
YfiBNR Mediates Cyclic di-GMP Dependent Small Colony Variant Formation and Persistence in
During long-term cystic fibrosis lung infections, Pseudomonas aeruginosa undergoes genetic adaptation resulting in progressively increased persistence and the generation of adaptive colony morphotypes. This includes small colony variants (SCVs), auto-aggregative, hyper-adherent cells whose appearance correlates with poor lung function and persistence of infection. The SCV morphotype is strongly linked to elevated levels of cyclic-di-GMP, a ubiquitous bacterial second messenger that regulates the transition between motile and sessile, cooperative lifestyles. A genetic screen in PA01 for SCV-related loci identified the yfiBNR operon, encoding a tripartite signaling module that regulates c-di-GMP levels in P. aeruginosa. Subsequent analysis determined that YfiN is a membrane-integral diguanylate cyclase whose activity is tightly controlled by YfiR, a small periplasmic protein, and the OmpA/Pal-like outer-membrane lipoprotein YfiB. Exopolysaccharide synthesis was identified as the principal downstream target for YfiBNR, with increased production of Pel and Psl exopolysaccharides responsible for many characteristic SCV behaviors. An yfi-dependent SCV was isolated from the sputum of a CF patient. Consequently, the effect of the SCV morphology on persistence of infection was analyzed in vitro and in vivo using the YfiN-mediated SCV as a representative strain. The SCV strain exhibited strong, exopolysaccharide-dependent resistance to nematode scavenging and macrophage phagocytosis. Furthermore, the SCV strain effectively persisted over many weeks in mouse infection models, despite exhibiting a marked fitness disadvantage in vitro. Exposure to sub-inhibitory concentrations of antibiotics significantly decreased both the number of suppressors arising, and the relative fitness disadvantage of the SCV mutant in vitro, suggesting that the SCV persistence phenotype may play a more important role during antimicrobial chemotherapy. This study establishes YfiBNR as an important player in P. aeruginosa persistence, and implicates a central role for c-di-GMP, and by extension the SCV phenotype in chronic infections.
Published in the journal:
. PLoS Pathog 6(3): e32767. doi:10.1371/journal.ppat.1000804
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1000804
Summary
During long-term cystic fibrosis lung infections, Pseudomonas aeruginosa undergoes genetic adaptation resulting in progressively increased persistence and the generation of adaptive colony morphotypes. This includes small colony variants (SCVs), auto-aggregative, hyper-adherent cells whose appearance correlates with poor lung function and persistence of infection. The SCV morphotype is strongly linked to elevated levels of cyclic-di-GMP, a ubiquitous bacterial second messenger that regulates the transition between motile and sessile, cooperative lifestyles. A genetic screen in PA01 for SCV-related loci identified the yfiBNR operon, encoding a tripartite signaling module that regulates c-di-GMP levels in P. aeruginosa. Subsequent analysis determined that YfiN is a membrane-integral diguanylate cyclase whose activity is tightly controlled by YfiR, a small periplasmic protein, and the OmpA/Pal-like outer-membrane lipoprotein YfiB. Exopolysaccharide synthesis was identified as the principal downstream target for YfiBNR, with increased production of Pel and Psl exopolysaccharides responsible for many characteristic SCV behaviors. An yfi-dependent SCV was isolated from the sputum of a CF patient. Consequently, the effect of the SCV morphology on persistence of infection was analyzed in vitro and in vivo using the YfiN-mediated SCV as a representative strain. The SCV strain exhibited strong, exopolysaccharide-dependent resistance to nematode scavenging and macrophage phagocytosis. Furthermore, the SCV strain effectively persisted over many weeks in mouse infection models, despite exhibiting a marked fitness disadvantage in vitro. Exposure to sub-inhibitory concentrations of antibiotics significantly decreased both the number of suppressors arising, and the relative fitness disadvantage of the SCV mutant in vitro, suggesting that the SCV persistence phenotype may play a more important role during antimicrobial chemotherapy. This study establishes YfiBNR as an important player in P. aeruginosa persistence, and implicates a central role for c-di-GMP, and by extension the SCV phenotype in chronic infections.
Introduction
Many bacterial pathogens are able to establish long-term chronic infections within their respective hosts. The strategies and mechanisms employed by pathogens to persist against attacks by the host immune system and the action of antimicrobial substances are still relatively poorly understood. An appreciation of these processes is needed to develop strategies that help to avoid complications with antibiotic resistance development and infection relapses associated with prolonged colonization of host tissue. The opportunistic pathogen Pseudomonas aeruginosa is responsible for chronic infections in the airways of cystic fibrosis (CF) patients and for much of the associated morbidity and mortality [1]. During long-term lung colonization, P. aeruginosa undergoes phenotypic and genetic adaptation resulting in the progressive loss of virulence and the development of increased persistence [2]. As a manifestation of this process, distinct adaptive colony morphotypes of P. aeruginosa appear in CF sputum sample isolates. These include mucoid colonies, which overproduce the exopolysaccharide alginate [1], and small colony variants (SCVs), slow-growing isolates characterized by several phenotypes including auto-aggregation, attachment to surfaces [3] and enhanced exopolysaccharide production [3],[4]. The appearance of P. aeruginosa SCVs in CF sputum samples correlates with antibiotic resistance [5],[6], poor lung function, and persistence [7]. Likewise, Burkholderia cepacia SCVs are associated with increased serum resistance and fatal systemic infections post lung transplantation [8]. These studies suggest that during the course of chronic lung infections, SCVs are selected for due to a fitness advantage in this unique environment, and that they might play an important role in the pathogenesis of P. aeruginosa lung infections.
Following early indications [9],[10], evidence has accumulated that the SCV morphotype is strongly linked to the second messenger cyclic-di-GMP (c-di-GMP) [11]–[13]. A recent study of CF patients showed clear links between c-di-GMP-related systems, and hence SCV, and chronic lung infection [2]. C-di-GMP is an ubiquitous bacterial second messenger that regulates the sessile-motile transition in a wide range of species [14],[15]. C-di-GMP is produced from GTP by di-guanylate cyclases (DGC), and degraded to pGpG by phosphodiesterases (PDE) [16]. These enzymes contain the conserved GGDEF and EAL domains, respectively [17]–[20]. GGDEF domains that harbor enzymatic activity generally contain two conserved motifs; the catalytic active (A) site, and the regulatory (I) site, required for non-competitive product inhibition [21]. In general, elevated levels of c-di-GMP are associated with a sessile, cooperative lifestyle. Phenotypes expressed in this state include biofilm formation [22],[23], enhanced exopolysaccharide production [24],[25], expression and production of attachment factors [9],[26], loss of various forms of motility [27],[28], and reduced virulence [29],[30]. Conversely, low levels of c-di-GMP promote a unicellular, free-swimming lifestyle.
The c-di-GMP regulatory system in P. aeruginosa is highly complex, with 33 predicted GGDEF and 17 EAL domain-containing proteins [15],[29]. These proteins are thought to modulate the intracellular c-di-GMP level and hence regulate the production of numerous outputs. These include the exopolysaccharides Pel, Psl and alginate [25],[31],[32], fimbrial adhesins [26], virulence and cytotoxicity factors [29],[33], pili [27],[34],[35] and flagella function [36]. Regulation of these output systems has been observed at the transcriptional [32] and post-translational levels [25],[31]. To date, the biological roles and mechanisms of action of several P. aeruginosa c-di-GMP signaling systems have been investigated. These include the Wsp system [12],[37],[38], comprising a Che-like chemosensory system with a DGC response-regulator output, WspR [12],[37]. Over-activation of WspR results in an SCV phenotype [9],[12], displaying strong attachment and increased expression of the pel and psl exopolysaccharide operons [12],[13]. Sad/RocARS [26],[33] is a two-component signaling system (RocS1) with two output proteins; a transcriptional regulator (RocA1) and an EAL domain containing protein (RocR). Sad/RocARS is required for biofilm maturation [33] and regulates cup fimbriae expression [26]. This system is further implicated in pathogenicity, with links to cyanide-mediated toxicity [39] and type-III secretion [33]. The transition between reversible and irreversible attachment in P. aeruginosa is regulated by the putative hydrolase SadB [40], which promotes Pel exopolysaccharide production and represses swarming motility [40],[41]. These processes are similarly co-regulated by the opposing enzymatic activities of SadC, a membrane-bound DGC [36] and the PDE BifA [42]. In addition, c-di-GMP has been associated with the response of P. aeruginosa to antibiotic [10],[43],[44] and detergent-mediated [45] stress, and with type IV pili production and control [27],[34].
To investigate the role of c-di-GMP in the generation of the P. aeruginosa SCV phenotype, we undertook a transposon mutagenesis screen in strain PA01 for genes whose disruption produced autoaggregative, Congo Red binding colonies. Three independent transposon insertions were found in yfiR, the first gene of the yfiBNR operon, which is highly conserved in many γ-proteobacteria. Here we characterize the yfiBNR genes, and report that this conserved system represents a tripartite signaling module that regulates c-di-GMP levels in P. aeruginosa in response to as-yet unknown environmental signals. YfiN acts as a membrane-integral DGC whose activity is tightly controlled by YfiR, a small periplasmic protein, and the OmpA/Pal-like outer-membrane lipoprotein YfiB. The primary downstream target for the cyclic-di-GMP produced by YfiBNR was identified as the Pel exopolysaccharide system, with transcription of pel genes being directly proportional to the inferred output level of YfiN. Disruption of the tight YfiN regulation via deletion of yfiR resulted in the generation of an SCV morphology in PA01. Consequently, the ΔyfiR genotype was used as a representative SCV in cell culture assays and in a mouse model of persistent infection. Compared to PA01, the ΔyfiR mutant exhibited strong, exopolysaccharide-dependent resistance to macrophage phagocytosis. In addition, the SCV phenotype was shown to effectively persist in a subcutaneous catheter infection model. Together this establishes the YfiBNR module as an important player in the persistence of P. aeruginosa and implicates a role for c-di-GMP in chronic infections.
Materials and Methods
Strains and growth conditions
Strains and plasmids used in this study are listed in Table S1. Primers are listed in Table S2. Unless otherwise stated, P. aeruginosa PA01 and all E. coli strains were grown at 37°C in Luria Bertani (LB) medium [46], solidified with 1.3% agar where appropriate. For P. aeruginosa, gentamycin was used at 30 µg/ml (E. coli 20 µg/ml), tetracycline at 50-100 µg/ml (E. coli 12.5 µg/ml), streptomycin at 200 µg/ml (E. coli 50 µg/ml), and carbenicillin at 200 µg/ml. For E. coli, kanamycin was used at 30 µg/ml and ampicillin at 100 µg/ml. Where required, chloramphenicol was added to conjugation plates at 10 µg/ml. Congo Red dye was added to a final concentration 0.04%. For inducible plasmids, vanillate was added to a final concentration 1.0 mM, arabinose to 0.2% and IPTG to 0.5 mM, as appropriate. J774 macrophages were cultured in 150 µl RPMI medium (Sigma) supplemented with 2% Glutamine and 2% FCS, and incubated at 37°C, 5% CO2.
Molecular biology procedures
Cloning was carried out in accordance with standard molecular biology techniques. pBBR5-yfiR was produced by ligation of the yfiR PCR fragment (amplified with primers A and B, from PA01 genomic DNA), between the HindIII and BamHI sites of pBBR-MCS5 [47]. pME-araC was constructed by ligation of the araC-pBAD promoter fragment of pBAD18s [48] between the BamHI and EcoRI sites of pME6032. pME-araC-yfiN was produced by ligation of the yfiN PCR fragment (amplified with primers C and D, from PA01 genomic DNA), between the SacI and BglII sites of pME-araC. Truncated and non-truncated yfiR-phoA fusion plasmids, and pME-yfiR-cherry were produced by ligation of SOE-PCR/PCR fragments generated with primers F-J, BJ and BK between the EcoRI and XhoI sites of pME-araC, and the EcoRI and BglII sites of pME6032 respectively. The yfiBNR complementation vectors were constructed by ligation of the relevant yfiBNR PCR fragments (amplified with primers A, D, and E from PA01 and ΔyfiR genomic DNA) between the HindIII and BamHI sites of pUC18T-mini-Tn7T-Gm [49]. A and I site yfiN mutants were constructed via SOE PCR [50], from template PCR fragments amplified with primers A, D, E, and K-N. The plasmid pME6032-yfiB was constructed by ligation of the yfiB PCR fragment (amplified with primers E and O, from PA01 genomic DNA), between the EcoRI and BamHI sites of pME6032 [51]. Plasmid pBBR4-wspR was constructed by ligation of the EcoRI-HindIII wspR fragment of plasmid pWspR12 [52] between the EcoRI and HindIII sites of pBBR-MCS4 [47].
To produce a chromosomal M2-tagged copy of yfiR, the PCR fragment amplified with primers A and E was ligated between the HindIII and BamHI sites of pMR20 [53] and E. coli DY330 [54] was transformed with the resulting plasmid. The PCR fragment amplified with primers P and Q from plasmid pSUB11 [55] was then used to produce an yfiR-M2 fusion in pMR20 by the method described by Yu et al. [54]. The yfiR-M2 fusion was ligated into the HindIII and BamHI sites of pUC18T-mini-Tn7T-Gm. The PCR fragment amplified with primers R and S was used to produce pAD6-Ω from plasmid pAD6 (A. Dürig, unpublished), by the method described in [54].
The plasmids pET42b-yfiN and yfiB were constructed by ligation of the relevant PCR fragments (amplified with primers T-W, from PA01 genomic DNA), between the XhoI and NdeI sites of pET42b (Novagen). Bacterial two-hybrid vectors were constructed by ligation of the relevant PCR fragments (amplified with primers D, E, and X-AK from PA01 genomic DNA) between the XbaI and BamHI sites of pUT18C and pKT25, and the HindIII and BamHI sites of pUT18 [56].
Plasmid pME6032-luxCDABE was constructed by ligation of the EcoRI-BamHI fragment of plasmid pSB417 [57] between the EcoRI and BamHI sites of pME6032. Lux fusions with the cupA and pel promoters were then constructed by ligation of EcoRI-excised promoter fragments from plasmids pMPFCAL [58] and p-pelA-lacZ [59] respectively into the EcoRI site of pME6032-luxCDABE. Lux fusions with the cupB, cupC, psl and yfi promoters were constructed by ligation of the relevant PCR fragments (amplified with primers AL-AS, from PA01 genomic DNA), between the XhoI and EcoRI sites of pME6032-luxCDABE.
Transduction with phage E79tv2
E79tv2 [60] transducing lysates were prepared as follows: 100 µl of a donor overnight culture were mixed with 100 µl of wild type phage (app. 107 to 105 PFU/ml), incubated for 10 min at 37°C and mixed with 3 ml top-agar. The mixture was poured onto LB plates and incubated at 37°C overnight. Top-agar with semi-confluent plaques was then scraped from the plates and re-suspended in 2 ml of TNM buffer (10 mM Tris HCl [pH 7.4], 150 mM NaCl, 10 mM MgSO4), vortexed and centrifuged at 20,000 g for 10 min, the supernatant containing the transducing phage was then transferred into a glass storage vial. For transduction, 500 µl of an overnight culture of recipient strain was re-suspended in 500 µl TNM buffer, mixed with 500 µl of UV-attenuated transducing lysate (5×108 PFU/ml, 15 sec. UV Stratalinker 2400) and incubated at 37°C for 15 min. Non-adsorbed phage was removed by washing twice in TNM buffer and samples were plated onto selective media.
Transposon mutagenesis
The plasmid pALMAR3 was introduced into PA01 via biparental mating with E. coli S17-1. Mariner transposon insertions yielding an SCV morphology were selected on LB agar containing tetracycline, chloramphenicol and Congo Red, and restreaked onto fresh plates. The location of the transposon in each SCV strain was determined by semi-random PCR [61], using primers AT-AW.
Deletion of the yfiBNR genes
The strains PA01 ΔyfiNR and ΔyfiBNR were constructed via an adaptation of the protocol described elsewhere [62]. Briefly, deletion constructs were produced by SOE-PCR using primers AX-BE, containing homologous flanking regions to the target genes and FLP-excisable gentamycin cassettes. These constructs were ligated into pEX18Ap between HindIII and KpnI. The resulting vectors were then used to delete yfiNR/BNR as described in [62]. PA01 ΔyfiR was deleted by two-step allelic exchange. A deletion construct containing the flanking regions of yfiR was produced by SOE-PCR using primers BF-BI and ligated between HindIII and KpnI of pEX18Ap. The resulting vector was then transformed into PA01. Following transformation, single crossovers were selected on carbenicillin and restreaked. Counterselection on 5% sucrose plates was used to force the resolution of double crossovers. In all cases, deletion strains were confirmed by colony PCR.
Attachment assays
Assays were adapted from [63]. 96 well plates containing 150 µl LB medium/well were inoculated with single colonies using sterile toothpicks, and incubated overnight at 37°C without shaking. Plates were washed three times with distilled water. Remaining cell material was then stained with 0.1% Crystal Violet solution (5% methanol, 5% isopropanol) before further washing to remove excess dye. Crystal Violet was re-dissolved in 20% acetic acid solution and absorbance measured at 600 nm. Assays were performed with 6 wells/strain and repeated independently at least once in each case.
Motility assays
Swarm agar was based on TB and solidified with 0.5% agar. After solidification, plates were dried overnight and then inoculated on the surface with colonies re-suspended in LB. Plates were incubated overnight at 37°C. Twitching motility was assayed using TB plates containing 1% agar. Samples were stabbed through the surface using toothpicks and plates incubated overnight at 37°C. Twitching cells were visualized by staining with 1% Crystal Violet following removal of the agar. Swimming motility was assayed using TB plates containing 0.3% agar. Colonies were stabbed into the agar using toothpicks and plates incubated overnight at 37°C. Assays were repeated three times independently.
Purification of N-terminal truncated YfiN and YfiB-His6 and production of antisera
200 ml of overnight cultures of BL21 pET42b-yfiN/yfiB were used to inoculate 2L LB medium supplemented with kanamycin. Cultures were grown for 2.5 hours at 37°C with shaking, His6-yfiN/B expression was induced with 100 µM IPTG, and cultures incubated for a further 2.5 hours at 30°C with shaking. Cells were harvested by centrifugation and lysed with 15 ml lysis buffer (250 mM NaCl, 10 mM Tris pH 8.0, 6.0 M urea). Insoluble cell debris was harvested by centrifugation (30,000xg, 15 min, 4°C), and truncated-YfiN-His6 and YfiB-His6 were purified from the supernatant by nickel-NTA (BioRad) affinity chromatography. Following binding to 0.5 ml nickel-NTA (4°C, 30 min, shaking) proteins were eluted with a stepwise increase in imidazole concentration, with both proteins eluting in 200 mM imidazole. Native truncated-YfiN-His6 was purified as described, but without 6.0 M urea and with the addition of 10% glycerol to the lysis buffer. A French Press was used to lyse the sample in place of urea. Purified proteins were separated on preparative SDS gels, stained with 4 M KCl, excised and sent to Laboratoire d'Hormonologie, Marloie, Belgium for polyclonal antisera production. Protein concentrations were assayed with a Protein Assay kit (BioRad).
Immunoblot analysis
Cell lysates from overnight cultures were separated on 15% Tris-HCl gels and blotted onto polyvinylidene difluoride (PVDF) membranes (Millipore). After overnight incubation in blocking solution (1 x PBS pH 7.4, 0.01% Tween20, 5% milk powder), proteins were detected with 1/5000 (α-M2, α-YfiB) or 1/500 (α-YfiN) specific antiserum and 1/10,000 rabbit anti-mouse (α-M2) or swine anti-rabbit (α-YfiB, α-YfiN) secondary antibody (DakoCytomation). Bound antibodies were visualized with ECL chemiluminescent detection reagent (Perkin-Elmer).
Diguanylate cyclase (DGC) activity assay
DGC activity was assayed as described previously [18],[21]. Assays were run in 50 µl running buffer containing approximately 25 µg purified truncated-YfiN-His6 and started by the addition of (final concentration) 100 µM GTP [18.5 kBq α33P-GTP] (Amersham Biosciences). Samples were removed at regular intervals and the reaction stopped with (final concentration) 250 mM EDTA. Purified DgcA [21] was included as a positive control.
Nucleotide extraction and analysis
Following protein purification, 100 µl of the truncated-YfiN-His6 elution fraction was mixed with 200 µl of 0.5 M formic acid, and nucleotides were extracted for 10 min at 4°C. Insoluble components were then pelleted, and the supernatant directly analyzed by chromatography after [21]. Nucleotides were extracted and separated on a 125/4 Nucleosil 4000-1 polyethyleneimine column (Macherey-Nagel) using the SMART System (Amersham Biosciences). The nucleotide peak corresponding to c-di-GMP was verified by co-elution with a chemically synthesized c-di-GMP standard.
Quantitation of cyclic-di-GMP by mass spectrometry
Log phase growing cultures of PA01 strains were cooled rapidly in iced water and 5-10 ml of cells were concentrated by centrifugation (5,300xg, 10 min, 4°C). Extracts were prepared essentially as described in [64]. C-di-GMP levels were measured by liquid chromatography-tandem mass spectrometry on an API 3000 triple quadrupole mass spectrometer (Applied Biosystems Inc, Foster City, CA, USA) coupled with a Series 200 HPLC System (Perkin Elmer Instruments, Norwalk, CT, USA). C-di-GMP was detected via selected reaction monitoring (SRM) in positive ionization mode. Liquid chromatography separation was achieved on a reversed-phase column using an ammonium acetate-methanol gradient (retention time for c-di-GMP: 8.6 min). Details of this method will be described elsewhere (Spangler C. et al., manuscript in preparation). Following nucleotide extraction, the pellet was dissolved in 800 µl 0.1 M NaOH by heating for 15 min at 95°C. The protein content of each pellet was then determined with a Protein Assay kit (BioRad). Measurements were repeated at least in triplicate and values were expressed as pmol c-di-GMP per mg protein.
In-vivo crosslinking assays
Overnight cultures of PA01 strains were washed once and re-suspended in 1 volume PBS. An aliquot was removed, and crosslinking started by the addition of 1% formaldehyde. Aliquots were removed after 20 minutes crosslinking at room temperature, and washed once to remove formaldehyde. Benzon nuclease was added to digest DNA (5 U, 30 min, RT) and samples were boiled (20 min, 95°C) where required. Samples were subsequently analyzed by Western blotting.
Subcellular localization of proteins
For the fractionation of soluble and membrane proteins, an overnight culture of PA01::yfiR-M2 was re-suspended in 0.2 volumes lysis buffer (20 mM Tris pH 8.0, 250 mM NaCl, protease inhibitor cocktail (Roche)). Cells were lysed by French Press and centrifuged to remove cell debris (10,000 g, 1 hour, 4°C). The soluble and insoluble cell fractions were then separated by ultracentrifugation (100,000 g, 3 hours, 4°C). Samples were subsequently analyzed by immunoblotting. For the fractionation of periplasmic proteins, overnight cultures were resuspended in 0.125 volumes osmotic shock buffer (50 mM Tris pH 8.0, 20% sucrose, 2 mM EDTA) and incubated at room temperature for 30 to 60 min, depending on the strain. Samples were centrifuged (15,000 g, 10 minutes, 4°C) and the supernatant and pellet collected. The pellet fraction was washed twice and resuspended in PBS. The supernatant (periplasmic fraction) was either subjected to alkaline phosphatase assays directly, or proteins were precipitated with 5% TCA, washed twice with di-ethyl ether and resuspended in PBS before immunoblot analysis.
Alkaline phosphatase assay
Alkaline phosphatase activity was assayed for YfiR-PhoA protein fusions by measuring the rate of p-nitrophenyl-phosphate hydrolysis according to the method described in [65]. Assays were carried out in triplicate for each protein fusion and the experiment was repeated independently.
Bacterial two-hybrid
Bacterial two-hybrid assays were carried out after [66]. E. coli MM337 cells were freshly transformed with constructs containing potential interaction partners. Transformants were then streaked onto MacConkey base and M9 plates, supplemented with 0.5% maltose, kanamycin and ampicillin. Plates were incubated for two days at 30°C and positive interactions distinguished by red coloration on MacConkey and growth on M9 plates.
Luminescence assay for promoter activity
Strains containing pME6032-promoter-lux fusion constructs were streaked onto LB plates containing tetracycline, incubated overnight and the resulting colonies used to inoculate LB cultures. These cultures were grown for 6 hours at 37°C and then two 150 µl aliquots were transferred to a black, clear-bottomed 96-well plate (Costar). Luminescence and OD600 were recorded for each sample using a Synergy 2 plate reader (Biotek). Samples were repeated in triplicate for each promoter fusion and the assay repeated independently at least twice.
Scanning electron microscopy
Cells were grown overnight at 37°C, in 2 ml LB in 24-well plates and in the presence of a hanging sterile glass slide. After growth, the glass slides were removed, rinsed gently with 1x PBS and fixed in 2.5% glutaraldehyde in 1x PBS for 2 h at RT. Glutaraldehyde was washed out with 1x PBS and subsequently with water, and the sample dehydrated with an acetone step gradient (30%, 50%, 70%, 90%, 100%; 10 min each). Samples were critical point-dried and sputter-coated with a 3–5 nm platinum layer. Micrographs were recorded on a Hitachi S-4800 field emission scanning electron microscope. Acceleration voltage was generally between 1.5 and 5 kV.
Nematode absorption assays
100 µL drops of PA01 overnight cultures were dried onto M9 plates with 0.4% glucose, and the plates incubated overnight at 37 °C. Approximately 20 adult Caenorhabditis elegans were added to each plate, and plates were incubated at room temperature for 72 hours. For GFP labeling experiments, PA01 strains containing pAD6-Ω were used and incubated with C. elegans for 3 hours before imaging.
Macrophage phagocytosis and NF-κB activation assays
J774 macrophages were incubated overnight in a black, clear-bottomed 96-well plate, at a concentration of 2×104 cells/well. Lysotracker red dye (Invitrogen) was added to each well 30 minutes prior to infection. PA01 strains containing pAD6-Ω were grown in LB for 4 hours at 37°C and then used to infect the mature macrophages (final MOI 5). Samples were vortexed thoroughly at every stage of the preparation process to minimize the proportion of cell aggregates in the inoculum. After 2 hours incubation at 37°C, 5% CO2, macrophages were fixed by the addition of 4% PFA solution. Wells were washed with PBS and stained with Hoechst 33342, then subjected to analysis with an ImageXpressMICRO microscope running MetaXpress 2.0 (Molecular Devices). For the NF-κB activation assay, the Lysotracker stain was omitted, and the macrophages permeabilised with Triton-X100 and stained with an anti-NF-κB p65 antibody (Santa Cruz Biotechnology) following infection. The Translocation enhanced analysis module of MetaXpress was used to calculate the ratio of cytosolic to nuclear p65 intensity at the single cell level. Six wells were inoculated per strain and each assay was repeated independently.
Siderophore and pyocyanin production
Siderophore and pyocyanin levels were measured in the supernatants of overnight cultures grown in M9 plus 0.4% glucose and 0.2% casamino acids (Pyoverdin, Pyochelin) and LB (Pyocyanin) using the methods described in [67].
LDH release assay for cytotoxicity
J774 macrophages were inoculated in a 24 well plate, at a concentration of 1×105 cells/well and incubated overnight. Overnight cultures of PA01 strains were inoculated into LB, grown for 6 hours at 37°C and used to infect the mature macrophages (final MOI 10). After 3 hours incubation (37°C, 5% CO2), LDH release was measured with a CytoTox96 kit (Promega) according to the manufacturer's instructions. Values were calculated as a percentage of the positive control (+ 0.5% TritonX100), and each sample was repeated in triplicate.
Infection models
C57BL/6 mice (10–14 weeks old) were obtained from RCC (Füllinsdorf, BL, Switzerland) and kept in the animal facility of the Department of Biomedicine, University Hospitals Basel; animal experimentation guidelines were followed in accordance with the regulations of Swiss veterinary law. Methods employed were approved by the review board of the Kantonales Veterinäramt Basel-Stadt (permit no. 1957). Mice were infected as described elsewhere [68], with minor modifications. Briefly, mice were anesthetized with 20 mg/kg Ketalar (Pfizer) and 4 mg/kg xylazinum (Graeub). A 3–4 mm incision was made 1–1.5 cm lateral to the spine, and a catheter segment (1 cm Vialon IV catheters, diameter of 2.1 mm; Becton Dickinson), was inserted subcutaneously. Next, 25 µl of pyrogen-free saline containing 10,000 cfu of PA01-GmR (wt), ΔyfiR or ΔyfiBNR bacteria grown to exponential phase in LB medium was injected into the beds of uncoated catheters, and the incision was closed with wound clips. Four and eight weeks after infection, mice were sacrificed and the catheter and surrounding tissue were aseptically removed and separated. Catheters were vortexed in saline, 0.15% EDTA, and 0.1% Triton X-100 and sonicated for 3 min at 130 W. Tissue samples were homogenized, dilutions from both preparations were plated onto LB agar plates, and colony-forming units were counted following overnight incubation at 37°C. For competition experiments, wt and ΔyfiR or ΔyfiBNR bacteria were injected at a 1∶1 ratio. Differences in colony morphology were used to distinguish between wild type and ΔyfiR mutant colonies. Statistically significant differences were determined using the Mann-Whitney test.
Antibiotic survival assay
Glass tubes containing 4 ml LB and tobramycin (0–2.0 µg/ml) were inoculated with 10,000 cfu of PA01-GmR (wt) or ΔyfiR. After 18 hours incubation at 37°C with shaking, dilutions were plated onto LB agar plates containing Congo Red. Colony-forming units were counted following overnight incubation at 37°C. Statistically significant differences between samples were determined using the Mann-Whitney test. Five replicates were run for each sample.
Results
The yfiBNR operon is an SCV-related locus in Pseudomonas aeruginosa
To begin our analysis of the SCV phenotype, we sought to identify those loci in P. aeruginosa PA01 whose disruption led to a characteristic SCV morphology and behavior. A comprehensive transposon mutagenesis screen was conducted, selecting for mutants that stably maintained an SCV phenotype on LB Congo Red agar plates (i.e. that could be maintained by serial re-streaking without losing the SCV phenotype). Three independent transposon insertions were mapped to yfiR (PA1121), the first gene of the predicted three-gene operon yfiBNR (Figure 1A). In silico analysis predicted that yfiN encodes a 47.5 kDa protein with two transmembrane helices flanking a periplasmic PAS-like domain, a HAMP domain and a GGDEF domain with a conserved GGDEF active-site motif. YfiB was predicted to be an 18.4 kDa outer-membrane lipoprotein with a conserved OmpA peptidoglycan binding domain [69], while yfiR encodes a 20.7 kDa protein with a probable signal peptide but no predicted tertiary structure [70]–[72](Figure 1B).

Given the established link between c-di-GMP production and SCV and the fact that YfiN has been suggested to function as a DGC [29], we hypothesized that the SCV phenotype of the yfiR transposon insertions was caused by up-regulation of YfiN activity. This would occur either via the release of repression by YfiR or as a consequence of polar effects from the Tn insertions on the yfiN and/or yfiB genes. The yfiR::Tn phenotype was successfully complemented in trans with a plasmid-borne wild type copy of yfiR (data not shown), and an in-frame deletion of yfiR yielded an SCV phenotype indistinguishable from that of the transposon mutants (Figure 1C). The yfiR transposon mutants (data not shown) as well as the ΔyfiR mutant (Figure 1D) showed an almost 10-fold increased propensity for surface attachment. Over-expression of PA5295, a PDE from P. aeruginosa [73], but not of an active site mutant (PA5295AAL), reduced ΔyfiR attachment to wild type levels and abolished the SCV phenotype (Figure 1D). Swimming, swarming, and twitching motility were severely impaired in ΔyfiR compared to wild type PA01 (Figure S1). Similarly to the SCV phenotype, these motility defects were fully complemented by expression of yfiR in cis or in trans (Figure S1D).
To determine the nature of the YfiBNR system output, a His6-tagged version of YfiN lacking the 182 residues corresponding to the transmembrane region (truncated-YfiN-His6) was purified and tested for DGC activity. DGC activity was previously suggested for YfiN of P. aeruginosa strain PA14 based on HPLC analysis of over-expression strains [29]. In accordance with these findings, truncated-YfiN-His6 generated c-di-GMP from GTP, confirming that YfiN functions as a DGC (Figure 2A). HPLC analysis of the effluent co-eluting with the purified truncated-YfiN-His6 fraction indicated the presence of large amounts of c-di-GMP (Figure 2B), consistent with the idea that the YfiN GGDEF domain contains a conserved high-affinity binding site utilized for allosteric product inhibition (I-site) [21]. These results indicated that the ΔyfiR SCV phenotype is a consequence of derepressed YfiN DGC activity. To confirm this, we determined the cellular concentration of c-di-GMP in wild type and mutant strains. In growing cells of PA01 wild type and a strain lacking YfiN (ΔyfiBNR, see below) the concentration of c-di-GMP was 3.54±0.67 pmol/mg protein and 2.39±0.33 pmol/mg, respectively. In contrast, a strain lacking YfiR (289.30±36.53 pmol/mg) or over-expressing yfiN from a plasmid (1835.70±235.93 pmol/mg) showed a marked increase of c-di-GMP. Together, these data strongly suggest that YfiR functions to negatively regulate YfiN, and that the ΔyfiR SCV phenotype results from de-repression of YfiN DGC activity, leading to increased levels of c-di-GMP.

YfiB and YfiR inversely control YfiN activity
In order to understand the regulatory interplay of the YfiB, YfiN and YfiR proteins, a series of epistasis experiments were performed. In addition to the ΔyfiR mutant, ΔyfiNR and ΔyfiBNR deletion strains were constructed. As expected, both strains displayed wild type colony morphology (data not shown) and reduced attachment (Figure 3A), indicating that YfiN contributes to surface attachment of PA01 wild type under the conditions tested, and that YfiN is the effector of the SCV phenotype in the ΔyfiR mutant. A series of yfiBNR alleles were then constructed and inserted into the att-Tn7 locus of ΔyfiBNR (a strain that harbors a deletion of the entire yfi operon). In this way, the effects of all combinations of yfiB, yfiN, and yfiR mutations could be tested. Replacement of the full yfiBNR operon at att-Tn7 yielded a phenotype indistinguishable from wild type PA01 (Figure 3A, B). Likewise, complementation with yfiBNΔR produced an SCV phenotype with an attachment level comparable to the ΔyfiR mutant (Figure 3A, B). These results demonstrated that the Tn7-complementation strains can be used to model the effects of yfiBNR disruptions in vivo.
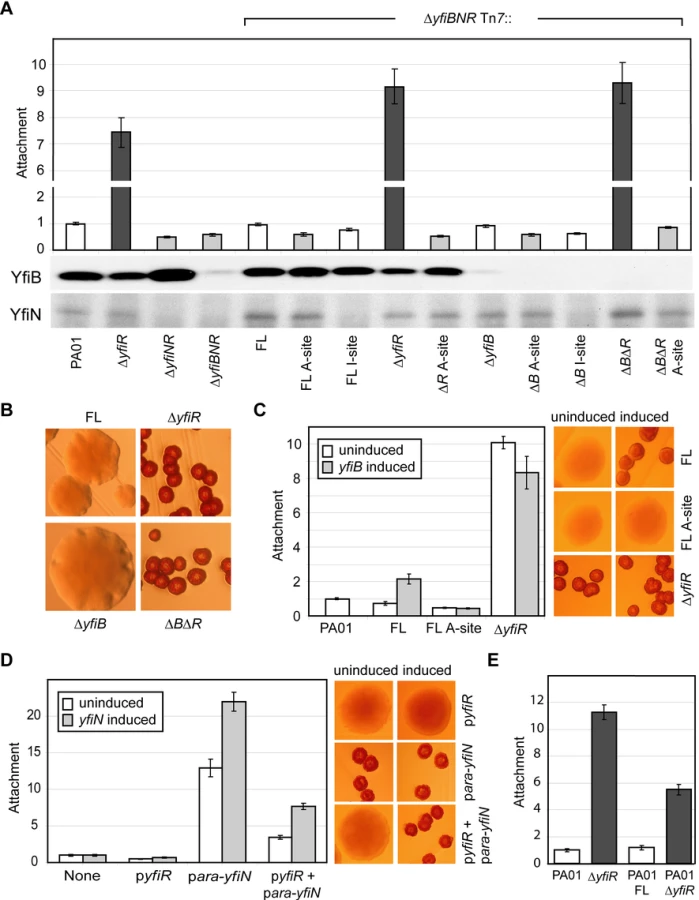
Comparison of the morphology and attachment of the Tn7-based complementation strains led to several observations (Figure 3A, B). Firstly, deletion of yfiN, or expression of an yfiN active site (A-site) mutant (yfiND330A) produced a wild type colony morphology and an attachment level around 60% of wild type, independent of the presence or absence of yfiR or yfiB. Secondly, deletion of yfiR produced the characteristic SCV morphology in any strain with a wild type version of yfiN. Deletion of yfiB did not affect attachment or colony morphology, and a ΔyfiRB double mutant behaved similarly to a ΔyfiR single mutant, suggesting that yfiR is epistatic over yfiB and that YfiB may function upstream of YfiR. Over-expression of yfiB in trans led to a two-fold increase in ΔyfiBNR Tn7::yfiBNR attachment (Figure 3C, ‘FL’). This effect was dependent upon the presence of YfiR, and on a functional copy of YfiN; no attachment increase was seen upon yfiB expression in either strain ΔyfiBNR Tn7::yfiBNΔR or in strain ΔyfiBNR Tn7::yfiBND330AR (Figure 3C, ‘ΔyfiR’ and ‘FL A-site’). These data support the hypothesis that YfiB functions upstream of YfiR. Thirdly, the stoichiometry of YfiR and YfiN appears to be critical for the tight control of the Yfi system (Figure 3D, E). Basal level expression of yfiN from the plasmid p-ara-yfiN lead to increased levels of YfiN relative to YfiR, and to a SCV phenotype in PA01 wild type. This SCV morphology was suppressed by the introduction of a plasmid expressing yfiR (p-yfiR). When yfiN expression in this strain was induced with arabinose, levels of YfiN once again dominated, leading to an SCV phenotype (Figure 3D). Similarly, adding a second copy of yfiN to the PA01 wild type chromosome (inserted into the att-Tn7 locus) produced an SCV phenotype with a lower level of attachment than ΔyfiR, accounting for the chromosomal copy of yfiR present, while the wild type phenotype was maintained when additional copies of both yfiN and yfiR were added (Figure 3E). Finally, an yfiN I-site mutation (yfiNR319A) only partially complemented wild type yfiN. Because immunoblot analysis showed reduced levels of the YfiN I-site mutant as compared to wild type (Figure 3A), this may be due to reduced stability of the mutant protein. Despite the reduced effectiveness of yfiNR319A, we were unable to introduce this allele into a strain that lacked yfiR, suggesting that loss of both YfiR-mediated inhibition of YfiN activity and YfiN feedback control produces a highly toxic situation, similar to that observed for feedback negative mutants of C. crescentus DgcA [21]. This in turn implies that YfiN, in addition to its regulation by YfiR, is subject to tight feedback control via I-site binding of c-di-GMP. Together, these data strongly indicate that the diguanylate cyclase YfiN is subject to tight negative control by YfiR and that YfiB, directly or indirectly, counteracts this inhibitory effect.
YfiR is a periplasmic protein
Epistasis experiments indicated that YfiR plays an important role in regulating YfiN activity, possibly by relaying information from YfiB in the outer membrane to YfiN in the cytoplasmic membrane. Such a mechanism would predict that YfiR is located in the periplasm. To test this and due to the absence of an effective YfiR antibody, yfiR triple-M2 tagged (yfiR-M2) reporter strains were constructed. Insertion of the yfiR-M2 allele into the att-Tn7 locus of a ΔyfiR mutant restored the wild type phenotype, indicating that YfiR-M2 is fully functional (data not shown). The subcellular location of YfiR-M2 was subsequently investigated upon fractionation of cell extracts by ultracentrifugation followed by immunoblot analysis (Figure 4A). Because YfiB is predicted to localize to the outer membrane [70],[72], it was included as a control. As expected, YfiB almost exclusively localized to the insoluble, membrane-associated fraction. YfiR-M2 was found exclusively in the soluble fraction (Figure 4A). Next, a strain expressing both yfiR-M2 and gfp (PA01 yfiR-M2 pAD6-Ω) was subjected to periplasmic extraction by osmotic shock, and the fractions were analyzed by immunoblotting. As expected for a cytosolic protein, GFP was detected in the whole-cell and spheroplast fractions only, confirming that cell fractionation did not result in spheroplast lysis. In contrast, YfiR-M2 was found exclusively in the periplasmic fraction (Figure 4B).

To provide further evidence for the periplasmic location of YfiR, an yfiR-phoA fusion was analyzed. This construct was able to complement the SCV morphology of a ΔyfiR mutant (Figure S2B), indicating that the YfiR section of the fusion protein was fully functional. PA01 wild type containing the full length YfiR-PhoA fusion displayed strong phosphatase activity, with a large proportion of this activity localized to the periplasmic fraction (Figure S2A). In contrast, an yfiR-phoA allele lacking the presumable YfiR export signal was unable to complement the ΔyfiR SCV morphology and showed little or no phosphatase activity (Figure S2A, B). Finally, an yfiR-Mcherry fusion was constructed and shown to fully complement the ΔyfiR SCV phenotype (data not shown). When protein localization was visualized by fluorescence microscopy in PA01, the red fluorescent YfiR fusion protein was found exclusively in the cell perimeter (Figure S2C). Together, these data strongly suggest that YfiR is indeed located primarily in the periplasm.
The genetic interaction between YfiB and YfiR together with their presumable location in the outer membrane and periplasm, respectively, prompted us to gather more direct evidence for a link between YfiB and YfiR function. In vivo cross-linking experiments were performed, in which YfiR-M2 containing strains were treated with formaldehyde followed by immunoblot analysis of the cross-linked lysates (Figure 4C). In a wild type background, formaldehyde treated samples showed a clear shift of YfiR-M2 from the monomeric state (19.8 kDa, without the signal sequence) to a multimer of approximately 40–42 kDa. This multimer was absent in a strain that lacked both YfiB and YfiN (ΔyfiBN yfiR-M2), but was present in a strain that only lacked YfiN (ΔyfiN yfiR-M2) (Figure 4C), indicating that YfiB, but not YfiN, is required for YfiR oligomerisation. Immunoblotting with YfiB antiserum failed to detect a band of similar size (data not shown). Whilst the nature of the YfiR multimer is currently unknown, its size is consistent with an YfiR-YfiR homodimer.
Bacterial-two hybrid analysis failed to provide evidence for direct interaction between YfiR and the isolated periplasmic or cytoplasmic domains of YfiN, and between YfiB and any other protein variant (Table S3). Given the different subcellular localization of these proteins some of these negative results might reflect limitations in the experimental procedure. A truncated variant of YfiR missing the predicted signal peptide (residues 1-34) [70] interacted strongly with itself (Figure 4D), consistent with the results obtained from cross-linking experiments (see above). Also, while no interaction was observed with the full-length YfiN (data not shown), a truncated version of YfiN missing its transmembrane region (residues 1-182) formed strong interactions with itself (Figure 4D), arguing that, like other DGCs [74]-[76], YfiN functions as a dimer and that the membrane spanning part of YfiN negatively controls YfiN dimerisation. The isolated HAMP domain, but not the isolated GGDEF domain of YfiN also interacted with the truncated YfiN variant, suggesting that the HAMP domain is involved in YfiN multimerization (Figure 4D). Together, these data suggest that YfiB and YfiR are localized outside of the cell and together control YfiN activity.
The YfiBNR system controls the Pel and Psl exopolysaccharide systems
Elevated levels of c-di-GMP have been linked to a number of biofilm-promoting systems including Pel and Psl exopolysaccharides [12] and the Cup fimbrial adhesins [9],[11],[26]. To identify potential downstream targets of YfiN that contribute to the SCV behavior, the ΔyfiR deletion allele was combined with disruptions in several of these c-di-GMP output systems. Disruption of pel (pelG::Tn) or psl (ΔpslAB) strongly attenuated the SCV phenotype of a ΔyfiR mutant, resulting in strains with altered colony morphologies and Congo Red binding on LB-agar plates (Figure 5A), as well as significantly reduced attachment (Figure 5B). Importantly, a pelG::Tn ΔpslAB ΔyfiR triple mutant exhibited a smooth colony morphology, a marked loss of Congo Red binding and complete abolishment of surface attachment. In contrast, mutational disruption of the CupA (cupA4::Tn), CupB (cupB4::Tn), or CupC (cupC2::Tn) fimbrial adhesins showed no discernable effects on the ΔyfiR SCV phenotype (Figure 5A, B). Over-expression of wspR19, which encodes a constitutively active DGC [52] produced high levels of attachment, comparable to the ΔyfiR mutant. However, in this case up-regulation of Pel alone seems to be responsible for increased attachment, as disruption of psl produced little effect (Figure 5B). Disruption of pel and psl in the ΔyfiR background failed to restore swimming and twitching motility and only partially restored swarming motility (Figure S1). This suggested that the impairment of cellular motility in the ΔyfiR mutant is not an indirect consequence of Pel and Psl exopolysaccharide overproduction and that additional, motility-related systems are affected.

To investigate the relationship between YfiBNR and its downstream targets in more detail, gene transcription in different Δyfi-backgrounds was probed using lux-promoter fusion constructs (Figure 5C). Compared to PA01, deletion of the entire yfiBNR operon led to a 40% drop in pel transcription level, which likely contributes to the reduced attachment level seen in ΔyfiBNR (Figure 3A). In contrast, in a ΔyfiR background, pel transcription was massively induced compared to PA01. Transcription of the psl and cupB operons was subject to a modest two-fold increase in ΔyfiR over wild type levels. Interestingly, cupA transcription was strongly inhibited in this strain under the conditions tested.
Electron micrographs of PA01 and ΔyfiR strains (Figure S3) showed cells embedded in a dense matrix of thin fibres. This matrix was markedly thicker for the ΔyfiR mutant, and the cells in this case appeared to form a more structured biofilm. In PA01, disruption of both pel and psl greatly reduced biofilm formation (Figure S3). In the ΔyfiR background, disruption of pel did not abolish biofilm formation, but the biofilm that was formed displayed few extracellular fibres, suggesting that Pel is a component of the extracellular matrix. ΔyfiR ΔpslAB produced both a biofilm and extracellular fibres, but the latter in this case were irregularly distributed and had a ‘ragged’ appearance, in contrast to the thick, even matrix seen with the parental ΔyfiR strain (Figure S3). Little or no biofilm formation was observed when both pel and psl were disrupted in either yfiR background (data not shown). Together, these data indicate that de-repression of YfiN leads to the activation of the Pel and Psl exopolysaccharide systems, and that most of the morphological and behavioral characteristics of the ΔyfiR SCV are mediated by the up-regulation of Pel and Psl.
YfiN-dependent up-regulation of Pel and Psl exopolysaccharide systems confers resistance against phagocytosis
The clinical SCV phenotype is associated with persistence in CF lung infections [7]. In addition, SCV is a highly aggregative phenotype associated with over-production of exopolysaccharide in both clinical [4] and lab strains [77]. To determine whether yfiBNR mutations give rise to SCVs in CF lung infections, ten SCV strains isolated from the sputum of CF patients were transformed with pMR20-yfiR-M2. The SCV phenotype of one of these strains, ClinSCV-110, was abolished by yfiR-M2 expression, similarly to the ΔyfiR SCV mutant (Figure S4). This strongly suggested that the SCV phenotype in this case arose as the consequence of a mutation in the yfi operon, and validates the use of the ΔyfiR mutant as a model SCV for subsequent in vivo analyses.
To assay the persistence behavior of the ΔyfiR mutant and to test the hypothesis that persistence was related to the exopolysaccharide components of the biofilm, we analyzed the interaction of the ΔyfiR mutant with both murine immune cells and external predators. Caenorhabditis elegans, incubated for 72 hours on minimal agar plates onto which drops of PA01 had been spotted grew to maturity with no apparent adverse effects (Figure 6A, panel 1). In contrast, nematodes incubated with ΔyfiR were starved, with many individuals in the dauer larva stage, a stage reflecting low food resources (Figure 6A, panel 2). When incubated for three hours on GFP-labeled cells, the gut of nematodes fed with PA01 was filled with green bacteria (Figure 6A, panel 1 inset), while nematodes fed on ΔyfiR showed almost no bacteria in the gut (Figure 6A, panel 2 inset). Disruption of either the pel or psl operon alone was insufficient to overcome the resistance of the ΔyfiR mutant to effective nematode scavenging (Figure 6A, panels 3-4). However, a ΔyfiR pelG::Tn ΔpslAB triple mutant was consumed readily; nematodes incubated with this strain grew to maturity in the same way as with wild type PA01 (Figure 6A, panel 5). These results demonstrate that the ΔyfiR SCV phenotype confers exopolysaccharide-dependent resistance to ingestion by C. elegans.

To investigate the interaction of ΔyfiR mutant cells with professional phagocytic cells of the immune system, J774 macrophages were incubated with GFP-labeled PA01 variants. When incubated for three hours with PA01 wild type, 71.9±4.8% of the macrophages had taken up at least one bacterium. In contrast, only 5.6±2.3% of the macrophages had phagocytosed ΔyfiR mutant cells, indicating that the ΔyfiR SCV phenotype confers substantial resistance to macrophage phagocytosis (Figure 6B). To investigate the contribution of the Pel and Psl systems to interference with internalization, the assay was repeated with ΔyfiR pelG::Tn and ΔyfiR ΔpslAB mutant strains. The percentage of macrophages that had taken up at least one bacterium rose to 42.8±6.0% for the ΔyfiR pelG::Tn, 27.0±4.5% for the ΔyfiR ΔpslAB, and to 75.6±5.3% for the ΔyfiR ΔpslAB pelG::Tn triple mutant (Figure 6B). The observation that phagocytosis of the ΔyfiR SCV mutant was fully restored upon disruption of the pel and psl loci not only provided further evidence that these exopolysaccharides represent one of the main cellular outputs for the YfiBNR system, but in agreement with previous studies [78],[79] also confirms the notion that matrix engulfment enables bacterial cells to escape the host immune response.
In addition to interfering with internalization, the different yfi, psl, and pel mutants displayed markedly different surface attachment phenotypes in this assay. Clusters of extracellular ΔyfiR (pel+ psl+) mutant cells observed in the macrophage phagocytosis assay showed a specific ‘spider-like’ organization, where cells spread out from the central region of aggregation along a number of defined trajectories (Figure 6C). The pattern for the ΔyfiR ΔpslAB mutant was markedly different in that cells randomly spread out from the central region in every direction, lacking the radial tracks seen for ΔyfiR spreading. In contrast, the ΔyfiR pelG::Tn mutant, although unable to form large clusters of aggregated cells, appeared to be organized into similar linear trajectories as the ΔyfiR SCV mutant (Figure 6C). No particular cell arrangement was observed for the ΔyfiR ΔpslAB pelG::Tn triple mutant, although few non-phagocytosed cells were seen for this strain (Figure 6B). These observations suggest that Pel and Psl not only contribute to cell adherence, biofilm formation, and protection against phagocytosis, but also mediate a specific architecture of surface attached microcolonies.
Given the clear differences in physical interaction between macrophages and SCVs, we sought to test whether these differences were mirrored in the internal response of the immune cells. To test whether the ΔyfiR SCV phenotype affected macrophage activation or cytotoxicity, nuclear NF-κB translocation (Figure S5) and LDH (lactate dehydrogenase) release (Figure S6) was determined in J774 macrophages infected with PA01 wild type and mutant cells, respectively. No significant difference in macrophage activation was seen between samples infected with PA01 or the ΔyfiR mutant. When siderophore production was measured for the ΔyfiR strain, increased levels of pyoverdin and pyochelin were found compared with wild type PA01 (Figure S7). An increase in the production of siderophores and other excreted ‘scavenger’ molecules would appear to be consistent with the persistence phenotype proposed for SCV strains. The ΔyfiR strain also showed increased production of the phenazine pyocyanin (Figure S7).
The ΔyfiR SCV phenotype persists in a murine subcutaneous catheter infection
Although the auto-aggregative and slow growing SCV morphotypes are at a considerable disadvantage under conditions that permit rapid growth, they are able to persist in vivo [7]. This phenomenon might be explained by conditions in the host environment (e.g. immune system attack or antimicrobial chemotherapy) that put an even higher burden on rapidly growing, non-adherent strains and thus provide selection for SCVs. To test this, we compared the competitive behavior of PA01 wild type and ΔyfiR SCV strains in vitro and in vivo. When grown in LB the ΔyfiR SCV strain was significantly outperformed by the wild type. Also, suppressors with wild type colony morphology quickly arose in every ΔyfiR sample, forming the majority of the cell population after 18 hours incubation (Figure 7A). The addition of increasing concentrations of tobramycin in the sub-inhibitory range led to a reduction of both wild type and ΔyfiR cell numbers. However, wild type growth decreased at a much steeper rate and at 1.5 µg/ml tobramycin, no significant difference in cfu numbers was observed between the two strains (Figure 7A). Likewise, in the presence of tobramycin the number of suppressors arising from ΔyfiR dropped sharply with no suppressors arising at concentrations above 0.5 µg/ml of the inhibitor. Thus, the fitness disadvantage of the ΔyfiR SCV is strongly reduced in the presence of sub-inhibitory concentrations of tobramycin. Two observations indicate that this effect is not linked to some form of antibiotic tolerance of the ΔyfiR SCV, but rather reflects converging fitness during slow growth under stressful conditions. Firstly, a similar relative increase of ΔyfiR fitness compared to wild type was observed when cells were grown at reduced temperatures (data not shown). Secondly, no differences in MIC were seen between PA01 and ΔyfiR for tobramycin or for any of the other antibiotics tested (Figure S8).

Next, we analyzed the ΔyfiR mutant in a mouse catheter model with respect to its in vivo persistence behavior. Total bacterial numbers and colony morphologies were scored in the catheters and in the surrounding tissue four and eight weeks after infection with PA01 wild type, ΔyfiR and ΔyfiBNR mutants, either individually or in competition. When strains were infected individually, total numbers of ΔyfiR mutants scored within the catheter were significantly lower after four weeks as compared to wild type (Figure 7B). In contrast, no significant difference was observed between the corresponding tissue samples (Figure 7B). Likewise, in four week competition experiments, the ΔyfiR mutant was strongly outperformed by wild type in the catheter but less so in the surrounding tissue (Figure 7C). After eight weeks of infection, viable counts of wild type cells were significantly reduced in the catheter compared with four-week infections. However, no significant drop in cfu/ml was noted for the ΔyfiR mutant between four and eight weeks, accounting for an improved relative performance of the SCV mutant (Figure 7B). In competition assays after eight weeks the wild type/ΔyfiR ratio decreased relative to the four-week results in both catheter and tissue samples. While a small (non-significant) disadvantage was still seen for the ΔyfiR mutant in the catheter, no such disadvantage was noted for the eight-week tissue samples (Figure 7C). The improved relative performance of the ΔyfiR mutant appeared to be due to a reduction in the number of wild type cfus, rather than an increase in SCVs. Importantly, in vivo no suppressors of the SCV morphology arose for any of the ΔyfiR mutant samples over the entire course of the experiment, in contrast to the rapid emergence of suppressors in vitro (Figure 7A). No significant differences were seen between wild type and the ΔyfiBNR strain after four weeks, either in single infections or competition experiments (data not shown).
Together this demonstrates that the slow-growing ΔyfiR SCV mutant, although suffering from a tremendous growth disadvantage, shows characteristic persistence behavior under antibiotic selection and during prolonged infection in vivo.
Discussion
In this work we identify and characterize YfiBNR, an important regulatory system involved in Pseudomonas aeruginosa biofilm formation and in vivo persistence. Our research suggests that YfiB and YfiR are upstream regulatory components of the YfiN diguanylate cyclase, which represents the cellular readout of the system. Activation of YfiN results in c-di-GMP production, and the activation of several downstream targets affecting cell motility and exopolysaccharide production. We identify YfiR as a key player in containing YfiN activity. Mutants lacking YfiR strongly activate YfiN leading to a characteristic SCV phenotype distinguished by slow growth, reduced motility, a highly aggregative and wrinkled colony morphology, strong attachment to surfaces, and persistence in vivo. Epistatic and biochemical analyses led to a model for YfiBNR function and control (Figure 4E), wherein YfiR acts as small periplasmic protein that is able to tightly repress the activity of the membrane-localized DGC YfiN. YfiB counteracts YfiR-mediated repression of YfiN and hence leads to increased c-di-GMP production. YfiB is a homolog of the Pal family of lipoproteins that are anchored either in the inner or outer membrane and directly interact with the peptidoglycan structure via a conserved peptidoglycan-binding site [69]. Because mutations of the tol-pal genes induce hypersensitivity to external stress-factors [80], it is possible that YfiB plays a role in sensing envelope stress, and, in response, stimulates an SCV response by relaying changes in the outer cell layer to YfiN via YfiR. The observation that DGCs like YfiN are active as dimers [74],[75], together with the possible YfiB-dependent dimerisation of YfiR is consistent with the idea that YfiR represses YfiN activity by interfering with its oligomerisation state. In this model, YfiB activates YfiN by promoting the oligomerisation of YfiR, freeing YfiN to form active dimers and thus produce c-di-GMP. An interesting alternative regulatory mechanism for YfiN has recently been proposed [81]. In this scheme, YfiN activity is controlled by de-phosphorylation of the periplasmic domain by the tyrosine phosphatase TpbA. This mechanism could in principle work in parallel with YfiR/YfiB-mediated control of YfiN activity, or as part of the same regulatory pathway. Further experiments are required to fully address the modes of action of YfiB and YfiR, and the regulatory hierarchy of the Yfi system.
While the signals that activate the Yfi system remain to be identified, several of the downstream targets are well known. Genetic experiments indicated that YfiN-mediated induction of the Pel and Psl exopolysaccharides plays a central role in the SCV morphotype. Disruption of either exopolysaccharide operon led to a partial phenotype, while disruption of both systems produced colonies with wild type morphology. Transcription of pel was massively increased in the ΔyfiR strain compared to wild type; much of the increase in Pel exopolysaccharide production in ΔyfiR may be as a consequence of this increased transcription. Despite the substantial contribution of Psl to the ΔyfiR phenotype, psl transcription was only modestly increased in this mutant, suggesting that c-di-GMP mediated Psl stimulation in ΔyfiR may be at a different regulatory level. In an yfiBNR deletion strain, pel transcription and surface attachment drop by approximately 40% when compared to wild type, arguing that the Yfi system contributes significantly to Pel and Psl-dependent biofilm formation under standard laboratory conditions. In contrast to pel and psl transcription, cupA transcription was strongly repressed in the ΔyfiR mutant under the conditions tested. Similar reciprocal relationships between exopolysaccharide and fimbrial gene transcription have been noted previously in P. aeruginosa [82] and E. coli [83]. The physiological relevance of this is unknown, although one may speculate that cupA repression is a compensatory response to the enhanced production of Pel and Psl in the SCV state. The patterns of biofilm formation and surface attachment observed suggested critical roles for Pel and Psl in the formation and organization of SCV colonies, respectively. The ΔyfiR (pel+ psl+) mutant strain formed clusters of aggregated cells, from which cells radiated along defined trajectories. Whereas disruption of the pel operon appeared to primarily affect cell-cell association, a psl mutation did not affect the number or size of cell clusters, but rather their organization, with cells spreading out from the central region in an apparently random fashion. While Pel appears to provide the principal structural element of the P. aeruginosa SCV biofilm, Psl may function more as a scaffold to mediate a specific biofilm architecture and to ensure organized and effective biofilm construction, in agreement with previous studies [84].
The control of Pel exopolysaccharide biosynthesis appears to be complex, as several c-di-GMP signaling components have been implicated with this process [12],[13],[25],[32],[36],[37],[40],[42],[45]. Recently, Starkey and co-workers [13] described two classes of lab-derived SCVs, which in many respects resemble the phenotype of the ΔyfiR mutant. While the molecular nature of class B SCVs is unclear, members of class A could be complemented by wspF, arguing that they were caused by mutations that activate the WspR DGC pathway [12]. Both classes of SCVs over-produce exopolysaccharides and display a transcriptional profile for pel and psl similar to that observed here for the ΔyfiR SCV. Likewise, CupA fimbrial adhesins do not play a role in the SCV phenotype of either sub-class [13]. Loss of function mutations in yfiR are thus prime candidates for class B SCVs. Clinical and lab-derived SCVs are physiologically similar, with a number of characteristic phenotypes in common [4],[13]. Because of the relatively high frequency of loss of function mutations, both wspF and yfiR might be important targets for genetic adaptations leading to clinical SCV development. In support of this, wspF mutations have already been identified in long-term P. aeruginosa CF isolates [2]. Similarly, the SCV phenotype of strain ClinSCV-110 appears to derive from an yfi mutation. In addition to ClinSCV-110, another potential candidate for a clinical yfiR SCV has also been isolated, although the genetic basis for this SCV has not yet been identified [11]. The clinical isolate SCV-20265 reverted to a wild type phenotype upon mutation of yfiN, strongly implicating the Yfi system in the generation of the SCV phenotype. Surprisingly, CupA adhesin is an important component of the SCV phenotype of SCV-20265, contrary to our findings for the ΔyfiR mutant [11]. A possible explanation for this is that the downstream targets of YfiN in SCV-20265 may differ from those seen in the laboratory strain.
The SCV phenotype caused by de-repression of the YfiN DGC conferred a marked resistance to nematode scavenging and phagocytosis by macrophages. This phenotype was shown to be strongly dependent on exopolysaccharide production, with concomitant disruption of the pel and psl operons restoring the ability of nematodes to scavenge, and macrophage phagocytosis levels to those of wild type PA01. There is evidence to suggest that this resistance to phagocytosis derives from the protection offered to each individual cell by the exopolysaccharide, rather than being a consequence of cell aggregation. First, macrophages are able to take up particles larger than many of the SCV cell aggregates [85]. Second, in contrast to wild type cells, many single cells of the SCV strain remained un-internalized even after several hours of co-incubation with macrophages (Figure 6C). This phenotype is in agreement with the role of mannose receptors as major phagocytic receptors for P. aeruginosa [86]. Exopolysaccharides may sterically hinder binding or alter macrophage surfaces and internalization via mannose receptors. Enhanced resistance to the immune system may help to explain the persistence of SCV infections in the subcutaneous catheter infection model. Although bacterial counts of the SCV strain were initially lower due to the severe growth impediment of this strain, SCV cell numbers remained constant over eight weeks in both catheter and neighboring tissue samples. In contrast, numbers of wild type cells rapidly declined during the same time window. The exact molecular basis for SCV persistence in vivo is currently unclear. However, given the resistance to macrophage phagocytosis seen in vitro, and in the absence of complicating external factors such as antibiotic stress, it is tempting to speculate that the competitive advantage of SCVs compared with wild type P. aeruginosa cells in long term infections is due to immune evasion. The better relative performance of the YfiN-mediated SCV strain in tissue compared with catheters would suggest that exopolysaccharide protection against phagocytosis plays a more important role in the surrounding tissue, consistent with an increased exposure to the vascularized immune system. Cells in a bacterial biofilm have been shown to be highly tolerant of antibiotic treatment [87], with tolerance attributed to characteristics including biofilm structure [88], persister cells [89] and slow growth rates [90]. Resistance to phagocytosis and clearance, especially following aggressive antibiotic therapy when the bacterial load of an infection is lower, and the relative risk of exposure to immune cells is correspondingly higher, might thus contribute to the SCV persistence phenotype in CF patients.
Although the persistence effect of the ΔyfiR SCV strain observed in these experiments was significant, even after eight weeks of infection wild type counts were still comparable with those of the mutant. Likewise, no SCVs arose in the animals from wild type samples, contrary to the situation in CF patients [5],[6]. Longer periods of infection, higher infection load, and external challenges (e.g. antibiotic stress) may increase the competitive advantage of SCVs, resulting in a higher proportion of SCVs and the evolution of new SCV morphotypes from the wild type population. In support of this hypothesis, exposure to sub-lethal concentrations of tobramycin was shown to significantly reduce the number of suppressors arising in an in vitro culture of the ΔyfiR SCV strain, and at higher concentrations, decreased the relative fitness advantage of wild type PA01 over SCV. In addition, it is possible that the comparatively large number of persister cells found in biofilms could further increase the advantage of SCV-generating mutations in the presence of antibiotic stress [89]. Work is ongoing to investigate these possibilities.
In this study, we have identified and characterized a novel c-di-GMP regulatory system, and investigated its role both in wild type P. aeruginosa and in the SCV morphotype. While homologous systems have been mentioned previously in the literature [91],[92], this work represents the first comprehensive experimental analysis of the YfiBNR system. The YfiN cyclase is tightly controlled by its upstream regulatory system, while loss of this control leads to the formation of a strong SCV phenotype. In the SCV form, P. aeruginosa adopts a hyper-adherent, aggregative lifestyle, with significant implications for immune evasion and ultimately for long term persistence of infection. Further research should determine the extent to which these findings are relevant to chronic colonization in patients with CF and implant-borne infections.
Supporting Information
Zdroje
1. GovanDVJR
1996 Microbial pathogenesis in cystic fibrosis: mucoid Pseudomonas aeruginosa and Burkholderia cepacia. Microbiol Rev 60 539 574
2. SmithEE
BuckleyDG
WuZ
SaenphimmachakC
HoffmanLR
2006 Genetic adaptation by Pseudomonas aeruginosa to the airways of cystic fibrosis patients. Proc Natl Acad Sci U S A 103 8487 8492
3. HausslerS
ZieglerI
LottelA
von GotzF
RohdeM
2003 Highly adherent small-colony variants of Pseudomonas aeruginosa in cystic fibrosis lung infection. J Med Microbiol 52 295 301
4. KirisitsMJ
ProstL
StarkeyM
ParsekMR
2005 Characterization of colony morphology variants isolated from Pseudomonas aeruginosa biofilms. Appl Environ Microbiol 71 4809 4821
5. HausslerS
TummlerB
WeissbrodtH
RohdeM
SteinmetzI
1999 Small-colony variants of Pseudomonas aeruginosa in cystic fibrosis. Clin Infect Dis 29 621 625
6. ReinhardtA
KohlerT
WoodP
RohnerP
DumasJL
2007 Development and persistence of antimicrobial resistance in Pseudomonas aeruginosa: a longitudinal observation in mechanically ventilated patients. Antimicrob Agents Chemother 51 1341 1350
7. HausslerS
2004 Biofilm formation by the small colony variant phenotype of Pseudomonas aeruginosa. Environ Microbiol 6 546 551
8. HausslerS
LehmannC
BreselgeC
RohdeM
ClassenM
2003 Fatal outcome of lung transplantation in cystic fibrosis patients due to small-colony variants of the Burkholderia cepacia complex. Eur J Clin Microbiol Infect Dis 22 249 253
9. D'ArgenioDA
CalfeeMW
RaineyPB
PesciEC
2002 Autolysis and autoaggregation in Pseudomonas aeruginosa colony morphology mutants. J Bacteriol 184 6481 6489
10. DrenkardE
AusubelFM
2002 Pseudomonas biofilm formation and antibiotic resistance are linked to phenotypic variation. Nature 416 740 743
11. MeissnerA
WildV
SimmR
RohdeM
ErckC
2007 Pseudomonas aeruginosa cupA-encoded fimbriae expression is regulated by a GGDEF and EAL domain-dependent modulation of the intracellular level of cyclic diguanylate. Environ Microbiol 9 2475 2485
12. HickmanJW
TifreaDF
HarwoodCS
2005 A chemosensory system that regulates biofilm formation through modulation of cyclic diguanylate levels. Proc Natl Acad Sci U S A 102 14422 14427
13. StarkeyM
HickmanJH
MaL
ZhangN
De LongS
2009 Pseudomonas aeruginosa rugose small colony variants have adaptations likely to promote persistence in the cystic fibrosis lung. J Bacteriol 191 3492 3503
14. JenalU
2004 Cyclic di-guanosine-monophosphate comes of age: a novel secondary messenger involved in modulating cell surface structures in bacteria? Curr Opin Microbiol 7 185 191
15. GalperinMY
2005 A census of membrane-bound and intracellular signal transduction proteins in bacteria: bacterial IQ, extroverts and introverts. BMC Microbiol 5 35
16. RossP
WeinhouseH
AloniY
MichaeliD
Weinberger-OhanaP
1987 Regulation of cellulose synthesis in Acetobacter xylinum by cyclic diguanylic acid. Nature 325 279 281
17. SchmidtAJ
RyjenkovDA
GomelskyM
2005 The ubiquitous protein domain EAL is a cyclic diguanylate-specific phosphodiesterase: enzymatically active and inactive EAL domains. J Bacteriol 187 4774 4781
18. PaulR
WeiserS
AmiotNC
ChanC
SchirmerT
2004 Cell cycle-dependent dynamic localization of a bacterial response regulator with a novel di-guanylate cyclase output domain. Genes Dev 18 715 727
19. ChristenM
ChristenB
FolcherM
SchauerteA
JenalU
2005 Identification and Characterization of a Cyclic di-GMP-specific Phosphodiesterase and Its Allosteric Control by GTP. J Biol Chem 280 30829 30837
20. RyjenkovDA
TarutinaM
MoskvinOV
GomelskyM
2005 Cyclic Diguanylate Is a Ubiquitous Signaling Molecule in Bacteria: Insights into Biochemistry of the GGDEF Protein Domain. J Bacteriol 187 1792 1798
21. ChristenB
ChristenM
PaulR
SchmidF
FolcherM
2006 Allosteric control of cyclic di-GMP signaling. J Biol Chem 281 32015 32024
22. GjermansenM
RagasP
SternbergC
MolinS
Tolker-NielsenT
2005 Characterization of starvation-induced dispersion in Pseudomonas putida biofilms. Environ Microbiol 7 894 906
23. SpiersAJ
BohannonJ
GehrigSM
RaineyPB
2003 Biofilm formation at the air-liquid interface by the Pseudomonas fluorescens SBW25 wrinkly spreader requires an acetylated form of cellulose. Mol Microbiol 50 15 27
24. SpiersAJ
KahnSG
BohannonJ
TravisanoM
RaineyPB
2002 Adaptive Divergence in Experimental Populations of Pseudomonas fluorescens. I. Genetic and Phenotypic Bases of Wrinkly Spreader Fitness. Genetics 161 33 46
25. LeeVT
MatewishJM
KesslerJL
HyodoM
HayakawaY
LoryS
2007 A cyclic-di-GMP receptor required for bacterial exopolysaccharide production. Mol Microbiol 65 1474 1484
26. KulasekaraHD
VentreI
KulasekaraBR
LazdunskiA
FillouxA
2005 A novel two-component system controls the expression of Pseudomonas aeruginosa fimbrial cup genes. Mol Microbiol 55 368 380
27. KazmierczakBI
LebronMB
MurrayTS
2006 Analysis of FimX, a phosphodiesterase that governs twitching motility in Pseudomonas aeruginosa. Mol Microbiol 60 1026 1043
28. SimmR
MorrM
KaderA
NimtzM
RomlingU
2004 GGDEF and EAL domains inversely regulate cyclic di-GMP levels and transition from sessility to motility. Mol Microbiol 53 1123 1134
29. KulasakaraH
LeeV
BrencicA
LiberatiN
UrbachJ
2006 Analysis of Pseudomonas aeruginosa diguanylate cyclases and phosphodiesterases reveals a role for bis-(3′-5′)-cyclic-GMP in virulence. Proc Natl Acad Sci U S A 103 2839 2844
30. TischlerAD
CamilliA
2005 Cyclic diguanylate regulates Vibrio cholerae virulence gene expression. Infect Immun 73 5873 5882
31. MerighiM
LeeVT
HyodoM
HayakawaY
LoryS
2007 The second messenger bis-(3′-5′)-cyclic-GMP and its PilZ domain-containing receptor Alg44 are required for alginate biosynthesis in Pseudomonas aeruginosa. Mol Microbiol 65 876 895
32. HickmanJW
HarwoodCS
2008 Identification of FleQ from Pseudomonas aeruginosa as a c-di-GMP-responsive transcription factor. Mol Microbiol 69 376 389
33. KuchmaSL
ConnollyJP
O'TooleGA
2005 A three-component regulatory system regulates biofilm maturation and type III secretion in Pseudomonas aeruginosa. J Bacteriol 187 1441 1454
34. HuangB
WhitchurchCB
MattickJS
2003 FimX, a multidomain protein connecting environmental signals to twitching motility in Pseudomonas aeruginosa. J Bacteriol 185 7068 7076
35. AlmRA
BoderoAJ
FreePD
MattickJS
1996 Identification of a novel gene, pilZ, essential for type 4 fimbrial biogenesis in Pseudomonas aeruginosa. J Bacteriol 178 46 53
36. MerrittJH
BrothersKM
KuchmaSL
O'TooleGA
2007 SadC Reciprocally Influences Biofilm Formation and Swarming Motility via Modulation of Exopolysaccharide Production and Flagellar Function. J Bacteriol 189 8154 8164
37. BantinakiE
KassenR
KnightCG
RobinsonZ
SpiersAJ
2007 Adaptive divergence in experimental populations of Pseudomonas fluorescens. III. Mutational origins of wrinkly spreader diversity. Genetics 176 441 453
38. GuvenerZT
HarwoodCS
2007 Subcellular location characteristics of the Pseudomonas aeruginosa GGDEF protein, WspR, indicate that it produces cyclic-di-GMP in response to growth on surfaces. Mol Microbiol 66 1459 1473
39. GallagherLA
ManoilC
2001 Pseudomonas aeruginosa PAO1 kills Caenorhabditis elegans by cyanide poisoning. J Bacteriol 183 6207 6214
40. CaiazzaNC
O'TooleGA
2004 SadB is required for the transition from reversible to irreversible attachment during biofilm formation by Pseudomonas aeruginosa PA14. J Bacteriol 186 4476 4485
41. CaiazzaNC
MerrittJH
BrothersKM
O'TooleGA
2007 Inverse regulation of biofilm formation and swarming motility by Pseudomonas aeruginosa PA14. J Bacteriol 189 3603 3612
42. KuchmaSL
BrothersKM
MerrittJH
LiberatiNT
AusubelFM
2007 BifA, a Cyclic-Di-GMP Phosphodiesterase, Inversely Regulates Biofilm Formation and Swarming Motility by Pseudomonas aeruginosa PA14. J Bacteriol 189 8165 8178
43. HoffmanLR
D'ArgenioDA
MacCossMJ
ZhangZ
JonesRA
2005 Aminoglycoside antibiotics induce bacterial biofilm formation. Nature 436 1171 1175
44. GotohH
ZhangY
DalloSF
HongS
KasaraneniN
2008 Pseudomonas aeruginosa, under DNA replication inhibition, tends to form biofilms via Arr. Res Microbiol 159 294 302
45. KlebensbergerJ
LautenschlagerK
BresslerD
WingenderJ
PhilippB
2007 Detergent-induced cell aggregation in subpopulations of Pseudomonas aeruginosa as a preadaptive survival strategy. Environ Microbiol 9 2247 2259
46. MillerJH
1972 Experiments in molecular genetics. 352 355 Cold Spring Harbor Laboratory, Cold Spring Harbor, New York
47. KovachME
ElzerPH
HillDS
RobertsonGT
FarrisMA
1995 Four new derivatives of the broad-host-range cloning vector pBBR1MCS, carrying different antibiotic-resistance cassettes. Gene 166 175 176
48. GuzmanLM
BelinD
CarsonMJ
BeckwithJ
1995 Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol 177 4121 4130
49. ChoiKH
GaynorJB
WhiteKG
LopezC
BosioCM
2005 A Tn7-based broad-range bacterial cloning and expression system. Nat Methods 2 443 448
50. HoSN
HuntHD
HortonRM
PullenJK
PeaseLR
1989 Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 77 51 59
51. HeebS
ItohY
NishijyoT
SchniderU
KeelC
2000 Small, stable shuttle vectors based on the minimal pVS1 replicon for use in gram-negative, plant-associated bacteria. Mol Plant Microbe Interact 13 232 237
52. AldridgeP
PaulR
GoymerP
RaineyP
JenalU
2003 Role of the GGDEF regulator PleD in polar development of Caulobacter crescentus. Mol Microbiol 47 1695 1708
53. RobertsRC
MohrCD
ShapiroL
1996 Developmental programs in bacteria. Curr Top Dev Biol 34 207 257
54. YuD
EllisHM
LeeEC
JenkinsNA
CopelandNG
2000 An efficient recombination system for chromosome engineering in Escherichia coli. Proc Natl Acad Sci U S A 97 5978 5983
55. UzzauS
Figueroa-BossiN
RubinoS
BossiL
2001 Epitope tagging of chromosomal genes in Salmonella. Proc Natl Acad Sci U S A 98 15264 15269
56. KarimovaG
UllmannA
LadantD
2001 Protein-protein interaction between Bacillus stearothermophilus tyrosyl-tRNA synthetase subdomains revealed by a bacterial two-hybrid system. J Mol Microbiol Biotechnol 3 73 82
57. WinsonMK
SwiftS
HillPJ
SimsCM
GriesmayrG
1998 Engineering the luxCDABE genes from Photorhabdus luminescens to provide a bioluminescent reporter for constitutive and promoter probe plasmids and mini-Tn5 constructs. FEMS Microbiol Lett 163 193 202
58. ValletI
DiggleSP
StaceyRE
CamaraM
VentreI
2004 Biofilm formation in Pseudomonas aeruginosa: fimbrial cup gene clusters are controlled by the transcriptional regulator MvaT. J Bacteriol 186 2880 2890
59. VentreI
GoodmanAL
Vallet-GelyI
VasseurP
SosciaC
2006 Multiple sensors control reciprocal expression of Pseudomonas aeruginosa regulatory RNA and virulence genes. Proc Natl Acad Sci U S A 103 171 176
60. MorganAF
1979 Transduction of Pseudomonas aeruginosa with a mutant of bacteriophage E79. J Bacteriol 139 137 140
61. O'TooleGA
KolterR
1998 Initiation of biofilm formation in Pseudomonas fluorescens WCS365 proceeds via multiple, convergent signalling pathways: a genetic analysis. Mol Microbiol 28 449 461
62. ChoiKH
SchweizerHP
2005 An improved method for rapid generation of unmarked Pseudomonas aeruginosa deletion mutants. BMC Microbiol 5 30
63. MerrittJH
KadouriDE
O'TooleGA
2005 Growing and analyzing static biofilms. Curr Protoc Microbiol Chapter 1: Unit 1B 1
64. RabinowitzJD
KimballE
2007 Acidic acetonitrile for cellular metabolome extraction from Escherichia coli. Anal Chem 79 6167 6173
65. MichaelisS
InouyeH
OliverD
BeckwithJ
1983 Mutations that alter the signal sequence of alkaline phosphatase in Escherichia coli. J Bacteriol 154 366 374
66. KarimovaG
PidouxJ
UllmannA
LadantD
1998 A bacterial two-hybrid system based on a reconstituted signal transduction pathway. Proc Natl Acad Sci U S A 95 5752 5756
67. WagnerT
SoongG
SokolS
SaimanL
PrinceA
2005 Effects of azithromycin on clinical isolates of Pseudomonas aeruginosa from cystic fibrosis patients. Chest 128 912 919
68. KristianSA
BirkenstockTA
SauderU
MackD
GotzF
2008 Biofilm formation induces C3a release and protects Staphylococcus epidermidis from IgG and complement deposition and from neutrophil-dependent killing. J Infect Dis 197 1028 1035
69. ParsonsLM
LinF
OrbanJ
2006 Peptidoglycan recognition by Pal, an outer membrane lipoprotein. Biochemistry 45 2122 2128
70. BendtsenJD
NielsenH
von HeijneG
BrunakS
2004 Improved prediction of signal peptides: SignalP 3.0. J Mol Biol 340 783 795
71. SchultzJ
MilpetzF
BorkP
PontingCP
1998 SMART, a simple modular architecture research tool: identification of signaling domains. Proc Natl Acad Sci U S A 95 5857 5864
72. SodingJ
BiegertA
LupasAN
2005 The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res 33 W244 248
73. DuerigA
AbelS
FolcherM
NicollierM
SchwedeT
2009 Second messenger-mediated spatiotemporal control of protein degradation regulates bacterial cell cycle progression. Genes Dev 23 93 104
74. PaulR
AbelS
WassmannP
BeckA
HeerklotzH
2007 Activation of the diguanylate cyclase PleD by phosphorylation-mediated dimerization. J Biol Chem 282 29170 29177
75. WassmannP
ChanC
PaulR
BeckA
HeerklotzH
2007 Structure of BeF3 - -modified response regulator PleD: implications for diguanylate cyclase activation, catalysis, and feedback inhibition. Structure 15 915 927
76. DeN
PirruccelloM
KrastevaPV
BaeN
RaghavanRV
2008 Phosphorylation-independent regulation of the diguanylate cyclase WspR. PLoS Biol 6 e67 doi:10.1371/journal.pbio.0060067
77. FriedmanL
KolterR
2004 Two genetic loci produce distinct carbohydrate-rich structural components of the Pseudomonas aeruginosa biofilm matrix. J Bacteriol 186 4457 4465
78. LeidJG
WillsonCJ
ShirtliffME
HassettDJ
ParsekMR
2005 The exopolysaccharide alginate protects Pseudomonas aeruginosa biofilm bacteria from IFN-gamma-mediated macrophage killing. J Immunol 175 7512 7518
79. ConwayBA
ChuKK
BylundJ
AltmanE
SpeertDP
2004 Production of exopolysaccharide by Burkholderia cenocepacia results in altered cell-surface interactions and altered bacterial clearance in mice. J Infect Dis 190 957 966
80. GodlewskaR
WisniewskaK
PietrasZ
Jagusztyn-KrynickaEK
2009 Peptidoglycan-associated lipoprotein (Pal) of Gram-negative bacteria: function, structure, role in pathogenesis and potential application in immunoprophylaxis. FEMS Microbiol Lett 298 1 11
81. UedaA
WoodTK
2009 Connecting quorum sensing, c-di-GMP, pel polysaccharide, and biofilm formation in Pseudomonas aeruginosa through tyrosine phosphatase TpbA (PA3885). PLoS Pathog 5 e1000483 doi:10.1371/journal.ppat.1000483
82. BurrowesE
BaysseC
AdamsC
O'GaraF
2006 Influence of the regulatory protein RsmA on cellular functions in Pseudomonas aeruginosa PAO1, as revealed by transcriptome analysis. Microbiology 152 405 418
83. TschowriN
BusseS
HenggeR
2009 The BLUF-EAL protein YcgF acts as a direct anti-repressor in a blue-light response of Escherichia coli. Genes Dev 23 522 534
84. MaL
ConoverM
LuH
ParsekMR
BaylesK
2009 Assembly and development of the Pseudomonas aeruginosa biofilm matrix. PLoS Pathog 5 e1000354 doi:10.1371/journal.ppat.1000354
85. CannonGJ
SwansonJA
1992 The macrophage capacity for phagocytosis. J Cell Sci 101 (Pt 4) 907 913
86. SpeertDP
WrightSD
SilversteinSC
MahB
1988 Functional characterization of macrophage receptors for in vitro phagocytosis of unopsonized Pseudomonas aeruginosa. J Clin Invest 82 872 879
87. GilbertP
DasJ
FoleyI
1997 Biofilm susceptibility to antimicrobials. Adv Dent Res 11 160 167
88. GordonCA
HodgesNA
MarriottC
1988 Antibiotic interaction and diffusion through alginate and exopolysaccharide of cystic fibrosis-derived Pseudomonas aeruginosa. J Antimicrob Chemother 22 667 674
89. SpoeringAL
LewisK
2001 Biofilms and planktonic cells of Pseudomonas aeruginosa have similar resistance to killing by antimicrobials. J Bacteriol 183 6746 6751
90. GilbertP
CollierPJ
BrownMR
1990 Influence of growth rate on susceptibility to antimicrobial agents: biofilms, cell cycle, dormancy, and stringent response. Antimicrob Agents Chemother 34 1865 1868
91. GirgisHS
LiuY
RyuWS
TavazoieS
2007 A comprehensive genetic characterization of bacterial motility. PLoS Genet 3 e154 doi:10.1371/journal.pgen.0030154
92. GiddensSR
JacksonRW
MoonCD
JacobsMA
ZhangXX
2007 Mutational activation of niche-specific genes provides insight into regulatory networks and bacterial function in a complex environment. Proc Natl Acad Sci U S A 104 18247 18252
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2010 Číslo 3
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Měli bychom postcovidový syndrom léčit antidepresivy?
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- Kaposi's Sarcoma-Associated Herpesvirus ORF57 Protein Binds and Protects a Nuclear Noncoding RNA from Cellular RNA Decay Pathways
- Endocytosis of the Anthrax Toxin Is Mediated by Clathrin, Actin and Unconventional Adaptors
- Perforin and IL-2 Upregulation Define Qualitative Differences among Highly Functional Virus-Specific Human CD8 T Cells
- Exoerythrocytic Parasites Secrete a Cysteine Protease Inhibitor Involved in Sporozoite Invasion and Capable of Blocking Cell Death of Host Hepatocytes