
Potent Dengue Virus Neutralization by a Therapeutic Antibody with Low Monovalent Affinity Requires Bivalent Engagement
Dengue virus (DENV) is a globally important mosquito-transmitted human pathogen for which there is no approved vaccine or antiviral therapy. In recent years, the number and severity of DENV human infections have increased due to the expanded geographic range of the virus. Neutralizing antibodies are a key component of a protective natural and vaccine-induced immune response against human DENV infections. One recently described monoclonal antibody (E106) protects mice against infection of DENV-1 when administered before or several days after virus infection. Because of these results, we investigated the mechanism of action of E106 using a combination of structural and functional approaches. E106 engaged an epitope on domain III of the viral envelope protein that is a composite of two previously described epitopes. Unexpectedly, and in contrast to the intact IgG, Fab fragments of E106 were ineffective at neutralizing virus; this was explained by their weak micromolar affinity for virus particles. Our results suggest that neutralization by E106, our most potently inhibitory and protective anti-DENV MAb, requires bivalent binding of adjacent DIII subunits on a single virion. Immunization strategies with intact virions that skew the selection of neutralizing antibodies to those with bivalently binding properties could augment the potency of antiviral humoral responses against DENV and other flaviviruses.
Published in the journal:
. PLoS Pathog 10(4): e32767. doi:10.1371/journal.ppat.1004072
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004072
Summary
Dengue virus (DENV) is a globally important mosquito-transmitted human pathogen for which there is no approved vaccine or antiviral therapy. In recent years, the number and severity of DENV human infections have increased due to the expanded geographic range of the virus. Neutralizing antibodies are a key component of a protective natural and vaccine-induced immune response against human DENV infections. One recently described monoclonal antibody (E106) protects mice against infection of DENV-1 when administered before or several days after virus infection. Because of these results, we investigated the mechanism of action of E106 using a combination of structural and functional approaches. E106 engaged an epitope on domain III of the viral envelope protein that is a composite of two previously described epitopes. Unexpectedly, and in contrast to the intact IgG, Fab fragments of E106 were ineffective at neutralizing virus; this was explained by their weak micromolar affinity for virus particles. Our results suggest that neutralization by E106, our most potently inhibitory and protective anti-DENV MAb, requires bivalent binding of adjacent DIII subunits on a single virion. Immunization strategies with intact virions that skew the selection of neutralizing antibodies to those with bivalently binding properties could augment the potency of antiviral humoral responses against DENV and other flaviviruses.
Introduction
Dengue virus (DENV) infection in humans causes symptoms ranging from a mild febrile illness to a severe and sometimes fatal disease. Over 3.6 billion people globally are at risk for DENV infection, with an estimated 390 million infections annually and no currently approved vaccine or antiviral therapy [1]. DENV belongs to the Flaviviridae family of medically important positive-stranded RNA viruses. Within the DENV serocomplex, there is significant diversity, including four serotypes (DENV-1, -2, -3, and 4) that differ at the amino acid level of the envelope (E) protein by ∼25 to 40 percent and multiple genotypes within a serotype that vary by up to ∼3 percent [2], [3].
A humoral response against DENV infection is believed to contribute to lifelong immunity against challenge by the homologous serotype. In comparison, protection against a heterologous DENV serotype infection is more transient (∼6 months to two years) [4], [5], allowing re-infection and disease to occur with a heterologous serotype in hyper-endemic areas of the world. Estimates suggest that greater than 90% of severe cases occur during secondary infection with a heterologous DENV serotype, possibly because sub-neutralizing amounts of cross-reactive antibody facilitate viral entry into myeloid cells expressing Fc-γ receptors, a phenomenon termed antibody-dependent enhancement of infection (ADE) [6]. Antibody-mediated protection against homologous DENV infection correlates with a neutralizing antibody response directed predominantly against the viral E protein [7]. The ectodomain of E is comprised of three domains: domain I (DI), a central nine-stranded β-barrel that connects domain II (DII), which contains the fusion peptide at its distal end, and an immunoglobulin-fold like domain III (DIII) [6]–[9]. Although neutralizing antibodies have been mapped to all three domains of the E protein, many potently inhibitory anti-DENV mouse MAbs map to DIII [10]–[17], specifically to the lateral ridge or A-strand epitopes, and block flavivirus infection at a post-attachment stage, likely by preventing E protein dimer-to-trimer transitions that are required for viral fusion [11], [15], [18].
We recently described a highly therapeutic MAb, DENV-1-E106 (hereafter termed E106), which neutralized infection of strains corresponding to all five DENV-1 genotypes and protected against lethal DENV-1 infection when administered as a single dose even four days after virus inoculation [13]. To understand the basis for the potency (plaque reduction neutralization titer (PRNT50) of 0.6 ng/ml) [13] and specificity of this MAb, we solved the crystal structure of E106 Fab in complex with DENV-1 E DIII. Our analysis revealed a small antibody-antigen interface with contact residues corresponding to two previously characterized DIII epitopes. Remarkably, a ∼20,000-fold disparity in neutralization by intact IgG and Fab correlated with distinct abilities to bind intact virions. Our results are consistent with a model in which our most potently inhibitory and therapeutic DENV-1 MAb requires bivalent binding through dual and simultaneous engagement of two antigen binding sites on a single virion to neutralize infection. E106 is therefore one of only a few unique antibodies described to date where effective neutralization requires a bivalent binding mechanism, and is the first such characterized MAb directed against flaviviruses.
Results
The crystal structure of E106 in complex with DIII reveals a small footprint
E106 is a sub-complex specific therapeutic MAb that binds to DENV-1 and DENV-4 infected cells and neutralizes infection of all five DENV-1 genotypes efficiently (EC50 ranging from 1 to 50 ng/ml), without neutralizing DENV-2, 3 and 4 serotypes or WNV [13]. The 2.45 Å crystal structure of E106 Fab bound to DIII revealed only nine contact residues, from the A-strand (K307, K310), the end of the B-strand (K325, Y326), and the connecting BC (E327, T329, D330) and DE (K361, E362) loops (Fig. 1A and Table 1); these results are consistent with prior mapping data by yeast surface display, which implicated five of these residues as essential recognition determinants (K310, T329, D330, K361 and E362) [13]. Yeast surface display results also implicated G328, P332, and P364 in E106 binding, and mutation of any of these residues would likely result in an altered presentation of the direct contact residues. Charge reversals at either E384 or K385 in the FG-loop also diminished E106 recognition, and this loop is adjacent to the primary E106 epitope. Overall, the contact residues contributed 24 van der Waal interactions, 14 hydrogen bonds, and 10 electrostatic interactions to the interface (Table S1). The E106 structural footprint represents a unique composite of previously identified DIII-specific neutralizing epitopes on flaviviruses including the lateral ridge (N-terminal region, BC, DE and FG loops) [19], [20] and A-strand epitopes [10], [21]. DENV-1 DIII was engaged by 11 heavy chain residues from CDR1 (I30, G31, Y32 and Y33), CDR2 (N52, E50, and R53), and CDR3 (R95, I196, N97 and W98) (Fig. 1B, top panel) and four light chain residues from CDR1 (D30, D32), CDR2 (E50), and CDR3 (L94) (Table S1).


A comparison of the DIII structure in complex with Fab versus unbound DIII revealed small conformational differences, with a root mean square displacement of 0.9 Å in the α-carbons over 98 residues. Of the DIII residues that directly interacted with the E106 Fab, the greatest differences in α-carbon positions involved residue T329 (1.2 Å); this was significant because a recently identified E106 MAb neutralization escape mutant showed a T to A amino acid change at position 329 (Fig. 1B, bottom panel) and [22]).
The E106 structural epitope on DIII was characterized by a high shape complementarity score (Sc = 0.73, with a perfect fit being 1) [23], which is greater than typical antibody-antigen interactions (Sc = 0.64–0.68) [23] but similar to anti-flavivirus MAb-E protein interactions (Table 2). The combined surface area buried by the DIII-E106 Fab complex was ∼1,243 Å2 (Fig. 1C and Table 2) [24] which is less than most antibody-antigen (1,400–2,300 Å2) [25], [26] and anti-flavivirus MAb-E protein interactions (Table 2). Typical of many antibody-antigen complexes, the majority (∼70%) of the DIII-E106 Fab interface was contributed by the heavy chain (Fig. 1C), with a combined buried surface of 877 Å2 (401 Å2 of the heavy chain and 476 Å2 of domain III). The light chain contributed the remaining buried surface (172 Å2 of the light chain and 194 Å2 of domain III).

All nine DIII contact residues were conserved in the five DENV-1 genotypes (K361 is replaced by the conservative substitution R361 in genotype 5 strains); this likely explains why E106 neutralized infection of all five DENV-1 genotypes efficiently (Fig. 1D and 1E), and [13]). In comparison, only one of the nine contact residues (Y326) was conserved in DENV-2, DENV-3, DENV-4 or WNV, a finding that is consistent with virological data showing that DENV-1-E106 MAb neutralizes infection in a serotype-specific manner [13]. E106 binds to DENV-4 but not to DENV-2 or DENV-3 [13]. The number of conserved contact residues did not correlate with DENV serotype binding specificity (DENV-4 has four whereas DENV-2 and DENV-3 each have five, Fig. 1E), which may instead be accounted for by other factors, including differential maturation [27] or relative virion dynamics [28].
E106 Fab binds DIII with micromolar monovalent affinity
To determine the significance of the small buried interface of our E106-DIII complex, we investigated E106 binding to DIII by surface plasmon resonance (SPR). Increasing concentrations of purified DENV-1 DIII monomer were flowed over immobilized E106 Fab (Fig. 2A). Equilibrium analysis surprisingly revealed a micromolar affinity (4.8±2.1 µM) for this interaction. A similar result was observed when increasing concentrations of DENV-1-DIII monomer was flowed over immobilized intact E106 MAb in the solid phase (3.2±0.8 µM, Fig. 2B); this experiment eliminates the possibility that papain cleavage and the removal of the E106 Fc region altered the monovalent binding parameters. Binding to the ectodomain of DENV-1 E (DI-DII-DIII) also appeared equivalently weak (1.1±0.1 µM). As an independent measurement of affinity, we performed isothermal titration calorimetry under similar experimental conditions as SPR by injecting DENV-1 DIII into a solution of E106 Fab. Using this method with completely unmodified proteins we again measured micromolar affinity for the E106 Fab/DIII interaction (KD = 0.7±2 µM) (Fig. 2C).

E106 Fab binds and neutralizes DENV-1 poorly
The micromolar monovalent affinity of the highly therapeutic E106 antibody was unanticipated in light of its picomolar inhibitory activity (4±2 pM or 0.6±0.3 ng/mL; our most potent neutralizing anti-flavivirus MAb isolated to date); as a comparison, our therapeutic DIII-specific anti-WNV MAb E16 (inhibitory activity 30 to 80 pM or 4–18 ng/mL), which has advanced to human clinical trials [29], has a monovalent affinity of 3.4 nM [30]. We hypothesized that while E106 MAb potently inhibited DENV-1 infection, Fabs should lack this activity. To test this, we compared the ability of Fab and intact IgG from E106 and E103, a lateral ridge DIII-specific DENV-1 neutralizing antibody [13], to inhibit infection. While monovalent E103 Fab showed a 114-fold decrease in neutralization potency compared to the intact bivalent IgG, the Fab of E106 Fab showed a remarkable 18,150-fold decrease in inhibitory activity when compared to intact IgG (Fig. 3A).

To investigate this observation further, we performed a virion-binding assay by ELISA. DENV-1 virions (strain 16007) were captured with humanized DIII A-strand-specific antibody (DENV-1-E50) [31] and then detected with Fab or intact IgG of E103, E106, or WNV E16 (negative control). Notably, the amount of virus detected with E106 IgG was indistinguishable from E103 IgG (Fig. 3B). In comparison, E106 Fab bound virions to a significantly lower level (P<0.0001) at all concentrations tested, than those derived from E103 (Fig. 3B). Thus, disparate neutralization of E106 MAb and Fab correlated with discordant binding to DENV-1 virions and was consistent with the biophysical measurements: monovalent binding by DENV-1-E106 is surprisingly inefficient given the potent inhibitory activity of intact antibody.
E106 MAb binds bivalently to DENV-1 virus particles
Based on these experiments, we hypothesized that efficient neutralization of DENV-1 infection by E106 required bivalent binding. Using bio-layer interferometry (BLI), we measured the affinity and kinetics of MAb and Fab binding to intact DENV-1 virus particles [32] (Fig. 4). E106 MAb (Fig. 4A) bound DENV-1 particles with an apparent affinity (avidity), KDapp of 13±2 nM. In contrast, E106 Fab had an affinity of KD>1 µM, with a rapid dissociation rate (t1/2<2 sec) that was at least 800-fold faster than E106 MAb (t1/2>400 sec) (Fig. 4B). These results contrast with more comparable binding of E103 MAb (KDapp of 0.8±0.1 nM) (Fig. 4C) and E103 Fab (KD of 7±1 nM) to DENV-1 particles (Fig. 4D). Importantly, the binding affinities of E106 Fab engaging isolated DIII measured by SPR and ITC is remarkably similar to that observed by BLI for the binding to DENV-1 virions, suggesting that our structurally defined DIII epitope corresponds to the entire virion surface recognized by a single Fab.

Mechanistic correlates of E106 MAb neutralization
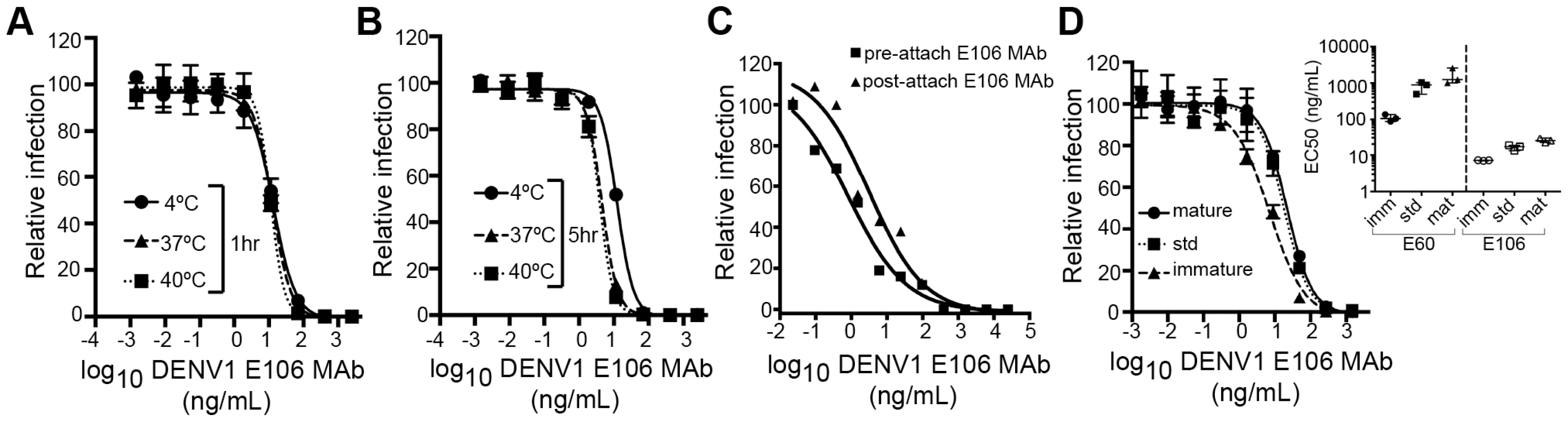
We next investigated the time - and temperature-dependence of E106 neutralization, as this analysis can provide information as to the relative accessibility of epitopes [28], [33]. Changes in the time or temperature of incubation did not appreciably affect E106 neutralization (Fig. 5A and B). By performing pre - and post-attachment neutralization assays, we found that, similar to several other potently neutralizing DIII-specific antibodies against flaviviruses [15], [30], [34], E106 can neutralize infection even after virus attaches to cells (Fig. 5C). Finally, we tested the neutralization of E106 MAb as a function of the maturation state of the virus. DENV virions are a heterogeneous mixture of immature, partially mature and fully mature virions, with immature virions being generally less or non-infectious. In this assay, E106 MAb neutralization proved to be independent of the maturation state of the virus (Fig. 5D). In comparison, neutralization by E60, a DII fusion-loop-specific MAb, was sensitive to virion maturation, as seen previously [27].
Modeling of E106 binding to virus
In contrast to non-enveloped viruses where a structural understanding of bivalent antibody binding has emerged [35]–[37], there currently is no such data for icosahedral enveloped viruses including flaviviruses. To address how E106 might recognize DENV-1 bivalently we docked our structure onto the cryo-EM derived model of the mature DENV virion (Fig. 6) [38]. While the E106 epitope is predominantly exposed on all 180 E protein monomers (Fig. 6A), unimpeded binding is readily apparent only on monomers in the 3-fold and 2-fold symmetry axes, similar to what we observed for E16 binding to WNV [20], [39]. However, minor reorientation of DIII subunits on the inner 5-fold symmetry axis would allow for up to two E106 Fabs to bind there at the same time as three Fabs could bind to the outer 5-fold (2-fold) related epitopes. We measured the distance separating the docked Fab CH1 domain C-termini with the expectation that distances greater than 50 Å would be unlikely spanned by 16 hinge residues [36], [37]. This analysis indicated the possibility for limited bivalent bridging, with the primary candidates being adjacent outer 5-fold epitopes (49 Å CH1 separation) (Fig. 6B and D) as well as adjacent inner and outer 5-fold epitopes (24 Å CH1 separation) (Fig. 6C and D). These epitopes are 85 Å and 79 Å apart, respectively, within the expected reach of a single IgG molecule (117–134±40 Å) [40]. We also examined the E106 epitope on the recent cryo-EM reconstruction of DENV-2 at 37°C [41], [42]; this ‘bumpy’ virion supports a similar model of bivalent binding to DIII on the 5-fold and 2-fold symmetry axes.

Discussion
Epitope mapping studies have enhanced our understanding of the mechanisms of virus neutralization and identified sites on the E protein of flaviviruses that are targeted by neutralizing antibodies [7]. These include the lateral ridge of DIII of WNV and JEV [19], [20], the A-strand of DIII of DENV [10], [21], the CC′ loop of DIII of DENV-1 [33], the fusion loop of DII of WNV and DENV [43], a DI epitope of DENV-4 [11], and a complex epitope centered at the hinge of DI and DII on WNV [44] and DENV [45], [46]. Here, we describe a composite epitope, comprised of regions of the lateral ridge and A-strand of DIII that is targeted by the therapeutic MAb E106. DIII residues contacted by E106 were highly conserved among DENV-1 genotypes but variable in other DENV serotypes. Consistent with this, E106 potently neutralized all five DENV-1 genotypes, but not other DENV serotypes nor WNV [13].
The E106 Fab-DIII complex was characterized by a small-buried interface, which correlated with an unexpectedly weak micromolar affinity, as determined both by SPR and isothermal calorimetry. We found no evidence for E106 binding to residues outside of DIII as the binding affinity to recombinant DIII appears to be very similar to the binding of Fab to E ectodomain protein or virions; consistent with this, neutralization escape studies only identified residues in DIII [22]. Monovalent E106 Fab poorly neutralized DENV-1 compared to intact E106 IgG, and this finding correlated with poor binding of Fab to intact virus. Although our structural models suggest that E106 can readily bind the isolated pre-fusion dimer and post-fusion trimer and possibly prevent the ∼70° transition of DIII that is associated with membrane fusion [47], [48], the inability of E106 Fab fragments to neutralize virus efficiently argues against this model. Our data are more consistent with a mechanism of neutralization that requires bivalent binding of E106 IgG to single virions, and cross-linking of E protein monomers in adjacent symmetry groups to prevent requisite E protein rearrangements during infection.
The measurement of micromolar monovalent affinity was unexpected given that E106 is our most potently neutralizing and protective anti-DENV-1 MAb (EC50 of 0.6 ng/ml against DENV-1 strain 16007), which is at least 10-fold more potent than our well-characterized DIII-lateral ridge-specific therapeutic MAb (E16) against WNV [20], [30]. Indeed, E106 had the lowest EC50 value of ∼500 anti-flavivirus MAbs (DENV-1, DENV-2, DENV-3, DENV-4, and WNV) generated to date in our laboratory [13], [15], [30], [49]. Is there a correlation between neutralization potency and E106 bivalent binding to single virions? The icosahedral arrangement of the E protein on the mature DENV virion displays 180 copies of the E protein. Our in silico modeling predicts that in one possible arrangement, up to 48 of these sites may be available for bivalent engagement by 24 intact E106 IgG. Since monovalent binding was insufficient for neutralization, bivalent binding to single virions could neutralize infection by inhibiting an essential stage of the virus lifecycle (attachment, entry, or fusion). Alternatively, bivalent binding across virions could neutralize DENV infection by aggregating virus. Our post-attachment studies suggest that E106 MAb was capable of neutralizing infection even after virus attached to the cell surface. Aggregation also appears less likely because the neutralization curves did not show a characteristic triphasic dose-response curve that was reported in inhibition studies describing antibody-virus aggregation [50]. We favor a model in which bivalent binding of E106 stabilizes and/or cross-links one or more E protein monomers in different symmetry groups, analogous to monovalent binding of WNV CR4354 MAb [44], and thus prevents radial expansion and rearrangement that is requisite for fusion of viral and host endosomal membranes [48], [51].
E106 is one of very few MAbs that have been shown to require bivalent binding for efficient virus neutralization [35], [36], [52], and the first one directed against a flavivirus. While antibodies can be multivalent, with the potential to bind to virus particles with high avidity, the relatively small number of bivalent binding MAbs described to date may be attributed to the following: (i) the limited number of epitopes displayed on a single particle for some viruses; (ii) the position and orientation of epitopes that are beyond the reach of a bivalent MAb, which is limited by torsional flexibility of its hinge; (iii) the radius of curvature of virions, which may restrict the accessibility of epitopes by MAb (iv) post-translational modification (e.g. N-linked glycans) of virions that may hinder bivalent MAb engagement; (v) immunization protocols that rely on isolated recombinant envelope proteins, rather than envelope proteins in the structurally unique form of the intact virion; and (vi) assays that do not screen for bivalent neutralizing MAbs. E106 was generated after priming and boosting with infectious DENV-1 [13]. The repetitive E antigens in the icosahedral orientation of the virion may have facilitated selection of low monovalent affinity yet high avidity antibodies. While some antibodies against HIV have been described as bivalent, they actually are bispecific, with each arm binding distinct epitopes [53]. This is likely due to the paucity of trimeric spikes on the surface of the virus (∼14) and their irregular spacing [54]. Although several MAbs have been proposed to require bivalent binding for efficient virus neutralization [55], [56], compelling evidence is presented only for the non-enveloped positive strand RNA viruses, specifically, human rhinovirus 2 [36] and 14 [35], [52] and the rabbit hemorrhagic disease calicivirus [37]. It may be that the quasi-icosahedral arrangement of the flavivirus envelope creates a landscape that permits limited bivalent MAb engagement.
Bivalent engagement of the virion by antibodies could be an important concept for DENV vaccine development. Immunity against DENV may not be achieved optimally using a subunit vaccine approach, as isolated E protein monomers may not induce antibodies that require bivalent binding for strong binding and neutralization. Analogously, some human MAbs against WNV bind a complex epitope at two independent positions on adjacent E protein monomers in different symmetry groups, which is only present on an intact WNV particle [44]. Human MAbs isolated from DENV-infected individuals are believed to recognize similar quaternary epitopes in E that are present only in the context of the intact DENV virion [45], [46]. Given that E106 MAb was our most potent and therapeutic anti-DENV-1 MAb, strategies that enhance the likelihood of generating and identifying neutralizing antibodies that function through bivalent binding mechanisms may improve the potency of inhibitory humoral responses against DENV and other flaviviruses. Regardless, an understanding of the structural and mechanistic basis for the neutralization activity of E106 MAb provides new insights into the humoral response against flaviviruses.
Materials and Methods
E106 Fab-DIII crystal structure determination
DENV-1 DIII (residues 293 to 399 of the E protein of strain 16007) was expressed in bacteria and re-folded oxidatively from isolated inclusion bodies as described previously [13]. Fab fragments of E106 were prepared using immobilized papain resin according to the manufacturer's instructions (Pierce). MAb (5 to 10 mg) was digested for 18 hours at 37°C, and passed over a protein A agarose resin to remove Fc fragments and undigested MAb and purified on a S-75 size exclusion chromatography column equilibrated in 20 mM HEPES pH 7.4 and 150 mM NaCl. Antibody–antigen complexes were formed by mixing papain-generated, gel filtered E106 Fab fragments with DIII at a ratio of 1.2∶1 and crystallized by the hanging drop vapor diffusion method at a total protein concentration of 14 mg/mL in a solution of 22% PEG 6,000, and 0.1 M MES pH 5.0 (final pH 5.7). Crystals (in 1 µL crystallization drops) were cryoprotected by the addition of 0.2 µL aliquots of cryobuffer (in 23.5% PEG 6,000, 0.1 M MES pH 5.0, final pH 5.7, and 20% glycerol), then transferred to a fresh drop of cryobuffer and rapidly cooled in liquid nitrogen. X-ray diffraction data were collected at ALS beamline 4.2.2 (Lawrence Berkeley Laboratories) at a wavelength of 0.976 Å at 100 K with a CCD detector, and indexed and scaled in HKL2000 [57]. The crystals diffracted to 2.45 Å resolution and belonged to the space group P212121 with unit cell dimensions of a = 82.7 Å, b = 91.8 Å, c = 92.6 Å, with one E106 Fab-DIII complex per asymmetric unit. Crystallographic phasing was obtained by molecular replacement using the program Phaser [58] and the coordinates of DENV-1 DIII (Protein Data Bank (PDB) 3EGP) and the Fab fragment of CTM01 IgG (PDB 1AD9). Iterative model building and refinement was performed using Coot [59] and Refmac [60] and later Phenix [61]. The final structure was refined to Rcryst = 18.9% and Rfree = 23.9%. The final model includes DENV-1 DIII amino acid residues 297 to 394, E106 heavy chain residues 1 to 214 (Chothia numbering), and light chain residues 1 to 213. The atomic coordinates and structure factors of E106 Fab bound to DENV-1 DIII (CSGID target number IDP00272) have been deposited in the Protein Data Bank (www.rcsb.org) under PDB accession number 4L5F. Structural figures were prepared using CCP4MG [62] and Pymol [63] (surface representation using 1.4 Å solvent probe) and where shown, spheres represent van der Waal radii, vdw * 1.1.
Surface plasmon resonance and isothermal titration calorimetry
Monovalent antibody affinity analysis was performed using SPR (BIAcore T100, GE Company) and ITC (VP-ITC instrument, Microcal) at 10°C in 50 mM HEPES, pH 7.5 and 100 mM NaCl. For SPR, E106 MAb or Fab was immobilized at low concentrations (∼500 Response Units) to a CM5 chip (GE healthcare) using amine-coupling chemistry. Bacterially-expressed DIII (residues 293–399) of DENV-1 (strain 16007) was injected at a flow rate of 65 µl/min at concentrations ranging from 0.2 µM to 500 µM for three minutes to saturate binding and then allowed to dissociate for seven minutes. The half-life of the monovalent interaction was short and did not require additional regeneration of the chip surface in preparation for the next DIII injection. The observed binding curves were double referenced to a non-reactive antibody (WNV E16) as well as buffer in the absence of DIII. Curves were analyzed by a steady-state fit for a 1∶1 interaction, and a nonlinear least squares fit was used to evaluate the fit of the curve to the observed data. Alternatively, 500 response units (RU) of DIII were immobilized onto a CM5 chip and E106 Fab fragments were injected to saturation. Affinity measurements of E106 for the DENV-1 E ectodomain (DI-DII-DIII) were performed such that insect-derived DENV-1 E glycoprotein (ProSpec-Tany TechnoGene Ltd.) was in the stationary phase and E106 Fab fragments were in the mobile phase, in order to conserve limited protein and avoid avidity affects. Additional regeneration was not necessary because of the short half-life for the interaction. WNV E ectodomain was used as a negative control for E106 binding. Analysis was performed as with the DIII described above. ITC experiments were performed such that 4 to 8 µL of DENV-1-DIII protein (90 to 110 µM) was injected into 1.4 mL of E106 Fab protein (6 to 7 µM) over a total of 36 injections. The titration data were integrated and normalized in Origin (Microcal) to determine the reaction stoichiometry, n, and equilibrium constant Ka ( = K−1d).
Neutralization assays and capture ELISA
Plaque reduction neutralization tests (PRNT) and pre - and post-attachment neutralization assays were performed with DENV-1 strain 16007 on Vero cells as previously described [13], [64]. Binding of intact MAbs or Fabs (E103, E106, and a negative control WNV E16) to DENV-1 virions (strain 16007) was detected by capture ELISA [13], [64]. Briefly, humanized DENV-1 E50 MAb (subcomplex DIII A-strand specific antibody) was coated at 2 µg/ml on MaxiSorp (Nunc) polystyrene 96-well microtiter plates in a sodium carbonate (pH 9.3) buffer. Plates were washed three times in wash buffer (PBS with 0.02% Tween 20) and blocked for one hour at 37°C with blocking buffer (DMEM with 10% FBS). DENV-1 virions (2.5×105 PFU) diluted in DMEM with 10% heat-inactivated FBS were captured on plates for two hours at 37°C. Wells were washed thrice with blocking buffer and DENV-1 MAb or Fab was then added at 100 µg/ml and 4-fold serial dilutions to duplicate wells and incubated for two hours at 37°C. Plates were washed five times and then sequentially incubated with goat anti-mouse (whole molecule) IgG-HRP (Sigma, St Louis, MO) and tetramethylbenzidine substrate (Dako). The reaction was stopped with the addition of 2 N H2SO4 to the medium, and emission (450 nm) was read using an iMark microplate reader (Bio-Rad).
Time and temperature-dependent neutralization of DENV-1
A plasmid expressing the C-prM-E genes of DENV-1 (strain 16007) was co-transfected into HEK-293T cells with a plasmid encoding a WNV replicon expressing GFP. Transfected cells were incubated at 30°C and RVP harvested at 72 and 96 hours post-transfection, filtered through a 0.2 µM filter, and stored aliquoted at −80°C. DENV-1 RVP were incubated with serial dilutions of MAb under conditions of antibody excess at 4°C, 37°C, or 40°C for one or five hours. Subsequently, MAb-RVP mixtures were added to Raji-DCSIGNR cells and incubated at 37°C for 48 hours. Infected cells were assayed for GFP expression using a BD FACSCalibur flow cytometer as described [28].
The kinetics of DENV-1 RVP binding to DENV-1 MAbs and Fabs
All bio-layer interferometry studies were performed in PBS buffer supplemented with 1 to 2 mg/ml BSA (PBS-B) at 25°C using an Octet Red biosensor system (ForteBio). DENV-1 reporter virus particles (RVPs) (Western Pacific 74 strain) were produced as previously described [65]. To purify virus particles, RVP production supernatant was harvested, clarified through a 0.22 µm filter (Corning), and PEG precipitated using 7.5% PEG 8000 (Sigma). The virus particles were further purified through two 20% sucrose cushions before resuspension in HBS. Samples were stored at −80°C and gently thawed prior to use. RVPs were loaded onto streptavidin (SA) biosensor tips using a human monoclonal antibody against DENV-1 (1H7.2, the anti-prM antibody, a gift from James Crowe), which was captured using a biotinylated goat anti-human polyclonal antibody (GAH Fc, Southern Biotech). Briefly, GAH Fc was diluted to 5 µg/ml in PBS-B and bound to SA sensor tips for five minutes. Following a brief rinse in PBS-B, 1H7.2 (5 µg/ml in PBS-B) was captured for five minutes. After another brief rinse, DENV-1 RVPs diluted to 10 µg/ml (or 50 µg/ml (E106 Fab)) were loaded for 45 minutes followed by a five-minute stabilization. Antibody association was measured for up to 10 minutes followed by dissociation for 20 minutes (E106) or 45 minutes (E103) in buffer. Non-specific binding was assessed using sensor tips without RVPs as well as using sensor tips loaded with retroviral pseudotypes (Lipoparticles) containing only endogenous cell surface receptors (no viral envelope protein). Data analysis was performed using Octet Data Analysis v6.4 (ForteBio). Binding kinetics were analyzed using a standard 1∶1 binding model.
Supporting Information
Zdroje
1. BhattS, GethingPW, BradyOJ, MessinaJP, FarlowAW, et al. (2013) The global distribution and burden of dengue. Nature 496 : 504–507 doi:10.1038/nature12060
2. Rico-HesseR (1990) Molecular evolution and distribution of dengue viruses type 1 and 2 in nature. Virology 174 : 479–493 doi:10.1016/0042-6822(90)90102-W
3. HolmesEC, TwiddySS (2003) The origin, emergence and evolutionary genetics of dengue virus. Infect Genet Evol 3 : 19–28 doi:S1567134803000042 [pii]
4. SabinAB (1952) Research on dengue during World War II. Am J Trop Med Hyg 1 : 30–50.
5. ReichNG, ShresthaS, KingAA, RohaniP, LesslerJ, et al. (2013) Interactions between serotypes of dengue highlight epidemiological impact of cross-immunity. J R Soc Interface 10 doi:10.1098/rsif.2013.0414
6. HalsteadSB (1989) Antibody, macrophages, dengue virus infection, shock, and hemorrhage: a pathogenetic cascade. Rev Infect Dis 11 Suppl 4: S830–9 doi:10.1111/j.1742-4658.2005.04870.x
7. PiersonTC, FremontDH, KuhnRJ, DiamondMS (2008) Structural insights into the mechanisms of antibody-mediated neutralization of flavivirus infection: implications for vaccine development. Cell Host Microbe 4 : 229–238 doi:S1931-3128(08)00260-6 [pii] 10.1016/j.chom.2008.08.004
8. ReyFA, HeinzFX, MandlC, KunzC, HarrisonSC (1995) The envelope glycoprotein from tick-borne encephalitis virus at 2 A resolution. Nature 375 : 291–298 doi:10.1038/375291a0
9. ModisY, OgataS, ClementsD, HarrisonSC (2003) A ligand-binding pocket in the dengue virus envelope glycoprotein. Proc Natl Acad Sci U S A 100 : 6986–6991 doi:10.1073/pnas.0832193100 0832193100 [pii]
10. CockburnJJ, Navarro SanchezME, FretesN, UrvoasA, StaropoliI, et al. (2012) Mechanism of dengue virus broad cross-neutralization by a monoclonal antibody. Structure 20 : 303–314 doi:S0969-2126(12)00005-6 [pii] 10.1016/j.str.2012.01.001
11. CockburnJJ, Navarro SanchezME, GoncalvezAP, ZaitsevaE, SturaEA, et al. (2011) Structural insights into the neutralization mechanism of a higher primate antibody against dengue virus. EMBO J 31 : 767–779 doi:emboj2011439 [pii] 10.1038/emboj.2011.439
12. GromowskiGD, BarrettAD (2007) Characterization of an antigenic site that contains a dominant, type-specific neutralization determinant on the envelope protein domain III (ED3) of dengue 2 virus. Virology 366 : 349–360 doi:S0042-6822(07)00379-0 [pii] 10.1016/j.virol.2007.05.042
13. ShresthaB, BrienJD, Sukupolvi-PettyS, AustinSK, EdelingMA, et al. (2010) The development of therapeutic antibodies that neutralize homologous and heterologous genotypes of dengue virus type 1. PLoS Pathog 6: e1000823 doi:10.1371/journal.ppat.1000823
14. WahalaWM, DonaldsonEF, De AlwisR, Accavitti-LoperMA, BaricRS, et al. (2010) Natural strain variation and antibody neutralization of dengue serotype 3 viruses. PLoS Pathog 6: e1000821 doi:10.1371/journal.ppat.1000821
15. Sukupolvi-PettyS, AustinSK, EngleM, BrienJD, DowdKA, et al. (2010) Structure and function analysis of therapeutic monoclonal antibodies against dengue virus type 2. J Virol 84 : 9227–9239 doi:JVI.01087-10 [pii] 10.1128/JVI.01087-10
16. RoehrigJT, BolinRA, KellyRG (1998) Monoclonal antibody mapping of the envelope glycoprotein of the dengue 2 virus, Jamaica. Virology 246 : 317–328 doi:S0042-6822(98)99200-5 [pii] 10.1006/viro.1998.9200
17. MidgleyCM, FlanaganA, TranHB, DejnirattisaiW, ChawansuntatiK, et al. (2012) Structural analysis of a dengue cross-reactive antibody complexed with envelope domain III reveals the molecular basis of cross-reactivity. J Immunol 188 : 4971–4979 doi:10.4049/jimmunol.1200227
18. ThompsonBS, MoeskerB, SmitJM, WilschutJ, DiamondMS, et al. (2009) A therapeutic antibody against west nile virus neutralizes infection by blocking fusion within endosomes. PLoS Pathog 5: e1000453 doi:10.1371/journal.ppat.1000453
19. WuKP, WuCW, TsaoYP, KuoTW, LouYC, et al. (2003) Structural basis of a flavivirus recognized by its neutralizing antibody: solution structure of the domain III of the Japanese encephalitis virus envelope protein. J Biol Chem 278 : 46007–46013 doi:10.1074/jbc.M307776200 M307776200 [pii]
20. NybakkenGE, OliphantT, JohnsonS, BurkeS, DiamondMS, et al. (2005) Structural basis of West Nile virus neutralization by a therapeutic antibody. Nature 437 : 764–769 doi:nature03956 [pii] 10.1038/nature03956
21. LokSM, KostyuchenkoV, NybakkenGE, HoldawayHA, BattistiAJ, et al. (2008) Binding of a neutralizing antibody to dengue virus alters the arrangement of surface glycoproteins. Nat Struct Mol Biol 15 : 312–317 doi:nsmb.1382 [pii] 10.1038/nsmb.1382
22. ShresthaB, AustinSK, DowdKA, PrasadAN, YounS, et al. (2012) Complex phenotypes in mosquitoes and mice associated with neutralization escape of a Dengue virus type 1 monoclonal antibody. Virology 427 : 127–134 doi:S0042-6822(12)00105-5 [pii] 10.1016/j.virol.2012.02.010
23. LawrenceMC, ColmanPM (1993) Shape complementarity at protein/protein interfaces. J Mol Biol 234 : 946–950 doi:S0022-2836(83)71648-7 [pii] 10.1006/jmbi.1993.1648
24. KrissinelE, HenrickK (2007) Inference of macromolecular assemblies from crystalline state. J Mol Biol 372 : 774–797 doi:S0022-2836(07)00642-0 [pii] 10.1016/j.jmb.2007.05.022
25. DaviesDR, PadlanEA, SheriffS (1990) Antibody-antigen complexes. Annu Rev Biochem 59 : 439–473 doi:10.1146/annurev.bi.59.070190.002255
26. SundbergEJ, MariuzzaRA (2002) Molecular recognition in antibody-antigen complexes. Adv Protein Chem 61 : 119–160.
27. NelsonS, JostCA, XuQ, EssJ, MartinJE, et al. (2008) Maturation of West Nile Virus Modulates Sensitivity to Antibody-Mediated Neutralization. PLoS Pathog 4: e1000060 doi:10.1371/journal.ppat.1000060
28. DowdKA, JostCA, DurbinAP, WhiteheadSS, PiersonTC (2011) A dynamic landscape for antibody binding modulates antibody-mediated neutralization of West Nile virus. PLoS Pathog 7: e1002111 doi:10.1371/journal.ppat.1002111
29. BeigelJH, NordstromJL, PillemerSR, RoncalC, GoldwaterDR, et al. (2010) Safety and Pharmacokinetics of Single Intravenous Dose of MGAWN1, a Novel Monoclonal Antibody to West Nile Virus. Antimicrob Agents Chemother 54 : 2431–2436 doi:10.1128/AAC.01178-09
30. OliphantT, EngleM, NybakkenGE, DoaneC, JohnsonS, et al. (2005) Development of a humanized monoclonal antibody with therapeutic potential against West Nile virus. Nat Med 11 : 522–530 doi:nm1240 [pii] 10.1038/nm1240
31. RodrigoWWSI, BlockOKT, LaneC, Sukupolvi-PettyS, GoncalvezAP, et al. (2009) Dengue virus neutralization is modulated by IgG antibody subclass and Fcgamma receptor subtype. Virology 394 : 175–182 doi:10.1016/j.virol.2009.09.024
32. Ansarah-SobrinhoC, NelsonS, JostCA, WhiteheadSS, PiersonTC (2008) Temperature-dependent production of pseudoinfectious dengue reporter virus particles by complementation. Virology 381 : 67–74 doi:S0042-6822(08)00525-4 [pii] 10.1016/j.virol.2008.08.021
33. AustinSK, DowdKA, ShresthaB, NelsonCA, EdelingMA, et al. (2012) Structural Basis of Differential Neutralization of DENV-1 Genotypes by an Antibody that Recognizes a Cryptic Epitope. PLoS Pathog 8: e1002930 doi:10.1371/journal.ppat.1002930
34. StiasnyK, KiermayrS, HolzmannH, HeinzFX (2006) Cryptic properties of a cluster of dominant flavivirus cross-reactive antigenic sites. J Virol 80 : 9557–9568 doi:10.1128/JVI.00080-06
35. SmithTJ, OlsonNH, ChengRH, ChaseES, BakerTS (1993) Structure of a human rhinovirus-bivalently bound antibody complex: implications for viral neutralization and antibody flexibility. Proc Natl Acad Sci USA 90 : 7015–7018 doi:10.1073/pnas.90.15.7015
36. HewatEA, BlaasD (1996) Structure of a neutralizing antibody bound bivalently to human rhinovirus 2. EMBO J 15 : 1515–1523.
37. ThouveninE, LaurentS, MadelaineM-F, RasschaertD, VautherotJ-F, et al. (1997) Bivalent binding of a neutralising antibody to a calicivirus involves the torsional flexibility of the antibody hinge. Journal of Molecular Biology 270 : 238–246 doi:10.1006/jmbi.1997.1095
38. ZhangW, ChipmanPR, CorverJ, JohnsonPR, ZhangY, et al. (2003) Visualization of membrane protein domains by cryo-electron microscopy of dengue virus. Nat Struct Mol Biol 10 : 907–912 doi:10.1038/nsb990
39. KaufmannB, NybakkenGE, ChipmanPR, ZhangW, DiamondMS, et al. (2006) West Nile virus in complex with the Fab fragment of a neutralizing monoclonal antibody. Proc Natl Acad Sci U S A 103 : 12400–12404 doi:0603488103 [pii] 10.1073/pnas.0603488103
40. SosnickTR, BenjaminDC, NovotnyJ, SeegerPA, TrewhellaJ (1992) Distances between the antigen-binding sites of three murine antibody subclasses measured using neutron and X-ray scattering. Biochemistry 31 : 1779–1786 doi:10.1021/bi00121a028
41. FibriansahG, NgT-S, KostyuchenkoVA, LeeJ, LeeS, et al. (2013) Structural changes in dengue virus when exposed to a temperature of 37°C. J Virol 87 : 7585–7592 doi:10.1128/JVI.00757-13
42. ZhangX, ShengJ, PlevkaP, KuhnRJ, DiamondMS, et al. (2013) Dengue structure differs at the temperatures of its human and mosquito hosts. Proc Natl Acad Sci USA 110 : 6795–6799 doi:10.1073/pnas.1304300110
43. CherrierMV, KaufmannB, NybakkenGE, LokSM, WarrenJT, et al. (2009) Structural basis for the preferential recognition of immature flaviviruses by a fusion-loop antibody. EMBO J 28 : 3269–3276 doi:emboj2009245 [pii] 10.1038/emboj.2009.245
44. KaufmannB, VogtMR, GoudsmitJ, HoldawayHA, AksyukAA, et al. (2010) Neutralization of West Nile virus by cross-linking of its surface proteins with Fab fragments of the human monoclonal antibody CR4354. Proc Natl Acad Sci U S A 107 : 18950–18955 doi:1011036107 [pii] 10.1073/pnas.1011036107
45. De AlwisR, SmithSA, OlivarezNP, MesserWB, HuynhJP, et al. (2012) Identification of human neutralizing antibodies that bind to complex epitopes on dengue virions. Proc Natl Acad Sci USA 109 : 7439–7444 doi:10.1073/pnas.1200566109
46. TeohEP, KukkaroP, TeoEW, LimAPC, TanTT, et al. (2012) The structural basis for serotype-specific neutralization of dengue virus by a human antibody. Sci Transl Med 4 : 139ra83 doi:10.1126/scitranslmed.3003888
47. KuhnRJ, ZhangW, RossmannMG, PletnevSV, CorverJ, et al. (2002) Structure of dengue virus: implications for flavivirus organization, maturation, and fusion. Cell 108 : 717–725 doi:S0092867402006608 [pii]
48. ModisY, OgataS, ClementsD, HarrisonSC (2004) Structure of the dengue virus envelope protein after membrane fusion. Nature 427 : 313–319 doi:10.1038/nature02165 nature02165 [pii]
49. BrienJD, AustinSK, Sukupolvi-PettyS, O'BrienKM, JohnsonS, et al. (2010) Genotype-specific neutralization and protection by antibodies against dengue virus type 3. J Virol 84 : 10630–10643 doi:JVI.01190-10 [pii] 10.1128/JVI.01190-10
50. ThomasAA, VrijsenR, BoeyeA (1986) Relationship between poliovirus neutralization and aggregation. J Virol 59 : 479–485.
51. KaufmannB, ChipmanPR, HoldawayHA, JohnsonS, FremontDH, et al. (2009) Capturing a flavivirus pre-fusion intermediate. PLoS Pathog 5: e1000672 doi:10.1371/journal.ppat.1000672
52. SmithTJ, OlsonNH, ChengRH, LiuH, ChaseES, et al. (1993) Structure of human rhinovirus complexed with Fab fragments from a neutralizing antibody. J Virol 67 : 1148–1158.
53. MouquetH, ScheidJF, ZollerMJ, KrogsgaardM, OttRG, et al. (2010) Polyreactivity increases the apparent affinity of anti-HIV antibodies by heteroligation. Nature 467 : 591–595 doi:nature09385 [pii] 10.1038/nature09385
54. ZhuP, LiuJ, BessJ, ChertovaE, LifsonJD, et al. (2006) Distribution and three-dimensional structure of AIDS virus envelope spikes. Nature 441 : 847–852 doi:nature04817 [pii] 10.1038/nature04817
55. SchofieldDJ, StephensonJR, DimmockNJ (1997) Variations in the neutralizing and haemagglutination-inhibiting activities of five influenza A virus-specific IgGs and their antibody fragments. J Gen Virol 78(Pt 10): 2431–2439.
56. KlassePJ, SattentauQJ (2002) Occupancy and mechanism in antibody-mediated neutralization of animal viruses. J Gen Virol 83 : 2091–2108.
57. Otwinowski Z, Minor W (1997) Processing of X-ray diffraction data collected in oscillation mode. In: C.WCarter J& RMS, editors. Methods in Enzymology. New York: Academic Press. pp. 307–326.
58. McCoyAJ, Grosse-KunstleveRW, AdamsPD, WinnMD, StoroniLC, et al. (2007) Phaser crystallographic software. J Appl Crystallogr 40 : 658–674 doi:10.1107/S0021889807021206
59. EmsleyP, CowtanK (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60 : 2126–2132 doi:S0907444904019158 [pii] 10.1107/S0907444904019158
60. MurshudovGN, VaginAA, DodsonEJ (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr 53 : 240–255 doi:10.1107/S0907444996012255 S0907444996012255 [pii]
61. AdamsPD, AfoninePV, BunkócziG, ChenVB, DavisIW, et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallographica Section D Biological Crystallography 66 : 213–221 doi:10.1107/S0907444909052925
62. PottertonL, McNicholasS, KrissinelE, GruberJ, CowtanK, et al. (2004) Developments in the CCP4 molecular-graphics project. Acta Crystallogr D Biol Crystallogr 60 : 2288–2294 doi:10.1107/S0907444904023716
63. DeLano WL (2008) The PyMOL Molecular Graphics System, version 1. Schrödinger, LLC (Oregon).
64. VogtMR, MoeskerB, GoudsmitJ, JongeneelenM, AustinSK, et al. (2009) Human monoclonal antibodies against West Nile virus induced by natural infection neutralize at a postattachment step. J Virol 83 : 6494–6507 doi:JVI.00286-09 [pii] 10.1128/JVI.00286-09
65. MattiaK, PufferBA, WilliamsKL, GonzalezR, MurrayM, et al. (2011) Dengue reporter virus particles for measuring neutralizing antibodies against each of the four dengue serotypes. PLoS ONE 6: e27252 doi:10.1371/journal.pone.0027252
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2014 Číslo 4
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Měli bychom postcovidový syndrom léčit antidepresivy?
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- The 2010 Cholera Outbreak in Haiti: How Science Solved a Controversy
- , , , Genetic Variability: Cryptic Biological Species or Clonal Near-Clades?
- Efficient Parvovirus Replication Requires CRL4-Targeted Depletion of p21 to Prevent Its Inhibitory Interaction with PCNA
- An Overview of Respiratory Syncytial Virus