250 175 215
10. SeilerP
AicheleP
OdermattB
HengartnerH
ZinkernagelRM
1997 Crucial role of marginal zone macrophages and marginal zone metallophils in the clearance of lymphocytic choriomeningitis virus infection. Eur J Immunol 27 2626 2633
11. FranzosoG
CarlsonL
PoljakL
ShoresEW
EpsteinS
1998 Mice deficient in nuclear factor (NF)-kappa B/p52 present with defects in humoral responses, germinal center reactions, and splenic microarchitecture. J Exp Med 187 147 159
12. WeihDS
YilmazZB
WeihF
2001 Essential role of RelB in germinal center and marginal zone formation and proper expression of homing chemokines. J Immunol 167 1909 1919
13. KoppelEA
WielandCW
van den BergVC
LitjensM
FlorquinS
2005 Specific ICAM-3 grabbing nonintegrin-related 1 (SIGNR1) expressed by marginal zone macrophages is essential for defense against pulmonary Streptococcus pneumoniae infection. Eur J Immunol 35 2962 2969
14. LanoueA
ClatworthyMR
SmithP
GreenS
TownsendMJ
2004 SIGN-R1 contributes to protection against lethal pneumococcal infection in mice. J Exp Med 200 1383 1393
15. AicheleP
ZinkeJ
GrodeL
SchwendenerRA
KaufmannSH
2003 Macrophages of the splenic marginal zone are essential for trapping of blood-borne particulate antigen but dispensable for induction of specific T cell responses. J Immunol 171 1148 1155
16. KoppelEA
LitjensM
van den BergVC
van KooykY
GeijtenbeekTB
2008 Interaction of SIGNR1 expressed by marginal zone macrophages with marginal zone B cells is essential to early IgM responses against Streptococcus pneumoniae. Mol Immunol 45 2881 2887
17. GorakPM
EngwerdaCR
KayePM
1998 Dendritic cells, but not macrophages, produce IL-12 immediately following Leishmania donovani infection. Eur J Immunol 28 687 695
18. ElorantaML
AlmGV
1999 Splenic marginal metallophilic macrophages and marginal zone macrophages are the major interferon-alpha/beta producers in mice upon intravenous challenge with herpes simplex virus. Scand J Immunol 49 391 394
19. CrockerPR
GordonS
1985 Isolation and characterization of resident stromal macrophages and hematopoietic cell clusters from mouse bone marrow. J Exp Med 162 993 1014
20. CarrascoYR
BatistaFD
2007 B cells acquire particulate antigen in a macrophage-rich area at the boundary between the follicle and the subcapsular sinus of the lymph node. Immunity 27 160 171
21. JuntT
MosemanEA
IannaconeM
MassbergS
LangPA
2007 Subcapsular sinus macrophages in lymph nodes clear lymph-borne viruses and present them to antiviral B cells. Nature 450 110 114
22. PhanTG
GrigorovaI
OkadaT
CysterJG
2007 Subcapsular encounter and complement-dependent transport of immune complexes by lymph node B cells. Nat Immunol 8 992 1000
23. ZiporiD
FriedmanA
TamirM
SilverbergD
MalikZ
1984 Cultured mouse marrow cell lines: interactions between fibroblastoid cells and monocytes. J Cell Physiol 118 143 152
24. EngwerdaCR
SmeltSC
KayePM
1996 An in vivo analysis of cytokine production during Leishmania donovani infection in scid mice. Exp Parasitol 84 195 202
25. Alter-KoltunoffM
GorenS
NousbeckJ
FengCG
SherA
2008 Innate immunity to intraphagosomal pathogens is mediated by interferon regulatory factor 8 (IRF-8) that stimulates the expression of macrophage-specific Nramp1 through antagonizing repression by c-Myc. J Biol Chem 283 2724 2733
26. IshidaY
HayashiT
GotoT
KimuraA
AkimotoS
2008 Essential involvement of CX3CR1-mediated signals in the bactericidal host defense during septic peritonitis. J Immunol 181 4208 4218
27. WillmentJA
BrownGD
2008 C-type lectin receptors in antifungal immunity. Trends Microbiol 16 27 32
28. WellsCA
Salvage-JonesJA
LiX
HitchensK
ButcherS
2008 The macrophage-inducible C-type lectin, mincle, is an essential component of the innate immune response to Candida albicans. J Immunol 180 7404 7413
29. PluddemannA
HoeJC
MakepeaceK
MoxonER
GordonS
2009 The macrophage scavenger receptor a is host-protective in experimental meningococcal septicaemia. PLoS Pathog 5 e1000297 doi:10.1371/journal.ppat.1000297
30. SanoH
IshinoM
KramerH
ShimizuT
MitsuzawaH
2007 The microtubule-binding protein Hook3 interacts with a cytoplasmic domain of scavenger receptor A. J Biol Chem 282 7973 7981
31. KrakowiakPA
WassifCA
KratzL
CozmaD
KovarovaM
2003 Lathosterolosis: an inborn error of human and murine cholesterol synthesis due to lathosterol 5-desaturase deficiency. Hum Mol Genet 12 1631 1641
32. ChakrabortyD
BanerjeeS
SenA
BanerjeeKK
DasP
2005 Leishmania donovani affects antigen presentation of macrophage by disrupting lipid rafts. J Immunol 175 3214 3224
33. RubA
DeyR
JadhavM
KamatR
ChakkaramakkilS
2009 Cholesterol depletion associated with Leishmania major infection alters macrophage CD40 signalosome composition and effector function. Nat Immunol 10 273 280
34. McKnightAJ
GordonS
1998 The EGF-TM7 family: unusual structures at the leukocyte surface. J Leukoc Biol 63 271 280
35. ShamirR
Maron-KatzA
TanayA
LinhartC
SteinfeldI
2005 EXPANDER–an integrative program suite for microarray data analysis. BMC Bioinformatics 6 232
36. ElkonR
LinhartC
SharanR
ShamirR
ShilohY
2003 Genome-wide in silico identification of transcriptional regulators controlling the cell cycle in human cells. Genome Res 13 773 780
37. HondaK
YanaiH
NegishiH
AsagiriM
SatoM
2005 IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature 434 772 777
38. ColinaR
Costa-MattioliM
DowlingRJ
JaramilloM
TaiLH
2008 Translational control of the innate immune response through IRF-7. Nature 452 323 328
39. HondaK
OhbaY
YanaiH
NegishiH
MizutaniT
2005 Spatiotemporal regulation of MyD88-IRF-7 signalling for robust type-I interferon induction. Nature 434 1035 1040
40. DiefenbachA
SchindlerH
DonhauserN
LorenzE
LaskayT
1998 Type 1 interferon (IFNalpha/beta) and type 2 nitric oxide synthase regulate the innate immune response to a protozoan parasite. Immunity 8 77 87
41. BogdanC
RollinghoffM
DiefenbachA
2000 The role of nitric oxide in innate immunity. Immunol Rev 173 17 26
42. HataN
SatoM
TakaokaA
AsagiriM
TanakaN
2001 Constitutive IFN-alpha/beta signal for efficient IFN-alpha/beta gene induction by virus. Biochem Biophys Res Commun 285 518 525
43. LefkirY
MalbouyresM
GotthardtD
OzinskyA
CornillonS
2004 Involvement of the AP-1 adaptor complex in early steps of phagocytosis and macropinocytosis. Mol Biol Cell 15 861 869
44. De Souza LeaoS
LangT
PrinaE
HellioR
AntoineJC
1995 Intracellular Leishmania amazonensis amastigotes internalize and degrade MHC class II molecules of their host cells. J Cell Sci 108 ( Pt 10) 3219 3231
45. LangT
HellioR
KayePM
AntoineJC
1994 Leishmania donovani-infected macrophages: characterization of the parasitophorous vacuole and potential role of this organelle in antigen presentation. J Cell Sci 107 ( Pt 8) 2137 2150
46. BlackwellJM
GoswamiT
EvansCA
SibthorpeD
PapoN
2001 SLC11A1 (formerly NRAMP1) and disease resistance. Cell Microbiol 3 773 784
47. SchleicherU
LieseJ
KnippertzI
KurzmannC
HesseA
2007 NK cell activation in visceral leishmaniasis requires TLR9, myeloid DCs, and IL-12, but is independent of plasmacytoid DCs. J Exp Med 204 893 906
48. LieseJ
SchleicherU
BogdanC
2007 TLR9 signaling is essential for the innate NK cell response in murine cutaneous leishmaniasis. Eur J Immunol 37 3424 3434
49. StagerS
AlexanderJ
KirbyAC
BottoM
RooijenNV
2003 Natural antibodies and complement are endogenous adjuvants for vaccine-induced CD8+ T-cell responses. Nat Med 9 1287 1292
50. TsagozisP
KaragouniE
DotsikaE
2005 Function of CD8+ T lymphocytes in a self-curing mouse model of visceral leishmaniasis. Parasitol Int 54 139 146
51. StagerS
KayePM
2004 CD8+ T-cell priming regulated by cytokines of the innate immune system. Trends Mol Med 10 366 371
52. Le BonA
ToughDF
2008 Type I interferon as a stimulus for cross-priming. Cytokine Growth Factor Rev 19 33 40
53. QuerecTD
AkondyRS
LeeEK
CaoW
NakayaHI
2009 Systems biology approach predicts immunogenicity of the yellow fever vaccine in humans. Nat Immunol 10 116 125
54. ReinerNE
NgW
McMasterWR
1987 Parasite-accessory cell interactions in murine leishmaniasis. II. Leishmania donovani suppresses macrophage expression of class I and class II major histocompatibility complex gene products. J Immunol 138 1926 1932
55. GoubauD
Romieu-MourezR
SolisM
HernandezE
MespledeT
2009 Transcriptional re-programming of primary macrophages reveals distinct apoptotic and anti-tumoral functions of IRF-3 and IRF-7. Eur J Immunol 39 527 540
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2010 Číslo 3
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Měli bychom postcovidový syndrom léčit antidepresivy?
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- Kaposi's Sarcoma-Associated Herpesvirus ORF57 Protein Binds and Protects a Nuclear Noncoding RNA from Cellular RNA Decay Pathways
- Endocytosis of the Anthrax Toxin Is Mediated by Clathrin, Actin and Unconventional Adaptors
- Perforin and IL-2 Upregulation Define Qualitative Differences among Highly Functional Virus-Specific Human CD8 T Cells
- Exoerythrocytic Parasites Secrete a Cysteine Protease Inhibitor Involved in Sporozoite Invasion and Capable of Blocking Cell Death of Host Hepatocytes