
Suboptimal Activation of Antigen-Specific CD4
Effector Cells Enables Persistence of In
Vivo
Adaptive immunity to Mycobacterium tuberculosis controls
progressive bacterial growth and disease but does not eradicate infection. Among
CD4+ T cells in the lungs of M.
tuberculosis-infected mice, we observed that few produced IFN-γ
without ex vivo restimulation. Therefore, we hypothesized that one mechanism
whereby M. tuberculosis avoids elimination is by limiting
activation of CD4+ effector T cells at the site of infection in
the lungs. To test this hypothesis, we adoptively transferred Th1-polarized
CD4+ effector T cells specific for M.
tuberculosis Ag85B peptide 25 (P25TCRTh1 cells), which trafficked
to the lungs of infected mice and exhibited antigen-dependent IFN-γ
production. During the early phase of infection, ∼10% of P25TCRTh1
cells produced IFN-γ in vivo; this declined to <1% as infection
progressed to chronic phase. Bacterial downregulation of fbpB
(encoding Ag85B) contributed to the decrease in effector T cell activation in
the lungs, as a strain of M. tuberculosis engineered to express
fbpB in the chronic phase stimulated P25TCRTh1 effector
cells at higher frequencies in vivo, and this resulted in CD4+ T
cell-dependent reduction of lung bacterial burdens and prolonged survival of
mice. Administration of synthetic peptide 25 alone also increased activation of
endogenous antigen-specific effector cells and reduced the bacterial burden in
the lungs without apparent host toxicity. These results indicate that
CD4+ effector T cells are activated at suboptimal
frequencies in tuberculosis, and that increasing effector T cell activation in
the lungs by providing one or more epitope peptides may be a successful strategy
for TB therapy.
Published in the journal:
. PLoS Pathog 7(5): e32767. doi:10.1371/journal.ppat.1002063
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1002063
Summary
Adaptive immunity to Mycobacterium tuberculosis controls
progressive bacterial growth and disease but does not eradicate infection. Among
CD4+ T cells in the lungs of M.
tuberculosis-infected mice, we observed that few produced IFN-γ
without ex vivo restimulation. Therefore, we hypothesized that one mechanism
whereby M. tuberculosis avoids elimination is by limiting
activation of CD4+ effector T cells at the site of infection in
the lungs. To test this hypothesis, we adoptively transferred Th1-polarized
CD4+ effector T cells specific for M.
tuberculosis Ag85B peptide 25 (P25TCRTh1 cells), which trafficked
to the lungs of infected mice and exhibited antigen-dependent IFN-γ
production. During the early phase of infection, ∼10% of P25TCRTh1
cells produced IFN-γ in vivo; this declined to <1% as infection
progressed to chronic phase. Bacterial downregulation of fbpB
(encoding Ag85B) contributed to the decrease in effector T cell activation in
the lungs, as a strain of M. tuberculosis engineered to express
fbpB in the chronic phase stimulated P25TCRTh1 effector
cells at higher frequencies in vivo, and this resulted in CD4+ T
cell-dependent reduction of lung bacterial burdens and prolonged survival of
mice. Administration of synthetic peptide 25 alone also increased activation of
endogenous antigen-specific effector cells and reduced the bacterial burden in
the lungs without apparent host toxicity. These results indicate that
CD4+ effector T cells are activated at suboptimal
frequencies in tuberculosis, and that increasing effector T cell activation in
the lungs by providing one or more epitope peptides may be a successful strategy
for TB therapy.
Introduction
Even though its etiologic agent was discovered over 125 years ago, tuberculosis remains a global scourge, killing 1.7 million people in 2009, at least ¾ of whom were immunocompetent [1]. Long-term persistence of Mycobacterium tuberculosis, which resides principally in phagocytic cells within the lungs, results in a chronic infection despite the presence of an apparently appropriate adaptive immune response. In mice infected with virulent M. tuberculosis, the early phase of infection proceeds with unchecked bacterial growth until day 17–21 post-infection, when adaptive immunity finally exerts control of bacterial growth in the lungs. Control of infection in both humans and mice critically depends on M. tuberculosis-specific CD4+ Th1 cell responses, which include production of IFN-γ [2], [3]; however adaptive immune responses do not eradicate the infection.
Several potential mechanisms may account for the failure of adaptive immune responses to eradicate the bacteria in tuberculosis. Generation of M. tuberculosis-specific CD4+ effector T cells is delayed compared with responses to other pathogens [2], [4]. In addition, certain individuals, or strains of mice, may develop inappropriate (e.g., Th2) [5], [6] or imbalanced effector phenotypes such as Th1/Th17 [7] in response to infection. However, even in humans or mice that develop Th1 responses, a failure of CD4+ effector T cells to recognize infected cells may preclude their optimal activation and limit induction of effector functions in the lungs. Prevention of effector T cell activation could result from impaired antigen presentation by lung APCs containing M. tuberculosis [8], [9], [10] or because the antigens that effector T cells recognize are not expressed or otherwise available in the lungs. Furthermore, host regulatory mechanisms that limit immune pathology, such as T regulatory cells [11], production of inhibitory cytokines [12], and, possibly, onset of T cell exhaustion [13], [14] may inhibit the activity of effector T cells at the site of infection. Finally, even when CD4+ effector T cells are activated, the efficacy of these responses may be limited by the impaired ability of infected cells to respond to IFN-γ [15], [16], [17], induce phagosome maturation [18], [19], or undergo apoptosis [9], [20], [21], [22]. Understanding the contribution of each of these potential mechanisms limiting adaptive immunity to M. tuberculosis is an essential prerequisite for vaccine design and other immunologic approaches to tuberculosis prevention and therapy.
Here, we report that CD4+ effector T cells are activated at submaximal and suboptimal frequencies in the lungs during M. tuberculosis infection, that this is due in part to bacterial modulation of antigen expression, and that increasing the availability of a single antigen results in improved immune control of M. tuberculosis.
Results
Prevalence of CD4+ T cells activated to produce IFN-γ in the lungs of M. tuberculosis-infected mice
We hypothesized that M. tuberculosis evades adaptive immunity by modulating the activation of CD4+ effector T cells at the site of infection in the lungs. Since in vitro studies have revealed evidence that M. tuberculosis modulates MHC class II antigen presentation [10], [23], [24], [25], [26], we focused on in vivo activation of CD4+ T cells in the lungs. We reasoned that, if M. tuberculosis-infected cells do not present antigens efficiently to effector T cells in the lungs, then the frequency of activation of effector functions of CD4+ cells would also be low at the site of infection. To test this, we used direct intracellular cytokine staining of lung cells from infected mice for IFN-γ, without ex vivo restimulation. We found that that the frequency of IFN-γ expression by CD4+ T cells in the lungs varied with the time of infection (Figure 1B). IFN-γ+ CD4+ cells were undetectable in the lungs at day 14, increased in frequency beginning by day 21 to a peak at day 35 post-infection, and then markedly decreased afterward; no more than 7% of the bulk population of CD4+ T cells expressed IFN-γ at any time point after infection, and fewer than 1% expressed IFN-γ during the chronic phase. Other studies investigating IFN-γ production by CD8+ T cells in vivo have used treatment of mice with brefeldin A or inclusion of brefeldin A during cell isolation and staining [27], [28]. However, we determined that these methods did not improve detection of intracellular IFN-γ by CD4+ T cells during M. tuberculosis infection (Figure S1). These data indicate that a small minority of polyclonal CD4+ T cells recruited to the lungs of M. tuberculosis-infected mice are activated to produce IFN-γ at a given time, and are consistent with defective antigen presentation, costimulation, and/or inhibition of effector T cell activation at the site of infection.

Quantitating antigen-specific effector Th1 cell responses in the lungs of M. tuberculosis-infected mice
Since the low frequency of CD4+ T cell expression of IFN-γ in the lungs of M. tuberculosis-infected mice could be due to the presence of effector cells that traffic to the lungs but are not specific for M. tuberculosis antigens, we performed the remainder of our studies using CD4+ TCR transgenic T cells that specifically recognize a well-characterized immunodominant M. tuberculosis antigen. To quantitate the frequency of activation of M. tuberculosis antigen-specific effector cells in the lungs, we prepared CD4+ Th1 effector cells (P25TCRTh1 cells) from transgenic mice with a TCR specific for peptide 25 (amino acids 240–254) of Ag85B. When P25TCRTh1 cells were incubated with irradiated splenocytes in the absence of peptide 25, <1.0% of the cells expressed IFN-γ as detected by intracellular staining and flow cytometry, whereas addition of peptide 25 in vitro induced IFN-γ expression in ∼90% of cells (Figure 2A). This result demonstrated that the frequency of IFN-γ staining in P25TCRTh1 cells can specifically assay antigen dependent stimulation of P25TCRTh1 cells.

P25TCRTh1 cells recognize antigen at low frequency in vivo
Since Day 21 post-infection corresponds to an acute stage of infection when adaptive immune effector mechanisms have been initiated and reduce the rate of bacterial population growth in the lungs, and since it resembles the stage of LCMV infection in which a high frequency of antigen-specific CD8+ T cell responses are observed [28], we chose this time point for initial characterization of P25TCRTh1 cell responses in vivo. We verified that adoptively transferred P25TCRTh1 cells traffic to the site of infection by examining sections of lungs from infected mice that had received CFP+ P25TCRTh1 cells. CFP+ cells were abundant in the lung parenchyma, and were concentrated in granulomas (Figure 2B). Furthermore, we determined that >85% of the transferred cells were protected from labelling by an i.v. injection of PerCP-labeled anti-CD4 antibody, indicating that adoptively transferred P25TCRTh1 cells efficiently migrate out of the lung vasculature into the parenchyma of infected lungs (Figure S2A).
To determine the frequency of activation of antigen-specific CD4+ effector T cells in the lungs early in infection, we adoptively transferred P25TCRTh1 cells on day 18 and harvested them on day 21 after infection of wild-type mice with wild-type M. tuberculosis H37Rv. The frequency of IFN-γ+ P25TCRTh1 cells isolated from the lungs was unexpectedly low at Day 21 post-infection (Figure 2C and 2D). Approximately 1–2% of the transferred P25TCRTh1 cells were stimulated to produce IFN-γ in vivo at that time point (Figure 2C), and this percentage was similar to the frequency of total endogenous lung CD4+ T cells expressing IFN-γ on day 21 post-infection (Figure 1B, 2D). Moreover, after intravenous injection of PerCP-labeled anti-CD4 antibody, the only IFN-γ+ P25TCRTh1 cells identified were PerCP negative (Figure S2B), indicating that the responding cells were those that had migrated out of the vasculature into the lung parenchyma and were protected from staining by the in vivo injection of antibody.
We verified that stimulation of P25TCRTh1 cells to express intracellular IFN-γ is due to recognition of Ag85B peptide 25 by transferring P25TCRTh1 cells into mice infected with an Ag85B-null strain of M. tuberculosis (ΔAg85B), which is equivalent to wild-type H37Rv in virulence [2]. A lower mean percentage (0.74%) of P25TCRTh1 cells isolated from ΔAg85B-infected mice expressed IFN-γ than those from H37Rv-infected mice (Figure 2C and 2D). This indicates that in vivo IFN-γ production by P25TCRTh1 cells is antigen-dependent and not the consequence of inflammatory cytokines present at the site of infection. We also evaluated several alternative approaches to detecting effector T cell activation in the lungs. P25TCRTh1 cells expressed both CD25 and CD44 prior to adoptive transfer, which excluded their use in evaluating effector cell activation in vivo. Surface expression of CD69 was induced after adoptive transfer of P25TCRTh1 effector cells into H37Rv-infected mice; however, we found similar induction of CD69 in mice infected with ΔAg85B, indicating that it did not specifically reflect antigen-dependent effector cell activation. This result, together with evidence that CD69 can be induced by costimulation and by certain cytokines present at the site of M. tuberculosis infection [29], [30], [31], indicates that expression of intracellular IFN-γ is the most accurate reporter of antigen specific Th1 effector cell activation in the lungs. Together, these results indicate that even though they traffic efficiently to the site of infection, Ag85B peptide 25-specific CD4+ effector cells are activated to execute their Th1 effector function at low frequency in the lungs of M. tuberculosis-infected mice.
P25TCRTh1 cells are capable of responding to antigen at the site of infection
Although IFN-γ production by P25TCRTh1 cells at day 21 was dependent on Ag85B, the frequency of IFN-γ+ cells was surprisingly low in H37Rv infected mice. One possible explanation for the low frequency of activation of effector cells is that their cognate antigen is not available for recognition at the site of infection. To test this hypothesis, we provided antigen in vivo by injecting peptide 25 intravenously into mice that had been infected 21 days earlier. When P25TCRTh1 recipient, H37Rv-infected mice received peptide 25 six hours prior to lung cell harvest, the frequency of IFN-γ+ P25TCRTh1 cells increased to 20–50% (Figure 2C and 2D). Similarly, peptide 25 injection stimulated a higher frequency of IFN-γ expression by endogenous CD4+ T cells from mice infected with H37Rv (Figure 2C and 2D), consistent with prior evidence that peptide 25 of Ag85B is a dominant antigen in C57BL/6 mice infected with M. tuberculosis [32], [33]. P25TCRTh1 cells transferred into ΔAg85B-infected recipients were also stimulated at a higher frequency after intravenous peptide 25 treatment, while endogenous CD4+ T cells from ΔAg85B-infected mice did not respond to peptide 25 with increased IFN-γ expression (Figure 2D). The failure of endogenous CD4+ T cells from ΔAg85B-infected mice to respond to peptide 25 injection reflects the absence of Ag85B peptide 25-specific effector T cells generated in response to this infection. These results indicate that the frequency of IFN-γ+ P25TCRTh1 cells is an accurate and specific measure of CD4+ effector T cell stimulation in response to presentation of Ag85B peptide 25 in vivo. The observation that in vivo IFN-γ responses to peptide 25 injection depend on the presence of previously-generated (endogenous or transferred) peptide 25-specific effector T cells indicates that the responses are not due to a nonspecific effect of the epitope peptide on costimulation or responses of CD4+ T cells with specificity for other antigens. In addition, they demonstrate that if antigen is made available to them, adoptively transferred P25TCRTh1 cells can respond to antigen in the infected lungs, and they provide evidence against an exclusive role for T regulatory cells and/or suppressive cytokines in limiting the activation of CD4+ effector cells at the site of M. tuberculosis infection in the lungs.
To further characterize the in vivo assay system, and to evaluate the possibility that low frequencies of P25TCRTh1 responses are attributable to either competition for antigen by endogenous CD4+ T cells and/or a dominant effect of T regulatory cells, we specifically ablated endogenous T cells from M. tuberculosis-infected CD4-DTR mice [34] prior to assaying P25TCRTh1 responses in vivo. Compared to untreated mice, DT treatment reduced the fraction of endogenous CD4+ T cells in the lung by an average of 48.9%, p = 0.0053 (Figure S3A). However, this had no effect on the percentage of P25TCRTh1 cells activated to produce IFN-γ (Figure S3B). These results strongly suggest that the low frequency of activation of P25TCRTh1 cells is caused neither by competition for peptide 25:MHC II complexes by endogenous CD4+ T cells, nor by the influence of T regulatory cells in the lungs. We therefore conclude that the response of adoptively transferred P25TCRTh1 cells is an accurate reflection of MHC II presentation of Ag85B peptide 25 by lung APCs during infection.
Dynamics of M. tuberculosis-specific CD4+ effector T cell responses during the course of infection
Adaptive immunity restricts progressive growth of M. tuberculosis, but it does not eliminate the bacteria from the lungs, which results in chronic infection in mice and latent infection in humans. To determine whether suboptimal activation of M. tuberculosis-specific T cells contributes to the ability of the bacteria to persist, we first asked whether activation of P25TCRTh1 cells in the lungs changes as infection progresses to a chronic phase. To compare the frequency of effector T cell stimulation at various stages of infection, we transferred P25TCRTh1 cells into H37Rv-infected mice on day 11, 18, 25, 32, or 39 post-infection. Lung cells were harvested 72 hours after transfer (day 14, 21, 28, 35, or 42 post-infection) and analyzed by flow cytometry for intracellular IFN-γ without ex vivo restimulation. The proportion of P25TCRTh1 cells producing IFN-γ was highest (∼10%) on day 14 (Figure 3A and 3C). These results indicate that during the acute stage of infection, adoptively transferred P25TCRTh1 cells are stimulated in the lungs at a frequency comparable to that of TCR transgenic CD4+ effector cells at the site of injection of a protein antigen and adjuvant [35]. In contrast, expression of IFN-γ by endogenous (CD45.2−) CD4+ cells was rare (<0.1%) at that time point (Figure 1B and 1C). The difference between transferred and endogenous cell responses on day 14 is consistent with our previous observation that initiation of adaptive immunity to M. tuberculosis is delayed until day 11–14 post-infection, and consequently, endogenous CD4+ effector T cells specific for M. tuberculosis antigens are first detected in the lungs on day 17 post-infection. [2].

The frequency of IFN-γ production by P25TCRTh1 cells progressively decreased from day 14 to day 42 post-infection, indicating a decrease in the efficiency of peptide 25-specific T cell stimulation as infection enters its chronic phase (Figure 3A and 3C). These results with TCR transgenic CD4+ effector cells closely mimic the results observed with endogenous polyclonal CD4+ T cells after day 14 post-infection (Figure 1B and 1C). Although Ag85B peptide 25-specific responses reached an earlier peak and decreased earlier than did those of endogenous polyclonal CD4+ T cell responses, the results with the two cell populations were similar, with endogenous CD4+ effector T cell responses also diminishing by day 42 post-infection.
To determine whether activation of naïve Ag85B peptide 25-specific CD4+ T cells is also diminished in the later stages of M. tuberculosis infection, we assayed the response of adoptively transferred naïve P25 TCR-Tg T cells in the lung-draining mediastinal lymph nodes of H37Rv-infected mice at various time points post-infection. 7 days after transfer, we harvested lymph node cells and measured in vivo T cell proliferation by flow cytometry using a CFSE dilution assay. The rate of naïve P25TCR-tg T cells was highest upon transfer into mice on day 18 post-infection, while fewer cells exhibited CFSE dilution at days 24 and 48 post-infection (Figure 3B). These results indicate that decreased stimulation of P25TCRTh1 effector cells is also accompanied by decreased generation of peptide 25 specific effector T cells from naive cells at later stages of infection.
Progressive decreases of P25TCRTh1 cell activation accompany decreased fbpB expression
Since treatment of infected mice with exogenous peptide 25 enhanced T cell responses, indicating that adoptively-transferred P25TCRTh1 cells are capable of responding to antigen stimulation in the lungs, we hypothesized that availability and/or presentation of antigen is a limiting factor in the activation of CD4+ effector T cells at the site of M. tuberculosis infection. To test this hypothesis, we first investigated whether changes in the expression of the M. tuberculosis gene that encodes Ag85B influence the frequency of activation of P25TCRTh1 effector cells. We found that the frequency of in vivo activation of P25TCRTh1 cells mimicked the temporal pattern of expression of fbpB (which encodes Ag85B) by M. tuberculosis in vivo (Figure 3C). This suggests that reduced expression of Ag85B contributes to the low frequency of activation of Ag85B-specific CD4+ effector cells in the lungs, thus resembling previously-reported observations with Salmonella FliC expression and FliC-specific CD4+ T cell responses [36].
Forced expression of fbpB induces greater P25TCRTh1 cell activation
To test the hypothesis that fbpB down-regulation contributes to the submaximal frequency of CD4+ effector cell activation and the limited efficacy of the Th1 response in vivo, we constructed a recombinant strain of M. tuberculosis to express fbpB at high levels during chronic infection. Using the ΔAg85B strain as a background, we introduced a wild-type fbpB allele under control of the hspX/acr/Rv2031c promoter to the M. tuberculosis chromosome via the pMV306 integrating vector. hspX is expressed at high levels during chronic phase infection in an expression pattern inverse to fbpB [37], [38]. This strain (hspXp:fbpB, termed “CPE85B” for chronic phase expressed Ag85B) exhibited higher fbpB expression compared to H37Rv in the lungs of mice after aerosol infection (Figure 4A). The expression of fbpB measured by RT-qPCR was approximately 10-fold higher (normalized for the abundance of 16S rRNA) at day 21 post-infection for CPE85B than for H37Rv. As the infection progressed to chronic phase (day 28–42 post-infection), fbpB expression from the native promoter declined by approximately 100-fold while fbpB expression driven by the hpsX promoter remained at nearly constant, higher levels (Figure 4A). Increased fbpB gene expression in the CPE85B strain was accompanied by markedly enhanced expression and secretion of Ag85B protein when the hspX promoter was induced in stationary liquid culture (Figure 4B).
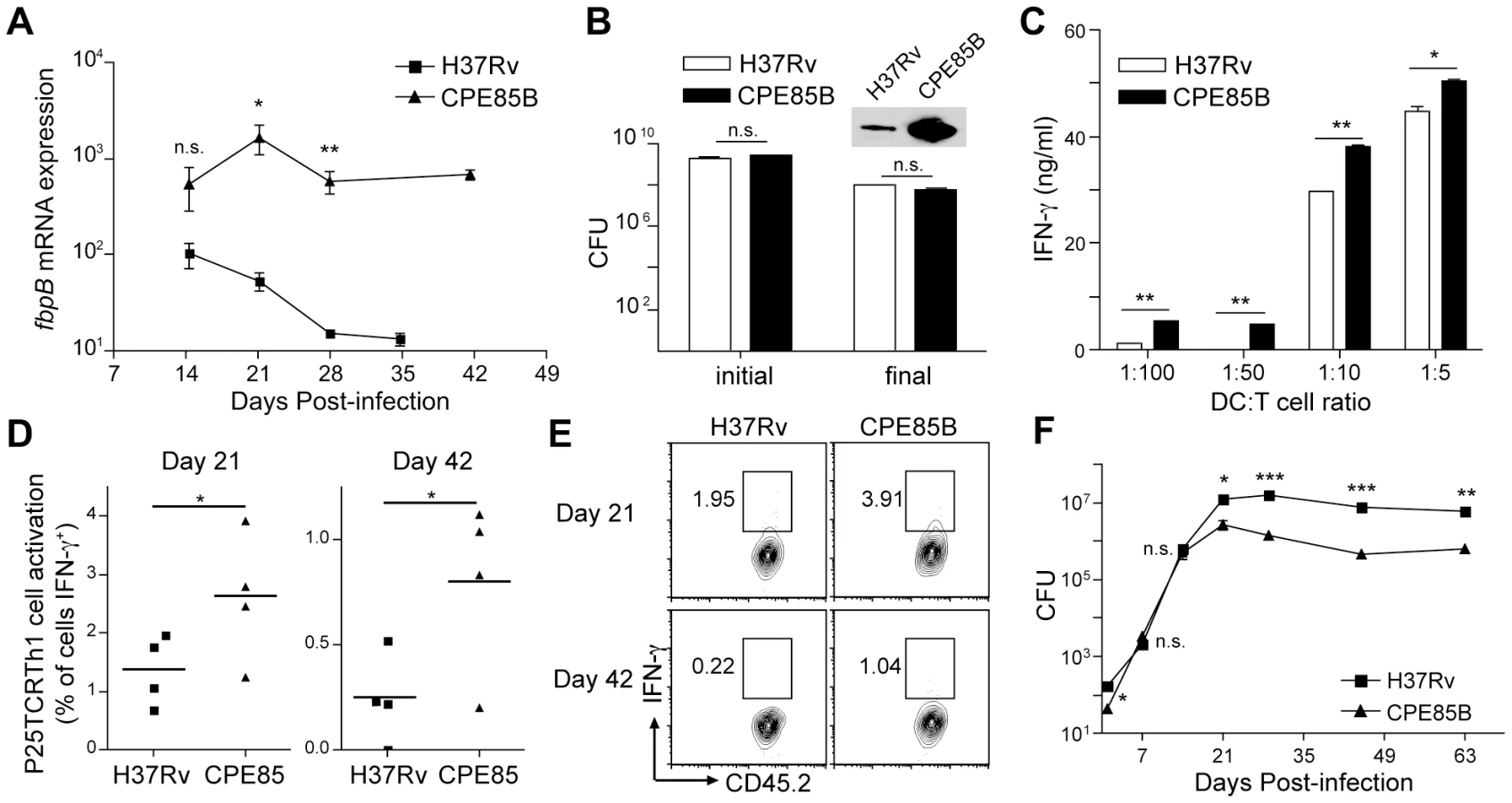
To determine whether forced expression of fbpB in M. tuberculosis results in increased presentation of Ag85B peptide 25 to CD4+ T cells, we infected bone marrow-derived dendritic cells (BMDC) with either H37Rv or CPE85B and compared their ability to activate P25TCRTh1 cells in culture. At all APC∶T cell ratios examined, DCs infected with CPE85B induced significantly greater amounts of IFN-γ secretion from P25TCRTh1 cells than did DCs infected with H37Rv (Figure 4C). To determine whether forced expression of fbpB can increase the frequency of P25TCRTh1 stimulation during H37Rv infection in vivo, we compared the frequency of P25TCRTh1 cell activation in the lungs of mice infected with either H37Rv or CPE85B. Compared to cells from H37Rv-infected recipients, P25TCRTh1 cells from CPE85B-infected mice produced IFN-γ with a 2-fold (day 21) to 5-fold (day 42) higher frequency (Figure 4D and E). These findings indicate that forced expression of fbpB by M. tuberculosis increases the proportion of P25TCRTh1 cells that are activated to produce IFN-γ in the lungs. By suppressing fbpB expression after the initial stages of infection, wild-type M. tuberculosis can reduce the frequency of activation of Ag85B-specific effector T cells. Although expression of fbpB was maintained at high levels from day 14 to day 42 post-infection, P25TCRTh1 cell stimulation in CPE85B-infected mice was only two - to five-fold higher than in mice infected with H37Rv, and decreased as infection progressed to chronic stage, indicating that other mechanisms, such as inhibition of antigen presentation and/or induction of regulatory T cells, exist to limit the activation of CD4+ effector T cells in the lung.
Forced expression of fbpB impairs bacterial persistence during chronic infection
We reasoned that, if diminishing fbpB expression during chronic infection limits effector T cell activation and thereby enables M. tuberculosis to evade adaptive immunity, then constitutive expression of fbpB throughout infection should improve immune control of infection. To test this hypothesis, we infected mice with either H37Rv or CPE85B and quantitated M. tuberculosis CFUs in the lungs throughout the course of infection. The rates of bacterial growth for the two strains were indistinguishable prior to day 14 post infection (Figure 4F), indicating that expression of fbpB by the hspX promoter does not attenuate M. tuberculosis in vivo during the innate immune stage of infection, prior to recruitment of CD4+ effector T cells to the lungs. Indeed, the in vivo generation time of the CPE85B strain (23.0 h) was slightly shorter than that of H37Rv (26.4 h) during days 1–14 of infection (these are not significantly different by nonlinear curve fit and F test). However, at times corresponding to the adaptive immune phase of infection, the bacterial burden of the CPE85B strain in the lungs was approximately 10-fold lower than that of H37Rv (Figure 4F). These results suggest that forced expression of fbpB partially overcomes the antigen deficit that limits the activation of CD4+ T cells in the lung during chronic infection and allows greater antimycobacterial efficacy of the adaptive immune response.
Chronic phase attenuation of CPE85B is dependent on CD4+ T cells
The observation that CPE85B demonstrates a growth pattern indistinguishable from H37Rv during the first two to three weeks of infection, prior to onset of adaptive immunity, suggested that CPE85B was not inherently attenuated for growth in vivo. However, we considered the possibility that over-expression of fbpB could cause attenuation of M. tuberculosis as a result of gene dysregulation or toxicity of an overabundant Ag85B protein. Notably CPE85B demonstrated a similar growth pattern to H37Rv during in vitro shaking culture. Furthermore, under conditions of hypoxic stationary culture, when Ag85B protein is strongly expressed by CPE85B compared to H37Rv, the survival of the CPE85B strain is not impaired compared with that of wild-type bacteria (Figure 4B). Taken together, these findings imply that impaired persistence of M. tuberculosis CPE85B in vivo is the consequence of increased antigen presentation and activation of CD4+ T cells, and not due to intrinsic attenuation of the CPE85B strain in vitro or in vivo. We reasoned that if the decreased lung bacterial burden of CPE85B compared with that of H37Rv is attributable to increased antigen presentation and recognition by CD4+ T cells, then the attenuated phenotype of CPE85B should be abrogated in mice lacking CD4+ T cells. Indeed, whereas wild type C57BL/6 mice infected with CPE85B survived significantly longer than those infected with H37Rv (median survival >300 and 239 days, respectively; p = 0.0062), MHCIIKO mice, which lack CD4+ T cells, exhibited indistinguishable susceptibility to infection with the CPE85B and H37Rv strains (median survival 79 and 81 days, respectively; p = 0.425), (Figure 5A), clearly establishing that in vivo attenuation of the CPE85B strain depends on MHC II antigen presentation and CD4+ T cell responses. These results also indicate that increased antigen expression, accompanied by increased antigen-specific T cell activation, can enhance control of M. tuberculosis without detectable detrimental effects, since wild-type mice infected with the CPE85B strain survived longer than mice infected with H37Rv.

Since MHC II-deficient mice are highly susceptible to M. tuberculosis infection, this could potentially mask any hypothetical CD4+ T cell-independent mechanisms of attenuation of the CPE85B strain. We reasoned that, if mechanisms other than increased CD4+ T cell recognition contribute to the lower burdens of CPE85B, then this strain would not recover and grow normally in the lungs when CD4+ T cells are depleted during the chronic phase of infection. We infected mice with H37Rv or CPE85B and allowed the infection to proceed for 28 days, when initial lung CFUs were measured for each group. As expected, bacterial CFUs for CPE85B were ∼3 fold lower than H37Rv at this time point (Figure 5B). The remaining mice in each infection group were then treated with monoclonal antibody GK1.5 every 6 days until day 50 post-infection to deplete CD4+ T cells. After an initial lag, in which neither bacterial strain expanded, both CPE85B and H37Rv resumed growth in the lungs at indistinguishable rates (Figure 5B). Taken together, these data provide strong evidence that improved control of the CPE85B strain is attributable to increased activation of Ag85B-specific CD4+ T cells, although we cannot exclude the possibility that other factors contribute to the lower lung burdens of CPE85B that appear after the development of adaptive immunity.
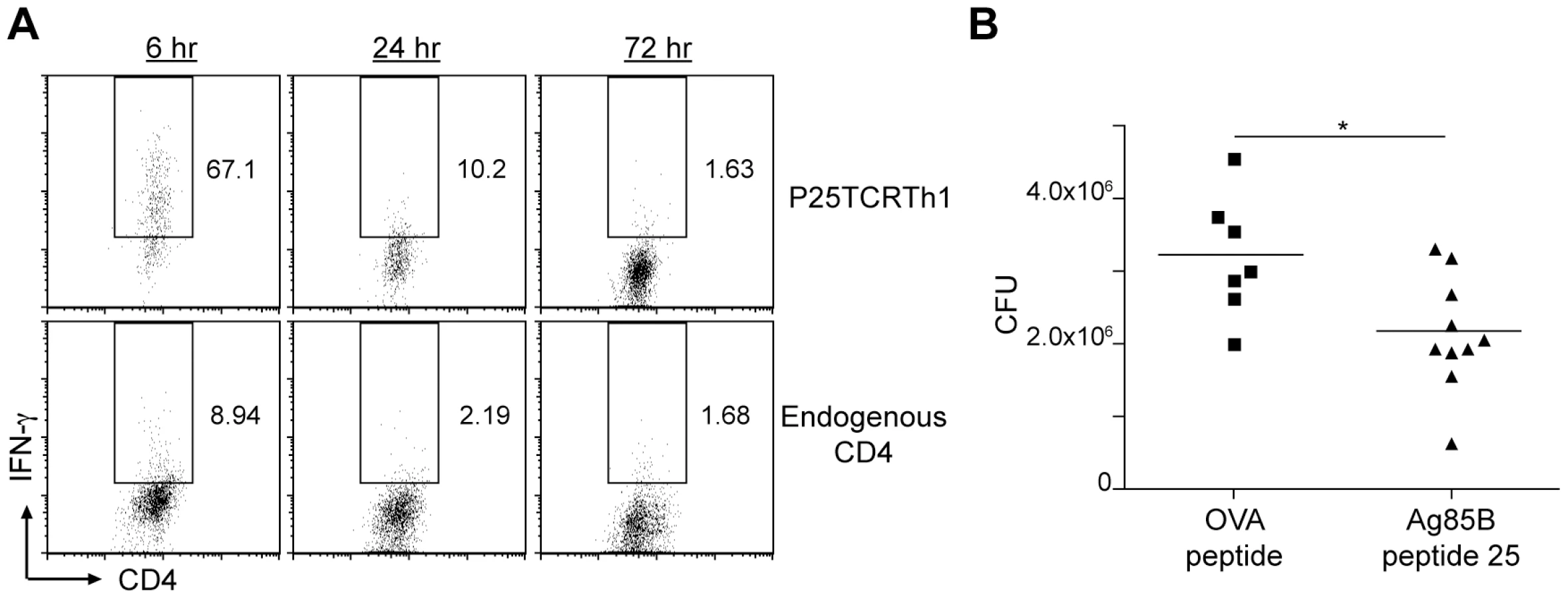
Treatment with intravenous peptide 25 during chronic infection reduces lung bacterial burden
Our observation that forced expression of fbpB increased the frequency of Ag85B peptide 25-specific CD4+ T cells and reduced the bacterial burden in the lungs (Figure 4D, 4E, and 4F), together with our observation that injection of peptide 25 also increased activation of CD4+ effector T cells at the site of infection (Figure 2B and 2C) suggested that providing antigen by injection of peptide 25 might also result in improved immune control of infection. We first determined the duration of increased IFN-γ production by adoptively transferred P25TCRTh1 cells or endogenous CD4+ T cells after peptide 25 injection. The frequency of IFN-γ cells was highest in both 6 hours after treatment, and decreased to approximately 20% of maximal levels by 24 hours after peptide injection for both endogenous CD4+ and P25TCRTh1 cells (Figure 6A). By 72 hours post-treatment, the frequency of IFN-γ+ cells returned to levels observed in the absence of peptide 25 injection, indicating that the activating effect of peptide 25 treatment is remarkably transient, entirely dissipating within 3 days of the treatment. Despite the transient nature of this effect, we found that treatment of M. tuberculosis H37Rv-infected mice with peptide 25 (in the absence of adoptively transferred P25TCRTh1 cells) every 2–3 days from day 28 to day 45 post-infection reduced lung bacterial burdens by 1.05±0.40×106 bacteria (p = 0.018) compared with that in mice treated with OVA peptide, an unrelated MHC II epitope (Figure 6B). Neither group of mice displayed any signs of toxicity, even after repeated peptide injections. These results indicate that during M. tuberculosis infection, CD4+ effector T cells are not stimulated at their maximum potential frequency at the site of infection in the lungs. Because effector T cell responses progressively decrease during chronic infection, and enhancing T cell responses with exogenous peptide antigen improves immune clearance of M. tuberculosis, we conclude that failure to optimally activate effector T cells at the site of infection is an important determinant of the limited efficacy of adaptive immunity in tuberculosis.
Discussion
M. tuberculosis evades adaptive immunity to persist in the lungs, often for the lifetime of the host. Here, we have characterized one mechanism by which this impressive feat of immune evasion is accomplished in vivo. We found that, of the large number of CD4+ effector T cells recruited to the lungs of infected mice, few are stimulated to produce IFN-γ (Figure 7A). While there are few precedents available for comparison, our findings are in stark contrast to those found in C57BL/6 mice infected with the Armstrong strain of LCMV [28]. In that context, which results in CD8+ T cell-dependent resolution of infection, >20% of virus-specific CD8+ T cells are activated to produce IFN-γ during the acute stage of infection when viral burdens and antigen availability are highest, and the frequency of in vivo-activated virus-specific CD8+ T cells does not decrease until the viral burden is reduced. We found that the initially low proportion of CD4+ T cells producing IFN-γ in the lungs of M. tuberculosis-infected mice diminishes further as infection progresses to chronic phase, even though the bacterial burden in the lungs remains high. Our studies using adoptively transferred Ag85B-specific P25TCRTh1 cells revealed that the decreasing responses of CD4+ effector cells are caused in part by decreasing expression of fbpB by M. tuberculosis. By reducing fbpB expression during chronic infection, M. tuberculosis restricts the availability of Ag85B, an immunodominant antigen, and thereby prevents infected APCs from optimally activating CD4+ effector T cells. Consistent with this model, we found that a recombinant strain of M. tuberculosis engineered to maintain the expression of fbpB at high levels during chronic infection (CPE85B) was attenuated during the chronic phase of infection in a strictly CD4+ T cell dependent manner, indicating that down-regulation of fbpB and limitation of antigen availability is important for evasion of adaptive immunity by M. tuberculosis. Treatment of infected mice with synthetic Ag85B peptide 25 also increased CD4+ effector T cell IFN-γ responses and significantly reduced the bacterial burden in the lungs. We conclude that suboptimal effector T cell activation enables M. tuberculosis to evade elimination by adaptive immunity during the chronic stage of infection, and that some of this suboptimal effector T cell activation is attributable to restricted antigen expression by the bacteria. In addition, other mechanisms that limit effector T cell activation, such as interference with the MHC class II antigen processing and presentation pathway and/or the action of regulatory T cells, likely contribute to the remarkable survival of M. tuberculosis in vivo.

Infection with M. tuberculosis induces a robust T cell response involving CD4+ and CD8+ T cells and the effector cytokines IFN-γ and TNF [3], which are all essential for control of infection [5], [39], yet adaptive immunity fails to eradicate M. tuberculosis. Mechanisms for the limited efficacy of the adaptive immune response in tuberculosis fall into two general (not mutually exclusive) categories: either the effector functions that T cells perform (e.g. IFN-γ production) are not effective because of failed responses by the infected cells targeted by effector T cells; or the T cells recruited to the site of infection do not optimally perform the effector functions required for immune clearance. Regarding the former, the ability of M. tuberculosis to resist and inhibit the TNF - and IFN-γ-induced microbicidal responses of the phagocytic cells it infects is one documented component of its immune evasion strategy in vivo [40]. However, our observation that only a small fraction of the CD4+ effector T cells in the lungs is activated to synthesize IFN-γ provides new support for the latter explanation. The potential causes of this mechanism include bacterial factors and host regulatory mechanisms that directly impair effector T cell function. As an example of a direct bacterial effect, mycobacterial cell wall glycolipids have been found to impair CD4+ T cell responses in vitro [41]. With regard to host regulatory mechanisms, during mouse infection, T regulatory cells limit the ability of adaptive immunity to restrict the bacterial population size in the lungs [11], [42]. Interleukin-10 (IL-10), whether expressed by myeloid cells or T cells, provides an additional host regulatory mechanism that inhibits T cell effector functions in tuberculosis, as transgenic over-expression of IL-10 in infected mice impaired T cell responses and caused an increase in bacterial CFUs [12], while deletion of IL-10 causes enhanced control of infection [43], indicating that T cell-directed suppressive factors can limit the success of the adaptive immune response to M. tuberculosis. On the other hand, CD4+ effector T cells at the site of infection may not recognize or become activated optimally by APCs bearing M. tuberculosis-derived peptide:MHC II complexes, a process that is required for IFN-γ production in peripheral tissues [35]. Recent observations using live imaging revealed that a small fraction of Leishmania major-infected macrophages interact with Leishmania-specific CD4+ T cells in vivo [44] indicating that in certain infections, effector T cells may not recognize infected cells efficiently, and this may contribute to slow clearance or persistence of infection. Suboptimal stimulation of CD4+ T cells could occur via direct targeting and inhibition of MHC II antigen presentation pathways in infected APCs, or as a result of the limited availability of peptide T cell epitopes, a consequence of bacterial suppression of antigen encoding genes, or a combination of these mechanisms.
In this study, we first determined that the frequency of endogenous polyclonal CD4+ T cells producing IFN-γ in the lungs was surprisingly low, and varied during the course of infection, with the highest responses during the acute stage and the lowest responses observed as infection reached the chronic stage. These reduced responses occur despite the presence of similar numbers of bacteria in the lungs during these stages of infection. To further understand the underlying mechanisms of the low frequency of effector T cell activation in the lungs, we quantitated CD4+ effector T cell responses to the peptide 25 epitope of M. tuberculosis Ag85B, a secreted protein targeted by a large number of M. tuberculosis-specific CD4+ T cells [45]. Ag85B is targeted by 5 of the 9 novel tuberculosis vaccine candidates currently in clinical trials [46], thus understanding its behavior and responses to it in vivo has considerable importance for TB vaccine development. The reduced expression of fbpB we observed is consistent with regulation by the state of bacterial growth, though it may be indirectly triggered by the onset of Th1 immunity, since expression of fbpB is maintained in mice lacking IFN-γ [38]. Because Ag85B is a cell wall biosynthesis enzyme, down-regulation of fbpB has been interpreted as a consequence of transition by M. tuberculosis into a relatively stationary state. Alternatively, fbpB suppression during chronic infection may also be an evolved bacterial immune evasion mechanism that enables long-term persistence of M. tuberculosis by limiting T cell activation. In support of this, we found that forced expression of fbpB by the CPE85B strain during chronic infection resulted in a higher proportion of P25TCRTh1 cells producing IFN-γ than in H37Rv-infected mice. Other studies have suggested but not directly examined the possibility that over-expression of certain M. tuberculosis proteins (including Hsp70 and ESAT-6) may cause attenuation of bacterial persistence by increased immune recognition [47], [48]. Our finding that polyclonal CD4+ effector T cell responses diminish in chronic infection suggests that this may be a general phenomenon in tuberculosis. Importantly though, the higher frequency of P25TCRTh1 cell activation observed in CPE85B-infected mice diminished at a later time point as it did in H37Rv infection, implying that other mechanisms, especially impairment of MHC II antigen presentation by M. tuberculosis, exist to limit effector T cell activation during chronic infection in vivo.
Several in vitro studies have found that M. tuberculosis subverts or impairs antigen presentation by the cells it infects, limiting the capability of infected APCs to activate antigen specific T cells [8], [10]. Initial observations include the finding that M. bovis BCG survives in primary human macrophages that CD4+ T cells fail to recognize [26] and that M. tuberculosis-infected THP-1 cells express low amounts of surface MHC II [25]. Several mechanisms for inhibition of MHC II antigen presentation have been characterized using a spectrum of mycobacterial strains and cell components. Among these, impaired phagosome maturation, a well-characterized component of the ability of M. tuberculosis to survive in phagocytic cells [19], has been found to limit activation of cathepsin D for efficient processing of mycobacterial antigens [24], while inducing autophagy with rapamycin was recently found to improve the efficacy of BCG and other live mycobacterial vaccines, by enhancing presentation of mycobacterial antigens [49]. Impaired expression of MHC II by macrophages after IFN-γ treatment was also observed after in vitro infection or treatment of macrophages with certain mycobacterial cell components [23], [50], [51], [52], [53]. This effect may involve prolonged signals received through bacterial pattern recognition receptors (PRRs) including TLR2, although we recently reported a TLR2 independent mechanism for impaired MHC II expression in response to IFN-γ [53], [54].
These and other in vitro studies are consistent with our present results and lend support for the hypothesis that APCs do not efficiently stimulate CD4+ effector T cells in the lungs during M. tuberculosis infection in vivo. Attempts to verify and explore the significance of these in vitro findings with in vivo infection models have been limited thus far, until the present paper. One study of mouse infection with GFP-expressing M. bovis BCG found a modest decrease in surface expression of MHC II on some populations of lung APC that harbored intracellular bacteria when compared to those that did not contain bacteria [55]. In contrast, in a low dose aerosol infection of mice with GFP-expressing H37Rv, we did not detect a difference in surface MHC II expression between infected and non-infected APCs at various time points post-infection; we also found that M. tuberculosis-infected APCs isolated from the lungs expressed high levels of the costimulatory molecules CD80 and CD86 [54]. Nonetheless, there is evidence that the activation of M. tuberculosis-specific T cell responses is impaired during in vivo infection, indicating that M. tuberculosis may specifically impair presentation of its antigens without decreasing overall surface expression of MHC II. One recent study found that mice provided with CD4+ TCR-transgenic effector T cells specific for the M. tuberculosis antigen ESAT-6 prior to infection can restrict bacterial population size to a lower level but cannot prevent establishment of infection [56]. Despite the presence of this effector T cell population in the lungs from the onset of infection, control of bacterial growth was delayed until 7 days post-infection. Likewise, despite mounting apparently normal anti-M. tuberculosis CD4+ T cell responses, infected mice and humans treated with anti-mycobacterial drugs to eliminate primary infection remain susceptible to reinfection [33], [57]. These studies indicate that susceptibility to persistent tuberculosis is more likely due to failure to activate antigen-specific effector T cells, rather than to insufficient development of antigen specific T cells in response to infection.
We observed increased survival of wild type, but not CD4+ T cell-deficient mice infected with the CPE85B strain when compared to those infected with H37Rv, highlighting the importance of enhanced T cell stimulation to the long-term outcome of infection, and indicating that enhanced effector T cell activation, through increased antigen availability, can be accomplished without detrimental effects. Moreover, our finding that sustained expression of Ag85B during the adaptive immune phase of infection was associated with a 2 - to 5-fold increase in antigen-specific CD4+ T cell activation, yet reduced the bacterial burdens approximately 10-fold implies that a massive increase in effector T cell activation is not necessary to significantly improve immune control of tuberculosis. Future efforts to develop tuberculosis therapies should therefore aim to bypass or overcome factors that limit effector T cell activation including direct T cell suppression, impaired antigen presentation, and bacterial gene regulatory mechanisms. For example, we found that the chronic phase antigen deficit resulting from bacterial suppression of fbpB could be overcome by systemic treatment of infected mice with synthetic peptide 25, which strongly but transiently enhanced CD4+ T cell responses specific for this epitope and reduced the bacterial burden. This result implies that the endogenous CD4+ T cells generated in response to infection with M. tuberculosis and recruited to the infected lungs can be stimulated to perform their effector functions if they are provided antigen, resulting in improved bacterial clearance (Figure 7B). The potential for anti-tuberculosis therapies that aim to enhance existing T effector cell responses in infected individuals with synthetically produced peptides encoding known T cell epitopes remains unexplored; however, given the steadily increasing prevalence of drug resistant M. tuberculosis, such immunotherapeutic approaches to tuberculosis are an attractive option. Although the consequences of increasing the activation of existing T cell responses have not been widely tested, in the context of certain highly monoclonal T cell responses, administration of epitope peptides has caused rapid mortality of infected or previously immunized mice [58], [59]. However, despite these findings and concerns about possible immunopathology induced by hyperactivation of effector T cells in tuberculosis [60], we observed no morbidity or mortality in infected mice repeatedly treated with peptide 25, a result that encourages the continued exploration of this therapeutic strategy. Future studies should also aim to determine the host and bacterial regulatory mechanisms that account for chronic phase suppression of fbpB and whether genes encoding other immunodominant M. tuberculosis antigens behave similarly. Identification of the elements of this host-pathogen interaction may lead to the development of therapies that target antigen gene suppression and inhibition of antigen presentation and provide a novel strategy for overcoming bacterial persistence in vivo, leading to better outcomes in M. tuberculosis-infected individuals.
Methods
Mice
C57BL/6, B6.SJL-Ptprca Pepcb/BoyJ (CD45.1+), and MHCII KO mice for aerosol M. tuberculosis infection experiments were either bred in the New York University School of Medicine Skirball animal facility or purchased from Taconic Farms, Inc. P25TCR-Tg mice, whose CD4+ T cells express a transgenic T-cell antigen receptor that recognizes the complex of peptide 25 (aa 240–254) of M. tuberculosis Ag85B and the mouse MHC II allele I-Ab were prepared on a C57BL/6 background, as previously described [2], [61]. All animal experiments were done in accordance with procedures approved by the NYU School of Medicine Institutional Animal Care and Use Committee and in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health under the Assurance of Compliance Number A3435-01.
M. tuberculosis in vitro growth and aerosol mouse infection
Wild type M. tuberculosis H37Rv was originally obtained from ATCC. Frozen stocks for aerosol infection and in vitro use were prepared and stored at −80°C. GFP-expressing H37Rv and Ag85B null (ΔAg85B) strains of M. tuberculosis were generated as previously described [2], [62]. M. tuberculosis cultures were grown in 10 mL Middlebrook 7H9 liquid medium supplemented with 10% v/v albumin dextrose catalase enrichment and incubated under shaking conditions at 37°C. Mice at 8–12 weeks of age were infected with ∼100 CFU of M. tuberculosis via the aerosol route using an Inhalation Exposure Unit (Glas-Col) as previously described [62]. To verify inoculum size, 3–5 infected mice were euthanized 24 hours after infection and lungs were homogenized and plated on Middlebrook 7H11 medium supplemented with 10% v/v albumin dextrose catalase enrichment. To determine bacterial population size at time points post-infection, lungs were homogenized, diluted in PBS+Tween-80 (0.5%), and added to 7H11 plates. Plates were incubated at 37°C for 3 weeks and single colonies were counted. To determine M. tuberculosis survival in stationary culture, 7H9 medium was inoculated with H37Rv or CPE85B, grown in shaking conditions to saturation (O.D.600>1.0), and initial CFUs were measured. Cultures were then placed in stationary incubator at 37°C for 17 days, and final CFUs were measured.
Fluorescent microscopy of frozen tissue sections
C57BL/6 mice were infected with M. tuberculosis H37Rv and on day 25 post-infection received 1×106 CFP+ P25TCRTh1 cells via adoptive transfer. On 28 post-infection, lungs were perfused and frozen in OCT before 5 µm sectioning and fixation in cold acetone. Sections were stained with DAPI to label nuclei and analyzed on a Leica DMRB fluorescent microscope (objective: Leica PL Fluotar 20×/0.50) equipped with a Spot RT digital camera. Separate images for DAPI and CFP fluorescence were acquired and merged using Spot software.
P25TCRTh1 CD4+ effector T cells
P25 TCR-Tg CD4+ Th1 effector cells were generated in vitro as follows: naïve CD4+ T cells were magnetically isolated from lymph node cell suspensions of P25 TCR-Tg mice (or for fluorescent microscopy, a P25TCR-Tg mouse expressing CFP under control of the ubiquitin promoter) using CD4 (L3T4) microbeads and an AutoMACS (Miltenyi Biotech). P25TCR-Tg CD4+ T cells were co-cultured with irradiated C57BL/6 splenocytes in the presence of mouse IL-12p70 (10 ng/ml), mouse IL-2 (5 ng/ml), anti-IL-4 neutralizing antibody (50 ng/ml), and synthetic peptide 25 (0.5 µM). Cells were cultured at 37°C with 5% CO2. On days 3 and 5 of culture, cells were split 1∶3 with fresh media containing IL-12p70, IL-2, and anti-IL-4, but no peptide 25. Cells were washed with PBS and counted on day 7 of culture before use for in vitro or in vivo assays. For in vitro restimulation, P25TCRTh1 cells were co-cultured with irradiated C57BL/6 splenocytes for 24 hours in RPMI-10 in the presence or absence of peptide 25 (0.5 µM) or bone marrow derived dendritic cells infected with M. tuberculosis (MOI: 0.1). Cells were collected and analyzed by flow cytometry for intracellular IFN-γ, or culture supernatants were analyzed for IFN-γ by ELISA. For in vivo experiments, 1×106 P25TCRTh1 cells were injected via tail vein or retro-orbital sinus into recipient mice at various time points post-infection. Cells were routinely isolated from lungs of recipient mice 72 hours after adoptive transfer and analyzed by flow cytometry.
Naïve P25TCR-Tg T cell proliferation
3×106 CFSE-labeled CD4+ T cells, harvested from the lymph nodes of P25TCR-Tg mice were adoptively transferred into infected recipients at various time points post-infection. 7 days after adoptive transfer, mediastinal lymph nodes were harvested from recipient mice and cells were analyzed for CFSE dilution by flow cytometry.
Generation of CPE85B strain of M. tuberculosis
The Ag85B null strain of M. tuberculosis (ΔAg85B), previously created by our lab from wild-type H37Rv [2], was used as a background strain for generating CPE85B. Both the hspX promoter sequence, consisting of 254 bp directly 5′ of the hspX start codon, as well as the fbpB open reading frame were amplified by PCR from H37Rv genomic DNA. Each of these fragments was ligated into the pMV306 integrating vector to create a recombinant construct, whose sequence was verified by Sanger sequencing performed by the NYU DNA sequencing facility. ΔAg85B was grown in 7H9 liquid media and transformed with this construct via electroporation. The reaction was plated on 7H11 plates containing 25 µg/ml kanamycin to select for bacteria incorporating the construct into the M. tuberculosis chromosome. Presence of the construct in kanamycin resistant colonies was verified by PCR. Expression and secretion of Ag85B by CPE85B was confirmed by SDS-PAGE and anti-Ag85B western blot of supernatants from 7H9 liquid medium after stationary culture. For stationary culture-induced expression of Ag85B by the CPE85B strain, 10 mL cultures were grown to late phase (OD600∼1.0) in normal shaking conditions, then flasks were sealed and transferred to a stationary incubator for >1 week before supernatants were collected.
RT-qPCR of bacterial mRNA from infected mouse lungs
To quantitate expression of M. tuberculosis genes during mouse infection, lungs of infected mice were rapidly placed into a solution of RNAlater (Ambion) and stored overnight at room temperature in accordance with manufacturer recommendations to allow permeation of the tissue. Thereafter, samples for RNA isolation were stored at −80°C. When comparing expression of genes at various time points, tissues were transferred to TRIzol (Invitrogen) and quickly homogenized using a Tissue Tearor homogenizer to disrupt mouse cells. Lung homogenates were centrifuged to pellet intact bacterial cells, and supernatants discarded. M. tuberculosis pellets were disrupted with zirconia/silica beads, RNA was extracted, and RT-qPCR was carried out as previously described [37] with fbpB copy number normalized to the constitutively expressed 16S rRNA and multiplied by a factor of 105. The following RT-qPCR primers were used in this study. 16S rRNA: RT 5-ATTACGTGCTGGCAACATGA-3, qPCR For 5-GCCGTAAACGGTGGGTACTA-3, qPCR Rev 5-TGCATGTCAAACCCAGGTAA-3; hspx/acr/Rv2031c: RT 5-GAATGCCCTTGTCGTAGGTG-3, qPCR For 5-AGATGAAAGAGGGGCGCTAC3, qPCR Rev 5-TAATGTCGTCCTCGTCAGCA3; fbpB/Rv1886c: RT 5-TCCTGGAACTTCAGGTTGCT-3, qPCR For 5-ACCCCCAGCAGTTCATCTAC-3, qPCR Rev 5-TTCCCGCAATAAACCCATAG-3.
Tissue processing and flow cytometry
To isolate cells from infected tissues for flow cytometry, mice were euthanized with CO2 followed by cervical dislocation. Tissues were removed and mechanically disrupted by mincing in RPMI as previously described [62] or using a gentleMACS dissociator (Miltenyi Biotec) in the manufacturer-recommended HEPES buffer. Lung suspensions were incubated in Collagenase D and DNase at 37°C with 5% CO2 for 30 minutes and cells were isolated by forcing suspensions through a 70 µM cell strainer. RBCs were removed by ACK lysis and live cells counted by trypan blue exclusion. Cell suspensions were stained using the following fluorescently-labeled antibodies (Biolegend, BD Pharmingen, or eBioscience): anti-CD3 PE, anti-CD4 (L3T4) FITC, anti-CD45.2 PerCP, anti-CD45.1 Pacific Blue, anti-IFN-γ (XMG1.2) APC, and rat IgG1 APC isotype control. Flow cytometry was performed using a FACSCalibur or LSR II (BD Biosciences) at the NYU Cancer Institute Flow Cytometry and Cell Sorting facility. Analysis of flow cytometry data was performed using FlowJo software.
Detection of IFN-γ-producing cells by direct intracellular cytokine staining
To detect intracellular IFN-γ produced by cells in vivo, a protocol was developed based on a previous study [28]. In contrast to this study, however, optimal detection of IFN-γ producing cells from the lungs of mice infected with M. tuberculosis did not require treatment of mice with i.v. brefeldin A or inclusion of brefeldin A in tissue processing buffers. Instead, after euthanasia, tissues were rapidly placed on ice and all cell isolation steps except collagenase/DNase digestion (37°C for 30 minutes) and ACK lysis (room temperature for 5 minutes) were carried out quickly and on ice. Cells were stained for surface markers at 4°C for 30 minutes followed by permeabilization and fixation with Cytofix/Cytoperm (BD Biosciences) at 4°C for 20 minutes. Finally, fixed cells were stained with anti-IFN-γ or a rat IgG1 isotype control at 4°C for 30 minutes. Flow cytometry dot plot gates for IFN-γ+ cells were set based on comparison with isotype control and unpermeabilized cells stained for IFN-γ.
Total CD4+ T cell depletion
Mice were treated with an intra-peritoneal dose of 500 µg of either monoclonal antibody GK1.5, which depletes CD4+ T cells, or a rat IgG2b isotype control (LTF-2) every 6 days from day 28 to Day 50 post-infection. Efficiency of CD4+ T cell depletion 6 days after GK1.5 treatment was determined to be >95% by flow cytometry of cell suspensions from lungs, spleen and blood. In mice treated with LTF-2 isotype control, no differences were observed in CD4+ T cell number or bacterial burden when compared to untreated mice.
Selective ablation of endogenous CD4+ T cells
To determine the influence of endogenous CD4+ T cells on the response of adoptively transferred P25TCRTh1 cells in vivo, a system was developed to deplete endogenous CD4+ T cells selectively from infected mice. Mice expressing Cre recombinase under control of the CD4 promoter were crossed with those carrying an inducible Diphtheria Toxin Receptor (iDTR) allele, whose baseline expression is prevented by a stop codon flanked by loxp sites [34]. Progeny of this cross (CD4-DTR) carry CD4+ T cells that are sensitive to Diphtheria Toxin mediated ablation. CD4-DTR mice were infected with H37Rv and received daily intraperitoneal doses of DT (100 ng) to ablate endogenous CD4+ T cells from day 21 to day 28 post-infection. The efficiency of CD4+ T cell ablation in the lungs was determined by flow cytometry to be 48.9%. P25TCRTh1 cells were adoptively transferred on day 25 post-infection and the frequency of IFN-γ production was assessed on day 28 post-infection.
Assessment of intravascular and extravascular location of adoptively-transferred P25TCRTh1 cells
On day 25 post-infection, wild-type mice infected with M. tuberculosis H37Rv received P25TCRTh1 cells via adoptive transfer. On day 28 post-infection, mice were treated intravenously with 800 ng (at 4.0 ng/µL) PerCP-labeled anti-CD4 (RM4-5). Fifteen minutes later, mice were euthanized and total lung cells were stained with FITC-labeled anti-CD4 (GK1.5). Lung cells stained by anti-CD4-PerCP were considered to be CD4+ T cells residing in the intravascular compartment at the time of antibody injection. Cells staining positive for anti-CD4-FITC and negative for PerCP were considered to be CD4+ T cells residing in an extravascular or parenchymal lung compartment protected from labeling with intravenous antibody. IFN-γ production in vivo was assessed by intracellular staining of all cells with APC-labeled anti-IFN-γ as previously described.
Systemic treatment of mice with synthetic peptides
Mice were intravenously treated with 100 µg of Ag85B peptide 25 (FQDAYNAAGGHNAVF) or OVA peptide control (ISQAVHAAHAEINEAGR) in 100 µl sterile PBS via tail vein or retro-orbital sinus. Peptides were synthesized by EZBiolab or Peptides International to a purity of >95%.
Statistical analyses
Data shown are representative of 2 or more experimental replicates. In all figures, error bars indicate mean ± SEM. To determine statistical significance when comparing experimental values from two groups of mice, one - or two-tailed student's t-tests were routinely used, each where appropriate. To compare the growth rate of H37Rv and CPE85B in vivo, a non-linear regression analysis (curve fit) with F-test was used to determine whether a single curve could account for both data sets. In mouse survival experiments, Logrank test was used to evaluate statistical significance when comparing survival of one mouse strain after infection with either of the two bacterial strains. * = p<0.05; ** = p<0.005; n.s = not significant.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. World Health Organization
2010
Global tuberculosis control 2010
Geneva, Switzerland
WHO
2. WolfAJDesvignesLLinasBBanaieeNTamuraT
2008
Initiation of the adaptive immune response to Mycobacterium
tuberculosis depends on antigen production in the local lymph node, not the
lungs.
J Exp Med
205
105
115
3. CooperAM
2009
T cells in mycobacterial infection and disease.
Curr Opin Immunol
21
378
384
4. ReileyWWCalayagMDWittmerSTHuntingtonJLPearlJE
2008
ESAT-6-specific CD4 T cell responses to aerosol Mycobacterium
tuberculosis infection are initiated in the mediastinal lymph
nodes.
Proc Natl Acad Sci U S A
105
10961
10966
5. FlynnJLGoldsteinMMChanJTrieboldKJPfefferK
1995
Tumor necrosis factor-alpha is required in the protective immune
response against Mycobacterium tuberculosis in mice.
Immunity
2
561
572
6. WangooASparerTBrownINSnewinVAJanssenR
2001
Contribution of Th1 and Th2 cells to protection and pathology in
experimental models of granulomatous lung disease.
J Immunol
166
3432
3439
7. ChenXZhangMLiaoMGranerMWWuC
2009
Reduced Th17 response in patients with tuberculosis correlates
with IL-6R expression on CD4+ T Cells.
Am J Respir Crit Care Med
181
734
742
8. BaenaAPorcelliSA
2009
Evasion and subversion of antigen presentation by Mycobacterium
tuberculosis.
Tissue Antigens
74
189
204
9. DivangahiMDesjardinsDNunes-AlvesCRemoldHGBeharSM
2010
Eicosanoid pathways regulate adaptive immunity to Mycobacterium
tuberculosis.
Nat Immunol
11
751
758
10. HardingCVBoomWH
2010
Regulation of antigen presentation by Mycobacterium tuberculosis:
a role for Toll-like receptors.
Nat Rev Microbiol
8
296
307
11. Scott-BrowneJPShafianiSTucker-HeardGIshida-TsubotaKFontenotJD
2007
Expansion and function of Foxp3-expressing T regulatory cells
during tuberculosis.
J Exp Med
204
2159
2169
12. TurnerJGonzalez-JuarreroMEllisDLBasarabaRJKipnisA
2002
In vivo IL-10 production reactivates chronic pulmonary
tuberculosis in C57BL/6 mice.
J Immunol
169
6343
6351
13. YiJSCoxMAZajacAJ
2010
T-cell exhaustion: characteristics, causes and
conversion.
Immunology
129
474
481
14. ReileyWWShafianiSWittmerSTTucker-HeardGMoonJJ
2010
Distinct functions of antigen-specific CD4 T cells during murine
Mycobacterium tuberculosis infection.
Proc Natl Acad Sci U S A
107
19408
19413
15. TingLMKimACCattamanchiAErnstJD
1999
Mycobacterium tuberculosis inhibits IFN-gamma transcriptional
responses without inhibiting activation of STAT1.
J Immunol
163
3898
3906
16. BanaieeNKincaidEZBuchwaldUJacobsWRJrErnstJD
2006
Potent inhibition of macrophage responses to IFN-gamma by live
virulent Mycobacterium tuberculosis is independent of mature mycobacterial
lipoproteins but dependent on TLR2.
J Immunol
176
3019
3027
17. PaiRKConveryMHamiltonTABoomWHHardingCV
2003
Inhibition of IFN-gamma-induced class II transactivator
expression by a 19-kDa lipoprotein from Mycobacterium tuberculosis: a
potential mechanism for immune evasion.
J Immunol
171
175
184
18. RohdeKYatesRMPurdyGERussellDG
2007
Mycobacterium tuberculosis and the environment within the
phagosome.
Immunol Rev
219
37
54
19. ClemensDLHorwitzMA
1995
Characterization of the Mycobacterium tuberculosis phagosome and
evidence that phagosomal maturation is inhibited.
J Exp Med
181
257
270
20. HincheyJLeeSJeonBYBasarabaRJVenkataswamyMM
2007
Enhanced priming of adaptive immunity by a proapoptotic mutant of
Mycobacterium tuberculosis.
J Clin Invest
117
2279
2288
21. MillerJLVelmuruganKCowanMJBrikenV
2010
The type I NADH dehydrogenase of Mycobacterium tuberculosis
counters phagosomal NOX2 activity to inhibit TNF-alpha-mediated host cell
apoptosis.
PLoS Pathog
6
e1000864
22. VelmuruganKChenBMillerJLAzogueSGursesS
2007
Mycobacterium tuberculosis nuoG is a virulence gene that inhibits
apoptosis of infected host cells.
PLoS Pathog
3
e110
23. NossEHPaiRKSellatiTJRadolfJDBelisleJ
2001
Toll-like receptor 2-dependent inhibition of macrophage class II
MHC expression and antigen processing by 19-kDa lipoprotein of Mycobacterium
tuberculosis.
J Immunol
167
910
918
24. SinghCRMoultonRAArmitigeLYBidaniASnuggsM
2006
Processing and presentation of a mycobacterial antigen 85B
epitope by murine macrophages is dependent on the phagosomal acquisition of
vacuolar proton ATPase and in situ activation of cathepsin
D.
J Immunol
177
3250
3259
25. HmamaZGabathulerRJefferiesWAde JongGReinerNE
1998
Attenuation of HLA-DR expression by mononuclear phagocytes
infected with Mycobacterium tuberculosis is related to intracellular
sequestration of immature class II heterodimers.
J Immunol
161
4882
4893
26. PancholiPMirzaABhardwajNSteinmanRM
1993
Sequestration from immune CD4+ T cells of mycobacteria
growing in human macrophages.
Science
260
984
986
27. HuffordMMKimTSSunJBracialeTJ
2010
Antiviral CD8+ T cell effector activities in situ are
regulated by target cell type.
J Exp Med
208
167
180
28. LiuFWhittonJL
2005
Cutting edge: re-evaluating the in vivo cytokine responses of
CD8+ T cells during primary and secondary viral
infections.
J Immunol
174
5936
5940
29. AbeRVandenberghePCraigheadNSmootDSLeeKP
1995
Distinct signal transduction in mouse CD4+ and CD8+
splenic T cells after CD28 receptor ligation.
J Immunol
154
985
997
30. SunSZhangXToughDFSprentJ
1998
Type I interferon-mediated stimulation of T cells by CpG
DNA.
J Exp Med
188
2335
2342
31. LahnMKalataradiHMittelstadtPPflumEVollmerM
1998
Early preferential stimulation of gamma delta T cells by
TNF-alpha.
J Immunol
160
5221
5230
32. YanagisawaSKoikeMKariyoneANagaiSTakatsuK
1997
Mapping of V beta 11+ helper T cell epitopes on
mycobacterial antigen in mouse primed with Mycobacterium
tuberculosis.
Int Immunol
9
227
237
33. JungYJRyanLLaCourseRNorthRJ
2005
Properties and protective value of the secondary versus primary T
helper type 1 response to airborne Mycobacterium tuberculosis infection in
mice.
J Exp Med
201
1915
1924
34. BuchTHeppnerFLTertiltCHeinenTJKremerM
2005
A Cre-inducible diphtheria toxin receptor mediates cell lineage
ablation after toxin administration.
Nat Methods
2
419
426
35. McLachlanJBCatronDMMoonJJJenkinsMK
2009
Dendritic cell antigen presentation drives simultaneous cytokine
production by effector and regulatory T cells in inflamed
skin.
Immunity
30
277
288
36. CummingsLABarrettSLWilkersonWDFellnerovaICooksonBT
2005
FliC-specific CD4+ T cell responses are restricted by
bacterial regulation of antigen expression.
J Immunol
174
7929
7938
37. BanaieeNJacobsWRJrErnstJD
2006
Regulation of Mycobacterium tuberculosis whiB3 in the mouse lung
and macrophages.
Infect Immun
74
6449
6457
38. ShiLJungYJTyagiSGennaroMLNorthRJ
2003
Expression of Th1-mediated immunity in mouse lungs induces a
Mycobacterium tuberculosis transcription pattern characteristic of
nonreplicating persistence.
Proc Natl Acad Sci U S A
100
241
246
39. MoguesTGoodrichMERyanLLaCourseRNorthRJ
2001
The relative importance of T cell subsets in immunity and
immunopathology of airborne Mycobacterium tuberculosis infection in
mice.
J Exp Med
193
271
280
40. JungYJLaCourseRRyanLNorthRJ
2002
Virulent but not avirulent Mycobacterium tuberculosis can evade
the growth inhibitory action of a T helper 1-dependent, nitric oxide
Synthase 2-independent defense in mice.
J Exp Med
196
991
998
41. MahonRNRojasREFultonSAFrankoJLHardingCV
2009
Mycobacterium tuberculosis cell wall glycolipids directly inhibit
CD4+ T-cell activation by interfering with proximal T-cell-receptor
signaling.
Infect Immun
77
4574
4583
42. ShafianiSTucker-HeardGKariyoneATakatsuKUrdahlKB
2010
Pathogen-specific regulatory T cells delay the arrival of
effector T cells in the lung during early tuberculosis.
J Exp Med
207
1409
1420
43. RedfordPSBoonstraAReadSPittJGrahamC
2010
Enhanced protection to Mycobacterium tuberculosis infection in
IL-10-deficient mice is accompanied by early and enhanced Th1 responses in
the lung.
Eur J Immunol
40
2200
2210
44. Filipe-SantosOPescherPBreartBLippunerCAebischerT
2009
A dynamic map of antigen recognition by CD4 T cells at the site
of Leishmania major infection.
Cell Host Microbe
6
23
33
45. RogersonBJJungYJLaCourseRRyanLEnrightN
2006
Expression levels of Mycobacterium tuberculosis antigen-encoding
genes versus production levels of antigen-specific T cells during stationary
level lung infection in mice.
Immunology
118
195
201
46. BarkerLFBrennanMJRosensteinPKSadoffJC
2009
Tuberculosis vaccine research: the impact of
immunology.
Curr Opin Immunol
21
331
338
47. StewartGRSnewinVAWalzlGHussellTTormayP
2001
Overexpression of heat-shock proteins reduces survival of
Mycobacterium tuberculosis in the chronic phase of
infection.
Nat Med
7
732
737
48. OholYMGoetzDHChanKShilohMUCraikCS
2010
Mycobacterium tuberculosis MycP1 protease plays a dual role in
regulation of ESX-1 secretion and virulence.
Cell Host Microbe
7
210
220
49. JagannathCLindseyDRDhandayuthapaniSXuYHunterRLJr
2009
Autophagy enhances the efficacy of BCG vaccine by increasing
peptide presentation in mouse dendritic cells.
Nat Med
15
267
276
50. GehringAJDobosKMBelisleJTHardingCVBoomWH
2004
Mycobacterium tuberculosis LprG (Rv1411c): a novel TLR-2 ligand
that inhibits human macrophage class II MHC antigen
processing.
J Immunol
173
2660
2668
51. PecoraNDGehringAJCanadayDHBoomWHHardingCV
2006
Mycobacterium tuberculosis LprA is a lipoprotein agonist of TLR2
that regulates innate immunity and APC function.
J Immunol
177
422
429
52. KincaidEZErnstJD
2003
Mycobacterium tuberculosis exerts gene-selective inhibition of
transcriptional responses to IFN-gamma without inhibiting STAT1
function.
J Immunol
171
2042
2049
53. FortuneSMSolacheAJaegerAHillPJBelisleJT
2004
Mycobacterium tuberculosis inhibits macrophage responses to
IFN-gamma through myeloid differentiation factor 88-dependent and
-independent mechanisms.
J Immunol
172
6272
6280
54. KincaidEZWolfAJDesvignesLMahapatraSCrickDC
2007
Codominance of TLR2-dependent and TLR2-independent modulation of
MHC class II in Mycobacterium tuberculosis infection in
vivo.
J Immunol
179
3187
3195
55. PecoraNDFultonSARebaSMDrageMGSimmonsDP
2009
Mycobacterium bovis BCG decreases MHC-II expression in vivo on
murine lung macrophages and dendritic cells during aerosol
infection.
Cell Immunol
254
94
104
56. GallegosAMPamerEGGlickmanMS
2008
Delayed protection by ESAT-6-specific effector CD4+ T cells
after airborne M. tuberculosis infection.
J Exp Med
205
2359
2368
57. ChiangCYRileyLW
2005
Exogenous reinfection in tuberculosis.
Lancet Infect Dis
5
629
636
58. KitamuraHSedlikCJacquetAZaragozaBDusseauxM
2010
Long peptide vaccination can lead to lethality through CD4+
T cell-mediated cytokine storm.
J Immunol
185
892
901
59. LiuFFeuerRHassettDEWhittonJL
2006
Peptide vaccination of mice immune to LCMV or vaccinia virus
causes serious CD8 T cell-mediated, TNF-dependent
immunopathology.
J Clin Invest
116
465
475
60. BarberDLMayer-BarberKDFengCGSharpeAHSherA
2011
CD4 T cells promote rather than control tuberculosis in the
absence of PD-1-mediated inhibition.
J Immunol
186
1598
1607
61. TamuraTArigaHKinashiTUeharaSKikuchiT
2004
The role of antigenic peptide in CD4+ T helper phenotype
development in a T cell receptor transgenic model.
Int Immunol
16
1691
1699
62. WolfAJLinasBTrevejo-NunezGJKincaidETamuraT
2007
Mycobacterium tuberculosis infects dendritic cells with high
frequency and impairs their function in vivo.
J Immunol
179
2509
2519
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2011 Číslo 5
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Měli bychom postcovidový syndrom léčit antidepresivy?
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- Crystal Structure and Functional Analysis of the SARS-Coronavirus RNA Cap 2′-O-Methyltransferase nsp10/nsp16 Complex
- Lymphoadenopathy during Lyme Borreliosis Is Caused by Spirochete Migration-Induced Specific B Cell Activation
- The OXI1 Kinase Pathway Mediates -Induced Growth Promotion in Arabidopsis
- : An Emerging Cause of Sexually Transmitted Disease in Women