
A Highly Conserved Haplotype Directs Resistance to Toxoplasmosis and Its Associated Caspase-1 Dependent Killing of Parasite and Host Macrophage
Toxoplasmosis is a ubiquitous parasitic infection causing a wide spectrum of diseases. It is usually asymptomatic but can lead to severe ocular and neurological disorders. The host factors that determine natural resistance to toxoplasmosis are yet poorly characterized. Among the animal models to study susceptibility to toxoplasmosis, rats develop like humans a subclinical chronic infection. The finding of a total resistance in the LEW rat strain has allowed genetic studies leading to the identification of Toxo1, a unique locus that controls the outcome of toxoplasmosis. In this report, a panel of recombinant inbred rat strains was used to genetically reduce the Toxo1 locus, on chromosome 10, to a limited region containing 29 genes. This locus is highly conserved among five resistant, by comparison to four susceptible, rat strains, indicating that refractoriness to toxoplasmosis could be predicted. The Toxo1-controlled refractoriness depends on the ability of macrophages to restrict parasite proliferation and the rapid death of both T. gondii and host macrophages in vitro. The NOD-like receptor NLRP1a/Caspase-1 pathway is the best candidate to mediate the parasite-induced cell death. Our data represent new insights towards the identification of a major pathway of innate immunity that protects from toxoplasmosis.
Published in the journal:
. PLoS Pathog 10(4): e32767. doi:10.1371/journal.ppat.1004005
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004005
Summary
Toxoplasmosis is a ubiquitous parasitic infection causing a wide spectrum of diseases. It is usually asymptomatic but can lead to severe ocular and neurological disorders. The host factors that determine natural resistance to toxoplasmosis are yet poorly characterized. Among the animal models to study susceptibility to toxoplasmosis, rats develop like humans a subclinical chronic infection. The finding of a total resistance in the LEW rat strain has allowed genetic studies leading to the identification of Toxo1, a unique locus that controls the outcome of toxoplasmosis. In this report, a panel of recombinant inbred rat strains was used to genetically reduce the Toxo1 locus, on chromosome 10, to a limited region containing 29 genes. This locus is highly conserved among five resistant, by comparison to four susceptible, rat strains, indicating that refractoriness to toxoplasmosis could be predicted. The Toxo1-controlled refractoriness depends on the ability of macrophages to restrict parasite proliferation and the rapid death of both T. gondii and host macrophages in vitro. The NOD-like receptor NLRP1a/Caspase-1 pathway is the best candidate to mediate the parasite-induced cell death. Our data represent new insights towards the identification of a major pathway of innate immunity that protects from toxoplasmosis.
Introduction
Toxoplasma gondii is a widespread obligate intracellular protozoan parasite. One preeminent aspect of its life cycle is the establishment of a chronic infection in humans and many other vertebrate hosts [1]. Toxoplasmosis is most often asymptomatic depending on the parasite's ability to elicit host protective immunity [1]. A serious threat to human health can occur under congenital infection or reactivation of a latent infection in immunodeficient patients [2]. Epidemiological studies have indicated that the phenotypic expression of toxoplasmosis depends on the genetic make-up of both the host and the parasite [3], [4]. Variations in the outcome of Toxoplasma infection after exposure to similar risk factors [5], [6] and twin studies [7] support a significant role of the human host genetic background in the susceptibility to toxoplasmosis. Nevertheless, genetic studies in human are hampered by both population heterogeneity and environment variability. In experimental conditions, genetic and environmental factors are under control. Rats, like humans, usually develop subclinical toxoplasmosis. This contrasts with the severity of the disease developed in most strains of mice. Interestingly, an unexpected refractoriness to T. gondii infection was found in the LEW rat strain [8]. Compared to susceptible BN rats, infected LEW indeed displayed negative serology and lack of cyst burden in their brain [9]. Refractoriness of LEW rats was found to be a dominant trait dependent on hematopoietic cells [9]. It is associated with the ability of macrophages to restrict parasite proliferation in vitro [10].
Further genetic studies using LEW resistant and BN susceptible rats and derived reciprocal congenic strains have allowed the mapping of a locus named Toxo1, which fully controls the refractoriness of LEW rats to toxoplasmosis. Toxo1 has been confined to 7.6 megabases, on rat chromosome 10 (Rn10q.24) [10]. Recently, hNlrp1 a major candidate gene present in the orthologous region to Toxo1 in the human genome (Hs 17p32.2-p13.1) has been associated with human congenital toxoplasmosis [6].
In the present work, we used genetic dissection with a panel of BN and LEW sub-congenic rats and haplotype analysis of chromosome 10 on nine inbred rat strains either susceptible or resistant to define the localization of the gene or set of genes at work in Toxo1 and to analyze the mechanisms of toxoplasmosis refractoriness. We were able to localize the Toxo1 locus in a 891 kb region highly conserved in all resistant strains of rat. Sequencing of this locus in these nine strains revealed a high concentration of resistant-restricted conserved mutations at the bottom border of Toxo1 around Nlrp1. Functional studies in ex vivo infected peritoneal macrophages indicate that the Toxo1-mediated restriction of parasite proliferation is associated with the coordinate death of both parasites and host macrophages. The parasite-induced macrophage cell death involved a caspase-1 dependent mechanism and exhibited pyroptosis-like features. The parasite-induced killing of infected macrophages could be blocked by the capase-1 inhibitor without restoration of parasite proliferation. Therefore, we concluded that if at work, the NLRP1a/caspase-1 pathway is not indispensable to restrict parasite proliferation in macrophages.
Results
Genetic dissection maps Toxo1 to a <1 Mb region
We previously demonstrated that the Toxo1 7.6 Mb interval fully controls the outcome of T. gondii infection independently of the genetic background. The refractoriness to infection conferred by the LEW origin of Toxo1 is characterized by the early elimination of the pathogen resulting in a barely detectable specific immune response and in the absence of brain cysts [10]. In vitro, this Toxo1-LEW mediated refractoriness is associated with the control of parasite proliferation within macrophages [10]. To refine the localisation of the gene(s) that control(s) these in vivo and in vitro phenotypes, we generated a unique panel of congenic sub-lines. Results from the genetic dissection are shown on Figure 1. The parasites were found able to proliferate within the macrophages from the congenic BN.LEWc10-Ce, -Cf, -Cga, -Ci and LEW.BNc10-F sub-lines but not within the macrophages from the congenic BN.LEWc10-Cg and -Ch sub-lines (Figure 1A). Thus within the 7.6 Mb of the Toxo1 locus a 891 kb region controls the in vitro proliferation of parasites within macrophages. We further investigated refractoriness or susceptibility to T. gondii infection in vivo in rats from the seven congenic sub-lines used for these in vitro studies as well as in rats from the BN and LEW parental strains. The control of refractoriness to T. gondii defined by both the absence or low specific antibody response (Figure 1B) and the absence of cyst burden in the brain (Figure 1C), was directed by the same 891 kb region (Figure 1D). Thus, the interval located between the D10GF49 (57.26 Mb) and D10GF55 (58.15 Mb) microsatellite markers contains the gene or the set of genes that controls the toxoplasmosis outcome.
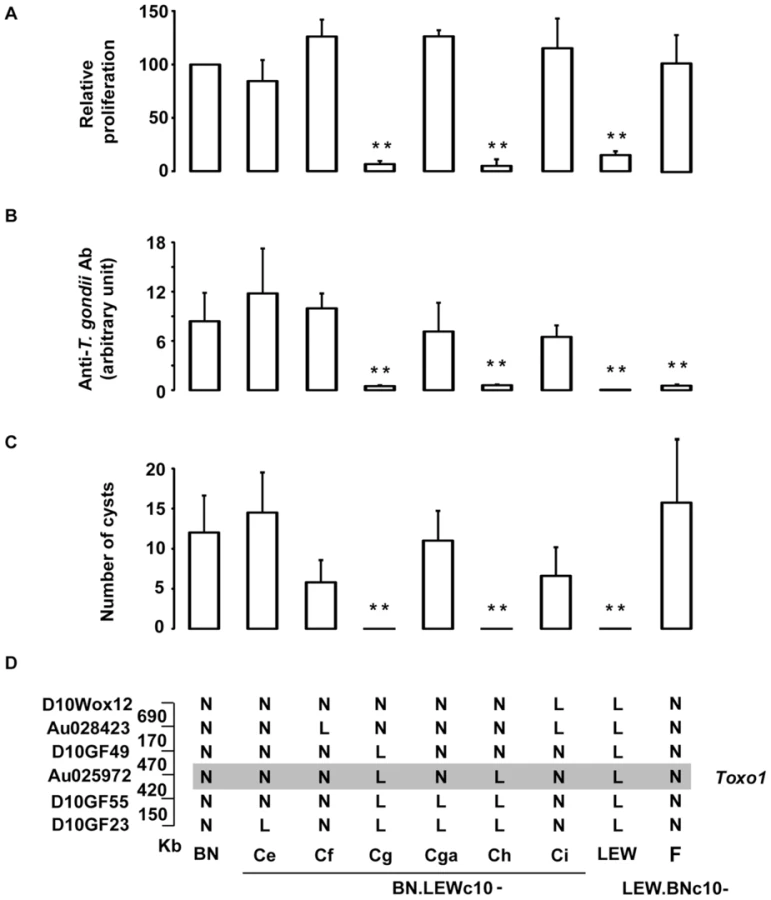
Innate refractoriness against T. gondii is a common trait of several inbred rat strains
We hypothesized that genetic variation(s) underlying Toxo1-mediated innate refractoriness against T. gondii infection result(s) from an ancestral polymorphism instead of independent newly acquired mutations in the LEW strain and thus could be identified in various inbred rat strains. To challenge this hypothesis, we investigated toxoplasmosis outcome in seven other inbred rat strains (LOU, WF, WK, BDIX, OM, F344 and DA), in comparison to BN and LEW. Following infection, rats exhibited a dichotomous phenotype either developing high titers of anti-toxoplasma antibodies and cerebral cysts or remaining refractory to parasite infection (Figure 2). Three strains (OM, DA and F344) displayed, like the BN rat, phenotypes associated to chronic infection with high anti-T. gondii antibody responses (≥5000 u.a) and the detection of brain cysts. Of note, the number of brain cysts was significantly different among these rat strains (Figure 2B) reflecting other regulatory mechanism(s) for cyst formation. By contrast, the four other rat strains (LOU, BDIX, WK and WF) showed the refractory phenotype to T. gondii infection of the LEW strain neither developing specific antibodies nor cerebral cysts (Figure 2). As expected, parasite proliferation within peritoneal macrophages was observed only in the BN and the three other susceptible strains but neither in LEW nor in the four other refractory strains (Figure 2C). Thus, refractoriness against T. gondii infection is a common trait of several inbred rat strains.
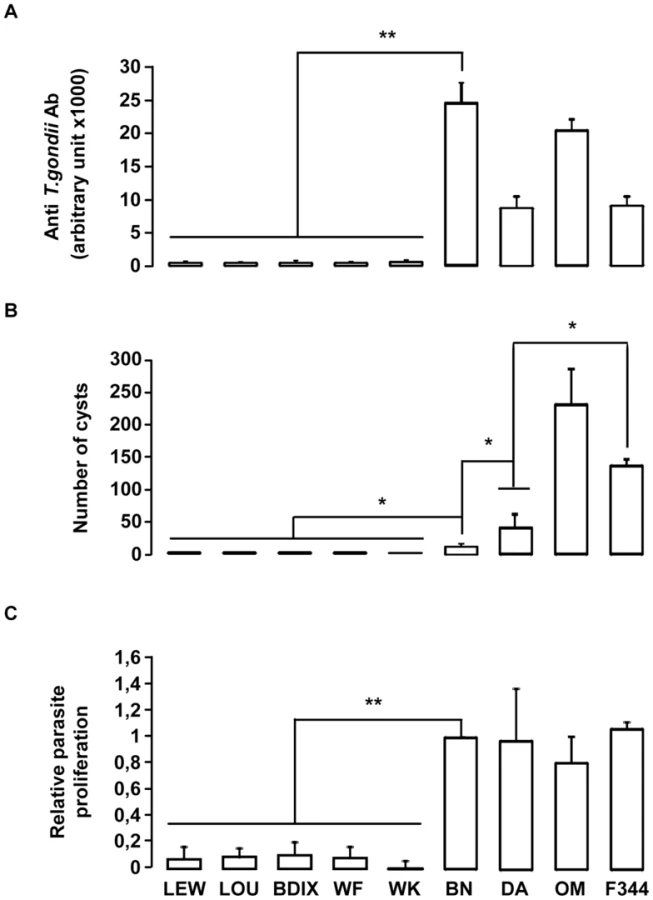
Toxo1 controls resistance against T. gondii infection in the LOU rats
To investigate the implication of the Toxo1 locus in resistance against T. gondii infection, we used a targeted method of genotyping to stratify (LOU x BN) F2 rat according to their genotype at Toxo1. Thirty five F2 rats were genotyped at the polymorphic marker D10GF41, located at the peak of the Toxo1 locus, and their anti-T. gondii Ab titers and brain cyst numbers were quantified following infection with T. gondii (Figure 3). All rats sharing the two LOU alleles (ll) showed no detectable or a weak anti-T. gondii Ab response (<10,000 a.u.). Conversely, all rats sharing the two BN alleles (nn) had high anti-T. gondii Ab titers (>20,000 a.u.). The apparent intermediate antibody response observed in the 15 heterozygous rats (nl) was not significantly different from that observed in the panel of rats sharing the LOU alleles (ll). In a similar way, brain cysts were observed in all the ten homozygous BN (nn) rats and in none of both the heterozygous BN/LOU (nl) and the nine homozygous LOU (ll). These results showed that Toxo1 directs in a dominant manner the toxoplasmosis outcome in LOU rats after T. gondii infection.

Toxo1-mediated refractoriness to toxoplasmosis is correlated with a high conservation of the LEW-genotype haplotype block
As Toxo1 controls resistance against T. gondii infection in the LEW and LOU rats, and given the similarities between the response of LEW, LOU, BDIX, WK and WF, we next examined if Toxo1 - allelic similarities are conserved among all those resistant animals. Considering that as a result of common ancestry, patterns of allelic similarities and differences among strains can be discerned for every variable locus [11], we conducted a haplotype study on the nine strains previously described. This analysis, based on allele size data for microsatellite markers, consisted in identifying among the different strains the chromosomal regions with LEW genotype. For this purpose, 41 polymorphic microsatellite markers on chromosome 10 were investigated on these nine strains (Table S1). The genotype of the nine strains for these markers showed the conservation of an haplotype block in the five resistant strains (LEW, LOU, WF, WK and BDIX) between D10Arb7 and D1Rat297 as compared to the four susceptible strains (BN, OM, F344 and DA) (Figure 4). This highly conserved haplotype block extends on 2.8 Mb between D10Arb7 and D1Rat297 and overlaps the entire 891 kb Toxo1 locus (Figure 4). Thus, the data clearly suggested that conserved genetic variations within the Toxo1 locus on chromosome 10 is a common cause of resistance against T. gondii infection in inbred rat strains.

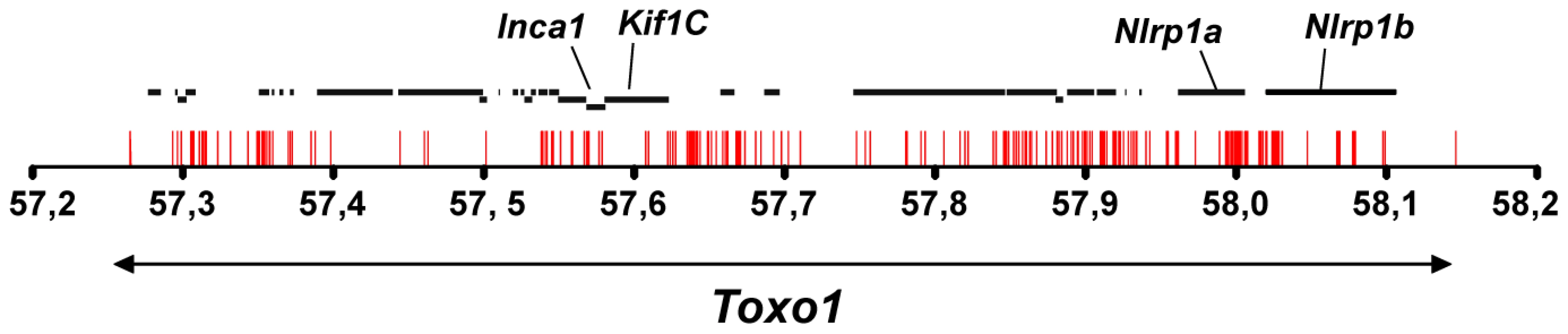
Sequencing of Toxo1 revealed a densification of resistant-restricted conserved mutations in the region of Nlrp1
According to the genome database (www.ensembl.org, RGSC3.4 version), the Toxo1-891 kb interval contains 29 genes. None of these 29 genes could be retained as a candidate on the basis of a significant difference in their level of expression between macrophages from resistant vs. macrophages from susceptible congenic lines (Table S2, TextS1). Therefore, the entire Toxo1 locus of the nine rat strains studied in the haplotype analysis was sequenced to identify resistance-correlated variations in coding - and non-coding sequences. A total of 373 SNPs and 21 insertions/deletions were found strictly conserved among the five resistant strains as compared to susceptible strains. The distribution of these mutations along the locus displays a gradient with a densification at the bottom of Toxo1 (Figure 5). We identified 23 SNPs of which 16 are missense and one is a deletion in the coding sequences leading to the selection of four candidate genes: Inca1 (1 SNP), Kif1C (1 ins/del), Nlrp1a (13 SNPs) and Nlrp1b (2 SNPs). Given that Inca1 and Nlrp1b mRNAs are undetectable in peritoneal macrophages (Table S2) and according to the number of mutations in Nlrp1a vs Kif1C coding sequences, Nlrp1a appeared as the major candidate gene. It encodes the NOD-like receptor (NLR) NLRP1 that acts as an intracellular pattern recognition receptor (PRR) [12]. Both the mouse Nlrp1b and its ortholog rat Nlrp1a have been described as implicated in the control of a cell death process called pyroptosis that is induced by the lethal toxin (LT) from Bacillus anthracis [13].
Early death of T. gondii within the parasitophorous vacuole is under the control of Toxo1
Based on NLRP1 known function, we investigated the impact of the Toxo1 locus on parasite and host cell fate after ex vivo infection of peritoneal macrophages. Following host-cell invasion, T. gondii replicates within a newly formed non-fusogenic compartment, the parasitophorous vacuole (PV). Using fluorescent parasites and vacuole staining with anti-GRA5 or -GRA3 antibodies which both stain the PV membrane, we compared the fate of intravacuolar parasites by immunofluorescence microscopy after allowing them to invade either non-permissive LEW or permissive BN naive peritoneal macrophages (Figure 6). The analyses were performed at 2 and 8 hours following invasion using transgenic-YFP2 fluorescent parasites. According to our previous work [10] we found similar rates of parasite invasion in both permissive (Toxo1-BN) (15%±5%) and non-permissive (Toxo1-LEW) (17%±6%) naive peritoneal macrophages. At 2 hours post-infection, most of intracellular YFP2-parasites were found within a compartment positive for the GRA5 PV membrane marker, in both LEW and BN macrophages (Figure 6A) indicating that parasites are able to enter efficiently into resistant macrophages. However, we observed a slight decrease in the percentage of YFP - and GRA5 - positive vacuoles within LEW (77%±4) as compared to BN (89%±6) macrophages (Figure 6B). At 8 hours post-infection, while YFP2-parasites started to divide within GRA5 positive vacuoles of permissive BN macrophages (Figure 6A), a dramatic drop of YFP staining was observed in GRA5-positive vacuoles of LEW (21%±4) as compared to BN (66%±6) macrophages (Figure 6A and B). The loss of YFP emission in GRA5-positive vacuoles was likely to reflect the death of parasites [14] although parasite egress could not be excluded. Interestingly, the difference was not so marked when the parasite surface was stained using anti-SAG1 antibodies. Indeed, in BN macrophages, the percentage of vacuoles containing SAG1-positive parasites (62.5%±1) was not statistically different from the percentage of vacuoles containing YFP2-parasites (66%±6). In contrast, in LEW macrophages the percentage of vacuoles containing SAG1-positive parasites (43%±3) was two-fold the percentage of vacuoles containing YFP2-parasites (21%±4), indicating that at least 2/3 of vacuoles in LEW macrophages still contained parasites (Figure 6C and D). Therefore, the decrease of YFP staining could be attributed, at least in majority, to parasite death resulting from the macrophage microbicidal activity, rather than to parasite egress. The parasite death within the PV was further examined using staining of small ubiquitin-related modifier (SUMO) as a read-out of the transcriptional activity of live parasites [15]. As shown in Figure 6E and 6F, 2 and 8 hours after infection, a dramatic drop in the percentage of SUMO positive vacuoles was observed in LEW macrophages while no difference was found in BN macrophages, thus supporting the early death of parasites within LEW macrophages. We finally examined whether this phenotype was under the Toxo1 control with our collection of sub-congenic rat strains. For this purpose, peritoneal macrophages from the four susceptible (BN.LEWc10-Ce, -Cf, -Cga, -Ci and LEW.BNc10-F) and the two refractory (BN.LEWc10-Cg and -Ch) sub-congenic lines were infected with T. gondii. In macrophages from the LEW and BN.LEWc10-Cg, -Ch lines in which Toxo1 is from LEW origin, the inhibition of parasite proliferation correlates with the induction of intracellular parasite death (LEW: 58±13%; BN.LEWc10-Cg: 69±2%; -Ch: 64±7%). Conversely, in macrophages from the BN.LEWc10-Ce, -Cf, -Cga and -Ci lines in which Toxo1 is from BN origin, the parasite proliferation was associated with a decrease of parasite death (BN.LEWc10-Ce: 17±11%; -Cf: 0±21%; -Cga: 3±7%; -Ci: 1±17%; LEW.BNc10-F: 0±16%) (Figure 6G). Altogether these results demonstrated that the Toxo1 locus from LEW origin mediates the T. gondii killing by infected peritoneal macrophages.

T. gondii triggers host macrophage death in a Toxo1-dependent way
We next examined the fate of infected host cells in a comparative way depending on the Toxo1 genotype. At 8 hours post-infection, we observed that most of LEW macrophages infected with YFP-negative vacuoles presented a condensed nucleus (Figures 6A, 6C, 6E). Indeed, when this phenomenon was quantified we found that 65% of LEW macrophages with YFP-negative vacuoles, but only 14% of BN susceptible macrophages, showed a condensed nucleus (Figure 7A). This observation indicating a parasite-induced killing of LEW macrophages was further investigated using propidium iodide (PI) uptake. At 6 hours post-infection, about 40% of LEW macrophages (39%±12) had lost membrane integrity as compared to less than 10% of BN macrophages (6%±8) (Figure 7B, p<0.05). The lack of PI uptake under incubation with lysed fibroblasts ruled out the possible triggering by unrelated pathogen-associated molecular pattern (PAMP) (Figure 7B). Tracking of both PI uptake and YFP loss in kinetic experiments indicated that the parasite-induced cell death of LEW macrophages is concomitant with the death of intracellular parasites. Indeed, the increase of PI uptake by macrophages and the loss of YFP by intracellular parasites started from 1 hour and increased steadily until 6 hours after infection (Figure 7C). Further quantification of PI and GRA5 PV marker positive LEW macrophages demonstrated that 88% of dying cells were parasite-invaded cells (Figure 7D). Finally, the implication of Toxo1 in the host cell death phenotype was validated using our panel of sub-congenic animals. Specific induction of host cell death after infection of non-permissive peritoneal macrophages was found in all resistant congenic rats (LEW: 30±10%; BN.LEWc10-Cg: 35±3%; -Ch: 24±3%). By contrast, peritoneal macrophages from all animals permissive to T. gondii proliferation failed to initiate such a cell death process after infection (BN.LEWc10-Ce: 11±1%; -Cf: 8±3%; -Cga: 10±4%; -Ci: 13±7% and LEW.BNc10-F: 17±5%) (Figure 7E). Altogether, these data demonstrated that T. gondii invasion of resistant macrophages is rapidly followed by the combined deaths of intracellular parasites and infected host macrophages using a mechanism under the control of Toxo1 locus.

The Toxo1-mediated death of T. gondii-infected macrophages is neither apoptosis nor autophagy
The rapid loss of membrane integrity of parasite-invaded LEW macrophages (Figure 7C) suggested that the T. gondii-induced cell death was not apoptotic. Accordingly, DNA from dying cells did not show the laddering resulting from chromatin fragmentation, observed in classical apoptotic cell death and pyroptosis (Figure 8A). Moreover, caspase-3 activation, which plays a central role in the executive phase of apoptosis, was not observed in infected LEW macrophages (Figure 8B). Additionally, when infected macrophages were incubated with FITC-labelled annexin V, phosphatidylserine exposure on the plasma membrane was not observed prior to the loss of membrane integrity as assessed by PI staining (Figure 8C). Finally, no difference in the number of acidic vacuoles was observed after lysotracker coloration between permissive BN and non-permissive infected LEW macrophages indicating that the death of infected LEW macrophages was not due to autophagy (Figure 8D).

The Toxo1-mediated death of T. gondii-infected macrophages is associated to ROS production and caspase-1 activation
Given the described role of NLRP1/Caspase-1 inflammasome pathway in the host response to pathogens [16], [17], we hypothesized that the T. gondii-induced cell death of infected resistant LEW macrophages could be associated with both ROS production and caspase-1 activation. The production of intracellular ROS by macrophages was monitored at two time points (15 min and 4 hours) by the dihydro-rhodamine 123. While T. gondii infection did not induce significant ROS production within permissive LEW.BNc10-F macrophages, a marked increase was recorded in infected resistant LEW macrophages (Figure 9A). We next examined caspase-1 activation within infected macrophages by using the fluorogenic activated caspase-1 specific staining (FLICA). At 4 hours post-infection, the percentage of parasite-induced FLICA positive cells was significantly higher in resistant LEW macrophages (29%±2) than in permissive LEW.BNc10-F macrophages (11%±1) (Figure 9B and C). The caspase-1 induction in infected LEW macrophages correlated with the increase of PI-positive cells indicating that caspase-1 was involved in the cell death induction process (Figure 9D). Consistent with these results, the processing of caspase-1 substrate IL-1β that could be prevented by the YVAD caspase-1 inhibitor was detected at 1 h and more evidently at 4 h post-infection in the culture supernatant of infected LEW macrophages and not in that of permissive LEW.BNc10-F macrophages (Figure 9E). The observed difference in the secretion of mature IL-1β was not due to a lack of pro-IL-1β expression since it was found to be induced in response to T. gondii infection within both resistant LEW and permissive LEW.BNc10-F macrophages (Figure 9E). By contrast, pro-IL-1β was not detectable in the cell lysate from LEW macrophages treated with YVAD (Figure 9E) indicating that the inhibition of caspase-1 activity resulted in the down-regulation of pro-IL-1β protein expression. Altogether, these results demonstrated that both ROS production and caspase-1/IL1β pathway are involved in the Toxo1-mediated cell death induction of resistant LEW macrophages.

The caspase-1 pathway controls the cell death induction of both host macrophages and intracellular parasites but not parasite proliferation
The role of caspase-1 in the resistance of LEW macrophages was further investigated using pharmacological inhibitors. Both caspase-1 and pan-caspase inhibitors were able to protect the resistant LEW macrophages from the parasite-induced cell death (Figure 10A). Consistent with our above observations that both parasites and host macrophages killing processes are connected, the two inhibitors also prevented the death of intracellular parasites (Figure 10B). By contrast, these inhibitors failed to restore significant parasite proliferation in non-permissive macrophages (Figure 10C). Altogether these experiments revealed that the resistance of macrophages bearing Toxo1-LEW alleles relies on two pathways, the first one controlling parasite proliferation and the second one controlling the death of both intracellular parasites and host macrophages, via caspase-1 dependent inflammasome activation.

Discussion
Forward genetics has proved to be a powerful tool to characterize novel biological pathways implicated in host resistance to infection [18], [19]. In rats, the Toxo1 locus located on chromosome 10 [10] controls the outcome of toxoplasmosis by a still poorly defined mechanism. In the present work, genetic dissection of Toxo1 with a panel of new congenic sub-lines together with haplotype mapping led us to identify a 891 kb interval of rat chromosome 10 that is highly conserved amongst resistant rat strains and that controls both T. gondii infection outcome in vivo and macrophage responses to infection in vitro. We further demonstrated that the Toxo1-mediated refractoriness of macrophages to T. gondii infection is associated with a caspase-1-dependent rapid T. gondii-induced death of both intracellular parasites and host macrophages.
The Toxo1-dependent death of infected macrophages displayed the chromatin condensation hallmark of apoptosis but neither caspase-3 activation nor DNA fragmentation were observed. In contrast to apoptosis which is usually a slow process characterized by membrane blebbing, the Toxo1-associated cell death was characterized by a rapid loss of plasma membrane integrity that occurs simultaneously with surface exposure of phosphatidylserine.
A non-apoptotic pathway triggered in T. gondii-infected macrophages has been described in mice [20]. It is mediated by IFN-γ-inducible immunity-related GTPases that trigger vacuole membrane disruption and the death of parasites, followed by the necrotic-like death of infected cells. While phenotypically, the IRG-dependent mouse T. gondii/macrophage deaths parallel the Toxo1-mediated T. gondii/macrophage deaths, major differences between the two in vitro models exist, providing strong evidences that mechanistically they are not identical. The mouse IRG-controlled mechanism requires IFN-γ stimulation and occurs in both fibroblasts and macrophages. In contrast, the Toxo1 effect in rats is strictly confined to hematopoeitic cells [9], [10] and does not depend on IFN-γ stimulation of macrophages in vitro. Moreover, the mouse IRG-controlled resistant system is genotypically restricted to non-virulent genotype II parasites, while in rats, both type I RH and type II PRU strains triggered the LEW T. gondii/macrophage deaths (Figure S1). In line with this, the effector of the LEW rat resistance is not the kinase parasite effector ROP 18 (data not shown) which, by phosphorylating IRGs, disrupts their association with the parasite-containing vacuole and thereby protects the parasite against elimination [21], [22]. Altogether, the rat Toxo1 locus-mediated resistance to toxoplasmosis does not operate via the IRG-dependent system.
Sequencing of the Toxo1 region in all five resistant rat strains (LEW, LOU, BDIX, WK and WF) and four susceptible strains (BN, OM, DA and F344) revealed a gradient in the conserved distribution of resistant-restricted mutations that peaks at the bottom of Toxo1 and particularly in the Nlrp1a coding sequence (23 SNPs). This highly conserved region has been previously demonstrated to be associated with the resistance of rats to the lethal Toxin (LT) of Bacillus anthracis [23]. Interestingly, while rat bearing the divergent Toxo1 alleles were susceptible to LT-mediated death, the highly conserved Toxo1-LEW alleles correlated with the total resistance of rats [23]. Moreover, similarly to what we found for the Toxo1 control of T. gondii infectivity, there was a perfect correlation between the in vivo phenotype (sensitivity vs resistance to LT) and the in vitro phenotype of macrophages, suggesting that the same gene or set of genes might be at work in the control of these two pathogens. In mice and rats, further genetic mapping associated genetic variants of both mNlrp1b and one of its rat ortholog rNlrp1a to macrophage sensitivity or resistance to LT by a mechanism dependent on a cell death process called pyroptosis, which is also triggered upon infection with intracellular pathogens such as Salmonella, Shigella or Listeria [24]–[26]. NLRP1 is part of the NLR family, which is known to act as an intracellular sensor for cytoplasmic danger signals [12]. After activation, the NLR form a multimeric protein complex called the inflammasome that provides a scaffold for the activation of caspase-1 and target death substrates by a still poorly understood mechanism [12]. The Toxo1 controlled-cell death induction that is triggered in peritoneal macrophages following T. gondii invasion, appeared to be also caspase-1 dependent and associated to IL-1β secretion and ROS production. Together, with the nuclear condensation, it thus features the hallmarks of the pyroptosis programmed-cell death which is uniquely dependent on caspase-1 and inherently proinflammatory. However, while pyroptosis is typically associated to DNA fragmentation [16], this later event was not observed in the T. gondii-induced cell death possibly due to a yet unexplained undetectable PARP (Poly ADP-Ribose Polymerase) expression in rat peritoneal macrophages (Figure S3, Text S1). Together these observations combined with the genetics studies tend to support that the NOD-like receptor NLRP1a/Caspase-1 pathway is the best candidate to mediate the Toxo1-dependent parasite-induced cell death.
Despite these remarkable genetic and biochemical similarities, LT - and T. gondii-induced phenotypes display striking major differences. First, while Toxo1-conserved LEW alleles are associated to the cell death induction of macrophages upon T. gondii infection, same allelic variants protect the macrophages from LT-mediated cell death [23]. Secondly, while the pharmacological inhibition of host cell death also prevents the concomitant death of parasites, it failed to restore the permissiveness of macrophages to parasite proliferation. Thus, although caspase-1 is induced following T. gondii infection, our data do not argue for its essential role in the macrophage control of parasite proliferation in vitro. They rather suggest that, like it is emerging in the case of intracellular bacteria, the Toxo1-directed resistance of macrophages to T. gondii proliferation may rather be a complex trait resulting from the combined activation of several pathways. For instance, the Naip5/Nlrc4-controlled restriction of Legionella pneumophila growth within non permissive murine macrophages is likely to involve both canonical pyroptosis and a yet undefined caspase-1 independent Naip5 pathway [27]. Very interestingly, in mouse macrophages infected by Shigella flexneri which display NLRC4 - and NLRP3-dependent activation of caspase-1, IL-1β/IL-18 processing and cell death [17], [26], it has been demonstrated that inflammasome might negatively regulate pathogen-induced autophagy [26]. Given that other NLRs may affect autophagy [17], inflammasome inhibition of autophagy might be a generalized mechanism. Moreover, Harris J. and al. demonstrated that autophagy controls IL1-β secretion by targeting pro-IL1-β for degradation [28]. In line with this, the lack of detectable pro-IL1-β in resistant LEW macrophages treated with caspase-1 inhibitor, suggested that inactivation of caspase-1 results in the down-regulation of pro-IL1-β protein expression. It is therefore possible that in our model, the inhibition of caspase-1 could block the negative regulation of inflammasome promoting thus the effect of other pathway(s) capable to restrict parasite proliferation without pyroptosis. Altogether, our work provide evidence that natural resistance of rat macrophages to T. gondii is a complex trait relying on a mechanism involving the combined activation of at least two pathways: (i) the classical NLRP1a/Caspase-1 pathway, mediating host and parasite cell death and (ii) a yet undefined pathway that would directly control parasite proliferation within the parasitophorous vacuole. The parasite effector(s) eliciting these pathways and their possible interconnections remain to be investigated.
Toxoplasma infection is naturally acquired by the oral route. Following transcytosis across the intestinal barrier [29], the tachyzoite stage encounters leukocytes in which it replicates to further disseminate into the organism using the migratory properties of infected macrophages and dendritic cells [30]. In rats bearing the LEW-type Toxo1 locus, the refractoriness to toxoplasmosis is evidenced by the absence of both local parasite burden and specific antibody response [9]. Thus resistance likely results from the rapid clearance of the parasite following a vigorous killing response at the site of infection. Our data led us to propose a model where the early death of infected macrophages impairs further dissemination of the parasite, hence constituting an efficient barrier against successful infection. How the Toxo1 locus controls dendritic cell and monocyte responses after infection remains to be characterized.
In conclusion, this work highlighted several novel aspects of the host-parasite gene interaction. We unambiguously mapped the Toxo1 locus to an 891 kb region which directs the outcome of toxoplasmosis in the rat. This locus controls parasite infectivity in vivo and is critically associated to macrophage-dependent restriction of parasite intracellular growth and cell death induction of both intracellular parasites and infected macrophages in vitro. It is included within a haplotype block remarkably subjected to strong selection pressure for resistant strains, in contrast to susceptible strains (Figure S2, Text S1). The robustness of Toxo1-mediated resistance in controlling parasite infectivity indicates that this resistance might have been conserved among naturally resistant species [11]. Hence, Transmission Disequilibrium Test studies revealed that hNlrp1, the human ortholog of rat rNlrp1a, has alleles associated with the susceptibility to human congenital toxopolasmosis [6]. In the same way, recent work demonstrated that, in mice, NLRP1 is an innate immune sensor for Toxoplasma infection inducing a host-protective innate immune response to the parasite [31]. Altogether, these data identified a genetically-controlled major pathway of innate immunity to toxoplasmosis allowing predicting resistance of individuals. The results open the way to further investigations towards the gene(s) and the mechanisms at work, and could be applied to human toxoplasmosis in regards to the conserved synteny of Toxo1 region between rat and human.
Materials and Methods
Ethics statement
Breeding and experimental procedures were carried out in accordance with national and international laws for laboratory animal welfare and experimentation (EEC Council Directive 2010/63/EU, September 2010). Experiments were performed under the supervision of M–F. C–D. (agreement 38 10 38) in the Plateforme de Haute Technologie Animale (PHTA) animal care facility (agreement n° A 38 516 10006 delivered by the Direction Départementale de la Protection des Populations) and were approved by the ethics committee of the PHTA (permits n° Toxo-PC-1 and n° Toxo-PC-2).
Rats and infection
Production and genotype analyses of congenic lines were as described previously [10]. The congenic lineages were maintained by regular brother-sister mating. LEW/OrlRj (LEW), BN/OrlRj (BN) male rats were obtained from Janvier Laboratory (Le Genest-Saint-Isle, France). WF/N (WF), WKY/NHsd (WK), BDIX/Han (BDIX), F344/Nhsd (F344), Lou/CNimrOlaHsd (LOU) and DA/OlaHsd (DA) male rats were obtained from Harlan Laboratory (Gannat, France). OM/Han (OM) rats were kindly supplied by the Hannover Medical School (Germany). F2 (LOU×BN) progenies were produced in our animal facilities under specific pathogen-free conditions. Cysts from the recombinant T. gondii Prugniaud strain were used to test in vivo the susceptibility to toxoplasma infection. Two-month-old Swiss mice (Janvier laboratory) were infected orally with 10 Prugniaud cysts. Their brains were collected 3 months later and ground in a Potter. Cysts were counted in a Thoma's cell and diluted in PBS. Rats were infected orally with 20 cysts. One month later, blood was collected from the retro-orbital sinus for detection of anti-toxoplasma Ab response by ELISA. Rats were euthanized 2 months after infection and brains were collected to determine number of cysts.
Parasites
Tachyzoites of Prugniaud type II strain, and RH, RH-YFP2 (kindly provided by B. Striepen, Athens) and RH-mcherry (kindly provided by A. Bougdour, Grenoble) type I T. gondii strains were maintained under standard procedures, by serial passage onto human foreskin fibroblast monolayers (HFFs) in D10 medium (DMEM supplemented with 10% heat-inactivated fetal bovine serum, 1 mM glutamine, 500 units.ml−1 penicillin and 50 µg.ml−1 streptomycin) at 37°C in a humidified atmosphere containing 5% CO2. The parasites were collected just before the experiment, centrifuged at 500× g for 7 min, suspended in serum-free medium (SFM, GIBCO) supplemented with 500 units.ml−1 penicillin and 50 µg.ml−1 streptomycin, and counted.
Peritoneal macrophages and infection
Rat resident peritoneal cells were obtained by injection of sterile PBS into the peritoneal cavity. Collected cells were centrifuged and resuspended in Serum Free Medium (SFM) (Life Technologies, Inc) and counted. Macrophages were obtained by adhering cells for 1 h at 37°C and 5% CO2. After 1 h, non-adherent cells were removed by gentle washing with SFM and parasites were added to macrophages settled on coverslips at a ratio of 3∶1. After incubation for 1 h at 37°C, wells were washed 3 times with SFM to remove extracellular parasites and cells fixed at different times post-infection in 4% formaldehyde.
Chemicals
Caspase-1 inhibitor VI (YVAD) and caspase inhibitor VI (pan-caspase, Z-VAD) were from Calbiochem (Merck Chemicals, France). Macrophages were incubated with 50 µM of Caspase-1 inhibitor or 100 µM of pan-caspase inhibitor for 2 h before infection and during all the infection.
Fluorescence microscopy
Infected macrophages were permeabilized with 0.002% saponine or 0.1% triton-×100 to detect SAG1 (mAb Tg05-54), GRA5 (mAb Tg17-113) or GRA3 (mAb Tg2H1). The rabbit anti-Tg small ubiquitin-like modifier (TgSUMO) polyconal antibody was kindly provided by M.A. Hakimi (Grenoble, France) [15]. To detect acidic vacuoles, infected macrophages were stained with 50 nM LysoTracker Red for 30 min prior to fixation. Cell death was analyzed by visualizing the uptake of Propidium Iodide (PI) (Molecular Probes). Alexa488 and Alexa594 antibody conjugates (Molecular Probes) were used as secondary antibodies. Coverslips were mounted in mowiol and observed and counted with a Zeiss Axioplan 2 microscope equipped for epifluorescence and phase-contrast. Kinetic experiments were performed on the IX2 Olympus microscope and analyzed with ScanR software.
Caspase-1 activation assay
For analysis of caspase-1 activation, we used a fluorescent caspase-1 activity assay, FLICA, from Immunochemistry Technologies (Bloomington, MN). The assay was performed in 24-well plates, with 5.105 cells per well. Cells were incubated with T. gondii and stained with FLICA reagent (FAM-YVAD-FMK) as recommended by the manufacturer. Fluorescence was measured on the IX2 Olympus microscope and analyzed with ScanR software.
Annexin V assay
The level of apoptosis of infected LEW macrophages was assessed with Annexin V-propidium iodide staining. Rat peritoneal exudent cells were treated with etoposide (Sigma Aldrich, 50 µM) or incubated with parasites in SFM at a MOI of 1∶3 at 37°C, 5% CO2. After 1 h, cells were collected by centrifugation and incubated 4 h at 37° after addition of fresh medium. Macrophages were stained by the addition of Annexin V (Santa Cruz biotechnologies, FL-319) and Alexa488 secondary antibody (Molecular Probes) for 30 min and 1 µg/mL of PI for 5 min. Data acquisition was performed by flow cytometry on 4-colours FACSCalibur (BD Biosciences) equipped with 488 nm argon laser and CellQuest Software.
DNA fragmentation analysis
Rat peritoneal macrophages (∼106) were infected with 3×106 parasites for 1 h, washed and returned to 37°C for 6 h, 12 h or 18 h. For positive control of apoptosis, macrophages were treated with 50 µM etoposide (Sigma Aldrich). Macrophages were treated in lysis-buffer (100 mM Tris, pH 8.0, 100 mM EDTA, 4% Sodium dodecyl sulfate (SDS) with 1 µg/ml RNAse (Roche) (30 min, 37°C) followed by treatment with 100 µg/ml proteinase K (Euromedex) (30 min, 55°C). After extraction with phenol/chloroform, DNA was recovered by precipitation and analysed on 1.8% agarose gels.
Radical oxygen species production by macrophages
Rat peritoneal exudent cells were incubated with parasites in SFM at a MOI of 1∶3 at 37°C, 5% CO2 for 15 min or 4 h and then incubated with 0.45 µM of dihydro-rhodamine 123 (DHR, Sigma Aldrich) 15 min at 37°C. At the end of incubation, FACS lysing buffer (BD Bioscience, Pont de Claix, France) was added to each sample and incubated for 15 min. The samples were then washed in PBS before analysis with a FACSCalibur flow cytometer (Becton Dickinson) and the CellQuest Pro software (Becton Dickinson).
Immunoblotting
Cells were lysed in cold RIPA (50 mM Tris-Hcl pH 7,4, 150 mM NaCl, 1% NP40, 0,25% Na-deoxycholate) buffer supplemented with protease inhibitors and centrifuged at 4°C and 13,000 g for 10 min. Supernatant of infected or uninfected macrophages (5.105 cells/well) were collected and precipitated with TCA (trihloroacetate) for 10 min at 4°C prior centrifugation at 16 000 g for 5 min. Pellets were washed two times in acetone then dried and resuspended in laemmli buffer. Protein extracts were subjected to electrophoresis on a 12% Tris-HCl SDS-PAGE and transferred to PVDF membranes (Amersham). Membranes were blocked for 1 h in TTBS (100 mM Tris-HCl, 0.9% NaCl, and 0.05% Tween 20) containing 5% skim milk before incubating overnight at 4°C with primary 1/500 anti-caspase 3 (Cell Signaling), 1/1000 anti-IL-1β (Millipore, AB1832P), 1/5000 anti-GAPDH (Santa Cruz) or 1/1000 anti-actin (Sigma Aldrich) antibodies followed by 1/10000 anti-rabbit secondary horseradish peroxidase (HRP)-linked antibodies (Jackson Immunoresearch). Visualization of signals was enhanced by luminol-based chemiluminescence (ECL, ThermoFisher Scientific).
T. gondii proliferation assay in peritoneal macrophages
The intracellular growth of T. gondii in rat peritoneal macrophages was monitored by selective incorporation of [3H]uracil as previously described [32]. Briefly, 5×105 macrophages were infected with 1.5×106 parasites for 1 h in SFM at 37°C and 5% CO2. After washing to eliminate extracellular parasites, cells were cultured for 20 h in the presence of [3H]uracil (5 µCi per well, Ci = 37 GBq). Monolayers were washed three times in PBS, disrupted with 500 µl of lysis/scintillation solution (Optiphase Supermix, Perkin Elmer) and radioactivity measured by liquid scintillation counting using a Wallac MicroBeta TriLux (Perkin Elmer).
Genetic markers and genotype analysis
Preparation of genomic DNA and genotyping were performed as described [33]. To genotype the 35 (LOU X BN) F2 rat progenies, six microsatellite markers (D10Rat49, D10Arb2, D10GF41, D10Rat27, D10Mgh4, D10Rat2) were selected to cover chromosome 10 with an average spacing of 20 Mb. For haplotype analysis, the polymorphism of 41 microsatellite sequences around the Toxo1 locus were analysed in the nine strains. Among these 41 markers, 17 newly identified microsatellite markers (Table S1) were used in addition to the 24 microsatellite markers selected from Rat Genome Database (RGD).
ELISA
The anti-Toxoplasma IgG response was measured by specific enzyme-linked immunosorbent assay (ELISA). Total Toxoplasma antigens were prepared as previously described [34]. Immuno plates Maxisorp (Nunc) were coated overnight at 4°C with Toxoplasma antigens at 20 µg/ml. After washing, saturation was 1 h at 37°C with PBS containing 5% of skim milk. Sera were diluted at 1/20 and 1/1000 in PBS-0.01% Tween 20 and incubated 1h30 at 37°C. Plates were then washed with PBS-0.01% Tween 20, and peroxydase-conjugated anti-rat IgG (KPL) secondary antibody diluted at 1/5000 was incubated 1 h at 37°C. Finally, after six washes, 100 µl of substrate TMB-hydrogen peroxyde (TMB Ultra 1 Step, ThermoScientific) solution was added to the wells. The color reaction was stopped adding 50 µl of 3 N HCl. Optical densities at 492 nm and 630 nm were measured using an ELx800 absorbance microplate reader (Bio TeK Instruments). Results were expressed as arbitrary units.
Cyst detection
Each rat brain was removed and homogenized in 16 ml of PBS. Brain suspensions were clarified by gentle incubation in proteinase K buffer (proteinase K 0.4 µg/ml, 10 mM Tris pH 8, EDTA 1 mM, sodium dodecyl sulfate 0.2%, sodium chloride 40 mM) for 15 min at 56°C. The reaction was stopped by incubation with PMSF 2 mM for 5 min at room temperature. Then, the suspension was washed with PBS and resuspended with FITC-Dolichos biflorus agglutinin (Vector laboratories, CA USA) 20 µg/ml for 30 min, at room temperature. After washing in PBS, rat brains were resuspended in 6 ml of PBS, distributed into six-well culture plates (1 ml per well) and cysts counted visually with an Axiovert 40 CFL inverted fluorescence microscope (Zeiss) [35].
Toxo1 sequencing
A custom-made SureSelect oligonucleotide probe library was designed by IntegraGen (Evry, France) to capture the chr10 region containing Toxo1 locus (chr10 : 57,200,000–58,200,000). The eArray web-based probe design tool was used for this purpose (https://earray.chem.agilent.com/earray). A total of 56,450 probes, covering a target of 6830692 bp, were synthesized by Agilent Technologies (Santa Clara, CA, USA). Library preparation, capture enrichment, sequencing, and variants detection and annotation, were performed by IntegraGen (Evry, France). Briefly, 3 µg of each genomic DNA were fragmented by sonication and purified to yield fragments of 150–200 bp. Paired-end adaptor oligonucleotides from Illumina were ligated on repaired DNA fragments, which were then purified and enriched by six PCR cycles. 500 ng of the purified libraries were hybridized to the SureSelect oligo probe capture library for 24 h. After hybridization, washing, and elution, the eluted fraction underwent 14 cycles of PCRamplification. This was followed by purification and quantification by qPCR to obtain sufficient DNA template for downstream applications. Each eluted-enriched DNA sample was then sequenced on an Illumina GAIIx as paired-end 75 bp reads. Image analysis and base calling was performed using Illumina Real Time Analysis (RTA) Pipeline version 1.10 with default parameters. Sequence reads were aligned to the reference rat genome (UCSC rn4) using commercially available software (CASAVA1.7, Illumina) and the ELANDv2 alignment algorithm. Sequence variation annotation was performed using the IntegraGen in-house pipeline, which consisted of gene annotation (RefSeq), detection of known polymorphisms (dbSNP 125) followed by mutation characterization (exonic, intronic, silent, nonsense etc.).
Statistical analyzes
For in vivo experiments, data are expressed as means ± SEM and the significance of differences found between groups was initially derived from a Kruskal-Wallis H test and subsequently confirmed by the Mann-Whitney test. For in vitro experiments, data are expressed as means ± SD and the significance of differences found between groups was determined using two-tailed Student's t test.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. HillD, DubeyJP (2002) Toxoplasma gondii: transmission, diagnosis and prevention. Clin Microbiol Infect 8 : 634–40.
2. CarruthersVB (2002) Host cell invasion by the opportunistic pathogen Toxoplasma gondii. Acta Trop 81 : 111–22.
3. MackDG, JohnsonJJ, RobertsF, RobertsCW, EstesRG, et al. (1999) HLA-class II genes modify outcome of Toxoplasma gondii infection. Int J Parasitol 29 : 1351–8.
4. SibleyLD, MordueDG, SuC, RobbenPM, HoweDK (2002) Genetic approaches to studying virulence and pathogenesis in Toxoplasma gondii. Philos Trans R Soc Lond B Biol Sci 357 : 81–8.
5. VillenaI, AncelleT, DelmasC, GarciaP, BrezinAP, et al. (2010) Congenital toxoplasmosis in France in 2007: first results from a national surveillance system. Euro Surveill 15 19600.
6. WitolaWH, MuiE, HargraveA, LiuS, HypoliteM, et al. (2011) NALP1 influences susceptibility to human congenital toxoplasmosis, proinflammatory cytokine response, and fate of Toxoplasma gondii-infected monocytic cells. Infect Immun 79 : 756–66.
7. CouvreurJ, DesmontsG, GirreJY (1976) Congenital toxoplasmosis in twins: a series of 14 pairs of twins: absence of infection in one twin in two pairs. J Pediatr 89 : 235–40.
8. KempfMC, Cesbron-DelauwMF, DesleeD, GrossU, HerrmannT, et al. (1999) Different manifestations of Toxoplasma gondii infection in F344 and LEW rats. Med Microbiol Immunol 187 : 137–42.
9. SergentV, CautainB, KhalifeJ, DesleeD, BastienP, et al. (2005) Innate refractoriness of the Lewis rat to toxoplasmosis is a dominant trait that is intrinsic to bone marrow-derived cells. Infect Immun 73 : 6990–7.
10. CavaillesP, SergentV, BisanzC, PapapietroO, ColaciosC, et al. (2006) The rat Toxo1 locus directs toxoplasmosis outcome and controls parasite proliferation and spreading by macrophage-dependent mechanisms. Proc Natl Acad Sci U S A 103 : 744–9.
11. CuppenE (2005) Haplotype-based genetics in mice and rats. Trends Genet 21 : 318–22.
12. FranchiL, Munoz-PlanilloR, NunezG (2012) Sensing and reacting to microbes through the inflammasomes. Nat Immunol 13 : 325–32.
13. MoayeriM, SastallaI, LepplaSH (2013) Anthrax and the inflammasome. Microbes Infect 14 : 392–400.
14. AndradeRM, WessendarpM, GubbelsMJ, StriepenB, SubausteCS (2006) CD40 induces macrophage anti-Toxoplasma gondii activity by triggering autophagy-dependent fusion of pathogen-containing vacuoles and lysosomes. J Clin Invest 116 : 2366–77.
15. BraunL, CannellaD, PinheiroAM, KiefferS, BelrhaliH, et al. (2009) The small ubiquitin-like modifier (SUMO)-conjugating system of Toxoplasma gondii. Int J Parasitol 39 : 81–90.
16. LabbeK, SalehM (2008) Cell death in the host response to infection. Cell Death Differ 15 : 1339–49.
17. SchroderK, TschoppJ (2010) The inflammasomes. Cell 140 : 821–32.
18. GruenheidS, GrosP (2010) Forward genetic dissection of innate response to infection in inbred mouse strains: selected success stories. Clin Exp Immunol 162 : 393–401.
19. JagodicM, ColaciosC, NohraR, DejeanAS, BeyeenAD, et al. (2009) A role for VAV1 in experimental autoimmune encephalomyelitis and multiple sclerosis. Sci Transl Med 1 : 10ra21.
20. ZhaoYO, KhaminetsA, HunnJP, HowardJC (2009) Disruption of the Toxoplasma gondii parasitophorous vacuole by IFNgamma-inducible immunity-related GTPases (IRG proteins) triggers necrotic cell death. PLoS Pathog 5: e1000288.
21. FentressSJ, BehnkeMS, DunayIR, MashayekhiM, RommereimLM, et al. (2010) Phosphorylation of immunity-related GTPases by a Toxoplasma gondii-secreted kinase promotes macrophage survival and virulence. Cell Host Microbe 8 : 484–95.
22. SteinfeldtT, Konen-WaismanS, TongL, PawlowskiN, LamkemeyerT, et al. (2010) Phosphorylation of mouse immunity-related GTPase (IRG) resistance proteins is an evasion strategy for virulent Toxoplasma gondii. PLoS Biol 8: e1000576.
23. NewmanZL, PrintzMP, LiuS, CrownD, BreenL, et al. (2010) Susceptibility to anthrax lethal toxin-induced rat death is controlled by a single chromosome 10 locus that includes rNlrp1. PLoS Pathog 6: e1000906.
24. CervantesJ, NagataT, UchijimaM, ShibataK, KoideY (2008) Intracytosolic Listeria monocytogenes induces cell death through caspase-1 activation in murine macrophages. Cell Microbiol 10 : 41–52.
25. FranchiL, AmerA, Body-MalapelM, KannegantiTD, OzorenN, et al. (2006) Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1beta in salmonella-infected macrophages. Nat Immunol 7 : 576–82.
26. SuzukiT, FranchiL, TomaC, AshidaH, OgawaM, et al. (2007) Differential regulation of caspase-1 activation, pyroptosis, and autophagy via Ipaf and ASC in Shigella-infected macrophages. PLoS Pathog 3: e111.
27. LamkanfiM, AmerA, KannegantiTD, Munoz-PlanilloR, ChenG, et al. (2007) The Nod-like receptor family member Naip5/Birc1e restricts Legionella pneumophila growth independently of caspase-1 activation. J Immunol 178 : 8022–7.
28. HarrisJ, HartmanM, RocheC, ZengSG, O'SheaA, et al. (2011) Autophagy controls IL-1beta secretion by targeting pro-IL-1beta for degradation. J Biol Chem 286 : 9587–97.
29. BarraganA, SibleyLD (2002) Transepithelial migration of Toxoplasma gondii is linked to parasite motility and virulence. J Exp Med 195 : 1625–33.
30. CourretN, DarcheS, SonigoP, MilonG, Buzoni-GatelD, et al. (2006) CD11c - and CD11b-expressing mouse leukocytes transport single Toxoplasma gondii tachyzoites to the brain. Blood 107 : 309–16.
31. EwaldSE, Chavarria-SmithJ, BoothroydJC (2014) NLRP1 is an inflammasome sensor for Toxoplasma gondii. Infect Immun 82 : 460–8.
32. PfefferkornER, PfefferkornLC (1977) Specific labeling of intracellular Toxoplasma gondii with uracil. J Protozool 24 : 449–53.
33. MasM, SubraJF, LagrangeD, Pilipenko-AppolinaireS, KermarrecN, et al. (2000) Rat chromosome 9 bears a major susceptibility locus for IgE response. Eur J Immunol 30 : 1698–705.
34. GodardI, DarcyF, DesleeD, DessaintJP, CapronA (1990) Isotypic profiles of antibody responses to Toxoplasma gondii infection in rats and mice: kinetic study and characterization of target antigens of immunoglobulin A antibodies. Infect Immun 58 : 2446–51.
35. AldebertD, HypoliteM, CavaillesP, TouquetB, FloriP, et al. (2011) Development of high-throughput methods to quantify cysts of Toxoplasma gondii. Cytometry A 79 : 952–8.
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2014 Číslo 4
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Měli bychom postcovidový syndrom léčit antidepresivy?
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- The 2010 Cholera Outbreak in Haiti: How Science Solved a Controversy
- , , , Genetic Variability: Cryptic Biological Species or Clonal Near-Clades?
- Efficient Parvovirus Replication Requires CRL4-Targeted Depletion of p21 to Prevent Its Inhibitory Interaction with PCNA
- An Overview of Respiratory Syncytial Virus